
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical characterization and sensory evaluation of a phenolic-rich melanoidin isolate contributing to coffee astringency†
Brianne M.
Linne
,
Edisson
Tello
,
Christopher T.
Simons
 and
Devin G.
Peterson
*
and
Devin G.
Peterson
*
Department of Food Science & Technology, The Ohio State University, 2015 Fyffe Rd., Columbus, OH 43210-1007, USA. E-mail: peterson.892@osu.edu; Tel: +1-614-688-2723
First published on 11th March 2025
Abstract
The tactile flavor sensations of coffee are an important indicator of product quality, yet the stimuli contributing to these percepts remain insufficiently defined. In the present work, compounds that contribute to astringency in coffee were investigated. A multi-dimensional sensory-guided fractionation method was employed whereby preparative-scale liquid chromatography was leveraged to separate the coffee brew into subfractions, and then followed by sensory evaluation to identify fractions with perceptual impact. This process, paired with high-resolution chemical characterization via Fourier-transform ion cyclotron resonance/mass spectrometry, revealed a complex, phenolic-dominant, melanoidin fraction. Further sensory recombination testing confirmed this fraction imparted a perceptible astringent sensation at coffee-relevant concentrations in both water and coffee matrices. We hereby provide the first evidence of melanoidins contributing to the complex tactile flavor profile of coffee.
Introduction
Coffee is the third most consumed beverage worldwide, following water and tea,1 and serves as an integral component of daily routines for many individuals. Global demand for coffee has risen by approximately 65% between 2000 and 2020, making it one of the most traded commodities worldwide, with an export value of $19 billion and a retail market value of $83 billion.2 Despite this existing (and growing) demand, only a fraction of green coffee beans goes on to produce coffee considered to be of premium or “specialty” quality. This, combined with increasing consumer discernment, provides abundant opportunities for coffee flavor optimization and quality improvement.Historically, research on coffee flavor has primarily focused on identifying and characterizing volatile compounds responsible for its distinctive aroma profile (for review, see ref. 3 and 4). More recently, efforts have emerged to elucidate the sensory relevance of the non-volatile coffee fraction, particularly by investigating compounds that contribute to or modulate bitterness,5–10 and that impact overall flavor quality.11–13 However, comparatively little research has examined chemical compounds contributing to coffee mouthfeel or “body”, despite recognition of its role in the overall sensory experience.14–17
Coffee “body” is one of ten attributes established by the Specialty Coffee Association (SCA) to contribute to coffee bean quality and value. The SCA defines “coffee body” as “the tactile sensation of liquid in the mouth”.15 Tactile perceptions are incorporated with olfactory and gustatory inputs (both centrally and peripherally) and play an important role in the multimodal sensation of flavor.18,19 Previous work ascertained a list of sub-attributes contributing to the tactile sensation in coffee.20 In the present work, we aimed to better understand drivers of tactile perception in coffee by pursuing chemical constituents specifically contributing to one of these attributes, astringency.
Astringency is described as a sensation of “puckering”, “drying”, or “roughness” often resulting from ingestion of polyphenol-rich foods or beverages.21,22 Multiple mechanisms have been proposed for how the sensation is imparted and perceived. The prevailing mechanism proposes that astringency-imparting phenolic compounds interact with proline-rich proteins in the saliva and promote aggregation and precipitation of these salivary proteins, which in turn disrupt the natural lubrication of the oral cavity.23–25 This reduction of lubrication is hypothesized to result in increased friction when oral surfaces are brought into contact with each other during oral processing, increasing both physical and perceived roughness as conveyed by mechanoreceptors in the oral cavity.23,24,26,27
In the present work, a sensory-guided fractionation methodology was implemented to identify chemical compounds in coffee responsible for the tactile perception of astringency. As the SCA definition of coffee “body” refers to the tactile sensation of the liquid in the mouth, understanding chemical compounds contributing to all tactile dimensions, including astringency, would therefore provide targets for prediction, modulation, and optimization of coffee “body” levels and, subsequently, coffee quality.
