 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual-selective polymerization: achieving chemoselectivity and stereoselectivity in a single catalytic system†
Hengxu Liu ,
Jiayun Jiang,
Xue Liang,
Wenli Wang,
Hongru Qiang,
Yuanzu Zhang and
Yunqing Zhu*
,
Jiayun Jiang,
Xue Liang,
Wenli Wang,
Hongru Qiang,
Yuanzu Zhang and
Yunqing Zhu*
Department of Polymeric Materials, School of Materials Science and Engineering, Tongji University, 4800 Caoan Road, Shanghai 201804, China. E-mail: 1019zhuyq@tongji.edu.cn
First published on 16th April 2025
Abstract
The precise synthesis of multifunctional block copolymers with tailored architectures remains a pivotal challenge in polymer chemistry, particularly when balancing chemoselectivity and stereoselectivity within a single catalytic system. To address this challenge, we report the dual chemoselective and stereoselective capabilities of a commercially available chiral thiourea catalyst, (S,S)-TUC, for the synthesis of well-defined block copolymers. By leveraging its dual selectivity, (S,S)-TUC enables distinct polymerization pathways dictated by monomer composition. In the TMC/rac-LA system, stereoselective ring-opening polymerization (ROP) of rac-LA preferentially consumes D-LA to form PDLA blocks, followed by simultaneous ROP of TMC and L-LA, yielding pentablock copolymers. Conversely, in the PA/PO/rac-LA system, alternating copolymerization of PA and PO precedes stereoselective ROP of rac-LA, generating pentablock architectures. Comprehensive characterization (NMR, SEC, in situ IR, CD spectroscopy) confirms the catalyst’s dual selectivity and adaptability. Notably, (S,S)-TUC operates under mild conditions, eliminates the need for multiple catalysts, and offers cost-effectiveness and low environmental toxicity. This work establishes a unified platform for synthesizing structurally complex copolymers, bridging the gap between precision polymerization and sustainable manufacturing. The methodology holds promise for applications in biodegradable materials, high-performance composites, and biomedical devices, where tailored polymer properties are critical.
Introduction
The precision synthesis of biodegradable polyesters with well-defined structures has emerged as a critical focus in the field of multicomponent polymerization.1–5 These materials are highly valued for their potential to address pressing environmental concerns while offering tunable properties for diverse applications, ranging from biomedical devices to sustainable packaging materials. Among biodegradable polyesters, polylactide (PLA) stands out as a particularly noteworthy candidate due to its excellent biodegradability, renewability, and versatility in structural design.6–8 Derived from lactide, a cyclic di-ester of lactic acid, PLA can be synthesized in various stereoisomeric forms, including isotactic, syndiotactic, and heterotactic configurations, depending on the choice of lactide enantiomers and the polymerization process employed.9 These stereochemical variations significantly influence the material’s properties, such as crystallinity, mechanical strength, and thermal behavior, making stereochemical control during polymerization an essential aspect of PLA synthesis.10The synthesis of advanced polyesters often involves ring-opening polymerization (ROP) or ring-opening copolymerization (ROCOP) of lactides, cyclic carbonates, epoxides, and anhydrides.11–16 These methods provide pathways to control polymer architectures, enabling the production of block copolymers with well-defined structures and tailored properties. Among these, chemoselective and stereoselective polymerization play pivotal roles in achieving precision synthesis.17–20 Chemoselective polymerization allows the selective incorporation of specific functional groups by controlling monomer reactivity, facilitating the construction of functional block copolymers. For instance, Li et al. demonstrated a self-switchable catalytic system utilizing t-BuP1 for the ring-opening polymerization of lactides, cyclic anhydrides, and epoxides. This system successfully achieved chemoselectivity.21 Similarly, Satoh and colleagues developed a dynamic catalytic system using alkali metal carboxylates to mediate sequential polymerization cycles, enabling the construction of multiblock copolymers.22 However, the hygroscopic nature of these metal salts introduced challenges, including the formation of byproducts and deviations in molecular weight from theoretical values. In contrast, stereoselective polymerization enables the controlled polymerization of stereochemically distinct monomers, producing polymers with specific stereochemical configurations. For example, Taton and Dove et al. developed a single-component thiourea-based Takemoto catalyst that incorporates both a thiourea group and a tertiary amino group.23–25 This catalyst operates through a bifunctional cooperative mechanism: the amino group activates the alcohol initiator, while the thiourea group activates the monomer, which achieves efficient control and high isoselectivity in the ring-opening polymerization (ROP) of rac-LA through kinetic resolution.26,27 Recently, the same group further refined this approach by combining the chiral Takemoto organocatalyst with t-BuP1 to establish a binary catalytic system. This novel system allows for rapid and stereoselective ROP of rac-LA at 25 °C, yielding highly isotactic PLA with a melting temperature (Tm) of 187 °C.28
Chemoselective and stereoselective polymerization strategies together provide unparalleled control over polymer structure and functionality.18–21,29–33 However, achieving both forms of selectivity simultaneously within a single catalytic system remains a significant challenge, despite considerable progress in optimizing these approaches independently. Existing methodologies often excel in one type of selectivity at the expense of the other. For instance, chemoselective polymerization allows for the precise incorporation of functional groups into block copolymers but falls short in controlling stereochemical configurations. Conversely, stereoselective polymerization excels in producing polymers with well-defined stereochemistry but lacks the capacity to discriminate between functional groups in multicomponent systems. This trade-off has hindered the development of structurally complex polymers that combine both functional versatility and stereochemical precision.
