
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequestration of acrylamide as amino acid-acrylamide adducts mitigates cellular stress in human gastrointestinal cell lines†
Axita
Patel
 a and
Bhaskar
Datta
*ab
a and
Bhaskar
Datta
*ab
aDepartment of Biological Sciences and Engineering, Indian Institute of Technology Gandhinagar, Gandhinagar, Gujarat, India. E-mail: bdatta@iitgn.ac.in
bDepartment of Chemistry, Indian Institute of Technology Gandhinagar, Gandhinagar, Gujarat, India
First published on 27th May 2025
Abstract
Acrylamide (ACR) present in starchy and cereal-based heat-processed foods raises a global health concern. While amino acids (AAs) have been suggested as effective scavengers of acrylamide, there is a dearth of information about the cellular response to amino acid-acrylamide (AA-ACR) adducts. In this work, we conducted a detailed comparison of the effects of ACR versus AA-ACR adducts on two human gastrointestinal cell lines, namely, Caco-2 and HCT-15. Adducts of lysine, glycine, cysteine, and methionine with ACR were prepared under optimised reaction conditions and characterized by a combination of ESI-MS and MS/MS. Exposure of Caco-2 and HCT-15 to these adducts resulted in significant reduction in the formation of reactive oxygen species (ROS) and the prevention of abnormal accumulation of the cells in the G1 phase. The analysis of apoptosis and necrosis in the cell lines treated with AA-ACR clearly indicated the ability of AAs to sequester ACR and block oxidative stress induction that would otherwise be observed, completely precluding apoptosis and necrosis. Sequestration of ACR with each of the four AAs prevented the loss of mitochondrial membrane potential and the induction of autophagy that would otherwise occur upon exposure to ACR. The hitherto untested behaviour of human gastrointestinal cells towards AA-ACR presented in this work supports the application of AAs for the mitigation of ACR in foods.
Introduction
Thermal processing of foods is closely correlated with the nutritional quality, microbiological safety and sensory properties of the prepared food products. Thermal processing is also commonly associated with the generation of undesirable chemicals in the food matrix.1,2 Acrylamide (ACR) is a notorious example of such a chemical and has been subject to intense scrutiny for more than two decades now.3 Prior to its emergence in food, ACR was a valuable building block in the industrial manufacture of plastics. ACR was sought as an important analyte in cigarette smoke and in the treatment of wastewater.1 ACR was also found in consumer products such as caulking, food packaging, and some adhesives.4 While ACR has been present for as long as people have been baking, roasting, or frying foods, the discovery of significant levels of ACR by Törnqvist and co-workers in foods exposed to heat treatments led to a substantive increase in the scrutiny of ACR in foods.1,5 Elevated levels of ACR, especially in starchy foods processed at high temperatures, were attributed to the Maillard reaction.6,7 The Maillard reaction is not a single reaction but a complex series of reactions involving the condensation of reducing sugars (glucose and fructose) with amino acids, mainly asparagine. The resulting N-glycosides usually rearrange to the Amadori rearrangement product, which in turn gets converted into melanoidin, where a further decarboxylation of the Schiff base leads to ACR formation.8–10 ACR is absent in raw foods and is formed favourably in low moisture-containing foods at temperatures of 120 °C or higher.11–13 The Maillard reaction is held responsible for the brown colour, crust, and characteristic sensory features of baked, fried, and toasted foods.14 The reducing sugars and asparagine can each behave as a limiting factor in different foods.15 Owing to the greater surface-localization of the reaction, ACR in bread is located mainly in the crust with very low amounts in the crumb.16 Worldwide concerns on the presence of ACR in heat-processed foods are based on its classification as a probable human carcinogen.17 While maximum limits for ACR in food have not been established, the World Health Organisation guideline for ACR in drinking water is 0.5 μg L−1. ACR levels in foods varies widely with higher levels associated with potato products such as Fries (300–2000 μg kg−1) and chips (200 to 2500 μg kg−1).18,19Numerous studies have sought to relate the consumption of fried and baked foods, containing high prospective levels of ACR, with the prevalence of cancer.20,21 ACR has been suggested as a multiorgan carcinogen in rats and mice, and as a potential carcinogenic hazard to humans.22,23 ACR is rapidly and completely absorbed by the gastrointestinal tract in rats and distributed to the peripheral tissues.24 The fate of ACR in humans is qualitatively similar to that in rats.24 Once absorbed, ACR metabolism can proceed via at least two main pathways. It could be conjugated to N-acetyl-S-(3-amino-3-oxopropyl)cysteine by glutathione-S-transferase (GST). It could also be converted into glycidamide, in a reaction catalysed by the cytochrome P450 enzyme complex (CYP450).25,26
While these pathways can provide direct detoxification mechanisms for the removal of ACR, excess exposure can overload the pathways rendering them less effective.27 The consumption of cysteine-rich foods has been suggested for lowering the risk of toxicity of ACR.27,28 The mercapturic acids of ACR and glycidamide represent the major metabolites of ACR metabolism, and their urinary excretion levels are suggested as biomarkers of ACR exposure.29 ACR and glycidamide can also form adducts with DNA and amino acids in hemoglobin, and these adducts also represent important biomarkers of ACR exposure.30 Mitigation strategies for ACR, especially from potato and cereal products, have assumed importance considering the serious long-term health risks associated with ACR ingestion.31 Such strategies include selection of potato varieties and identification of suitable time of their harvest that results in low content of reducing sugars.32–34 A variety of plant-derived bioactive products have proved effective for mitigating ACR toxicity. These include sesamol, vitamin E, chrysin and quercetin.35–38 Among whole plants and their parts, the use of rosemary extracts and bamboo leaves leads to lower levels of ACR formation.35 Several strategies have been proposed for controlling ACR levels in processed foods. Apart from using ingredients with low concentrations of reducing sugars, these approaches include the application of functional food ingredients such as amino acids for lower ACR formation. Kobayashi et al. found lysine- and cysteine-mediated ACR content reduction in aqueous solutions below 120 °C.39 Koutsidis et al. reported the suppression of ACR formation using proline, tryptophan, glycine, and cysteine.40 Pre-treatment of potato slices with specific concentrations of amino acids results in a significant lowering of ACR formation.41 The amino acid-facilitated elimination of ACR occurs via the Michael-type addition reaction.40,42,43 A rigorous examination of adduct structures and their associated anti-toxic or hazardous effects is yet to be reported. Similarly, there is a need to optimise the experimental protocols that facilitate adduct formation along with the assessment of the mitigating effects accompanying such adducts.
In this work, we examined the sequestration of ACR as AA-ACR adducts as a robust approach for the mitigation of ACR-induced cellular stress on two gastrointestinal cell lines, namely, HCT-15 and Caco-2. While the ability of amino acids to scavenge ACR is scattered across different reports, there have been no investigations into the response of human cells to the AA-ACR. Our results describe the ability of amino acids lysine (Lys), glycine (Gly), cysteine (Cys) and methionine (Met) to form stable adducts with ACR, which when exposed to gastrointestinal cell lines prevents the harmful cellular response associated with exposure to ACR. To the best of our knowledge, this is the first comprehensive evaluation of cellular response to AA-ACR.
Experimental
Materials
Amino acids L-lysine monohydrate (Lys, ≥98%), L-glycine (Gly, ≥99%), L-cysteine (Cys, ≥99%), L-methionine (Met, ≥99%), L-asparagine (Asn, ≥99%), L-aspartic acid (Asp, ≥98%) and L-glutamic acid (Glu, ≥98%), and HPLC-grade methanol and acetonitrile were purchased from Finar Chemicals (Ahmedabad, India). Acrylamide standard (≥99.8%, catalogue no. 23701) was procured from Sigma-Aldrich Chemical Company (Bangalore, India). Standard solutions of ACR were prepared in 80% methanol, and all solutions were stored at 4 °C in the dark. DTNB (5,5′-dithio-bis-[2-nitrobenzoic acid]) (Cas no.: 69-78-3), 2,4,6-trinitrobenzene sulfonic acid (TNBSA) (2508-19-2) and o-phthalaldehyde (CAS: 643-79-8) were purchased from Sigma Aldrich (Bangalore, India). Anti-LC3 antibody (Novus Biologicals, NBP2-46892), and secondary antibody Alexa-Flour™ 488 goat anti-mouse IgG (ThermoFischer Scientific-A11001) were purchased from ThermoFischer Scientific (Bangalore, India).3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 2′7′-dichlorofuorescin diacetate (DCFH-DA), JC-1 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbo-cyanine iodide (JC-1), YO-PRO-1-Iodide, and Propidium Iodide (PI) were purchased from Thermo Fisher scientific (Ahmedabad, India). RPMI-1640 medium, Dulbecco's modified Eagle's medium (DMEM), foetal bovine serum (FBS) and penicillin–streptomycin were purchased from Gibco (Mumbai, India). Two potato samples (Raw potato and Frozen potato powder) were purchased from local supermarket (Gujarat, India), Amylase from human saliva (A1031), pepsin from porcine gastric mucosa (P6887), pancreatin from porcine pancreas (P7545) and acrylamide-d3 standard (500 μg mL−1) (122775-19-3) were purchased from Sigma-Aldrich (Bangalore, India).
Cell lines HCT-15 (Human colon cancer cell line-large intestinal) and Caco-2 (Human colorectal adenocarcinoma-small intestinal) were procured from NCCS (Pune, India).
Instrumentation
High-Performance Liquid Chromatography was performed using a Shimadzu Prominence HPLC-DAD-RID system-20A, Japan. Ultra-high-performance liquid chromatography-mass spectrometry was performed using a UHPLC-MS by LCQ Fleet Dionex Ultimate 3000 of ThermoScientific. Samples were imaged using a Laser Scanning Confocal microscope (Leica SPI8) with a 63× oil immersion objective at 405 nm, 488 nm, 525 nm, and 594 nm wavelengths. Flow cytometry was conducted using a BD FACS Aria Fusion, USA, with lasers at 405 nm, 488 nm, 525 nm, and 594 nm wavelengths. UV-visible spectra were recorded using a JASCO-V-750, Tokyo, Japan, and using a BioTek Cytation Hybrid Multimode Reader (Agilent Technologies International Pvt. Ltd, Manesar, India).Scavenging capacity of amino acids for acrylamide
The scavenging ability of seven amino acids for ACR was studied for the purpose of optimizing the reaction conditions. First, 4 mL of reaction mixture containing 50 mM of a single amino acid species (lysine, glycine, methionine, cysteine, asparagine, aspartic acid and glutamic acid) and 10 mM ACR aqueous solution were placed in 10 mL glass tubes and heated in a water bath at 80 °C for 2 h. The concentrations used here were followed by few studies.42 The samples were then cooled in an ice bath for stopping the reaction and analysed using a high-performance liquid chromatography-diode array detector (HPLC-DAD) system based on a previously reported method.44 Briefly, 10 μL of the filtered sample was injected into a Shimadzu Shim-pack GIST column (4.6 mm × 250 mm, 5 μm, Spinco Biotech Pvt. Ltd, Ahmedabad, India) and isocratically eluted by methanol/water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :98, v/v) solution at a flow rate of 0.4 mL min−1 at 40.0 °C. The ACR residue was measured at 205 nm and quantified using an external standard curve with the standards prepared above.
:98, v/v) solution at a flow rate of 0.4 mL min−1 at 40.0 °C. The ACR residue was measured at 205 nm and quantified using an external standard curve with the standards prepared above.
The reaction products were further identified by HPLC-MS/MS analysis based on the method reported previously.45 HPLC was performed based on the above-mentioned parameters, and MS spectrum was acquired in the positive-ion mode over a range of m/z 50–500 at a source temperature of 300 °C, a desolvation temperature of 250 °C and a capillary voltage of 4.0 kV. The collision energy was set at 20.0 eV for product ion scans.