Materials and methods
Materials
Food grade formic acid and optima grade methanol and acetone were purchased from Fisher Scientific (Waltham MA, USA). A Barnstead nano-diamond system (Thermo Fisher, Waltham, MA, USA) was used to filter nanopure water. Oasis HLB cartridges (6 g) were purchased from Waters Co. (Milford, MA, USA). For pH adjustment during recombination model preparation, FCC grade NaOH (Fisher Scientific, Waltham MA, USA) and FCC grade HCl (Sigma Aldrich, St Louis MO, USA) were utilized. Methylparaben (methyl 4-hydroxybenzoate) was purchased for use as an internal standard (Sigma Aldrich, St Louis MO, USA). Two commercial drip coffee samples assigned a high body (HB) score of 7.5 out of 10 and a low body (LB) score of 4.8 out of 10, respectively by consensus of 4 licensed Q Arabica graders (Coffee Quality Institute, Aliso Viejo, CA, USA) from Keurig Green Mountain Coffee Co. were used for sensory and chemical analyses. Coffees were brewed using an automatic drip coffee machine (Keurig K475, Keurig Green Mountain, Waterbury VT, USA) as previously reported20 and pre-portioned coffee brew pods were stored at −40 °C prior to analysis.Sample preparation and preparative liquid chromatography-mass spectrometry (Prep LC-MS) fractionation
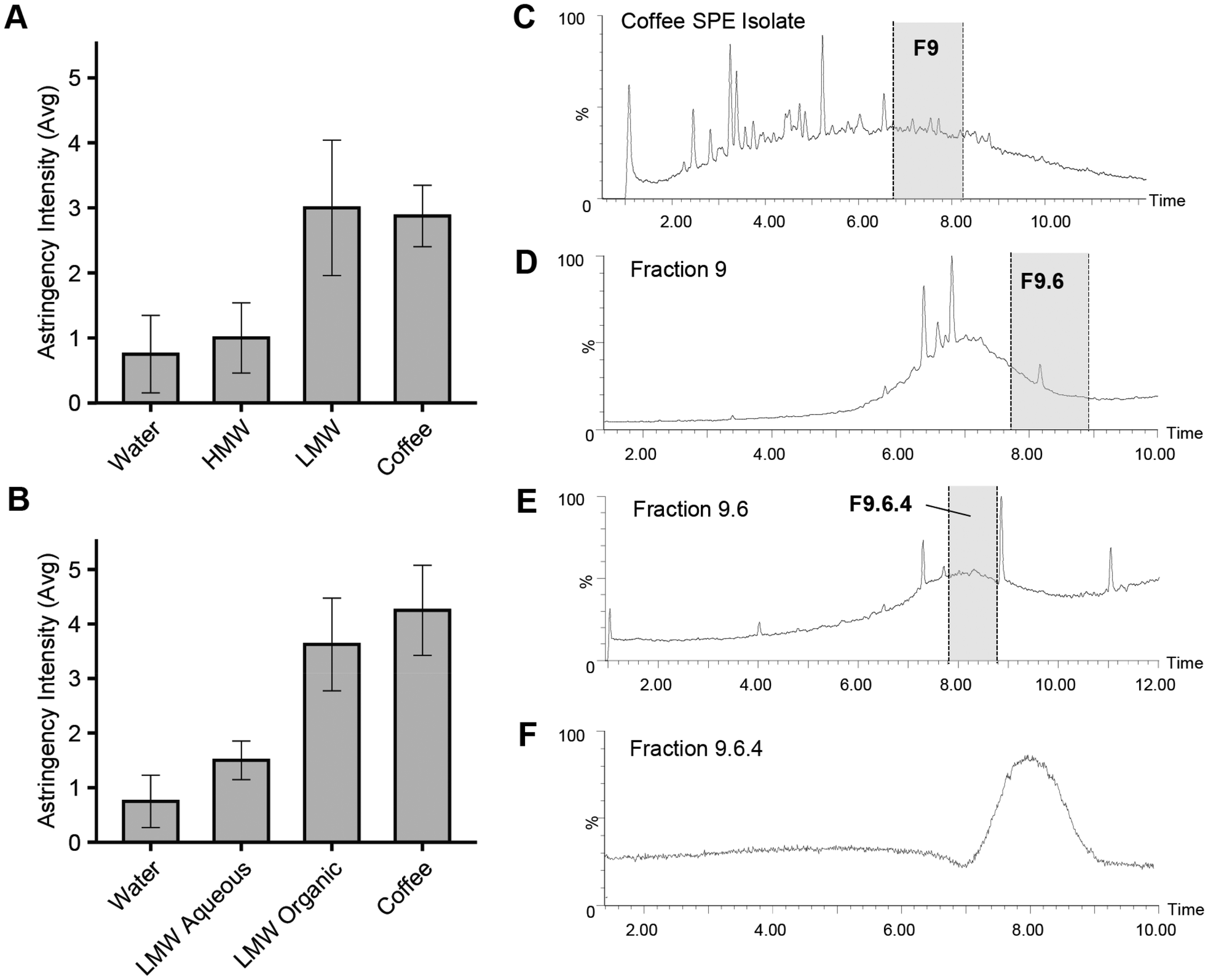 | ||
| Fig. 1 Sensory-guided analysis of astringency in coffee isolates. (A and B) Astringency intensity imparted by fractions resulting from ultrafiltration (UF) and solid-phase extraction (SPE) alongside water and coffee controls (average ± SEM). UF samples were divided into high (HMW, >5 kDa) and low (LMW, <5 kDa) molecular weight fractions (A) and SPE further separated the LMW fraction into organic (95% MeOH) and aqueous (5% MeOH) fractions (B). Astringency intensity was collected on a 0–10 scale, mean values displayed (n = 4, in duplicate). (C–F) UPLC-QToF chromatograms of first, second and third LC dimensions of sensory-guided fractionation in pursuit of compounds imparting astringency. Fraction 9 (C), Fraction 9.6 (D), and Fraction 9.6.4 (E) highlighted in grey (ESI−, total ion chromatograms). | ||
All dimensions of fractionation took place using a preparative-scale LC-MS system (AutoPurification System, Waters Co.) coupled to a triple quadrupole mass spectrometer (Quattro Micro API, Waters Co.) and a UV/Vis detector (Waters Co.). Settings for the mass spectrometer were as follows: ionization mode = ESI−, capillary voltage = 2.0 kV, cone voltage = 30 V, cone gas flow rate = 50 L h−1, source temperature = 150 °C, desolvation gas temperature = 300 °C, desolvation gas flow = 500 L h−1. Data were acquired in full scan mode (50–1250 m/z) with a scan time of 1s. UV/Vis data were simultaneously collected at absorbance wavelengths of 280 and 310 nm. For the LC, a mobile phase flow rate of 100 mL min−1 was used. Solvent composition, stationary phase, and chromatographic gradient conditions were selected independently for each of the three chromatographic separation dimensions to maximize orthogonality to the previous separation and are detailed below. After each LC chromatographic dimension of fractionation, solvent was evaporated from collected fractions (Genevac Rocket evaporator, SP Scientific, Warminster, PA, USA) which were then freeze-dried twice (to remove solvent) and reconstituted in nanopure water at a dosage level equivalent to that in coffee (based on UPLC/QToF peak area comparison) in preparation for sensory evaluation.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 v/v methanol/water (nanopure) solution and filtered using a PVDF syringe filter (0.45 μm) in preparation for injection onto preparative-LC system. The LC system was equipped with a phenyl hexyl column (Xselect, 5 μm particle size, 50 mm × 100 mm, Waters Co.) and a binary solvent system consisting of nanopure water (solvent A) and methanol (solvent B), each supplemented with formic acid (0.1% v/v). Separation was performed using the following gradient: 0–1 min, 5% B; 1–2 min, 5–50% B; 2–12 min, 50% B (isocratic); 12–18 min, 50–95% B; 18–21 min, held at 95% B; then returned to initial conditions for re-equilibration (21–23 min). The eluent was collected in a total of 6 fractions separated based on time (Fig. 1D).
