 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ measurements of gas–particle partitioning of organic compounds in Fairbanks†
Amna
Ijaz‡
 a,
Brice
Temime-Roussel
a,
Julien
Kammer
a,
Jingqiu
Mao
b,
William
Simpson
b,
Kathy S.
Law
c and
Barbara
D’Anna
a
a,
Brice
Temime-Roussel
a,
Julien
Kammer
a,
Jingqiu
Mao
b,
William
Simpson
b,
Kathy S.
Law
c and
Barbara
D’Anna
a
aAix-Marseille Université, CNRS, LCE, Marseille, France
bDepartment of Chemistry and Biochemistry, Geophysical Institute, University of Alaska, Fairbanks, AK, USA
cSorbonne Université, UVSQ, CNRS, LATMOS-IPSL, Paris, France
First published on 6th December 2024
Abstract
Organic compounds were measured in both the gas and particle phases in Fairbanks, Alaska, using a real-time, high-resolution proton transfer reaction-time of flight mass spectrometer (PTR-ToF MS) during a wintertime campaign. The organic aerosol (OA) was dominated by semi-volatile organic compounds (SVOCs), followed by compounds in the low-volatile bin (LVOCs). Due to the persistently cold conditions, both heavy and highly oxygenated compounds showed a limited shift in partitioning with temperature change. In contrast, some semi-volatile compounds, such as methoxy phenols from wood combustion, presented some partitioning to the particle phase at lower temperatures. Laboratory studies or theoretical efforts rarely explore gas–particle partitioning at extremely low temperatures, and thus, their applicability under complex meteorological conditions remains to be assessed. A comparison of the observed and estimated volatilities at temperatures from 5 to −33 °C revealed a clear disagreement, with higher estimated volatility for light molecules (m/z below 120) and lower volatilities for heavier compounds (m/z above 300) with respect to the observed ones. Our findings from the Fairbanks winter campaign stress the need to extend the breadth of environmentally relevant conditions under which phase partitioning of organic compounds is generally explored.
1. Introduction
Organic compounds are ubiquitously present in the troposphere, where they influence the climate and air quality, as well as human and ecosystem health. They associate with either the gas or particulate phases depending on their volatility, which is measured from their vapour pressure or saturation mass concentrations.1–3 Therefore, the volatility of organic compounds is a key physicochemical property that determines the mass concentrations, chemical composition, growth, and fate of atmospheric aerosol by controlling their partitioning between the two phases.4–6 The ambient air consists of thousands of molecular species with wide-ranging volatilities, reflected in saturation mass concentrations of less than 3 × 10−4 μg m−3 for the least volatile to greater than 3 × 106 μg m−3 for the most volatile compounds.7,8 The entire volatility spectrum has been divided into the volatility basis set (VBS), whereby organic compounds are categorised into bins separated by a factor of 10.5,9 The VBS provides a simplified outlook on both measurements10–12 and theoretical estimations.13,14Volatility of organic compounds can be quantified using multiple theoretical or experimental approaches or a combination of both. For instance, advanced high-resolution mass spectrometry measurements help delineate the molecular formula of individual ions in complex aerosol mixtures that can then be used to estimate the saturation mass concentrations of individual ions using the elemental composition, i.e., the number of certain atoms (C, H, O, N, S),7,8,15,16 or by estimating the vapour pressure by accounting for the contributions of different functional groups on a molecule.17,18 Good agreement has been observed between the elemental composition-based parameterisations by Li et al.8 and the chemical functional group-contribution method, SIMPOL,17 for estimated volatility of organic compounds.8,19–21 In other studies, much larger uncertainties were associated with these parameterisations of volatility due to a lack of detailed structural information in the elemental composition-based methods, as compared to SIMPOL-driven estimations.19,22 However, the accuracy of these theoretical methods in field settings remains unclear due to insufficient testing using in situ data. Testing these theoretical approaches under diverse environmental conditions can be useful and provide comprehensive information to better constrain volatility estimations and reduce uncertainties. In addition, the gas–particle partitioning of organics is well-established to be sensitive to concentrations of organic aerosol,2 sulphate,23 relative humidity,24 and temperature,9,25–27 but the partitioning at sub-zero temperatures is still unknown.