Optimization of reaction conditions between acrylamide and amino acids
Mixtures (4 mL) containing 10 mM ACR and different amounts (5, 10, 15, 20, 30, 40, 50, and 60 mM) of each of the four amino acids, namely, lysine, cysteine, methionine, and glycine, were reacted in sealed glass tubes at 80 °C for 2 h. Subsequently, the test tubes were cooled in an ice bath. The reaction mixtures were analyzed by HPLC and quantified by LC-MS. For the optimization of temperature and time of incubation, mixtures (4 mL) containing 10 mM ACR and previously optimized concentration of amino acids were reacted for different incubation times (0–120 min), and at different temperatures (80 °C, 120 °C, 160 °C, and 180 °C). The characterisation of adduct formation was performed as described above by HPLC and LC-MS.Kinetics of the reaction between acrylamide and amino acids
The kinetic evaluation consisted of 4 different reactions,1L-lysine (50 mM, 100 ml), (2) L-glycine (40 mM, 100 ml), (3) L-cysteine (60 mM, 100 ml), and (4) L-methionine (50 mM, 100 ml), with ACR (1 mM, 100 ml). The reactions were performed at 80.0, 120.0, 160.0 and 180.0 °C in a reflux system for 120 min. Solutions were prepared in PBS (60.0 mM, pH 7.4). These conditions were adopted based on the optimizations performed in the previous section. Then, 1 ml aliquots were taken from the reactions and each aliquot was mixed with 5 ml of PBS (60.0 mM, pH 7.4), 1 ml of DTNB (5,5′-dithio-bis-[2-nitrobenzoic acid]) (5.0 mM) prepared in PBS (60.0 mM, pH 7.4), 1 ml of 2,4,6-trinitrobenzene Sulfonic Acid (TNBSA or TNBS) (0.01% (w/v)) prepared fresh in 0.1 M sodium bicarbonate, pH 8.5, and 1 ml of OPDA according to the amino acid to be analysed. The mixtures were analysed using a UV-VIS Spectrophotometer (JASCO-V750) (Tokyo, Japan). The concentration of each amino acid was evaluated from the standard calibration plots.The pseudo-first-order rate constant (Kobs) for the amino acids was calculated according to eqn (1) and (2):
| Ln ([SH]/[SH]0) = −Kobs × t | (1) |
| Ln ([NH2]/[NH2]0) = −Kobs × t | (2) |
Purification of amino acid-acrylamide adducts
AA-ACR adducts were synthesised based on the method reported by Zhao et al. with suitable modifications and following the optimised reaction conditions developed in the initial part of the current work (see Table S1†).46 Briefly, 100 mL of the aqueous solution containing a different ratio of ACR and amino acids was incubated at the optimised temperature under constant stirring for 120 min. The concentrations of amino acids reacting with ACR (10 mM) are as follows: lysine and methionine: 50 mM; glycine: 40 mM; and cysteine: 60 mM. At the end of the incubation time, the products were concentrated using a vacuum rotary evaporator (Eyela N-1300, Tokyo, Japan), and the adducts in the concentrate were purified using a C18 reverse silica gel column (Gly-ACR and Lys-ACR) monitored by HPLC (UltiMate 3000, Thermo Fisher, Germany). The elution solution used was methanol:water (2:98 v/v), and the fractions containing more than 80% of the target adducts were collected and further purified by semi-preparative liquid chromatography.25 Separation was performed using a Shimadzu QC-C18 column (250 × 10 mm, 5 μm, Shimadzu Analytical, India). The HPLC data for the four adducts is provided in Table S2.† The purified adducts were evaporated using a rotary vacuum evaporator at 55 °C, followed by freeze-drying using a vacuum freeze-dryer (FreeZone Plus 4.5 Liter Cascade Benchtop Freeze Dry System, Labconco, Kansas, USA).
Cell culture and reagents
Caco-2 and HCT-15 cells were obtained from the National Centre for Cell Science (Pune, India). Caco-2, the immortalized cell line of human colorectal adenocarcinoma epithelial cells, was cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (Gibco), penicillin (100 units per mL, Gibco), and streptomycin (100 μg mL−1, Gibco). HCT-15 (Human colon cancer cell line) cells were cultured in a Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% fetal bovine serum (Gibco), penicillin (100 units per mL, Gibco), and streptomycin (100 μg mL−1, Gibco). Cells were then incubated in a humidifier at 37 °C with 5% CO2 to achieve 70%–80% confluency.The cells were washed every three days with 0.1 M PBS (pH 7.4) and the medium was renewed. Confluent cells were detached from the culture with 0.25% Trypsin (37 °C, 10 min) according to the manufacturer's instructions, suspended in sterile PBS, and gently aspirated off the plastic flask. The cell suspension was centrifuged (1000g, 5 min) and decanted, and the pellet was re-suspended in a fresh DMEM. The Caco-2 cells were ready for use after a cell count was performed using a hemo-cytometer. Cells were passaged every week, and passages 7–12 were used for all experiments.
Incubation with adducts and reagents
ACR and the targeted adducts (AA-ACR), namely, lysine-adduct (Lys-ACR), glycine-adduct (Gly-ACR), methionine-adduct (Met-ACR), and cysteine-adduct (Cys-ACR) were prepared as 3.4 mM and 3.78 mM stock solutions, for HCT-15 and Caco-2 cell lines, respectively.Identification of acrylamide and adducts in potato tuber samples
The selected potato tubers and frozen potato tubers were subjected to blanching treatment. The method of blanching and soaking/pre-treatment under optimised amino-acids conditions were followed according to a previously reported method with a slight modification.52 Briefly, potato tubers obtained from a local market were washed under running water, hand peeled and cut into round slices (5 cm diameter, 2 mm thickness). Blanching was done by immersing the potato slices in distilled water (control sample), L-cysteine (0.06 M), L-glycine (0.04 M), L-lysine (0.05 M) and L-methionine (0.05 M). Samples were removed after 10 min. All blanching steps were performed at 65 °C. Blanched potato samples fried in sunflower oil (500 g) were placed in a stainless-steel pan of electric fryer and heated at 190 °C ± 5 °C for 6 min.In vitro digestion of potato samples
The in vitro digestion of fresh ground food samples was carried out in triplicate following the INFOGEST protocol described by Egger et al. (2016),53 with minor modifications. Five grams of each sample system treated with the respective amino acids were measured into polypropylene tubes, and Milli-Q water was added to adjust the total weight to 5 g. This step ensured consistent acrylamide content and enzyme concentrations across all digestions. Instead of incorporating lipase in the intestinal phase, the samples were sonicated to enhance lipid accessibility for subsequent digestion steps.54 The digestion began with the addition of 0.5 mL of α-amylase solution, adjusted to a concentration of 75 U mL−1 as per the standardized protocol. The samples were incubated at 37 °C for 2 min with gentle agitation. Subsequently, 10 mL of pepsin and 5 μL of 0.3 M CaCl2 were added, and the pH was adjusted to 3.0 using 1 M HCl. The pepsin concentration in the final mixture was standardized to 2000 U mL−1. The mixtures were then incubated at 37 °C for 2 hours with continuous mild agitation. For the intestinal phase, the pH was increased to 7.0 using 1 M NaOH, and 20 mL of a solution containing pancreatin, bile salts, and 40 μL of 0.3 M CaCl2 was added. The pancreatin concentration was adjusted to 13.37 mg mL−1, while bile salts were set to 10 mM in the final mixture. These preparations were incubated at 37 °C for 2 hours under gentle and continuous agitation before being rapidly frozen in liquid nitrogen to terminate enzymatic reactions and stored at −20 °C. Frozen potato samples were thawed and centrifuged at 4 °C at 3000g for 45 min. The supernatant (bio-accessible fraction) and pellet (non-soluble residual fraction) were carefully separated. Blank digestions were prepared using 5 mL of Milli-Q water, following the complete digestion protocol.The limit of quantification (LOQ; signal-to-noise ratio = 10; 10.5 μg kg−1) and limits of detection (LOD; signal-to-noise ratio = 3; 5 μg kg−1) were calculated by injecting lower concentrations of standards. No suppression of acrylamide by the sample matrix was observed. All analyses were performed twice. All the peak concentrations were considered based on the respective standards including ACR-AA adducts.
Determination of acrylamide in potato samples
The homogenized sample (5 g) was extracted with 50 mL of methanol (5%) and shaken by hand for 30 min. After centrifugation for 30 min at 10000g, the supernatant solution was transferred to a 200 mL separating funnel and the aqueous layer was collected and used for analysis. The aqueous layer was centrifuged at 3000g for 5 min. The pooled supernatant was filtered through a 0.2 μ PVDF syringe filter. The C18 SPE column (Supelclean™ LC-18 SPE Tube 57054) was conditioned with 5 mL methanol and the extract (2 mL) was loaded with 5 mL of water onto the column. The extracts were eluted with 2 mL of water. The eluent was collected for HPLC analysis. The samples were analysed using a HPLC (Shimadzu Prominence HPLC-DAD-RID system-20A, Japan) with a C18 column (250 × 4.6 nm, 5 μ purity C18). The injected volume was 20 μL. The separation was carried out using 80% methanol/water (v/v) at a flow rate 1 mL min−1 and the effluent of the column was continuously monitored using a UV detector at 254 nm.55 The concentrations of acrylamide and the corresponding adducts in the respective sample were analysed using the standard curves (Table S3†).