:90 v/v methanol/water (nanopure) solution and filtered using a PVDF syringe filter (0.45 μm) in preparation for injection onto preparative-LC system. The LC system was equipped with a phenyl hexyl column (Xselect, 5 μm particle size, 50 mm × 100 mm, Waters Co.) and a binary solvent system consisting of nanopure water (solvent A) and methanol (solvent B), each supplemented with formic acid (0.1% v/v). Separation was performed using the following gradient: 0–1 min, 5% B; 1–2 min, 5–50% B; 2–12 min, 50% B (isocratic); 12–18 min, 50–95% B; 18–21 min, held at 95% B; then returned to initial conditions for re-equilibration (21–23 min). The eluent was collected in a total of 6 fractions separated based on time (Fig. 1D).
Compositional analysis of fraction 9.6.4 using Fourier-transform ion cyclotron resonance/mass spectrometry (FT-ICR MS)
The chromatogram of F9.6.4 presented as a broad hump with no distinct peaks and a diverse mass spectrum with few to no dominant ions (Fig. 1F). Due to its complexity and irresolvability, F9.6.4 was further examined compositionally and sensorially as the whole isolate. To characterize the chemical makeup, a high-resolution mixture analysis was conducted on the entire fraction using Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR MS). Analyses were conducted using both positive and negative electrospray ionization (ESI) modes on a Bruker 15 T solariX FT-ICR instrument (Bruker, Billerica MA, USA). Prior to infusion, F9.6.4 was prepared at a concentration of 1 mg L−1 in 50/50 acetonitrile/water. Resulting high resolution complex spectral information was visualized using a van Krevelen diagram and Kendrick mass defect plots to enable compositional inferences.28 The van Krevelen diagram visualization was produced by using the molecular formulas generated by FT-ICR MS to plot molecular ratio of hydrogen/carbon (H/C) on the y-axis against molecular ratios of oxygen/carbon (O/C) on the x-axis for each ion detected within F9.6.4. Kendrick mass defect plots were generated for mass differences of CH2 and C9H6O3, corresponding to methylation/demethylation and caffeic acid additions/removals, respectively. Kendrick mass diagrams were generated using the method described by Hughey et al.29Quantification of astringent coffee isolates
The concentration of F9.6.4 (Fig. 1F) in the coffee was determined by standard addition of F9.6.4 extracted from the HB coffee. F9.6.4 did not contain any prominently visible individual peaks in either positive or negative ESI modes, however direct MS infusion revealed distinct ions that were then selected for multiple reaction monitoring (MRM) method development using a Waters TQ-XS triple quadrupole (QqQ) mass spectrometer (Waters Co, Waltham MA). Evaluation of the specific MRM transition m/z 770.3 → 385.9 ESI− demonstrated good linearity when increasing amounts of F9.6.4 were added to water. Six 1 mL aliquots of HB coffee were prepared in triplicate using three biological replicates with increasing volumes from 100–500 μL of the 100 mg L−1 F9.6.4 solution, in addition to 20 μL each of internal standard (methylparaben, prepared at 1000 mg L−1). All samples were then made up to a final volume of 1520 μL through addition of nanopure water. SPE was undertaken using an Oasis HLB 96-well plate (30 mg bed) (Waters Co.) that was conditioned and equilibrated with 500 μL of 95% MeOH and 5% MeOH, respectively. The entirety of each 1520 μL coffee sample aliquot (plus standard addition and internal standard) was loaded onto independent SPE plate wells, washed with 5% MeOH (500 μL) (discarded), and then eluted with 95% acetonitrile (500 μL). Eluent was then made up to a final volume of 1000 μL through the addition of 500 μL each of nanopure water.Samples were analyzed with a UPLC-QqQ/MS on a Waters TQ-XS mass spectrometer paired with an Acuity H Class UPLC system (Waters Co, Waltham MA). For each run, an injection volume of 2 μL was loaded onto a phenyl hexyl column (1.7 μM particle size, 2.1 × 100 mm, Agilent Technologies, Santa Clara, CA). A binary solvent system using nanopure water (solvent A), and acetonitrile (solvent B) was used for separation, each dosed with 0.1% v/v formic acid. The following gradient was used for separation at a flow rate of 0.5 mL min−1: 0–0.5 min, 20% B; 0.5–11 min 20%–40% B, 11–12 min, 40–95% B, 12–13 min, held at 95% B, returned to initial conditions for 1 min (13–14 min). Settings for the mass spectrometer were as follows: ionization mode = ESI−, capillary voltage = 1.0 kV, cone voltage = 20 V, cone gas flow rate = 150 L h−1, source temperature = 150 °C, desolvation gas temperature = 600 °C, desolvation gas flow = 1000 L h−1. Data was acquired in multiple reaction monitoring (MRM) mode using the following optimized transitions and collision energies (CE): F9.6.4, ESI−m/z 770.3 → 385.9 (CE 28 eV); methylparaben, ESI−m/z 151 → 92 (CE 18 eV). Waters TargetLynx and MassLynx 4.1 software were used for analysis. The 6-point standard addition calibration curve showed good linearity (R2 = 0.994).