While Takemoto’s thiourea-based catalyst has been extensively studied for its outstanding stereoselective polymerization capabilities, especially in the synthesis of isotactic PLA,28 its potential for chemoselective polymerization has not been fully explored. This gap has hindered progress in designing multifunctional block copolymers with both chemical and stereochemical precision. In this work, we reveal, for the first time, that the chiral Takemoto catalyst (S,S)-TUC not only exhibits stereoselectivity in the polymerization of rac-LA but also demonstrates chemoselectivity in the ring-opening copolymerization (ROCOP) of lactides and other monomers, such as cyclic carbonates and cyclic anhydrides/epoxides. By investigating the polymerization kinetics and analyzing the resulting polymer structure, it is discovered that (S,S)-TUC can selectively initiate the polymerization of specific monomers while maintaining stereochemical control. These findings provide new insights into the dual selectivity of Takemoto catalysts, expanding their applicability in the synthesis of complex, well-defined block copolymers. This study underscores the potential of (S,S)-TUC to bridge the gap between chemoselective and stereoselective polymerization, paving the way for more sophisticated polymer design in the future.
Results and discussion
The ability of the (S,S)-TUC thiourea catalyst to achieve both chemoselective and stereoselective polymerization was evaluated through one-pot polymerization of rac-lactide (rac-LA) and trimethylene carbonate (TMC) at 40 °C. Using cyclohexanediol (CHD) as the chain transfer agent, the polymerization feed ratio was set to be [rac-LA]/[TMC]/[(S,S)-TUC]/[CHD] = 50/50/5/1 (Fig. 1A and Table S1†). | ||
| Fig. 1 (A) Scheme of the ring-opening copolymerization mechanism of rac-LA and TMC by using (S,S)-TUC as a catalyst. (B) The 1H NMR spectra of crude aliquots taken from the reaction system for monitoring the conversion of rac-LA and TMC and the formation of the block copolymer PTMC-b-P(LLA-ran-TMC)-b-PDLA-b-P(LLA-ran-TMC)-b-PTMC in CDCl3. (C) Time-dependent plots of monomer conversion rates, determined from 1H NMR spectra. (D) SEC traces at different reaction times. | ||
The polymerization process was found to occur in two distinct stages. Initially, 1H NMR spectroscopy revealed the disappearance of characteristic rac-LA peaks at 5.00–4.85 ppm (blue triangle in Fig. 1B), accompanied by the emergence of PLA-specific peaks at 5.32–4.96 ppm and 1.76–1.42 ppm. By 12 h, over 50% of rac-LA had been consumed. Importantly, no PTMC signals (4.32–4.16 ppm) were observed during this stage (red triangle in Fig. 1B), indicating that TMC polymerization had not yet commenced.
The second stage began with the simultaneous ring-opening polymerization (ROP) of TMC and the remaining ∼50% rac-LA, which was presumed to be predominantly L-LA due to the stereoselectivity of (S,S)-TUC.26 The L-LA monomer was fully consumed by ∼72 h, while TMC polymerization proceeded more gradually, reaching ∼90% conversion after ∼336 h (Fig. 1C). During this phase, a random copolymer block, P(LLA-ran-TMC), was most likely formed, as indicated by the conversion profiles in Fig. 1B and C.
Polymerization kinetics were analyzed to further confirm the sequential nature of the reaction. As shown in Fig. 2A, the polymerization rate during the first stage was higher (k1 = 0.06318) than in the second stage (k2 = 0.04437), with a rate ratio of approximately k1/k2 ≈ 1.42. The observed reduction in polymerization rate after 12 h aligns with the preferential consumption of D-LA during the initial stage.
 | ||
| Fig. 2 (A) The kinetic curves for the ring-opening copolymerization of rac-LA and TMC. Reaction conditions: CHD/(S,S)-TUC/rac-LA/TMC = 1/5/50/50, at 40 °C, in toluene. (B) Time resolved in situ IR spectra during the polymerization of mixtures of rac-LA and TMC. Reaction conditions: CHD/(S,S)-TUC/rac-LA/TMC = 1/5/50/50, 40 °C, in toluene. | ||
In situ infrared (IR) spectroscopy further confirmed the polymerization process (Fig. 2B, S1 and S2†). During the first stage, the PLA-specific C–O stretching peak at 1086 cm−1 increases significantly. However, tracking monomer consumption (rac-LA and TMC) proved challenging due to overlapping absorption bands with polymeric products. The transition to the second stage was marked by the appearance of a new peak at 1313 cm−1, characteristic of PTMC. These observations suggest that the second stage likely involves TMC copolymerization with residual L-LA, forming P(LLA-ran-TMC).