Cell viability assay
We measured cell viability towards ACR and amino acid-ACR adducts via an MTT assay. Approximately 104 Caco-2 and HCT-15 cells were seeded in each well of a 96-well plate with a complete culture medium. The cells were incubated overnight at 37 °C under 5% CO2. Next, the medium was aspirated and ACR and Acrylamide Adducts (AA-ACR) in respective media were added to achieve the final tested concentrations [mM]: 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 3.5, 4, 4.5 and 5. Six replicates of the samples were created. The negative controls contained only cells in DMEM without ACR and AA-ACR. Cells were incubated at 37 °C under 5% CO2 for 24 h. After incubation, the medium with ACR and AA-ACR was removed from each well and MTT (0.5 mg mL−1) was added and incubated at 37 °C under 5% CO2 for 3 h. After that time, MTT was removed, and formazan crystals were solubilized by adding DMSO. The absorbance was measured at 550 nm with a reference filter of 620 nm, using a BioTek Cytation Hybrid Multimode Reader (Agilent Technologies International Pvt. Ltd, Manesar, India).Cytotoxicity and half maximal inhibitory concentration (IC50)
The absorbance/fluorescence of the control sample (untreated cells) represented 100% cell viability. Cell viability (%) was calculated as follows:and cytotoxicity (%) was determined as 100 of cell viability (%). The results are presented as mean ± standard deviation (SD). Experiments were conducted with the same cell population. The IC50 value was determined from curves based on the reported method.47

Measurement of reactive oxygen species (ROS)
The intracellular ROS formation was measured using 2′,7′-dichlorofuorescin diacetate (DCFH-DA). Treatment of cells with ACR and AA-ACR for 24 h was provided based on the suitable concentrations identified via the MTT assay. The cultured cells were washed with PBS, and incubated with DCFH-DA, at a final concentration of 5 μM for an additional 20 min at 37 °C in the dark. The positive control contained 4 mM H2O2, and negative control comprised cells without any treatment. The cells that were stained with DCFH-DA were separated from the medium by centrifugation at 500g for 1 min. The removal of additional DCFH-DA from the medium was achieved by washing the cells twice with PBS. The fluorescence intensity of the cell suspensions was measured by flow cytometry, and the results were analyzed using the FlowJo software.48 The results were expressed as fluorescence intensity per 104 cells. Confocal Microscopy was performed to observe the ROS generation in the cells.Cell cycle distribution using flow cytometry
The HCT-15 and Caco-2 cells were trypsinized, fixed with ice-cold ethanol (70%), and centrifuged at 3000g for 5 min. After washing with ice-cold PBS, the pellets were re-distributed in a FxCycle™ PI/RNase staining solution (Thermo Fischer Scientific, catalog no. F10797). HCT-15 and Caco-2 were incubated for 20 min at room temperature and directly used for flow cytometry. The percentage of G0/G1, S and G2 phases of the cells were assessed based on the reported method.49Assessment of apoptosis and necrosis
Apoptosis and necrosis in cells were measured using a Membrane Permeability/Dead Cell Apoptosis Kit (Thermo Fisher Scientific catalogue no. V13243), along with YO-PRO-1 iodide and PI for flow cytometry. Cell apoptosis was induced during sample preparations by the addition of ACR or amino acid-ACR adducts. For the positive control, 1 μL of 4 mM of H2O2 (4 μM in final concentration) was added to one sterile centrifuge tube (1.5 mL) containing 1 mL warm cell culture medium (∼37 °C) for 20 min. All the samples were washed by the addition of 2 mL of warm PBS (∼37 °C) to each tube, followed by centrifugation at 400g for 5 min at 25 °C. The supernatant was removed, and cell pellet was re-suspended in 300 μL of fresh cell culture medium (∼37 °C). Flow cytometry and confocal microscopy on at least 3 replicates of samples were performed immediately thereafter. Viable, early apoptotic, late apoptotic and necrotic cells were counted following the manufacturer's instructions. HCT-15 and Caco-2 cells were analysed for the percentage of live, apoptotic, and necrotic cells.Measurement of mitochondrial membrane potential collapse
The mitochondrial membrane potential was monitored by the JC-1 dye staining method. The cells were seeded on a cell culture flask, cultured in a warm culture medium and maintained at 37 °C with 5% CO2 exposure. The culture medium was changed every alternate day until the cells reached 70%–80% confluency. Briefly, the medium was removed from the culture vessel by aspiration and then sufficient trypsin or trypsin/EDTA solution was dispensed into the culture vessel(s) to cover the monolayer of cells completely, followed by incubation at 37 °C for ∼6 min. The trypsin or trypsin/EDTA solution was removed and added to a sterile centrifuge tube (15 mL) containing an equal volume of warm cell culture medium (∼37 °C), followed by centrifugation at 125g for 7 min at 25 °C. The medium was removed by aspiration, and the cells were washed by addition of 2 mL of warm PBS (∼37 °C) to each tube and centrifuged at 400g for 5 min at 25 °C. After the removal of the supernatant, the cell pellet was suspended again in 1 mL of fresh cell culture medium or PBS (∼37 °C). The JC-1 dye (2 μM in final concentration) was added and cells were incubated at 37 °C, 5% CO2 for 15–30 min. For confocal fluorescence microscopy, absorption/emission wavelengths of 510/527 nm for the green monomeric form and 585/590 nm for the aggregate red form were used.50 The red-to-green fluorescence intensity ratio is only dependent on the membrane potential but not affected by other parameters such as shape, mitochondrial size and density. Fluorescent dye accumulation in the mitochondria was optically detected by flow cytometry and confocal microscopy.51Measurement of autophagy and autophagic marker across the cell cycle stages
The measurement of autophagy was performed using an Autophagy Assay Kit (Catalogue no. MAK138, Sigma Aldrich, Bengaluru, India) following the manufacturer's instructions. The kit provides a simple procedure for measuring autophagy using a proprietary fluorescent autophagosome marker (λex = 333/λem = 518 nm). HCT-15 and Caco-2 cells were seeded at a density of ∼1 × 104 in a 96-well flat bottom black plate with clear bottom. The cells were treated with ACR (IC50), Rapamycin (200 nM), Bafilomycin A1 (100 nM), Lys-ACR, Gly-ACR, Met-ACR and Cys-ACR, prior to staining with the dye in the kit. The manufacturer's instructions were followed for staining and washing steps. The fluorescence intensity measurements were made using a plate reader.The primary antibody used for analysing autophagy markers was LC3B (Novus Biologicals, NBP2-46892) at a concentration of 1:500. The cells were incubated with the secondary antibody Alexa-flour™ 568 goat anti-rabbit (H + L) (ThermoFischer Scientific-A11011) or Alexa-FlourTM 488 goat anti-mouse IgG (ThermoFischer Scientific-A11001) at a dilution of 1:200. To monitor autophagy across the cell cycle by flow cytometry, cells were cultured in 60 mm dishes, with different extracts as described in above sections and one well untreated. After treatment, cells were harvested through trypsinization and centrifuged at 200g for 5 min. The pelleted cells were washed with PBS and resuspended in 200 μL of permeabilization buffer. Permeabilization was expected to selectively release the unbound LC3-I protein while retaining the autophagosome-bound LC3-II. The cells were centrifuged at 1000g for 5 min, washed with PBS, and then fixed by resuspending in a 4% formaldehyde solution for 20 min. Following fixation, the cells were centrifuged and washed with PBS. Staining with a primary anti-LC3 antibody (diluted 1:500 in 2% BSA in PBS) was performed for 30 min to label autophagosomes, followed by washing and staining with a secondary Alexa Fluor 488-conjugated antibody. Finally, 5 μg mL−1 of propidium iodide and 200 μg mL−1 of RNase A were added followed by incubation for 30 min at room temperature. The samples were analyzed using a flow cytometer, detecting fluorescence signals for LC3-II (Alexa Fluor/FITC) and DNA content (propidium Iodide) to determine autophagosome levels across different cell cycle phases (G0/G1, S, and G2/M).
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 7.00 (GraphPad Software, San Diego, CA, USA). All the experiments were performed three times. The results were expressed as mean ± standard deviation (SD) and statistically analysed by a one-way ANOVA followed by Tukey's post hoc test to compare all the groups. P < 0.05 was considered statistically significant.Results and discussion
Scavenging capacity of amino acids for acrylamide
A few amino acids have been previously reported to form adducts with ACR.39,40,42,56,57 Considering the variability in conditions deployed in such reports and the absence of studies that systematically investigate amino acids, we first screened the ability of seven different amino acids to scavenge ACR. The reaction products between these amino acids and ACR were analysed by HPLC followed by ESI-MS. The reaction of ACR with four amino acids, lysine, glycine, cysteine and methionine, resulted in a measurable decrease in the HPLC peak of ACR and was accompanied by the emergence of new peaks (Fig. S1†). The reaction products of ACR with these four amino acids were subjected to ESI-MS, and distinctive m/z peaks corresponding to the respective AA-ACR adducts were observed in each case (Fig. S2†). The most abundant ion observed for the lysine + ACR reaction was observed at m/z 289 and is attributed to the conjugation of α-NH2 and ε-NH2 of lysine with two ACR molecules. Similarly, the most abundant ion for the product of cysteine + ACR reaction was observed at m/z 264 and can be attributed to the successful conjugation of the side chain –SH and α-NH2 on cysteine with two ACR moieties.39,40,42,56 The initial screening demonstrated the ability of amino acids Lys, Gly, Cys and Met to form adducts with ACR, albeit to differing extents. Factors that are likely to directly influence AA-ACR adduct formation include the concentration of reagents, temperature of the reaction, duration of incubation and pH of the reaction medium. Previous reports on AA-ACR product formation do not provide a systematic assessment of the influencing parameters. Thus, we next sought to experimentally identify suitable reaction parameters for product formation between the four amino acids and ACR. Reactions between ACR and each of the amino acids were performed with variations in amino acid concentration, temperature and duration of incubation, and the amount of ACR remaining was estimated via HPLC in each case (Fig. S3 & S4, see ESI†). Based on these experiments, the optimum conditions for AA-ACR adduct formation were identified and are listed in Table S1.† The greater proportion of amino acids versus ACR required for product formation reflects the nature of adducts formed and the differences in reactivities among the amino acids.Since the objective of our present work was to investigate the in cellulo effects of AA-ACR adducts, an investigation into the differences in reactivities of the amino acids towards ACR will be reported separately. The ACR adducts of lysine, glycine, cysteine and methionine were purified as described in the Experimental section and characterized by ESI-MS. The ESI-MS of purified Lys-ACR, Gly-ACR, Cys-ACR and Met-ACR showed prominent m/z peaks at 289.1, 218.1, 264.0 and 221.1, respectively (Fig. 1). MS/MS was conducted on these precursor ions for each AA-ACR, and the resulting fragment ions were observed (Fig. S5†). The fragment ions observed for Lys-ACR included m/z 142.0, 114.0 and 97.92, which correspond to the loss of the lysine side chain and putative ring formation (Table S4†).56 The fragment ions observed for Gly-ACR included m/z 129.83, 100.92, 87.83, and 71.83, which correspond to sequential losses within the glycine–acrylamide structure.57,58 While MS/MS of Cys-ACR and Met-ACR has not been analysed previously, fragmentation in the proximity of sulfur is responsible for peaks observed at 103.83 and 90.83, respectively. For each of the AA-ACR adducts, a combination of the m/z peaks and the MS/MS results support the adduct structures shown in Fig. S6.†
 | ||
| Fig. 1 ESI-MS of purified Lys-ACR-AA (top left), Gly-ACR-AA (top right), Cys-ACR-AA (bottom left), and Met-ACR-AA (bottom right). | ||
The acrylamide-amino acid adducts are formed via the Michael-type addition, where specific nucleophiles on the amino acids react with the α,β-unsaturated carbonyl group of acrylamide (Fig. S7†). Cysteine reacts via its thiol (–SH) group, while lysine and glycine react through their amine (–NH2) groups. The differences in nucleophilic strength and steric effects influence the reaction rates. To compare the kinetics of acrylamide-amino acid product formation, we conducted kinetic assays using the amino acids (lysine, glycine, cysteine, and methionine) with acrylamide under controlled reaction conditions (Fig. S8†).
The reaction rates were determined by monitoring the depletion of amino groups (via 2,4,6-trinitrobenzenesulfonic acid (TNBSA) assay) and sulfhydryl groups (via 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) assay) over time, and pseudo-first-order rate constants (Kobs) were calculated for each amino acid. Our results indicate that the kinetics of acrylamide-amino acid product formation varies depending on the nucleophilicity of the amino acid. Cysteine exhibited the highest reaction rate with acrylamide, followed by methionine, lysine, and glycine. The activation energy (Ea) values calculated from the Arrhenius plot further support this trend (Fig. S9†). The lower Ea value of cysteine corresponds with a higher acrylamide scavenging efficiency.