The concentration of F9.6.4 in LB coffee was determined through gravimetric comparison of a simultaneous extraction of F9.6.4 from 1L of each HB and LB coffee samples. The final amount of F9.6.4 in the LB coffee was finally determined based on the weight ratio of HB to LB and the known amount of F9.6.4 in the HB coffee determined by LC/MS/MS analysis as reported above.
Sensory evaluation
Sensory evaluation occurred over five days with either one or two 20-minute sessions per day. On the first day, panelists were reminded of the astringency definition, reference, and evaluation procedure, and were informed that this attribute enabled the initial selection of this fraction. Then, panelists were given the opportunity to taste F9.6.4 in water as a “warm-up” sample and make note of any differences perceived in comparison to a water control, using astringency as a non-restrictive guideline. For evaluation, a signal detection-based same/different methodology was employed. This entailed presentation of the respective reference (either water or LB coffee) paired with a three-digit code-blinded sample (either water/coffee with F9.6.4 added or a water/coffee blind control), after which panelists were asked if the blinded sample was “the same” or “different” from the reference. Following the same/different evaluation, they were then asked to indicate the certainty of their selection (“Sure”; “Unsure”; or “Guess”). If a panelist responded that they believed the samples to be different, they were also asked to describe the perceived difference. Both water and coffee evaluations utilized nine panelists in triplicate (27 total evaluations per sample); presentation order of samples was randomized across panelists and replicates, and nose clips were worn throughout to minimize olfactory interference.
Data analysis
Both fraction screening and recombination sensory evaluations were collected using Compusense Cloud consumer testing software (Compusense Inc, Guelph, CA). For fraction screening, statistical significance was determined using binomial probabilities for a directional (one-tailed) test with 50% chance probability and a p-value ≤ 0.172 (7 out of 10) was taken as sufficient for further examination via astringency intensity ratings.For sensory validation, signal detection results were processed using R-index analysis. “R-index” values were calculated based on pooled comparison of the same/different and certainty responses according to the response matrix and equation provided in the ESI (eqn (S1) and Table S1†), and can effectively be understood as the probability of a subject correctly discriminating between a stimulus and a control sample.30 Statistical significance of R-indices was determined using the “Rcriti” function in RStudio,31 which determined the r-critical value that must be exceeded to enable rejection of the null hypothesis for a given number of evaluations based on a Wald-type test statistic.32–34R-indices were therefore interpreted as indicative of discrimination if they exceeded chance discrimination by more than the corresponding R-critical value (α = 0.05, one-tailed).
Results
Sensory screening of fractions resulting from UF, SPE, and Prep-LC separations
The HB coffee underwent multiple sample preparation methods and LC dimensions of separation followed by sensory evaluation in pursuit of identifying astringent compounds (Fig. 1A–F). Initially, the coffee was separated into HMW (>5 kDa) and LMW (<5 kDa) isolates via ultrafiltration (UF) (Fig. 1A) and subsequently reconstituted in water and tasted alongside water and the HB coffee control. The average astringency intensity assigned to the LMW fraction sample was nearly identical to that of the HB coffee, while, on the contrary, the HMW fraction was rated similarly to water. When the LMW fraction was divided further using solid-phase extraction (SPE), the LMW organic phase (eluted with 95% methanol) reported similar astringency to the coffee control, while the LMW aqueous phase reported noticeably lower levels (Fig. 1B). The LMW organic phase was subsequently chosen for further sensory-guided liquid chromatography separations to identify astringent compounds.Following multi-dimensional LC separation and sensory-guided fractionation, fractions 9 (F9), 9.6 (F9.6), and then 9.6.4 (F9.6.4) were pursued from each dimension, based on their observed contribution to astringency (Fig. 1C–F). Finally, fraction 9.6.4 was selected as being higher in astringency than the control sample during 9 out of 10 paired-comparison presentations (p = 0.01) and was assigned an average astringency intensity rating 1.4 points greater than the control. Compositional analysis was thereby undertaken on F9.6.4 (Fig. 1F).
Further chromatographic separation of F9.6.4 proved challenging. The composition of F9.6.4 was thus further investigated with MALDI-TOF MS and FT-ICR MS analyses to define the molecular composition. The limited resolvability of F9.6.4 (Fig. 1F) suggested either the presence of large molecular weight compounds (<5 kDa) that were poorly separated on the LC column and/or that it contained many similarly structured compounds that were unable to be resolved due to similar stationary and mobile-phase affinities. MALDI-TOF MS analysis, which utilizes soft ionization ideal for revealing higher molecular weight components within the fraction, did not reveal the presence of any high molecular weight constituents (between 1.8–5 kDa, data not shown), contradicting the former possibility. Subsequently, FT-ICR MS analysis indicated the fraction was dominated instead by multiple lower molecular weight structurally related compounds. The FT-ICR MS spectrum of F9.6.4 (ESI− mode) revealed a complex landscape of compounds with masses ranging from m/z 200–1200 (Fig. 2). When the resulting mass spectrum was examined closely, a repeating mass difference of m/z 0.036 was observed throughout (Fig. 2, inlay) which is a notable mass difference commonly observed in a mixture of condensed phenolics indicating the loss of an oxygen atom and addition of CH4 (or vice versa). This mass difference was observed throughout F9.6.4, suggesting the presence of a variety of related condensed phenolics.