To this point, the preferential polymerization of D-LA over L-LA by (S,S)-TUC has been inferred based on previous literature and the observed polymerization kinetics. While the kinetic curves and in situ IR data provided indirect evidence of the sequential consumption of D-LA and L-LA during the copolymerization of rac-LA and TMC (Fig. S3†), direct confirmation of this stereoselective behavior was necessary. To validate these findings and unequivocally demonstrate the stereoselective preference of (S,S)-TUC, circular dichroism (CD) spectroscopy was employed.
As shown in Fig. 3A, the CD spectra of D-LA and L-LA exhibited opposing signals at 220 nm, reflecting their differential absorption of circularly polarized light. During the copolymerization of rac-LA and TMC (Fig. 3B), the spectra evolved over time, indicating preferential consumption of D-LA in the early stages of the reaction. As polymerization progressed, the characteristic absorption associated with L-LA became dominant, corroborating the stereoselective preference of (S,S)-TUC. Moreover, the stereochemical regularity of the PLA block was assessed through homonuclear decoupled 1H NMR analyses. The calculated Pm = 0.82 confirmed a high degree of stereoregularity, while the homonuclear decoupling spectra revealed a prominent mmm tetrad peak (Fig. S4†), further confirming the isotactic nature of the PLA segment.
 | ||
| Fig. 3 Circular dichroism (CD) spectroscopy results. (A) D-LA and L-LA monomers; (B) crude aliquots collected at different time points during the copolymerization of rac-LA and TMC. Reaction conditions: [CHD]/[(S,S)-TUC]/[rac-LA]/[TMC] = 1/5/100/100, 40 °C, in toluene. | ||
Size exclusion chromatography (SEC) provided additional insights into the molecular weight distribution of the resultant polymers (Fig. 1D). SEC traces retained a single-peak distribution throughout the reaction, indicating the successful formation of a well-defined block copolymer architecture. During the first stage of polymerization, corresponding to the selective ROP of D-LA, the molecular weight distribution was narrow (Đ < 1.10). As the reaction progressed into the second stage, the molecular weight distribution slightly broadened, likely due to transesterification side reactions.
These findings confirm that the copolymerization process occurs in distinct stages. Initially, (S,S)-TUC selectively polymerizes the D-LA enantiomer, forming a PDLA block. Subsequently, the simultaneous ROP of L-LA and TMC takes place, resulting in the formation of the P(LLA-ran-TMC) block. In the final stage, the ROP of TMC continues, producing a PTMC block. This sequential polymerization ultimately yields a well-defined pentablock copolymer with the structure PTMC-b-P(LLA-ran-TMC)-b-PDLA-b-P(LLA-ran-TMC)-b-PTMC. These results observed in the rac-LA/TMC system suggests that the selectivity exhibited by (S,S)-TUC is primarily governed by its enantioselective preference for D-LA. Given the well-documented kinetic resolution mechanism of Takemoto-type catalysts, it is reasonable to propose that (S,S)-TUC first catalyzes the rapid polymerization of D-LA due to stronger hydrogen bonding interactions and preferential activation. Once D-LA is depleted, the system transitions to a nonselective copolymerization of L-LA and TMC. This polymerization pathway provides a new perspective on sequence-controlled copolymerization and highlights the potential of chiral thiourea catalysts in achieving tailored microstructures in polyesters.
To confirm the structure of the synthesized pentablock copolymer, the precipitated polymer was analyzed using 1H NMR and 13C NMR spectroscopy (Fig. S5–S7†). As shown in Fig. S5,† characteristic signals corresponding to the methylene protons of PLA were observed in the range of 5.3–5.0 ppm, while the PTMC block exhibited distinct signals at 4.32–4.16 ppm. These observations confirm the presence of both PLA and PTMC blocks within the copolymer. The 13C NMR spectra further validated the copolymer structure by the consistent absorption signals with PLA and PTMC homopolymers (Fig. S6 and S7†). In addition, diffusion-ordered spectroscopy (DOSY) analysis provided insights into the connectivity of the copolymer blocks.34 The results showed that the PTMC-b-P(LLA-ran-TMC)-b-PDLA-b-P(LLA-ran-TMC)-b-PTMC copolymer exhibited a single diffusion coefficient (Fig. 4A), confirming that the blocks are covalently linked rather than existing as a physical blend. In contrast, when a physical blend of PLA and PTMC homopolymers with similar molecular weights was analyzed, two distinct diffusion coefficients were observed (Fig. 4B). These findings unequivocally demonstrate the successful synthesis of the pentablock copolymer PTMC-b-P(LLA-ran-TMC)-b-PDLA-b-P(LLA-ran-TMC)-b-PTMC, confirming that it is a single macromolecular entity rather than a mere mixture of homopolymers.