Acrylamide content in potato tubers
The doses of acrylamide employed in our experiments were carefully selected to align with realistic dietary exposure levels. We evaluated the acrylamide concentrations in untreated potato samples and included these assessments in Table 1. The acrylamide levels in fresh potatoes and frozen potatoes were estimated to be 2302 μg kg−1 and 2520 μg kg−1, respectively.59 These amounts are consistent with levels reported in fried or processed potato products, as documented by EFSA (2015).59 Next, to establish physiological relevance, we calculated the potential acrylamide concentrations in the human gut lumen based on typical dietary consumption.| Sample system | Acrylamide (μg kg−1) concentration before in vitro digestion | Acrylamide (μg kg−1) concentration after in vitro digestion |
|---|---|---|
| Control fresh potato | 2302 ± 231.1 | 2703 ± 331.6 |
| Lysine-treated fresh potato | 528 ± 102.4 | 634 ± 120.3 |
| Glycine-treated fresh potato | 1138 ± 140.3 | 1688 ± 150.2 |
| Cysteine-treated fresh potato | 40.2 ± 13.4 | 66.2 ± 12.4 |
| Methionine-treated fresh potato | 788 ± 16.2 | 705 ± 26.2 |
| Control frozen potato | 2520 ± 132.2 | 2889 ± 132.2 |
| Lysine-treated frozen potato | 645 ± 16.7 | 643 ± 1.7 |
| Glycine-treated frozen potato | 1695 ± 143.4 | 1705 ± 13.7 |
| Cysteine-treated frozen potato | 55.3 ± 14.6 | 56.3 ± 18 |
| Methionine-treated frozen potato | 890 ± 45.3 | 945 ± 27.3 |
Consumption of 150 g of fried potato containing 2.5 mg acrylamide per kg results in an intake of approximately 375 μg of acrylamide. Assuming a gut fluid volume of ∼1 L, this corresponds to a luminal concentration of 0.375 mg L−1 (375 μg L−1), which is within the concentration range tested in our study. This calculation reflects realistic exposure scenarios to the acrylamide concentrations used in the present work. Direct data on acrylamide adduct concentrations are scarce. After the in vitro digestion of the fresh and frozen potatoes, the acrylamide levels were estimated as 2302 μg kg−1 and 2520 μg kg−1, respectively. The in vitro digestion step simulates a physiologically relevant digestion event on the consumed potato products. These measures are consistent with EFSA-reported acrylamide levels in processed potato products (100–4000 μg kg−1).59 The estimated dietary intake of acrylamide from fried and baked potato products (up to 1 μg kg−1 body weight per day, EFSA 2015) translates to a total intake of 70 μg day−1 for a 70 kg individual.59 This corresponds to consuming approximately 30 g of untreated fresh potatoes or 28 g of untreated frozen potatoes in our study, which is realistic given typical portion sizes of fries or chips.
Further, the acrylamide concentration in potatoes post treatment with amino acid cysteine is 40.2 μg kg−1 and 66.2 μg kg−1, respectively, with and without in vitro digestion of samples. The amino acid treatment is thus capable of lowering the level of acrylamide to well below the lower limit of EFSA-reported range for processed foods.59 Even with a 150 g serving, the resulting acrylamide intake would be only 6 μg (40.2 μg kg−1 × 0.15 kg), far below the 70 μg day−1 estimated dietary intake for a 70 kg individual (1 μg kg−1 body weight per day, EFSA 2015).59 Similarly, lysine-treated fresh potatoes reduced acrylamide levels to 528 μg kg−1, comparable to levels found in less or moderately processed foods. These results highlight the potential for amino acid treatments to lower acrylamide exposure to physiologically relevant and safe levels while maintaining dietary relevance.
Effect of amino acid-acrylamide adducts on viability of HCT-15 and Caco-2 cells
We first investigated the effect of AA-ACR adducts on the viability of two different human cell lines, HCT-15 and Caco-2, via an MTT assay. Challenges in culturing and consistency in the performance of normal large or small intestinal cell lines preclude their use as effective models. HCT-15 cells are a viable model for studying the toxicity of ACR considering the absorption of the latter in the gastrointestinal tract and the origin of HCT-15 from human colon adenocarcinoma. HCT-15 is widely used in cancer research for studying various aspects of colon cancer biology, including tumour growth, invasion, metastasis, and drug resistance. The cell line is suitable for comparative analysis of ACR with ACR-adducts, thereby helping in the holistic in cellulo evaluation of the adducts. The Caco-2 cell line, derived from human colon adenocarcinoma, is extensively used in biomedical research for studying intestinal absorption, drug transport, and metabolism. These cells spontaneously differentiate into enterocyte-like cells, forming tight junctions and expressing various enzymes and transporters characteristic of the intestinal epithelium. Considering the absorption of ACR in the gastrointestinal tract, Caco-2 cells provide a relevant and effective system for investigating ACR toxicological effects and its comparative study with the effects of AA-ACR adducts. HCT-15 and Caco-2 cells are especially suited for assessing the cellular toxicity of AA-ACR adducts vis-à-vis ACR based on their relevance to human colorectal cells, which are among the primary targets for ACR exposure through diet.In this work, we sought to focus on the acute cellular responses to acrylamide toxicity and the mitigating effects of amino acid treatments from a mechanistic perspective. These responses are well-characterized in proliferative, undifferentiated Caco-2 cells, which provide a suitable model to investigate pathways of interest.60 Notably, the undifferentiated state of Caco-2 cells enabled an examination of the direct effects of acrylamide and its derivatives on cellular metabolism, oxidative stress, and other early-stage toxicological endpoints, which are less prominent or altered in fully differentiated cells.61
As shown in Fig. 2, ACR did not perturb the viability of HCT-15 and Caco-2 cells up to concentrations of 2 mM and 1 mM, respectively. ACR exerted a dose-dependent decrease in cell viability above these concentrations, with IC50 of 3.40 mM and 3.78 mM in HCT-15 and Caco-2, respectively. Our observed cytotoxic effects of ACR on HCT-15 and Caco-2 cells are consistent with previous reports.51,62–64 The cytotoxic and anti-proliferative activities of ACR were demonstrated for several cancer and normal cell lines.51,65–71 Notably, the adducts of Lys, Cys, Gly and Met with ACR showed no toxic effects on the HCT-15 and Caco-2 cells.
 | ||
| Fig. 2 Comparison of cytotoxicity of ACR and AA-ACR adducts evaluated by the MTT assay in (A) HCT-15 and (B) Caco-2 cells. Cells were treated with different AA-ACR adducts and acrylamide for 24 h. All data points were considered significant at p < 0.05. | ||
Based on our observations in Fig. 2A and B, the cellular viability, as inferred from the MTT assay, shows a concentration-dependent decrease in the range of 0.5–2.5 mM ACR, with a more pronounced reduction at 2.5 mM. This pattern of change in cellular viability is consistent with previous reports.51 This leaves the initial apparent increase in cellular viability between the absence of ACR and exposure to 0.5 mM ACR as a surprising observation, albeit one deserving explanation. The MTT assay used to investigate the effect of AA-ACR (and ACR) on HCT-15 and Caco-2 cells is really a sensitive assay measuring the relative metabolic activity inside cells.72 Interestingly, antioxidant enzymes in the gastrointestinal cell lines such as Caco-2 were reported to show a small but statistically significant increase in activity after confluence.73 Thus, at low ACR concentrations (<2.0 mM), it is likely that the cells experience a slight increase in metabolic activity compared to the absence of treatment. This slight increase in metabolic activity is captured by the MTT assay and appears to indicate a greater cellular viability at the low concentrations of ACR exposure. Interestingly, the AA-ACR adducts were successful in not only retaining the viability of cells but also encouraging cell growth in some instances. Several mechanisms have been implicated in the harmful cellular effects of ACR, including the interference with kinesin proteins and the disruption of spindle formation during cell division, thereby inhibiting cell proliferation and initiating ROS formation by disrupting the mitochondrial function.65,74–76 The relation between ACR-induced toxicity and oxidative stress has been reported previously.50 Our results indicate the AA-ACR adduct formation as an effective strategy for removing the cytotoxic effects of ACR.
In cellulo ROS formation upon treatment with AA-ACR adducts
ACR-induced ROS formation has been identified as a key contributor to oxidative stress. ACR depletes reduced glutathione, leading to excessive ROS accumulation, which disrupts cellular redox homeostasis and contributes to oxidative damage.74–76 Additionally, ACR activates MAPK signalling, further promoting ROS generation and apoptosis.74We investigated the oxidative stress in HCT-15 and Caco-2 cells upon treatment with ACR and the AA-ACR adducts, via flow cytometry and confocal fluorescence microscopy using DCHF-DA. DCFH-DA is hydrolysed inside cells to the nonfluorescent DCFH via the action of intracellular esterase, which in the presence of ROS can be rapidly oxidized to the highly fluorescent 2,7-dichlorofluorescein (DCF). Exposure of HCT-15 and Caco-2 cells to ACR and AA-ACR adducts was compared alongside the positive control containing 4 mM H2O2 and negative control comprising cells without any treatment. As shown in Fig. 3, the exposure of HCT-15 cells to ACR led to a substantive increase in ROS levels, comparable to the H2O2 positive control.
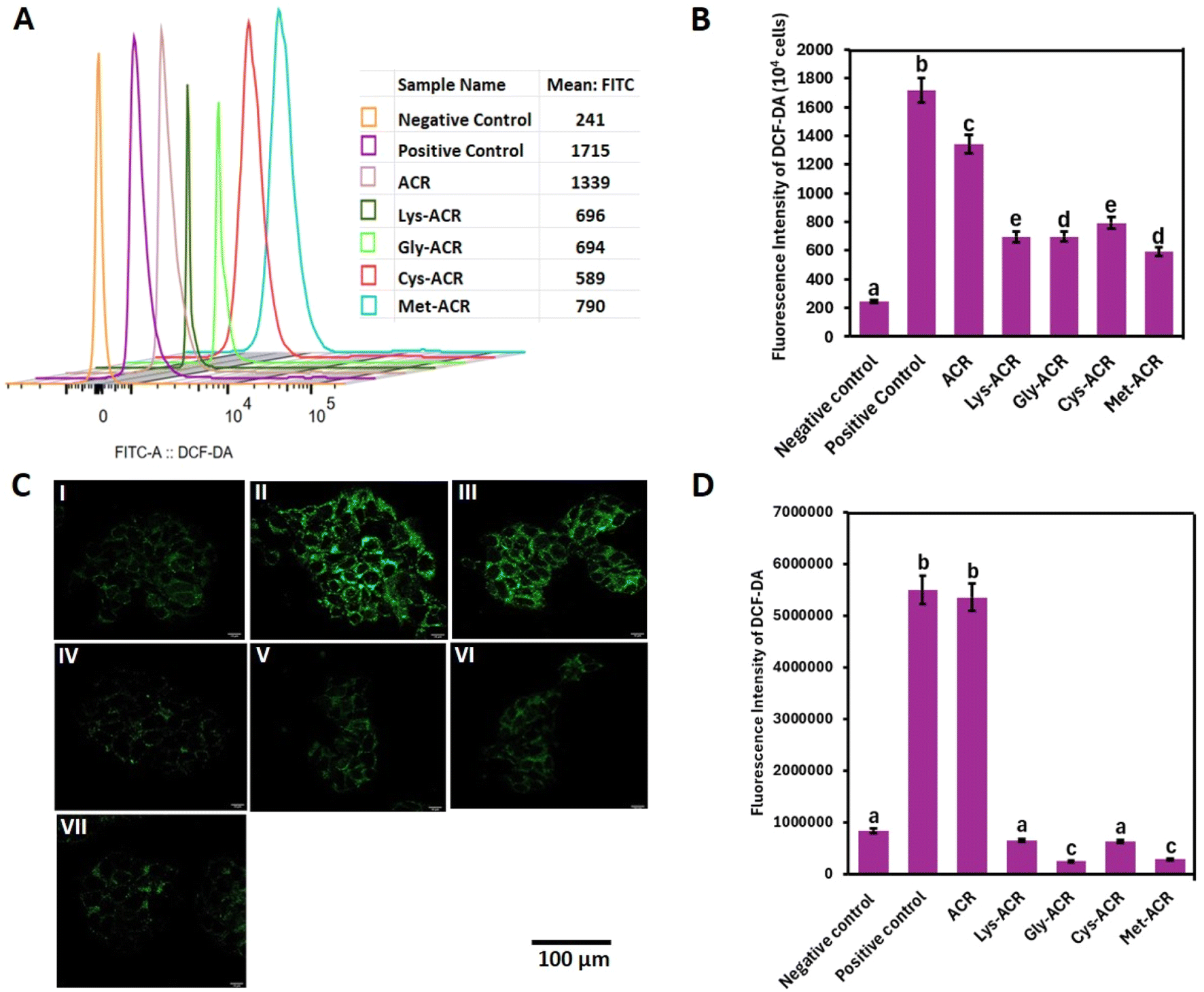 | ||
| Fig. 3 Effect of ACR and AA-ACR adducts on ROS accumulation in HCT-15 cells, assessed via (A) flow cytometric quantification of DCHF-DA; (B) comparison of DCHF-DA fluorescence intensity; (C) confocal microscopy: (I) negative control (II) positive control, (III) ACR, (IV) Lys-ACR, (V) Gly-ACR, (VI) Cys-ACR, and (VII) Met-ACR; and (D) quantification of DCHF-DA fluorescence. ACR was used at 3.40 mM and positive control (H2O2) at 4 mM. All values are expressed as mean ± SD (n = 3). Alphabets on bar graphs indicate significant differences (P < 0.05) among different treatment groups. | ||
Treatment with all the AA-ACR adducts resulted in a significant reduction in ROS formation in HCT-15. While few amino acids have been reported for their ability to scavenge ACR, to the best of our knowledge, there are no reports on the cellular effects of the resulting adducts.39,40,42,62,63 The difference in ROS production in Caco-2 cells, upon exposure to ACR and AA-ACR adducts, was more remarkable than that observed in HCT-15 cells. As shown in Fig. 4, the behaviours of AA-ACR adduct treatment were akin to the negative control, as reported by both flow cytometry and confocal microscopy. On a related note, amino acids have been investigated for their ability to scavenge acrolein and formaldehyde. The cytotoxicity of the products of such reactions varied in gastric epithelial and human intestinal epithelial cells and indicated the need for further evaluation of the strategy.77,78 The uniform absence of cytotoxic effects of all the AA-ACR adducts observed in our experiments, and the significant reduction in the in cellulo ROS formation accompanying their use across two different cell types, highlights the efficacy of sequestering ACR as AA-ACR adducts towards preventing the deleterious effects of ACR.