 | ||
| Fig. 2 FT-ICR MS spectra (ESI−) of coffee LC fraction 9.6.4. Inlay shows one (of many) sections of the spectrum in which a m/z 0.036 mass difference pattern was observed. This mass difference corresponds to a loss of an oxygen and addition of a CH4 (or vice versa), a common pattern observed when looking at a mixture condensed phenolics. | ||
The FT-ICR MS data analysis revealed 308 distinct molecules within the fraction and the elemental compositions were calculated for each with high mass accuracy (top most abundent based on relative intensity are shown in ESI, Table S2†). Observed mass to charge ratios ranged from m/z 227 to m/z 1170 with an average of m/z 444. The 308 molecules were further sorted by ion intensity (semi-quantitative) to establish relative compound concentrations,36 grouped into quartiles, and then plotted using a van Krevelen diagram (Fig. 3A) to characterize the chemical composition of the isolate. The van Krevelen diagram format allows visualization of structural patterns within complex chemical mixtures using atomic ratios of hydrogen to carbon (H/C) and oxygen to carbon (O/C), enabling a holistic understanding of the constituents.28 For example, if a cluster of data points fall at approximately H/C = 2, this cluster can be inferred to include saturated, lipid-like compounds. Alternatively, if a cluster falls within H/C = 1 to 1.5 and O/C = 0.3 to 0.7, this grouping can be attributed to condensed, aromatic, polyphenolic compounds.28 Based on the resulting plot, F9.6.4 contains compounds that can be divided into lipid-like (terpenes, fatty acids) and polyphenolic regions of the van Krevelen plot (Fig. 3A). However, examination of the compounds with the highest relative intensity (the 75–100th percentile) revealed a majority of the high intensity compounds (60 out of 79) were specifically within the polyphenolic region of the plot, highlighting the dominance of this class of compounds within this fraction (Fig. 3A and Table S2†). This distribution is consistent with the repeated m/z 0.036 mass difference observed within the mass spectrum characteristic of condensed phenolics (Fig. 2). Additionally, 14 of the 308 compounds detected in F9.6.4 were previously reported by Jaiswal et al.1 when utilizing FT-ICR MS to analyze the chemical composition of a heated mixture of chlorogenic acids (coffee roast model). Thus, at least some of the polyphenolics observed in F9.6.4 appeared to overlap with chlorogenic acid roasting derivatives.
 | ||
| Fig. 3 Chemical composition of LC fraction 9.6.4 though visualization of high-resolution FT-ICR MS outut. (A) Van Krevelen diagram of compounds detected in F9.6.4 plotted as hydrogen to carbon (H/C) vs. oxygen to carbon (O/C) ratios and compounds are broken into 4 color-coded quartiles (Q1–Q4) based on observed ion intensity (e.g., Q4 (yellow) encompasses the top 25% most intense ions observed). Ellipses indicate typical elemental ratio boundaries for select chemical classes as determined by Kuhnert et al.36 (B) Kendrick mass defect analysis for methylation (+CH2) of compounds found within the van Krevelen polyphenol ellipse. Green points highlight a sample of the homologous compound families related by CH2 additions and blue points indicate select homologous families of hydrogenation/dehydrogenations (±H2). Kendrick mass defect analysis at the C9H6O3 (C and D) reveal additional families of C9H6O3 (C) and C3H2O (D) additions, indicated by line segment connections. | ||
The chemical composition of F9.6.4 was further explored using Kendrick mass defect analysis, an approach complimentary to van Krevelen analysis.29,36 Using Kendrick analysis, complex mixtures can be normalized to a mass corresponding to a particular molecular addition (typically CH2), and then a series of compounds differing only in this substructure addition can be observed based on their identical Kendrick mass defect values. Two Kendrick mass defect plots (Fig. 3B–D) were produced for all compounds which fell within the condensed phenolic region of the van Krevelen diagram (Fig. 3A), and each were normalized to two specific mass additions CH2 (Fig. 3B) and C9H6O3 (Fig. 3C and D) which correspond to additions of methyl and caffeic acid groups, respectively. These two mass differences have been observed to be prevalent in both roasted mixtures of chlorogenic acids and in arabica coffee melanoidin extracts.1,36
In the Kendrick mass defect plot normalized to CH2 additions (Fig. 3B), visualization of multiple families of up to eight compounds differing only in methylation (CH2 additions) can be observed due to their identical mass defect values and regular mass increments forming a line parallel to the x-axis. Additionally, perpendicular to these lines, periodic hydrogenation reactions can also be observed. Often the compounds that fall within homologous CH2 addition/loss families also fall into perpendicular families of H2 additions/losses (green points) suggesting occurrence of complex, multi-directional derivatization during roasting. The compounds within the condensed phenolic cluster were explored further using another Kendrick mass defect plot normalized this time for caffeic acid additions corresponding to a mass difference of ±C9H6O3 (Fig. 3C and D). This plot revealed multiple pairs of C9H6O3 additions (>20) (Fig. 3C) and drew attention to an even more prolific number of homologous pairs of C3H2O additions (>50, Fig. 3D). The interconnected network of multiple moiety additions (CH2, C9H6O3, C3H2O) reveals an observable compound homology and apparent derivative nature of compound formation within F9.6.4.