 | ||
| Fig. 4 1H NMR DOSY spectra of (A) PTMC-b-P(LLA-ran-TMC)-b-PDLA-b-P(LLA-ran-TMC)-b-PTMC and (B) a blend of PLA and PTMC homopolymer, in CDCl3. | ||
Previous studies have highlighted the slow polymerization kinetics of TMC, thus prompting an investigation into the effect of elevated temperature on the reaction rate. One-pot solution polymerization of rac-LA and TMC using the (S,S)-TUC catalyst and CHD as the chain transfer agent were conducted at 80 °C with a feed ratio of [rac-LA]/[TMC]/[(S,S)-TUC]/[CHD] = 50/50/5/1. 1H NMR and SEC analyses (Table S2 and Fig. S8†) revealed that increasing the reaction temperature significantly accelerated TMC polymerization, with a rate constant k = 0.01032 (Fig. S9†). At 80 °C, TMC conversion reached approximately 90% within 8 days, whereas the same level of conversion required ∼13 days at 40 °C. However, the molecular weight distribution became broader than that observed in the reaction system at 40 °C. This change could be attributed to the reactions being conducted at a higher temperature, which promotes both inter- and intramolecular transesterification reactions.
To evaluate the stereoselectivity of (S,S)-TUC at 80 °C, 1H NMR and 13C NMR analyses were performed. The stereoregularity of the PDLA blocks was calculated from homonuclear decoupled 1H NMR spectra after deconvolution (Pm = 0.78, Fig. S10†). This indicates that higher temperatures are disadvantageous to the formation of highly isotactic PDLA segments. These results suggest that while stereoselectivity is maintained at elevated temperatures, the tacticity of the resulting polymer blocks is compromised. This could be attributed to previous studies indicating that side transesterification reactions typically occur at temperatures above 85 °C, especially when rac-LA conversion exceeds 90%.26
To address these limitations, a temperature-switching polymerization strategy was implemented. In the first stage, stereoselective polymerization of rac-LA was carried out at 40 °C to preserve the high tacticity of the PLA blocks and avoid unnecessary transesterification reactions. Upon depletion of the rac-LA monomer, the temperature was increased to 80 °C to accelerate the polymerization of TMC. This approach was validated by experimental results (Table S3 and Fig. S11†), which confirmed that TMC polymerization was significantly faster at the elevated temperature (Fig. S12 and S18†).
Furthermore, a slightly higher Pm value (0.79, Fig. S13†) was achieved compared to polymers synthesized at 80 °C. Thermal and mechanical properties of pentablock copolymers synthesized under different temperature conditions were analyzed (entries 8–10 in Table 1 and Fig. S14†). The absence of a clear Tm of PLA in the DSC thermograms is attributed to the presence of the atactic P(LLA-ran-TMC) block, which likely inhibits the crystallization of the isotactic PDLA segments, while a distinct melting peak can be observed in the polymer obtained from the homopolymerization of rac-LA catalyzed by the (S,S)-TUC (Fig. S15†). Variations in the glass transition temperature (Tg) were found to correlate with changes in polymer tacticity. The Pm parameter indicates the stereoregularity of PLA chains, with higher values reflecting more ordered conformations, which results in a higher Tg that increases from 11.9 °C to 25.7 °C.35,36
| Entry | [rac-LA]/[TMC]/[(S,S)-TUC]/[CHD] | T (°C) | t (h) | Conv.rac-LAb (%) | Conv.TMCb (%) | Mn,theoc (kg mol−1) | Mn,SECd (Đ) (kg mol−1) |
|---|---|---|---|---|---|---|---|