 | ||
| Fig. 4 Effect of ACR and AA-ACR adducts on ROS accumulation in Caco-2 cells, assessed via (A) flow cytometric quantification of DCHF-DA; (B) comparison of fluorescence intensity of DCHF-DA; (C) confocal microscopy: (I) negative control (II) positive control, (III) ACR, (IV) Lys-ACR, (V) Gly-ACR, (VI) Cys-ACR, and (VII) Met-ACR; and (D) quantification of DCHF-DA fluorescence. ACR was used at 3.78 mM and positive control (H2O2) at 4 mM. All values are expressed as mean ± SD (n = 3). Alphabets on bar graphs indicate significant differences (P < 0.05) among different treatment groups. | ||
Effects of acrylamide and amino acid-acrylamide adducts on cell cycle distribution
Cell populations experiencing stress are known to accumulate in the G1 phase.79 The arrest of cell cycle in G1 suggests extensive or irreparable DNA damage triggering prolonged checkpoint activation. This can lead to a cascade of stress responses that hinder normal cell proliferation and tissue regeneration, thereby disrupting healthy tissue homeostasis.80Flow cytometric data of cell cycle distribution of HCT-15 (Fig. 5) and Caco-2 (Fig. 6) cells showed that exposure to ACR and H2O2 induced the accumulation of populations in the G1 phase.64 While the disruptive effect of ACR on cell cycle distribution has been studied previously, there are no previous studies for AA-ACR adducts.64,81,82
 | ||
| Fig. 5 Effect of ACR and AA-ACR adducts on cell cycle distribution of HCT-15 cells. (A) Negative control, (B) positive control, (C) ACR, (D) Lys-ACR, (E) Gly-ACR, (F) Cys-ACR, (G) Met-ACR and (H) bar graph showing cell cycle distribution for different phases for different AA-ACR adducts. All values are expressed as mean ± SD (n = 3). Alphabets on bar graphs indicate significant differences (P < 0.05) among different treatment groups. | ||
 | ||
| Fig. 6 Effect of ACR and AA-ACR adducts on the cell cycle distribution of Caco-2 cells. (A) Negative control, (B) positive control, (C) ACR, (D) Lys-ACR, (E) Gly-ACR, (F) Cys-ACR, (G) Met-ACR and (H) bar graph showing cell cycle distribution for different phases for different AA-ACR adducts. All values are expressed as mean ± SD (n = 3). Alphabets on bar graphs indicate significant differences (P < 0.05) among different treatment groups. | ||
In the negative control group for HCT-15 cells, nearly 30.6% of the cell population were in G1, 34.6% in S, and 14% in G2M. Exposure to ACR resulted in an increase in G1 population to 43%, with 20.3% in S phase and 6.81% in G2M phase (Fig. 5). These populations are comparable to the ones observed upon exposure of cells to positive control H2O2. In contrast, the exposure of HCT-15 cells to ACR adducts of lysine, cysteine, glycine and methionine resulted in cell populations that were comparable to the negative control. Notably, treatment of HCT-15 cells with the Gly-ACR adduct appears to induce a greater entry of treated cells to the S phase than the other adducts.83
For Caco-2 cells, the negative control group had 50.5% of the cell population in the G1 phase, 40.0% in the S phase and 11.3% in the G2M phase (Fig. 6). Exposure to ACR resulted in an increase in G1 population to 68.3%. Analogous to HCT-15 cells, the exposure of Caco-2 cells to ACR adducts of amino acids resulted in cell populations that were comparable to the negative control. These results further support the ability of AAs to contain the harmful effects of ACR via adduct formation.
Apoptosis and necrosis upon exposure of cells to acrylamide and amino acid-acrylamide adducts
We next assessed the effect of ACR and amino acid-ACR adducts on induction of apoptosis and necrosis in HCT-15 and Caco-2 cells, via YO-PRO-Iodide-1/PI-staining followed by flow cytometry. The method relies on YO-PRO-Iodide-1, which is a green-fluorescent, cell-impermeant nucleic acid stain that can be used to stain dead or fixed cells, and PI staining of DNA of necrotic or late-stage apoptotic cells.51,64ACR is known to induce oxidative stress by depleting reduced glutathione (GSH) and altering antioxidant enzyme activities, such as increasing superoxide dismutase (SOD) activity.50,71,84,85 The excessive accumulation of ROS can trigger cellular damage and activate signalling pathways that regulate apoptosis. One such pathway involves mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38. These play crucial roles in apoptosis regulation by modulating pro-apoptotic and anti-apoptotic proteins.86 Exposure to ACR has been reported to increase phosphorylation of p53, a key tumour suppressor protein involved in the intrinsic apoptotic pathway. The phosphorylation of p53 leads to mitochondrial dysfunction, cytochrome c release, and activation of caspase-9 and caspase-3, ultimately resulting in cell death.87,88 Additionally, ACR-induced ROS generation can upregulate Bax, a pro-apoptotic member of the Bcl-2 family, while down-regulating Bcl-2, further promoting mitochondrial-mediated apoptosis. ACR-induced apoptosis is primarily mediated through oxidative stress-driven activation of MAPK and p53-dependent pathways, contributing to cellular dysfunction and cytotoxicity.
As shown in Fig. 7, the percentage of live HCT-15 cells in the ACR-treated group displayed a significant reduction to 72.4% compared to the negative control (97.9%). The ACR-treated cells also display a significant increase in the population of late apoptotic cells, and a small but definitive increase in necrotic cells that are comparable to the positive control (H2O2 treatment).
 | ||
| Fig. 7 Flow cytometric evaluation of apoptosis and necrosis in HCT-15 cells. (A) Negative control, (B) positive control (H2O2), (C) ACR, (D) Lys-ACR, (E) Gly-ACR, (F) Cys-ACR, (G) Met-ACR, and (H) quantification, indicating the % live cells in respective treated samples. The lower left square (Q4) shows the percentage of live cells with FITC- and PI-. The lower right square (Q3) shows the percentage of early apoptotic cells with FITC+ and PI−, the upper right square (Q2) displays the percentage of late apoptotic cells with FITC+ and PI+, and the top left square (Q1) shows necrotic cells with FITC- and PI+. All values are expressed as mean ± SD (n = 3). Alphabets on bar graphs indicate significant differences (P < 0.05) among different treatment groups. | ||
All the AA-ACR-treated cells showed between 97% and 99% live cells, highlighting their non-toxic effect on HCT-15 cells. Analogous results were obtained for Caco-2 cells with the ACR-treated cells displaying reduced live cell count (79.8%) and a substantial rise in late apoptotic cell count compared to the negative control (Fig. 8). Once again, all the AA-ACR adducts resulted in similar levels of live Caco-2 cells compared to the negative control, clearly indicating the stability of the AA-ACR adducts that facilitate blocking of stress-induction by ACR.
 | ||
| Fig. 8 Flow cytometric evaluation of apoptosis and necrosis in caco-2 cells. (A) Negative control, (B) positive control (H2O2), (C) ACR, (D) Lys-ACR, (E) Gly-ACR, (F) Cys-ACR, (G) Met-ACR, and (H) quantification, indicating the % Live cells in respective treated samples. The lower left square (Q4) shows the percentage of live cells with FITC- and PI-. The lower right square (Q3) shows the percentage of early apoptotic cells with FITC+ and PI−, the upper right square (Q2) displays the percentage of late apoptotic cells with FITC+ and PI+, and the top left square (Q1) shows necrotic cells with FITC- and PI+. All values are expressed as mean ± SD (n = 3). Alphabets on bar graphs indicate significant differences (P < 0.05) among different treatment groups. | ||
Effect of amino acid-acrylamide adducts on mitochondrial membrane potential
Apoptosis is accompanied by several major events in the mitochondria, the most significant of which is the loss of mitochondrial transmembrane potential.40 ACR-induced oxidative stress has been shown to regulate mitochondrial dysfunction and contribute to neurotoxicity.74,75 The accumulation of mtROS, which is primarily generated at mitochondrial respiratory chain complexes I and III, severely impairs electron transport chain, disrupting mitochondrial respiration and depolarization the mitochondrial membrane potential.68,89,90 We evaluated changes in the mitochondrial membrane potential upon treatment of HCT-15 and Caco-2 cells with ACR and AA-ACR adducts using the JC-1 staining method. The method relies on the ability of the JC-1 dye (5,5,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimi-dazoylcarbocyanine iodide) to enter healthy cells and accumulate into J aggregates inside the mitochondria. The concentration dependent formation of J-aggregates inside mitochondria by JC-1 results in a red shift in their fluorescence emission with a maximum at ∼590 nm. In contrast, in unhealthy cells, the JC-1 dye enters the mitochondria to a significantly lesser degree due to loss of electrochemical potential and the consequent membrane depolarization. The inability of the JC-1 dye to reach a sufficient concentration to trigger the formation of J aggregates results in retention of its typical green fluorescence. The red/green fluorescence ratio of JC-1 in the mitochondria provides a direct assessment of the state of mitochondrial membrane polarization. A higher membrane potential will result in a higher ratio, while the loss of membrane potential results in a reduction in the red-to-green fluorescence ratio. We exposed HCT-15 and Caco-2 cells to ACR, AA-ACRs and H2O2 for 20 min and followed by administration of JC-1 in all the samples. For HCT-15 cells, exposure to ACR resulted in green fluorescence of JC-1 like that observed for the H2O2 positive control (Fig. 9). | ||
| Fig. 9 Effect of ACR and AA-ACR adducts on mitochondrial damage in HCT-15 cells assessed by JC-1 dye staining. For confocal laser scanning microscopy, the monomer and J-aggregate forms are excited simultaneously by 488 nm argon-ion laser sources. The J-aggregate form is excited selectively using the 568 nm argon-krypton laser line. All values are expressed as mean ± SD (n = 3). Alphabets on bar graphs indicate significant differences (P < 0.05) among different treatment groups. | ||
The exposure of HCT-15 cells to Lys-ACR and Cys-ACR adducts resulted in intense red fluorescence and nearly complete absence of green fluorescence, suggesting a normal mitochondrial membrane polarization. Exposure to Gly-ACR and Met-ACR adducts resulted in only slightly lower red fluorescence than that observed with the Lys-ACR and Cys-ACR. Although some green fluorescence was evident upon treatment with these adducts, the loss of membrane polarization is significantly weaker than that of the ACR-treated cells. While analogous results were observed for Caco-2 cells, two aspects are worth noting. First, a sharper difference is observed between the ACR and AA-ACR adduct treated cells in terms of the relative intensity of the green and red fluorescence (Fig. 10).