Based on standard addition and gravimetric comparison, the concentrations of F9.6.4 in the HB and LB coffee was determined to be 15.7 and 7.9 mg L−1, respectively.
 | ||
| Fig. 4 Sensory impact of coffee fraction (F9.6.4) in water and coffee recombination models. Discrimination probabilities (expressed as R-index) of low (LB) and high body (HB) concentrations of F9.6.4 in water compared to water control (left) and addition of F9.6.4 to LB coffee compared to LB coffee control (right). Dashed line indicates critical value which must be exceeded for sample to be considered discriminable from control based on an α of 0.05 (62.54) (n = 9, 27 evaluations per sample; **p ≤ 0.01, ***p ≤ 0.001, one-tailed). | ||
Discussion
Compounds located within the condensed phenolic cluster were examined further using a Kendrick mass defect analyses and visualizations standardized to both CH2 and C9H6O3 mass differences (Fig. 3B–D). This analysis revealed that a majority belonged to one or more homologous families of CH2, H2, C9H6O3 and/or C3H2O additions, illustrating the diverse yet functionally derivative nature of the compounds contained within this fraction. Additions of CH2 and C9H6O3 groups correspond to periodic methylations and caffeic acid additions, respectively, reactions both known to occur during roasting of chlorogenic acids.36 Quinic acids can be esterified up to 4 times and cinnamic acids can also undergo multiple methylations. Single and double methylations of caffeoylquinic acid effectively result in feruloylquinic and dimethoxycinnamoyl acids, respectively.36 For F9.6.4, CH2 homologue families were observed to contain up to 8 molecules differing only in periodic CH2 additions with one of the longest visible CH2 homologous series comprised of a horizontal line parallel to the x-axis all with Kendrick mass defect values of 0.264 (Fig. 3B). For illustration, this series starts with a compound with m/z 287 and ends with m/z 385 and depicts a series of 7 methylation/demethylation reactions indicated by periodic CH2 additions between these, resulting in detection of intermediate masses m/z 301, 315, 329, 343, 357 and 371. The proposed molecular formula for the lowest mass within the series, m/z 287, is C16H15O5 (M–H) and it has been observed before in FT-ICR MS analysis of a model roasted mixture of chlorogenic acids found in coffee,1 suggesting this entire homologous series to result from derivatization of chlorogenic acid thermal reaction products during roasting. Multiple series of hydrogenation/dehydrogenation (±H2) and C3H2O additions are also observed, indicating the astringent F9.6.4 was dominated by a grid of homologous compounds differing in additions and removals of (at least) the four functional group patterns identified (CH2, H2, C9H6O3 and/or C3H2O).
During thermal processing of plant-based food materials such as the roasting of coffee beans, multiple reactions occur (repeatedly), ultimately resulting in the transformation of limited numbers of plant primary and secondary metabolites into numerous derivatives. The reported structural interrelatedness of compounds contained within the isolated fraction likely contributed to the observed chromatographic separation challenges of the individual components. In many foods, numerous compounds are anticipated to be unresolved by chromatographic analysis and could have the potential to contribute sensory and/or nutritional benefits. Other researchers have similarly employed FT-ICR MS techniques in order to understand the composition of complex food matrices including tea, cocoa, and coffee melanoidins and have found each of these matrices to contain as many as 10000, 30000, and 2000 distinct molecular components, respectively.36 Consequently, without examination of these complex mixtures, vast segments of processed food complexity could be overlooked. This risk is highlighted by the distinct astringency of fraction F9.6.4, which consists of a diverse array of similarly structured compounds, underscoring the importance of not disregarding complex segments that may hold meaningful chemical information. The findings of this study support the contribution of a complex fraction dominated by a subset of structurally related compounds to coffee astringency and advocate for further investigation into the contribution of other complex chemical populations to flavor perception.
Multiple studies have attempted to chemically characterize melanoidins, however a distinct challenge encountered is the difficulty of separating “melanoidin” from “non-melanoidin” material. Bekedam et al.35 specifically examined the “low molecular weight” melanoidin fraction of coffee. They defined low molecular weight as <12 kDa and quantified the melanoidin content based on absorption at 405 nm using a spectrophotometer and an extinction coefficient calculation using the law of Lambert–Beer.35,42 During these experiments, coffee brew was separated into high and low molecular weight portions using a 12–14 kDa size membrane filter. A non-trivial amount of the total coffee brew melanoidins (29%) were observed to partition into the LMW fraction, and, subsequently, into 40%, 60%, and 100% methanol eluents during solid-phase extraction (each contributing 25%, 34%, and 5% of the mass of the LMW melanoidins, respectively).
In the present study, the initial 5 kDa LMW cutoff resulted in a fraction reported to be the primary astringent component compared to its HMW counterpart (Fig. 1A). Further, during 1st dimension preparative-LC separation the fraction observed to be most astringent (F9, Fig. 1C) eluted between 50–60% MeOH, likely overlapping with the fraction that Bekedam et al.35 determined to contain the highest quantity of LMW melanoidin material (the 60% MeOH eluent). The researchers similarly found this isolate to be composed of large quantities of phenolic groups (41% w/w), hypothesizing this to be driven by incorporation of chlorogenic acids into these melanoidins. Thus, the melanoidin chemical composition described by Bekedam et al. was likely similar to the LMW phenolic-rich astringent isolated reported in the current study.
Despite the known role of phenolics in astringency perception and the acknowledged role of phenolics in melanoidin formation, melanoidins have not been discussed in prior literature as imparting astringency. While melanoidins have been associated with various biological activities—including antioxidant, anti-tumor, nitrosamine inhibition, antimicrobial, bacterial suppression, anti-carcinogenic, prebiotic functions, and amine binding41—research on their role in flavor perception remains limited. In coffee, melanoidins have been identified to bind with key odorants and alter the aroma profile over time after brewing.43,44 Our results indicate a more multifaceted role of melanoidins on flavor perception and therefore support further examination of this complex and prevalent class of compounds in both coffee and beyond.