| a Polymerization conditions: in toluene under a N2 atmosphere.b Determined by 1H NMR analysis of the obtained polymer in CDCl3.c Determined on the basis of (MWrac-LA) × (Conv.% of rac-LA)/[CHD] + (MWTMC) × (Conv.% of TMC)/[CHD].d Determined by SEC analysis of the obtained polymer in THF using polystyrene calibration.e Increase the temperature to 80 °C once the rac-LA conversion rate exceeds 99%. | |||||||
| 1 | 50/50/5/1 | 40 | 12 | 47.8 | 0 | 3.5 | 7.4 (1.11) |
| 2 | 50/50/5/1 | 40 | 24 | 76.8 | 10.6 | 6.0 | 9.3 (1.11) |
| 3 | 50/50/5/1 | 40 | 288 | 99 | 93.2 | 11.8 | 12.0 (1.29) |
| 4 | 50/50/5/1 | 40/80e | 12 | 58.2 | 0 | 4.2 | 4.6 (1.10) |
| 5 | 50/50/5/1 | 40/80e | 288 | 99 | 94.3 | 11.9 | 10.1 (1.33) |
| 6 | 50/50/5/1 | 80 | 8 | 56.1 | 4.0 | 4.2 | 6.4 (1.10) |
| 7 | 50/50/5/1 | 80 | 288 | 99 | 93.2 | 11.8 | 11.6 (1.49) |
| 8 | 100/100/5/1 | 40 | 336 | 99 | 65.6 | 21.0 | 14.4 (1.31) |
| 9 | 100/100/5/1 | 40/80e | 336 | 99 | 77.1 | 22.2 | 18.4 (1.38) |
| 10 | 100/100/5/1 | 80 | 336 | 99 | 85.6 | 23.0 | 12.7 (1.41) |
| 11 | 50/50/5/1 + 50 [rac-LA] | 80 | 336 + 72 | 99 | 90.1 | 18.2 | 13.4 (1.27) |
While increasing the reaction temperature can enhance the polymerization rate, the results demonstrate that this approach compromises enantioselectivity and promotes side transesterification reactions, particularly at high monomer conversions. This trade-off underscores the challenge of balancing polymerization efficiency with stereocontrol. To further optimize this approach, future investigations will focus on incorporating co-catalysts to enhance the polymerization of TMC while preserving stereoselectivity. Fine-tuning catalyst systems and reaction conditions is expected to improve both the polymerization efficiency and structural precision in multiblock polyester synthesis.
The SEC results demonstrate that the number-average molecular weight (Mn) of the polymers synthesized using this method can be precisely controlled by adjusting the monomer conversion rate and the molar ratio between the monomers and the chain transfer agent (Tables S1–S3†). Importantly, the theoretical Mn values closely correspond to those measured by SEC. To further verify that this polymerization system exhibits characteristics of living polymerization, a sequential monomer addition experiment was conducted. After 14 days of reaction (entry 11, Table 1), by which time TMC conversion had reached >90%, an additional 50 equiv. of rac-LA monomer was introduced into the reaction solution. During this second stage of the copolymerization, 1H NMR, SEC and DOSY analyses confirmed the reinitiation of ROP (entry 11, Table 1, Fig. S16 and S17†). This sequential addition led to the successful synthesis of ACBABCA-type heptablock copolyesters, demonstrating that block copolymers with complex architectures can be produced simply by introducing monomers in a controlled sequence.
These findings provide robust evidence that the (S,S)-TUC thiourea catalyst exhibits dual-selective capabilities, enabling both chiral-selective polymerization of rac-LA and chemoselective polymerization of TMC. The versatility of the dual selective polymerization catalyzed by the monocomponent thiourea-based (S,S)-TUC catalyst was evaluated using alternating copolymerization of anhydrides with epoxide monomers (Scheme 1). Specifically, the copolymerization of propylene oxide (PO) and phthalic anhydride (PA) was investigated in the presence of two optical isomers, D-LA and L-LA, respectively, to preliminarily assess the chemical selectivity of (S,S)-TUC. Characterization using 1H NMR, 13C NMR, SEC analyses and in situ IR confirmed that all copolymerization reactions exhibited excellent controllability and selectivity, with well-defined polymer architectures being achieved.