 | ||
| Fig. 10 Effect of ACR and AA-ACR adducts on mitochondrial damage in Caco-2 cells. For confocal laser scanning microscopy, the monomer and J-aggregate forms are excited simultaneously by 488 nm argon-ion laser sources. The J-aggregate form is excited selectively using the 568 nm argon-krypton laser line. All values are expressed as mean ± SD (n = 3). Alphabets on bar graphs indicate significant differences (P < 0.05) among different treatment groups. | ||
In other words, the Caco-2 cells manifest the loss of mitochondrial membrane potential better than the HCT-15 cells. Second, the AA-ACR adducts display a similar pattern of effects across the HCT-15 and Caco-2 cells. While the Lys-ACR and Cys-ACR treatment results in high red fluorescence in both cell types, the Gly-ACR is least effective in facilitating normal mitochondrial membrane potential. Several studies have previously described the disruptions in mitochondrial structure and function by ACR. However, there is a dearth of literature on the chemical sequestration of ACR, preventing the deleterious effects towards mitochondrial membrane polarization. The present results support the ability of all the AAs to block the ACR-induced disruption of mitochondrial membrane polarization, while suggesting nuances in their relative effects possibly due to differences in the uptake and stability of the respective AA-ACR adducts.
Effect of acrylamide and amino acid-acrylamide adducts on autophagy induction
Autophagy is a well-studied outcome of ACR-induced hepatoxicity and corresponds to the process of wrapping damaged organelles and proteins in the cytoplasm in a double-membrane structure called the autophagosome.91–93 The autophagosomes fuse with free lysosomes to form autolysosomes in the cytoplasm, which are degraded and induce new autophagy.94 We used an Autophagy Assay Kit as described in the Experimental for comparing the results of treatment of HCT-15 and Caco-2 cells with ACR and amino acid-ACR adducts, with the autophagy inducer Rapamycin and the autophagy inhibitor Bafilomycin A1. As shown in Fig. 11, Rapamycin treatment resulted in increased autophagic activity, as indicated by a nearly 1.5-fold increase in fluorescence intensity compared to the negative control. Treatment with Bafilomycin A1 resulted in nearly 1.6-fold and 1-fold increases in the fluorescence intensity for Caco-2 and HCT-15 cells (Fig. 11). Bafilomycin A1 is expected to inhibit the fusion of autophagosomes with lysosomes. The resulting accumulation of autophagosomes without their degradation is likely to result in increased fluorescence intensity. This increase is due to the blockade of autophagic flux in contrast to enhanced autophagic activity, as observed upon treatment with Rapamycin. Notably, treatment with Lys-ACR and Gly-ACR adducts resulted in fluorescence intensity that is at par with the negative control, suggesting negligible autophagic flux. Cys-ACR and Met-ACR treatment resulted in a slightly greater fluorescence intensity than the negative control. Nevertheless, the autophagic flux in these cases was significantly lower than the positive control and upon treatment with ACR. These results clearly indicate the conversion of ACR into AA-ACR adducts as an effective strategy for preventing induction of autophagy. | ||
| Fig. 11 Comparison of autophagy induction upon treatment of HCT-15 and Caco-2 cells with various agents. All values are expressed as mean ± SD (n = 3). Alphabets on bar graphs indicate significant differences (P < 0.05) among different treatment groups. | ||
Further, the assessment of autophagosome formation across cell cycle stages allowed the quantitative measurement of LC3-II levels in correlation with the DNA content, providing a dynamic and cell-cycle-resolved perspective on autophagic activity. This choice of experiment for additional validation was based on the ability of FACS to provide single-cell resolution analysis allowing us to determine how autophagy is regulated across distinct phases of the cell cycle. We were especially keen on probing the interplay between autophagy and cell cycle regulation with respect to key regulators such as p53, p21, and cyclin D1. The tumour suppressor p53 plays a crucial role in modulating both cell cycle arrest and autophagy in response to cellular stress. The activation of p53 can lead to the upregulation of p21, a cyclin-dependent kinase inhibitor that promotes G1 arrest, while also influencing cyclin D1, which regulates G1/S transition. Given that autophagy can act as both a pro-survival and a pro-death mechanism depending on the context, the FACS-based analysis provides direct insights into how autophagic activity varies throughout the cell cycle, which is essential for understanding the benefits of sequestering ACR as AA-ACR adducts. Our results show that AA-ACR adduct-treated cells exhibit an LC3-II staining pattern like the negative control, validating a reduction in acrylamide-induced autophagy. ACR treatment results in significant elevation of LC3-II levels, indicative of autophagic activation likely as a stress response. The observed differences further support the hypothesis that AA-ACR adduct formation is a suitable approach for mitigating acrylamide-induced cellular stress, leading to a more controlled autophagy response (Fig. S10†).
Conclusion
The formation of ACR in thermally processed foods poses health risks based on its potential carcinogenic behaviour and the unfettered increase in the consumption of such products. The Maillard reaction responsible for ACR formation is suppressed in food products containing high proportions of proteins. Foods that are rich in sulphur-containing amino acids also result in the moderation of ACR levels. In this context, individual amino acids have been examined for their ability to enter a Michael-type addition reaction with ACR, thereby diverting the ACR from exerting harmful effects. Strategies that lower the ACR content in foods prior to consumption are likely to be more effective, considering the facile absorption of ACR from the gastrointestinal tract and distribution to peripheral tissues. In this work, we have addressed a gap in knowledge pertaining to the cellular response towards amino acid-ACR adducts. We prepared ACR adducts of several amino acids and selected four AA-ACR adducts based on their ease of product formation and yield of reactions, for the detailed investigation of cellular response. Our results show that the ACR adducts of Lys, Gly, Cys and Met are stable and non-toxic towards HCT-19 and Caco-2 cells, especially at concentrations where bare ACR displays significant cytotoxicity. The trapping of ACR as AA-ACR adducts effectively mitigates the harmful cellular response associated with exposure to ACR, resulting in lower ROS generation in the intestinal cell lines, preventing the accumulation of cells in the G1 phase and the induction of apoptosis and necrosis. The sequestration of ACR as AA-ACR adducts also prevents mitochondrial membrane depolarization and induction of autophagy. Modest differences in the cellular responses observed across the ACR adducts of Lys, Gly, Cys and Met may originate from the differences in uptake behaviour. However, such differences are not significant compared to the adverse effects observed upon treatment of the intestinal cells with ACR. The conversion of ACR into AA-ACR adducts is a robust strategy for mitigating ACR toxicity based on the prevention of the induction of oxidative stress and ROS generation that would result from exposure to ACR. A clarification is necessary regarding the attribution of observed effects throughout our work. The experimental design of our present work was not intended to (1) investigate the ability of AA-ACR adducts to directly counter ACR-induced cellular stress, or to (2) examine active cytoprotective effects of AA-ACR adducts. These objectives require additional lines of experimentation, and should not be conflated with the present study. Notably, previous efforts for the reduction of acrylamide by use of amino acids have focused only on the performance of single amino acids.95,96 The dearth of quantitative reports on acrylamide reduction upon use of multiple amino acids could reflect the variability in optimum conditions underlying adduct formation with acrylamide by different amino acids, and the complex interplay of kinetics of such reactions. The substantive stress-mitigating effects of AA-facilitated ACR sequestration reported in the current work support the application of multiple amino acids towards the mitigation of ACR in foods, and are currently being pursued in our laboratory.Data availability
The data supporting this article have been included as part of the manuscript and the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
B. D. acknowledges financial support from the Food Safety & Standards Authority of India (FSSAI) through Sanction Order-SO/Adv.(S&S)/112/2021-22 under scheme of NetSCoFAN. A. P. acknowledges a Director's Fellowship of IIT Gandhinagar. The authors are grateful to the Central Instrumental Facility (CIF) and the DSIR-IITGN Common Research & Technology Development Hub (CRTDH) of IIT Gandhinagar for their support of this work.References
- E. Tareke, P. Rydberg and P. Karlsson, et al., Analysis of acrylamide, a carcinogen formed in heated foodstuffs, J. Agric. Food Chem., 2002, 50(17), 4998–5006 CrossRef CAS PubMed.
- G. Tucker and S. Featherstone, Essentials of Thermal Processing, Wiley-Blackwell, Hoboken, NJ, 2010, p. 288 Search PubMed.
- A. Hamzalıoğlu, B. A. Mogol and V. Gökmen, Acrylamide: an overview of the chemistry and occurrence in foods, Encycl. Food Chem., 2019, 492–499 Search PubMed.
- D. Sharp, Acrylamide in food, Lancet, 2003, 361(9355), 361–362 CrossRef PubMed.
- A. P. Arisseto, M. C. Toledo and Y. Govaert, et al., Determination of acrylamide levels in selected foods in Brazil, Food Addit. Contam., 2007, 24(3), 236–241 CrossRef CAS PubMed.
- E. Dybing, P. B. Farmer and M. Andersen, et al., Human exposure and internal dose assessments of acrylamide in food, Food Chem. Toxicol., 2005, 43(3), 365–410 CrossRef CAS PubMed.
- Y. Zhang, Y. Ren and Y. Zhang, New research developments on acrylamide: analytical chemistry, formation mechanism, and mitigation recipes, Chem. Rev., 2009, 109(9), 4375–4397 CrossRef CAS PubMed.
- D. R. Lineback, J. R. Coughlin and R. H. Stadler, Acrylamide in foods: a review of the science and future considerations, Annu. Rev. Food Sci. Technol., 2012, 3(1), 15–35 CrossRef CAS PubMed . Rifai and Saleh 99.
- D. V. Zyzak, R. A. Sanders and M. Stojanovic, et al., Acrylamide formation mechanism in heated foods, J. Agric. Food Chem., 2003, 51(16), 4782–4787 Search PubMed.
- R. H. Stadler, I. Blank and N. Varga, et al., Acrylamide from Maillard reaction products, Nature, 2002, 419(6906), 449–450 Search PubMed.
- T. Krishnakumar and R. K. T. Visvanathan, Acrylamide in food products: a review, J. Food Process. Technol., 2014, 5(7), 1–10 Search PubMed.
- N. G. Halford, T. Y. Curtis and N. Muttucumaru, et al., The acrylamide problem: a plant and agronomic science issue, J. Exp. Bot., 2012, 63(8), 2841–2851 Search PubMed.
- J. S. Ahn, L. Castle, D. B. Clarke, A. S. Lloyd, M. R. Philo and D. R. Speck, Verification of the findings of acrylamide in heated foods, Food Addit. Contam., 2002, 19(12), 1116–1124 CrossRef CAS PubMed.
- D. S. Mottram, B. L. Wedzicha and A. T. Dodson, Acrylamide is formed in the Maillard reaction, Nature, 2002, 419(6906), 448–449 Search PubMed.