Conclusions
A brown-colored isolate from coffee brew that primarily contained a complex mixture of structurally related melanoidin- or melanoidin precursor-associated condensed phenolics was reported to impart astringency in both water and coffee at coffee-relevant concentrations. This is the first evidence of a direct sensory impact of melanoidins in coffee and provides new avenues for expansion of our broader understanding of coffee quality.These findings also underscore the potential flavor impacts of complex, derivative fractions resulting from food processing reactions such as roasting, heating, and fermentation. Though such fractions may traditionally be overlooked due to chromatographic resolution challenges, their contributions should not be ignored for a better understanding of the chemical underpinnings of flavor perception.
Author contributions
Brianne M. Linne: methodology, investigation, validation, data curation, formal analysis, visualization, writing – original draft. Edisson Tello: supervision, validation, writing – review & editing. Christopher T. Simons: conceptualization, methodology, validation, project administration, supervision, writing – review& editing. Devin G. Peterson: conceptualization, methodology, validation, project administration, supervision, funding acquisition, resources, writing – review& editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors would like to thank Dr Arpad Somogyi and the Ohio State Campus Chemical Instrumentation Center for execution of MALDI-TOF and FT-ICR MS compositional analyses. This study served in partial fulfillment of the dissertation requirements for BML. The authors gratefully acknowledge the financial support from the Flavor Research and Education Center at The Ohio State University and its supporting members. This work is also supported in part by the USDA National Institute of Food and Agriculture, Hatch Project (OHO101231).References
- R. Jaiswal, M. F. Matei, A. Golon, M. Witt and N. Kuhnert, Understanding the fate of chlorogenic acids in coffee roasting using mass spectrometry based targeted and non-targeted analytical strategies, Food Funct., 2012, 3(9), 976–984 Search PubMed.
- M. Bozzola, S. Charles, T. Ferretti, E. Gerakari, H. Manson and N. Rosser, et al., The Coffee Guide, International Trade Centre, Geneva, Switzerland, 2021 Search PubMed.
- W. B. Sunarharum, D. J. Williams and H. E. Smyth, Complexity of coffee flavor: A compositional and sensory perspective, Food Res. Int., 2014, 62, 315–325, DOI:10.1016/j.foodres.2014.02.030.
- R. A. Buffo and C. Cardelli-Freire, Coffee flavour: An overview, Flavour Fragrance J., 2004, 19(2), 99–104 CrossRef CAS.
- O. Frank, G. Zehentbauer and T. Hofmann, Bioresponse-guided decomposition of roast coffee beverage and identification of key bitter taste compounds, Eur. Food Res. Technol., 2006, 222(5–6), 492–508 CrossRef CAS.
- O. Frank, S. Blumberg, G. Krumpel and T. Hofmann, Structure Determination of 3- O -Caffeoyl- epi - γ -quinide, an Orphan Bitter Lactone in Roasted Coffee, J. Agric. Food Chem., 2008, 9581–9585 CrossRef CAS.
- M. Ginz and U. H. Engelhardt, Identification of proline-based diketopiperazines in roasted coffee, J. Agric. Food Chem., 2000, 48(8), 3528–3532 Search PubMed.
- O. Frank, S. Blumberg and C. Kunert, Structure Determination and Sensory Analysis of Bitter-Tasting 4-Vinylcatechol Oligomers and Their Identification in Roasted Coffee by Means of LC-MS/MS, J. Agric. Food Chem., 2007, 55, 1945–1954 Search PubMed.
- C. Gao, E. Tello and D. G. Peterson, Identification of coffee compounds that suppress bitterness of brew, Food Chem., 2021, 350, 129225, DOI:10.1016/j.foodchem.2021.129225.
- C. Gao, E. Tello and D. G. Peterson, Identification of compounds that enhance bitterness of coffee brew, Food Chem., 2023, 415, 135674, DOI:10.1016/j.foodchem.2023.135674.
- F. Wei, K. Furihata, T. Miyakawa and M. Tanokura, A pilot study of NMR-based sensory prediction of roasted coffee bean extracts, Food Chem., 2014, 152, 363–369 Search PubMed.
- S. Sittipod, E. Schwartz, L. Paravisini, E. Tello and D. G. Peterson, Identification of Compounds that Negatively Impact Coffee Flavor Quality Using Untargeted Liquid Chromatography/Mass Spectrometry Analysis, J. Agric. Food Chem., 2020, 68, 10424–10431 Search PubMed.
- S. Sittipod, E. Schwartz, L. Paravisini and D. G. Peterson, Identification of flavor modulating compounds that positively impact coffee quality, Food Chem., 2019, 301, 125250, DOI:10.1016/j.foodchem.2019.125250.
- L. Navarini, R. Cappuccio, F. Suggi-Liverani and A. Illy, Espresso coffee beverage: Classification of texture terms, J. Texture Stud., 2004, 35(5), 525–541 CrossRef.
- Specialty Coffee Association. Protocols & Best Practices—Specialty Coffee Association, 2020. Available from: https://sca.coffee/research/protocols-best-practices Search PubMed.
- World Coffee Research. World Coffee Research Sensory Lexicon, 2nd ed, 2017, p. 54, Available from: https://worldcoffeeresearch.org/media/documents/20170622_WCR_Sensory_Lexicon_2-0.pdf Search PubMed.
- S. D. Williams, D. de Andrade and L. Liu, Coffee is more than flavor, the creation of a coffee character wheel, J. Sens. Stud., 2023, 38(6), e12886 CrossRef.
- Small and Green- a proposed model of flavor modality.
- D. M. Small and J. Prescott, Odor/taste integration and the perception of flavor, Exp. Brain Res., 2005, 166(3–4), 345–357 Search PubMed.
- B. M. Linne, E. Tello, C. T. Simons and D. G. Peterson, Characterization of the impact of chlorogenic acids on tactile perception in coffee through an inverse effect on mouthcoating sensation, Food Res. Int., 2023, 172, 1–10 Search PubMed.