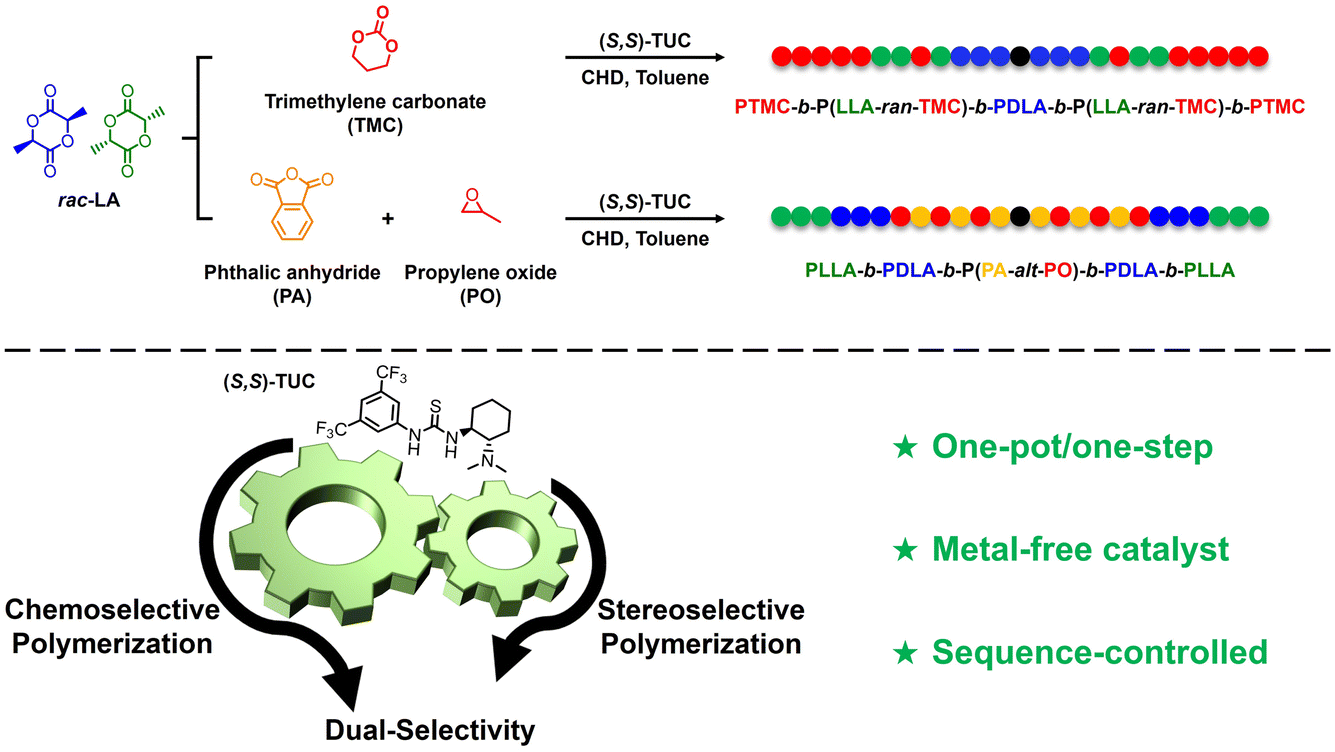 | ||
| Scheme 1 Chemoselective and stereoselective ring-opening polymerization of mixed epoxides, cyclic anhydrides, cyclic carbonates and cyclic esters using (S,S)-TUC as the catalyst. | ||
For the PA/PO/D-LA terpolymerization system, 1H NMR analysis revealed that the reaction also proceeds in two distinct stages. The initial stage involved the alternating copolymerization of PA and PO, resulting in the formation of the P(PA-alt-PO) block. This was followed by the ROP of D-LA to generate the next block (Table S4 and Fig. S19†). Similarly, the PA/PO/L-LA terpolymerization system exhibited analogous behavior. The alternating copolymerization of PA and PO occurred first, forming P(PA-alt-PO) block segments, followed by the ROP of L-LA (Table S5 and Fig. S20†). These results confirm that the (S,S)-TUC catalyst effectively affords chemoselective polymerization processes, maintaining precise control over the sequence of monomer incorporation. Additionally, in situ IR monitoring was also conducted for the D-LA/PA/PO and L-LA/PA/PO systems, which similarly exhibited two distinct polymerization stages (Fig. S22†). These results further validate the chemoselectivity of (S,S)-TUC, demonstrating its ability to catalyze complex polymerization sequences with both chemical and stereochemical precision.
The ability of the (S,S)-TUC catalyst to achieve both chemoselectivity and stereoselectivity was further investigated in the copolymerization system comprising propylene oxide (PO), phthalic anhydride (PA), and rac-lactide (rac-LA). Monitoring via 1H NMR revealed that no characteristic signals corresponding to PLA (5.30–5.00 ppm) were detected prior to 96 h, during which the conversion rate of PA reached approximately 85%. The polymerization of rac-LA began only in the second stage, around 360 h, when PA and PO had been fully consumed (Table S6 and Fig. S21†). The SEC analysis of the copolymers showed a discrepancy between the theoretical molecular weight (Mn,theo) and the molecular weight determined by SEC (Mn,sec). This difference can be attributed to the fact that Mn,sec represents the relative molecular weight measured against a polystyrene standard and alternating polyesters usually exhibit lower Mn,sec than their true values.37–40
In situ IR spectroscopy provided further evidence supporting the selective polymerization process. The copolymerization of rac-LA, PA, and PO exhibited two distinct stages. During the first stage, alternating copolymerization of PA and PO occurred, as indicated by a decrease in the intensity of the PA absorption peak at 895 cm−1. Simultaneously, the absorption peak for P(PA-alt-PO) at 1721 cm−1 increased significantly (Fig. 5). The second stage of polymerization commenced once the anhydride was fully consumed (∼64 h), as evidenced by the attenuation of the rac-LA absorption peak at 1241 cm−1. The continued increase of the absorption peak of P(PA-alt-PO) during the second stage is most likely due to its overlap with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching characteristic band of PLA at 1750 cm−1.41,42 These findings confirm the sequential nature of the polymerization process, with (S,S)-TUC first catalyzing the chemoselective alternating copolymerization of PA and PO, followed by the stereoselective ring-opening polymerization (ROP) of rac-LA.