- R. H. Stadler, The formation of acrylamide in cereal products and coffee, in Acrylamide and Other Hazardous Compounds in Heat-Treated Foods, ed. K. Skog and J. Alexander, Woodhead Publishing Limited, England, 2006, pp. 23–40 Search PubMed.
- R. V. HedegAcrrd, K. Granby and H. Frandsen, et al., Acrylamide in bread. effect of prooxidants and antioxidants, Eur. Food Res. Technol., 2008, 227(2), 519–525 CrossRef.
- K. A. Wong, Screening for Acrylamide Levels in French Fries Using Portable Vibrational Spectrometers, The Ohio State University, Columbus, OH, 2017 Search PubMed.
- Joint FAO/WHO Food Standards Programme Codex Committee, Food Additives and Contaminants. Thirty-sixth Session, WHO, Geneva, Switzerland, 2003 Search PubMed.
- Agenda Item 6 (b) Joint FAO/WHO Food Standards Programme Codex Committee. Paper presented at: Food Additives and contaminants: working document for information and support to the discussion on the general standard for food additives, Thirty-eighth Session; April 24–28, 2006; The Hague, the Netherlands.
- L. A. Mucci, P. W. Dickman, G. Steineck, H. O. Adami and K. Augustsson, Dietary acrylamide and cancer of the large bowel, kidney, and bladder: absence of an association in a population-based study in Sweden, Br. J. Cancer, 2003, 88(1), 84–89 Search PubMed.
- C. Pelucchi, S. Franceschi, F. Levi, D. Trichopoulos, C. Bosetti, E. Negri and C. La Vecchia, Fried potatoes and human cancer, Int. J. Cancer, 2003, 105(4), 558–560 CrossRef CAS PubMed.
- K. A. Johnson, S. J. Gorzinski, K. M. Bodner, R. A. Campbell, C. H. Wolf, M. A. Friedman and R. W. Mast, Chronic toxicity and oncogenicity study on acrylamide incorporated in the drinking water of Fischer 344 rats, Toxicol. Appl. Pharmacol., 1986, 85(2), 154–168 Search PubMed.
- M. A. Friedman, L. H. Dulak and M. A. Stedham, A lifetime oncogenicity study in rats with acrylamide, Fundam. Appl. Toxicol., 1995, 27(1), 95–105 Search PubMed.
- T. R. Fennell and M. A. Friedman, Comparison of acrylamide metabolism in humans and rodents, Adv. Exp. Med. Biol., 2005, 561, 109–116 Search PubMed.
- M. J. Miller, D. E. Carter and I. G. Sipes, Pharmacokinetics of acrylamide in fisher-334 rats, Toxicol. Appl. Pharmacol., 1982, 63(1), 36–44 CrossRef CAS PubMed.
- S. C. Sumner, T. R. Fennell and T. A. Moore, et al., Role of cytochrome P450 2E1 in the metabolism of acrylamide and acrylonitrile in mice, Chem. Res. Toxicol., 1999, 12(11), 1110–1116 Search PubMed.
- K. I. Ubaoji and V. U. Orji, A review on acrylamide in foods: sources and implications to health, Mgbakoigba: J. Afr. Stud., 2016, 6(1), 1–17 Search PubMed.
- D. Kraus, D. Rokitta and U. Fuhr, et al., The role of human cytochrome P450 enzymes in metabolism of acrylamide in vitro, Toxicol. Mech. Methods, 2013, 23(5), 346–351 CrossRef CAS PubMed.
- M. I. Boettcher, T. Schettgen and B. Kütting, et al., Mercapturic acids of acrylamide and glycidamide as biomarkers of the internal exposure to acrylamide in the general population, Mutat. Res., 2005, 580(1–2), 167–176 CAS.
- H. W. Vesper, N. Slimani and G. Hallmans, et al., Cross-sectional study on acrylamide hemoglobin adducts in subpopulations from the European perspective investigation into cancer and nutrition (EPIC) study, J. Agric. Food Chem., 2008, 56(15), 6046–6053 CrossRef CAS PubMed.
- B. J. Petersen and N. Tran, Exposure to acrylamide: placing exposure in context, Adv. Exp. Med. Biol., 2005, 561, 63–76 CrossRef CAS PubMed.
- K. Fiselier and K. Grob, Legal limit for reducing sugars in prefabricates targeting 50 μg/kg acrylamide in French fries, Eur. Food Res. Technol., 2005, 220(5–6), 451–458 CrossRef CAS.
- N. Muttucumaru, S. J. Powers and J. S. Elmore, et al., Acrylamide-forming potential of potatoes grown at different locations, and the ratio of free asparagine to reducing sugars at which free asparagine becomes a limiting factor for acrylamide formation, Food Chem., 2017, 220, 76–86 CrossRef CAS PubMed.
- D. Kumar, B. P. Singh and P. Kumar, An overview of the factors affecting sugar content of potatoes, Ann. Appl. Biol., 2004, 145(3), 247–256 CrossRef.
- Y. Zhang, T. Ying and Y. Zhang, Reduction of acrylamide and its kinetics by addition of antioxidant of bamboo leaves (AOB) and extract of green tea (EGT) in asparagine-glucose microwave heating system, J. Food Sci., 2008, 73(2), C60–C66 CAS.
- S. Mehri, H. Veis Karami and F. Vahdati Hassani, et al., Chrysin reduced acrylamide-induced neurotoxicity in both in vitro and in vivo assessments, Iran. Biomed. J., 2014, 18(2), 101–106 CAS.
- C. Uthra, S. Shrivastava, A. Jaswal, N. Sinha, M. S. Reshi and S. Shukla, Therapeutic potential of quercetin against acrylamide induced toxicity in rats, Biomed. Pharmacother., 2017, 86, 705–714 CrossRef CAS PubMed.
- R. Maslanka, R. Zadrag-Tecza and K. Kwolek, et al., The effect of berry juices on the level of oxidative stress in yeast cells exposed to acrylamide, J. Food Biochem., 2016, 40(5), 686–695 CrossRef CAS.
- A. Kobayashi, S. Gomikawa, A. Yamazaki, S. Sato and T. Konishi, Elimination of acrylamide by moderate heat treatment below 120 C with lysine and cysteine, Food Sci. Technol. Res., 2014, 20(5), 979–985 CrossRef CAS.
- G. Koutsidis, S. P. Simons, Y. H. Thong, Y. Haldoupis, J. Mojica-Lazaro, B. L. Wedzicha and D. S. Mottram, Investigations on the effect of amino acids on acrylamide, pyrazines, and Michael addition products in model systems, J. Agric. Food Chem., 2009, 57(19), 9011–9015 Search PubMed.
- A. A. El-Sayed, S. M. A. El-Maaty and M. M. Abdelhady, Acrylamide reduction in potato chips as functional food product via application of enzymes, baker's yeast, and green tea powder, Sci. Afr., 2023, 20, e01698 CAS.
- M. Yu, S. Ou, D. Liumengzi, C. Huang and G. Zhang, Effect of ten amino acids on elimination of acrylamide in a model reaction system, Afr. J. Food Sci., 2013, 7, 329–333 CrossRef.
- M. Friedman and C. Levin, Review of methods for the reduction of dietary content and toxicity of acrylamide, J. Agric. Food Chem., 2008, 56, 6113 CrossRef CAS PubMed.
- S. Ou, Q. Lin, Y. Zhang, C. Huang, X. Sun and L. Fu, Reduction of acrylamide formation by selected agents in fried potato crisps on industrial scale, Innovative Food Sci. Emerging Technol., 2008, 9, 116–121 CrossRef CAS.
- J. Hu, K. Jiang, C. Huang, J. Zheng, H. Zhou and J. Ou, et al., Glycine and serine markedly eliminate methylglyoxal in the presence of formaldehyde via the formation of imidazole salts, Food Chem., 2022, 369, 130952 CrossRef CAS PubMed.
- Q. Zhao, Y. Zou, C. Huang, P. Lan, J. Zheng and S. Ou, Formation of a hydroxymethylfurfural rats fed with lysine-biofortified rice. NA. next? in caco-2 cells, J. Agric. Food Chem., 2017, 65, 9902–9908 CrossRef CAS PubMed.
- S. Adler et al. , OECD Guidelines for the Testing of Chemicals, Section 4. Test No. 442D: In Vitro Skin Sensitisation ARE-Nrf2 Luciferase Test Method. Available online: https://ntp.niehs.nih.gov/iccvam/suppdocs/feddocs/oecd/oecd-tg442d-508.pdf (accessed on 19 November 2019).
- P. Huang, Y. H. Zhang, X. W. Zheng, Y. J. Liu, H. Zhang and L. Fang, et al., Phenylarsine oxide (PAO) induces apoptosis in HepG2 cells via ROS-mediated mitochondria and ER-stress dependent signaling pathways, Metallomics, 2017, 9(12), 1756–1764 CrossRef CAS PubMed.
- M. Baeeri, S. Mohammadi-Nejad, M. Rahimifard, S. Moeini-Nodeh, R. Khorasani and M. Abdollahi, Molecular and biochemical evidence on the protective role of ellagic acid and silybin against oxidative stress-induced cellular aging, Mol. Cell. Biochem., 2018, 441, 21–33 CrossRef CAS PubMed.
- W. Chen, Y. Shen, H. Su and X. Zheng, Hispidin derived from Phellinus linteus affords protection against acrylamide-induced oxidative stress in Caco-2 cells, Chem.-Biol. Interact., 2014, 219, 83–89 CrossRef CAS PubMed.
- A. Nowak, M. Zakłos-Szyda, D. Żyżelewicz, A. Koszucka and I. Motyl, Acrylamide decreases cell viability, and provides oxidative stress, DNA damage, and apoptosis in human colon adenocarcinoma cell line Caco-2, Molecules, 2020, 25(2), 368 CrossRef CAS PubMed.
- S. A. M. A. Ismial, R. F. M. Ali, M. Askar and W. M. Samy, Impact of pre-treatments on the acrylamide formation and organoleptic evolution of fried potato chips, Am. J. Biochem. Biotechnol., 2013, 9(2), 90 CrossRef CAS.
- L. Egger, O. Ménard, C. Delgado-Andrade, P. Alvito, R. Assunção, S. Balance, R. Barberá, A. Brodkorb, T. Cattenoz, A. Clemente, I. Comi, D. Dupont, G. Garcia-Llatas, M. Jesús Lagarda, S. Le Feunteun, L. JanssenDuijghuijsen, S. Karakaya, U. Lesmes, A. R. Mackie, C. Martins, A. Meynier, B. Miralles, B. S. Murray, A. Pihlanto, G. Picariello, C. N. Santos, S. Simsek, I. Recio, N. Rigby, L.-E. Rioux, H. Stoffers, A. Tavares, L. Tavares, S. Turgeon, E. K. Ulleberg, G. E. Vegarud, G. Vergères and R. Portmann, The harmonized INFOGEST in vitro digestion method: From knowledge to action, Food Res. Int., 2016, 88, 217–225 CrossRef CAS.
- M. Minekus, M. Alminger, P. Alvito, S. Ballance, T. Bohn, C. Bourlieu, F. Carrière, R. Boutrou, M. Corredig, D. Dupont, C. Dufour, L. Egger, M. Golding, S. Karakaya, B. Kirkhus, S. Le Feunteun, U. Lesmes, A Macierzanka, A. Mackie, S. Marze, D. J. McClements, O. Ménard, I. Recio, C. N. Santos, R. P. Singh, G. E. Vegarud, M. S. J. Wickham, W. Weitschies and A. Brodkorb, A standardised static in vitro digestion method suitable for food–an international consensus, Food Funct., 2014, 5(6), 1113–1124 RSC.