- C. J. Thomas and H. T. Lawless, Astringent subqualities in acids, Chem. Senses, 1995, 20(6), 593–600 CAS.
- C. B. Lee and H. T. Lawless, Time-course of astringent sensations, Chem. Senses, 1991, 16(3), 225–238 CrossRef . Available from: https://academic.oup.com/chemse/article/16/3/225/312646.
- M. R. Bajec and G. J. Pickering, Astringency: mechanisms and perception, Crit. Rev. Food Sci. Nutr., 2008, 48(9), 858–875 CrossRef CAS PubMed . Available from: https://www.ncbi.nlm.nih.gov/pubmed/18788010\nISI:000259145500004.
- H. L. Gibbins and G. H. Carpenter, Alternative mechanisms of astringency - What is the role of saliva?, J. Texture Stud., 2013, 44(5), 364–375 Search PubMed.
- C. A. Lee, B. Ismail and Z. M. Vickers, The Role of Salivary Proteins in the Mechanism of Astringency, J. Food Sci., 2012, 77(4), 381–387 Search PubMed.
- B. Linne and C. T. Simons, Quantification of oral roughness perception and comparison with mechanism of astringency perception, Chem. Senses, 2017, 42(7), 525–535 CAS.
- S. Ployon, M. Morzel, C. Belloir, A. Bonnotte, E. Bourillot and L. Briand, et al., Mechanisms of astringency : Structural alteration of the oral mucosal pellicle by dietary tannins and protective effect of b PRPs, Food Chem., 2018, 253, 79–87, DOI:10.1016/j.foodchem.2018.01.141.
- S. Kim, R. W. Kramer and P. G. Hatcher, Graphical Method for Analysis of Ultrahigh-Resolution Broadband Mass Spectra of Natural Organic Matter, the Van Krevelen Diagram, Anal. Chem., 2003, 75(20), 5336–5344 CAS.
- C. A. Hughey, C. L. Hendrickson, R. P. Rodgers, A. G. Marshall and K. Qian, Kendrick mass defect spectrum: A compact visual analysis for ultrahigh-resolution broadband mass spectra, Anal. Chem., 2001, 73(19), 4676–4681 CAS.
- M. O'Mahony, Understanding Discrimination Tests: a User–Friendly Treatment of Response Bias, Rating and Ranking R–Index Tests and Their Relationship To Signal Detection, J. Sens. Stud., 1992, 7(1), 1–47 Search PubMed.
- R. Core Team. R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, 2021. Available from: https://www.R-project.org/ Search PubMed.
- J. Bi and M. O’Mahony, R-index critical value, J. Sens. Stud., 2020, 35(4), e12591 Search PubMed.
- J. Bi and M. O'Mahony, Table for Testing the Significance of the R–Index, J. Sens. Stud., 2007, 10(4), 713–720 CrossRef.
- J. Bi and M. O'Mahony, Table for Testing the Significance of the R–Index, J. Sens. Stud., 1995, 10(4), 341–347 CrossRef.
- E. K. Bekedam, E. Roos, H. A. Schols, M. A. J. S. Van Boekel and G. Smit, Low molecular weight melanoidins in coffee brew, J. Agric. Food Chem., 2008, 56(11), 4060–4067 CrossRef CAS PubMed.
- N. Kuhnert, F. Dairpoosh, G. Yassin, A. Golon and R. Jaiswal, What is under the hump? Mass spectrometry based analysis of complex mixtures in processed food-lessons from the characterisation of black tea thearubigins, coffee melanoidines and caramel, Food Funct., 2013, 4(8), 1130–1147 Search PubMed.
- A. S. P. Moreira, F. M. Nunes and M. A. Coimbra, Melanoidins, in Production, Quality and Chemistry, ed. A. Farah, Royal Society of Chemistry, Coffee, 2019, pp. 662–678 Search PubMed.
- F. M. Nunes and M. A. Coimbra, Melanoidins from Coffee Infusions. Fractionation, Chemical Characterization, and Effect of the Degree of Roast, J. Agric. Food Chem., 2007, 55, 3967–3977 CrossRef CAS.
- E. K. Bekedam, H. A. Schols, M. A. J. S. Van Boekel and G. Smit, Incorporation of chlorogenic acids in coffee brew melanoidins, J. Agric. Food Chem., 2008, 56(6), 2055–2063 CrossRef CAS PubMed.
- E. K. Bekedam, M. J. Loots, H. A. Schols, M. A. J. S. Van Boekel and G. Smit, Roasting Effects on Formation Mechanisms of Coffee Brew Melanoidins, J. Agric. Food Chem., 2008, 56, 7138–7145 CrossRef CAS PubMed.
- F. M. Nunes and M. A. Coimbra, Role of hydroxycinnamates in coffee melanoidin formation, Phytochem. Rev., 2010, 9(1), 171–185 CrossRef CAS.
- E. K. Bekedam, H. A. Schols, M. A. J. S. Van Boekel and G. Smit, High molecular weight melanoidins from coffee brew, J. Agric. Food Chem., 2006, 54(20), 7658–7666 CAS.
- T. Hofmann, M. Czerny, S. Calligaris and P. Schieberle, Model studies on the influence of coffee melanoidins on flavor volatiles of coffee beverages, J. Agric. Food Chem., 2001, 49(5), 2382–2386 CrossRef CAS PubMed.
- M. Gigl, O. Frank, A. Gabler, T. Koch, H. Briesen and T. Hofmann, Key odorant melanoidin interactions in aroma staling of coffee beverages, Food Chem., 2022, 392, 133291 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fo04934a |
| This journal is © The Royal Society of Chemistry 2025 |