O stretching characteristic band of PLA at 1750 cm−1.41,42 These findings confirm the sequential nature of the polymerization process, with (S,S)-TUC first catalyzing the chemoselective alternating copolymerization of PA and PO, followed by the stereoselective ring-opening polymerization (ROP) of rac-LA.
 | ||
| Fig. 5 Time-resolved in situ IR spectra during the polymerization of mixtures of PA, PO, and rac-LA. Reaction conditions: CHD/(S,S)-TUC/PA/PO/rac-LA = 1/5/100/200/200, 80 °C, and toluene as the solvent. | ||
To further confirm the stereochemical regularity of the resulting copolymer, the stereoregularity parameter Pm was determined through homonuclear decoupled 1H NMR spectra after deconvolution. The calculated Pm = 0.75 confirmed the relatively good stereochemical regularity (Fig. S23†). Besides, DSC analysis of the copolymer sample revealed a melting transition at Tm = 183 °C (Fig. S24†). These results suggest that during the second stage, PDLA-b-PLLA formed as the second and third blocks. These results underscore the strong stereoselectivity of the (S,S)-TUC catalyst for rac-LA monomers during the second stage of the copolymerization process.
It is noteworthy that the dual selective behavior of (S,S)-TUC in the PA/PO/rac-LA system contrasts sharply with its mechanism in the rac-LA/TMC copolymerization system. In the PA/PO/rac-LA system, (S,S)-TUC chemoselectively catalyzes the alternating copolymerization of anhydride and epoxide monomers (PA and PO) first, followed by the stereoselective polymerization of rac-LA monomers. The structure of the obtained pentablock copolyester, PLLA-b-PDLA-b-P(PA-alt-PO)-b-PDLA-b-PLLA, was confirmed by 1H NMR and 13C NMR analyses (Fig. S25 and S26†). These results revealed the successful formation of the copolymer, with distinct signals corresponding to each block. Additionally, DOSY analysis showed a single diffusion coefficient (Fig. S27†), providing conclusive evidence that the blocks are covalently linked and not a simple physical blend.
Conclusion
In summary, this study highlights an advancement in the field of selective and precise polymerization by demonstrating the dual chemoselective and stereoselective capabilities of the commercially available chiral thiourea catalyst (S,S)-TUC. Unlike previous approaches that focused on either chemical or stereochemical selectivity in isolation, our work establishes (S,S)-TUC as a versatile and efficient catalyst capable of achieving both selectivities within a single catalytic system. This dual functionality was systematically validated through comprehensive analyses, including NMR, SEC, in situ IR, and CD spectroscopy.The results demonstrate the catalyst’s adaptability across different polymerization systems. In the TMC/rac-LA system, stereoselective polymerization of rac-LA occurs first, with D-LA being preferentially consumed to form PDLA blocks, followed by the simultaneous ring-opening polymerization of TMC and L-LA to yield the pentablock copolymer PTMC-b-P(LLA-ran-TMC)-b-PDLA-b-P(LLA-ran-TMC)-b-PTMC. In contrast, the PA/PO/rac-LA system exhibits an inverse sequence, where the alternating copolymerization of PO and PA occurs first to form aromatic polyester P(PA-alt-PO) blocks, followed by the stereoselective ROP of rac-LA to produce PLLA-b-PDLA-b-P(PA-alt-PO)-b-PDLA-b-PLLA. These distinct pathways highlight the ability of (S,S)-TUC to adapt its selectivity based on the monomer feed composition, providing a robust platform for the synthesis of complex and well-defined block copolymers. By leveraging the dual selectivity of (S,S)-TUC, a unified strategy for the precise synthesis of multifunctional, multi-block copolymers without the need for additional catalysts or complex reaction conditions has been established. Moreover, as a commercially available catalyst, (S,S)-TUC offers notable advantages, including low cost and reduced environmental toxicity, making it a more sustainable and scalable option for industrial and biomedical applications. This approach paves the way for the design and development of advanced polymeric materials with tailored structures and properties, addressing critical challenges in areas such as biodegradable polymers, functional coatings, and high-performance composites.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Liu, Jiang and Zhu conceived the idea, designed the experiments and analyzed the data collaboratively. Liang and Wang participated in polymerization test and DOSY NMR analysis. Qiang and Zhang carried out the experiments on polymer synthesis. All authors contributed to the scientific discussion and proofread the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (22175131), the National Key R&D Program of China (2022YFC2402901-4), Interdisciplinary Collaborative Research Project (2023-2-YB-03) and Tongcheng Youth Research and Development Fund (CPCIF-RA-0104).References
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2017, 9, 171–178 CrossRef CAS.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- G. Polymeropoulos, G. Zapsas, K. Ntetsikas, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2017, 50, 1253–1290 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- J. Gao, D. Zhu, W. Zhang, G. A. Solan, Y. Ma and W.-H. Sun, Inorg. Chem. Front., 2019, 6, 2619–2652 RSC.