- Z. Geng, P. Wang and A. Liu, Determination of acrylamide in starch-based foods by HPLC with pre-column ultraviolet derivatization, J. Chromatogr. Sci., 2011, 49(10), 818–824 CAS.
- D. Li, F. Xian, J. Ou, K. Jiang, J. Zheng, S. Ou, F. Liu, Q. Rao and C. Huang, Formation and identification of six amino acid-acrylamide adducts and their cytotoxicity toward gastrointestinal cell lines, Front. Nutr., 2022, 9, 902040 CrossRef PubMed.
- R. Zamora, R. M. Delgado and F. J. Hidalgo, Model reactions of acrylamide with selected amino compounds, J. Agric. Food Chem., 2010, 58(3), 1708–1713 CrossRef CAS PubMed.
- J. Liu, F. Chen, Y. Man, J. Dong and X. Hu, The pathways for the removal of acrylamide in model systems using glycine based on the identification of reaction products, Food Chem., 2011, 128(2), 442–449 CrossRef CAS PubMed.
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), Scientific Opinion on acrylamide in food, EFSA J., 2015, 13(6), 4104 Search PubMed.
- M. Puerto, S. Pichardo, Á. Jos and A. M. Cameán, Comparison of the toxicity induced by microcystin-RR and microcystin-YR in differentiated and undifferentiated Caco-2 cells, Toxicon, 2009, 54(2), 161–169 CrossRef CAS PubMed.
- C. M. Thompson, Y. Fedorov, D. D. Brown, M. Suh, D. M. Proctor, L. Kuriakose, L. C. Haws and M. A. Harris, Assessment of Cr(VI)-induced cytotoxicity and genotoxicity using high content analysis, PLoS ONE, 2012, e42720 CrossRef CAS PubMed.
- K. Saito, T. Xu and H. Ishikita, Correlation between C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrational frequency and p K a shift of carboxylic acids, J. Phys. Chem. B, 2022, 126(27), 4999–5006 CrossRef CAS PubMed.
O stretching vibrational frequency and p K a shift of carboxylic acids, J. Phys. Chem. B, 2022, 126(27), 4999–5006 CrossRef CAS PubMed. - W. Chen, L. Feng, Y. Shen, H. Su, Y. Li, J. Zhuang, L. Zhang and X. Zheng, Myricitrin inhibits acrylamide–mediated cytotoxicity in human Caco–2 cells by preventing oxidative stress, BioMed Res. Int., 2013, 1, 724183 Search PubMed.
- E. Mahdizade, M. Baeeri, M. Hodjat, M. Rahimifard, M. Navaei-Nigjeh, H. Haghi-Aminjan, S. Moeini-Nodeh, S. Hassani, G. Dehghan, M. A. Hosseinpour-Feizi and M. Abdollahi, Impact of acrylamide on cellular senescence response and cell cycle distribution via an in vitro study, Iran. J. Pharm. Res., 2021, 20(4), 165 CAS.
- S. Kacar, D. Vejselova, H. M. Kutlu and V. Sahinturk, Acrylamide-derived cytotoxic, anti-proliferative, and apoptotic effects on A549 cells, Hum. Exp. Toxicol., 2018, 37(5), 468–474 CrossRef CAS PubMed.
- V. Sahinturk, S. Kacar, D. Vejselova and H. M. Kutlu, Acrylamide exerts its cytotoxicity in NIH/3T3 fibroblast cells by apoptosis, Toxicol. Ind. Health, 2018, 34, 481–489 CrossRef CAS PubMed.
- S. Kacar, V. Sahinturk and H. M. Kutlu, Effect of acrylamide on BEAS-2B normal human lung cells: Cytotoxic, oxidative, apoptotic and morphometric analysis, Acta Histochem., 2019, 121, 595–603 CrossRef CAS PubMed.
- J.-H. Chen, C.-H. Yang, Y.-S. Wang, J.-G. Lee, C.-H. Chenga and C.-C. Chou, Acrylamide-induced mitochondria collapse and apoptosis in human astrocytoma cells, Food Chem. Toxicol., 2013, 51, 446–452 CrossRef CAS PubMed.
- K. Attoff, D. Kertika, J. Lundqvist, S. Oredsson and A. Forsby, Acrylamide affects proliferation and differentiation of the neural progenitor cell line C17.2 and the neuroblastoma cell line SH-SY5Y, Toxicol. In Vitro, 2016, 35, 100–111 CrossRef CAS PubMed.
- Z. Liu, G. Song, C. Zou, G. Liu, W. Wu, T. Yuan and X. Liu, Acrylamide induces mitochondrial dysfunction and apoptosis in BV-2 microglial cells, Free Radicals Biol. Med., 2015, 84, 42–53 CrossRef CAS PubMed.
- V. Mallepogu, P. Jayasekhar Babu, M. Doble, B. Suman, V. Nagalakshmamma, P. V. Chalapathi and K. Thyagaraju, Effects of acrylamide on cervical cancer (HeLa) cells proliferation and few marker enzymes, Austin J. Biotechnol. Bioeng., 2017, 4, 1087 Search PubMed.
- M. Ghasemi, T. Turnbull, S. Sebastian and I. Kempson, The MTT assay: utility, limitations, pitfalls, and interpretation in bulk and single-cell analysis, Int. J. Mol. Sci., 2021, 22(23), 12827 CrossRef CAS PubMed.
- S. S. Baker and R. D. Baker, Antioxidant enzymes in the differentiated Caco-2 cell line, In Vitro Cell. Dev. Biol.: Anim., 1992, 28, 643–647 CrossRef PubMed.
- X. Pan, L. Zhu, H. Lu, D. Wang, Q. Lu and H. Yan, Melatonin Attenuates Oxidative Damage Induced by Acrylamide in Vitro and in Vivo, Oxid. Med. Cell. Longevity, 2015, 703709 Search PubMed.
- O. O. Adewale, J. M. Brimson, O. A. Odunola, M. A. Gbadegesin, S. E. Owumi, C. Isidoro and T. Tencomnao, The Potential for Plant Derivatives against Acrylamide Neurotoxicity, Phytother. Res., 2015, 29, 978–985 CrossRef CAS PubMed.
- Y.-S. Luo, T.-Y. Long, L.-C. Shen, S.-L. Huang, S.-Y. Chiang and K.-Y. Wu, Synthesis, Characterization and Analysis of the Acrylamide- and Glycidamide-Glutathione Conjugates, Chem.-Biol. Interact., 2015, 237, 38–46 CrossRef CAS PubMed.
- E. Radi, P. Formichi, C. Battisti and A. Federico, Apoptosis and Oxidative Stress in Neurodegenerative Diseases, J. Alzheimer's Dis., 2014, 42, S125–S152 Search PubMed.
- J. Ou, J. Hu and S. Ou, Cytotoxicity of a novel compound produced in foods via the reaction of amino acids with acrolein along with formaldehyde, J. Agric. Food Chem., 2022, 70(49), 15583–15592 CrossRef CAS PubMed.
- T. Sumizawa and H. Igisu, Apoptosis induced by acrylamide in SH-SY5Y cells, Arch. Toxicol., 2007, 81, 279–282 CrossRef CAS PubMed.
- J. Y. Wang and S. K. Cho, Coordination of repair, checkpoint, and cell death responses to DNA damage, Adv. Protein Chem., 2004, 69, 101–135 CrossRef CAS PubMed.
- J. H. Chen, T. C. Tsou, I. M. Chiu and C. C. Chou, Proliferation inhibition, DNA damage, and cell-cycle arrest of human astrocytoma cells after acrylamide exposure, Chem. Res. Toxicol., 2010, 23(9), 1449–1458 Search PubMed.
- P. Gassner and I. D. Adler, Induction of hypoploidy and cell cycle delay by acrylamide in somatic and germinal cells of male mice, Mutat. Res., Genet. Toxicol., 1996, 367(4), 195–202 Search PubMed.
- R. Hannuniemi and S. S. Oja, Uptake of leucine, lysine, aspartic acid, and glycine into isolated neurons and astrocytes, Neurochem. Res., 1981, 6, 873–884 CrossRef CAS PubMed.
- M. Yousef and F. El-Demerdash, Acrylamide-induced oxidative stress and biochemical perturbations in rats, Toxicology, 2006, 219, 133–141 CrossRef CAS PubMed.
- M. Naruszewicz, D. Zapolska-Downar, A. Kośmider, G. Nowicka, M. Kozłowska-Wojciechowska and A. S. Vikström, et al., Chronic intake of potato chips in humans increases the production of reactive oxygen radicals by leukocytes and increases plasma C-reactive protein: a pilot study, Am. J. Clin. Nutr., 2009, 89, 773–777 CrossRef CAS PubMed.
- Y. Nakagawa-Yagi, D.-K. Choi, N. Ogane, S.-i. Shimada, M. Seya and T. Momoi, et al., Discovery of a novel compound: insight into mechanisms for acrylamide-induced axonopathy and colchicine-induced apoptotic neuronal cell death, Brain Res., 2001, 909, 8–19 CrossRef CAS PubMed.
- T. Okuno, M. Matsuoka, T. Sumizawa and H. Igisu, Involvement of the extracellular signal-regulated protein kinase pathway in phosphorylation of p53 protein and exerting cytotoxicity in human neuroblastoma cells (SHSY5Y) exposed to acrylamide, Arch. Toxicol., 2006, 80, 146–153 CrossRef CAS PubMed.
- R. S. Morrison, Y. Kinoshita, M. D. Johnson, W. Guo and G. A. Garden, p53-dependent cell death signaling in neurons, Neurochem. Res., 2003, 28, 15–27 CrossRef CAS PubMed.
- M. Zhao, P. Wang, Y. Zhu, X. Liu, X. Hu and F. Chen, The Chemoprotection of a Blueberry Anthocyanin Extract against the Acrylamide-Induced Oxidative Stress in Mitochondria: Unequivocal Evidence in Mice Liver, Food Funct., 2015, 6, 3006–3012 RSC.
- S. G. Kunnel, S. Subramanya, P. Satapathy, I. Sahoo and F. Zameer, Acrylamide Induced Toxicity and the Propensity of Phytochemicals in Amelioration: A Review, Cent. Nerv. Syst. Agents Med. Chem., 2019, 19, 100–113 CrossRef CAS PubMed.
- G. Filomeni, D. De Zio and F. Cecconi, Oxidative stress and autophagy: The clash between damage and metabolic needs, Cell Death Differ., 2015, 22(3), 377–388 CrossRef CAS PubMed.
- J. P. Gray, M. N. Uddin and R. Chaudhari, et al., Directed evolution of cyclic peptides for inhibition of autophagy, Chem. Sci., 2021, 12(10), 3526–3543 RSC.
- P. Mukhopadhyay, N. Eid and M. A. Abdelmegeed, et al., Interplay of oxidative stress, inflammation, and autophagy: Their role in tissue injury of the heart, liver, and kidney, Oxid. Med. Cell. Longevity, 2018, 2018, 2090813 CrossRef PubMed.
- M. P. Nelson, T. E. Tse and D. B. O'Quinn, et al., Autophagy-lysosome pathway associated neuropathology and axonal degeneration in the brains of alpha-galactosidase A-deficient mice, Acta Neuropathol. Commun., 2014, 2, 20 CrossRef PubMed.
- W. L. Claeys, K. De Vleeschouwer and M. E. Hendrickx, Effect of amino acids on acrylamide formation and elimination kinetics, Biotechnol. Prog., 2005, 21(5), 1525–1530 CrossRef CAS PubMed.
- C. T. Kim, E. S. Hwang and H. J. Lee, Reducing acrylamide in fried snack products by adding amino acids, J. Food Sci., 2005, 70(5), C354–C358 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5fo00301f |
| This journal is © The Royal Society of Chemistry 2025 |