- C. N. Jadrich, V. E. Pane, B. Lin, G. O. Jones, J. L. Hedrick, N. H. Park and R. M. Waymouth, J. Am. Chem. Soc., 2022, 144, 8439–8443 CrossRef CAS PubMed.
- B. Lin and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 1645–1652 CrossRef CAS PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- N. Nomura, J. Hasegawa and R. Ishii, Macromolecules, 2009, 42, 4907–4909 CrossRef CAS.
- H.-Y. Ji, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2018, 20, 641–648 RSC.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- J. Li, Y. Liu, W.-M. Ren and X.-B. Lu, J. Am. Chem. Soc., 2016, 138, 11493–11496 CrossRef PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC.
- S. Paul, C. Romain, J. Shaw and C. K. Williams, Macromolecules, 2015, 48, 6047–6056 CrossRef CAS.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 CrossRef CAS.
- H. Y. Ji, B. Wang, L. Pan and Y. S. Li, Angew. Chem., Int. Ed., 2018, 57, 16888–16892 CrossRef CAS.
- X. Xia, R. Suzuki, T. Gao, T. Isono and T. Satoh, Nat. Commun., 2022, 13, 163 CrossRef PubMed.
- Y. Hoashi, T. Okino and Y. Takemoto, Angew. Chem., Int. Ed., 2005, 44, 4032–4035 CrossRef PubMed.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef PubMed.
- T. Okino, S. Nakamura, T. Furukawa and Y. Takemoto, Org. Lett., 2004, 6, 625–627 CrossRef CAS PubMed.
- B. Orhan, M. J. L. Tschan, A.-L. Wirotius, A. P. Dove, O. Coulembier and D. Taton, ACS Macro Lett., 2018, 7, 1413–1419 CrossRef PubMed.
- J. Q. Shi, H. Y. Yin, Q. M. Sun, R. X. Teng, Y. Q. Zhu and J. Z. Du, Chem. Mater., 2024, 36, 5422–5435 CrossRef.
- M. S. Zaky, A.-L. Wirotius, O. Coulembier, G. Guichard and D. Taton, Chem. Commun., 2021, 57, 3777–3780 RSC.
- J. Chen and E. Y. X. Chen, Israel J. Chem., 2015, 55, 216–225 CrossRef.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef PubMed.
- C. Li, R. J. Sablong and C. E. Koning, Angew. Chem., Int. Ed., 2016, 55, 11572–11576 CrossRef PubMed.
- M. L. McGraw, R. W. Clarke and E. Y. X. Chen, J. Am. Chem. Soc., 2020, 142, 5969–5973 CrossRef PubMed.
- L. You and J. Ling, Macromolecules, 2014, 47, 2219–2225 CrossRef.
- P. Groves, Polym. Chem., 2017, 8, 6700–6708 RSC.
- A. Mi, Z. Li, M. Wu, W. Duan, C. Qiao, J. Yao and Q. Liu, J. Appl. Polym. Sci., 2025, 142, e56493 CrossRef.
- L. Xu, L. Yang, Z. Guo, N. Liu, Y.-Y. Zhu, Z. Li and Z.-Q. Wu, Macromolecules, 2019, 52, 5698–5706 CrossRef.
- B. Dong, G. Xu, R. Yang, X. Guo and Q. Wang, Macromolecules, 2023, 56, 10143–10152 CrossRef.
- V. T. Lipik, L. K. Widjaja, S. S. Liow, M. J. M. Abadie and S. S. Venkatraman, Polym. Degrad. Stab., 2010, 95, 2596–2602 CrossRef.
- N. Zhao, C. Ren, Y. Shen, S. Liu and Z. Li, Macromolecules, 2019, 52, 1083–1091 CrossRef.
- Y. Zhu, C. Romain, V. Poirier and C. K. Williams, Macromolecules, 2015, 48, 2407–2416 CrossRef.
- M. Socka, A. Duda, A. Adamus, R. A. Wach and P. Ulanski, Polymer, 2016, 87, 50–63 CrossRef.
- S. Zhu, Y. Wang, W. Ding, X. Zhou, Y. Liao and X. Xie, Polym. Chem., 2020, 11, 1691–1695 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and polymer characterization data. See DOI: https://doi.org/10.1039/d5fd00039d |
| This journal is © The Royal Society of Chemistry 2025 |