Experimentally, volatility has been determined by studying volumetric changes in aerosol over time, but this can be done feasibly in chamber studies only.28,29 In fact, a major hurdle in studying gas–particle partitioning in field settings is the real-time measurement of organic compounds across a wide volatility range, while ensuring their molecular integrity. Recently, combination of thermal evaporation or desorption of aerosol with a variety of ionisation and mass spectrometric setups for detection has been used. A popular example is the thermal desorption of an aerosol sample by a filter inlet for gases and aerosols (FIGAERO) and subsequent detection of ions by a chemical ionisation mass spectrometer (CIMS)22,30–32 for characterisation of the volatility of organic compounds using the gas/particle partitioning theory.1,9 However, the thermal decomposition of labile compounds during desorption is associated with significant bias in the volatility estimation from direct gas and particle-phase measurements.22,33 Studies using FIGAERO-CIMS do report a volatility range from 0.1 to 100 μg m−3 for effective saturation mass concentrations (C*).34,35
An alternative sampling and evaporation method is the chemical analysis of aerosol online (CHARON) inlet, which also uses thermal desorption, coupled with proton-transfer-reaction time-of-flight mass spectrometry (PTR-ToF-MS).36,37 The PTR-ToF-MS ensures minimal fragmentation that can potentially be corrected38 and provides quantitative molecular information. It also allows measurement of both gas and particle phases with fewer artefacts associated with particle collection and thermal desorption compared to traditional techniques using thermal desorption.39 It has given promising outcomes from real-time information on chemical composition in several laboratory, ground-based and aircraft studies37,40–42 although its application in gas–particle partitioning investigations is relatively new.39,42,43
We deployed a CHARON PTR-ToF MS in Fairbanks (64.84064°N, 147.72677°W; 136 m above sea level), Alaska, during the wintertime to alternatively measure gas and particle-phase organic compounds. Organics accounted for 66% of the total non-refractory submicron aerosol (NR-PM1) in Fairbanks,44 creating a unique opportunity to investigate several under-explored aspects of scientific interest: (i) the gas–particle partitioning of organic compounds spanning a wide range of oxidation states and volatilities in Fairbanks, (ii) the influence of extreme meteorological conditions, particularly the effects of ambient temperatures as low as −33 °C on the partitioning behaviour and (iii) the comparison with predicted partitioning metrics from absorptive partitioning instantaneous equilibrium theory.
2. Experimental
2.1. Field campaign and sampling site
The Alaskan Layered Pollution and Chemical Analysis (ALPACA) field experiment is a collaborative study designed to improve the understanding of pollution sources and chemical processes during the winter under cold conditions. Measurements were collected at a site in downtown Fairbanks, located at the Community and Technical College (CTC) of the University of Alaska, Fairbanks (64.84064°N, 147.72677°W; 136 m above sea level) from January 20 to February 26, 2022. This site is in an area that can be classified as an urban locality due to its proximity to major commercial and residential activities. A detailed description of the site and the overall goals and objectives of the ALPACA campaign are provided in Simpson et al.45 An overview of the meteorology and atmospheric aerosol during the sampling period is presented in Fig. S1.† The air temperature ranged from a minimum of −33 °C to a maximum of 5 °C during the field study period. Wind speed was generally low and ranged from 0 to 12 m s−1, but the average wind speed was 1.2 m s−1.2.2. Instrumentation and data collection
A trailer equipped with a suite of instruments acquiring high-temporal-resolution data (i.e., 10 seconds to 2 minutes) was deployed at the CTC parking lot.45 As detailed in our recent study,44 major equipment referred to in this study included two online mass spectrometers: a proton transfer reaction-time of flight mass spectrometer (PTR-ToF-MS 6000 X2, Ionicon Analytik GmbH, Austria) coupled with a CHARON inlet (PTRCHARON that detects particles of 150–1000 nm), and a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF AMS, Aerodyne or simply, AMS, which detects particles of 70–1000 nm).46 Both mass spectrometers were connected to the same inlet placed 3.5 meters above the ground level to sample air through a short stainless tube of ≈1 m with a 1/2′′ outer diameter. The instrumental background was measured regularly twice a week by placing a HEPA filter upstream of the inlet. Atmospheric temperatures at 3 and 23 m,47 the aerosol liquid water content, and pH (ref. 48) were obtained from previous studies associated with the ALPACA campaign.In total, 324 and 336 ions were selected above the S/N for gas- and particle-phase measurements with the PTRCHARON. Reference spectra of several common biomass-burning tracers (e.g., levoglucosan, vanillic acid, acetosyringone, and coniferylaldehyde) were investigated in the laboratory to assess their fragmentation patterns. For those compounds, the total mass was retrieved by summing the intensity of all identified fragments and attributed to the same parent ion. For example, in the case of levoglucosan, fragments at m/z 85, 127, and 145, were well-correlated with the parent ion at m/z 163; their signals were thus assigned to the parent compound. Additionally, to ensure credible information on partitioning, hourly signal counts below 3% of the campaign-average signal were eliminated. Further data processing to correct instrumental artefacts is summarised in Section S1.†
2.3. Observed phase partitioning and volatility estimation
The observed particle phase fraction (Fp,i) of a given species, i, was calculated as the ratio of its signal in the particle phase to the sum of the signal in both gas and particle phases (eqn (1)). It was then used to estimate the corresponding effective saturation mass concentration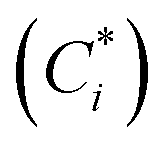 at temperatures measured in Fairbanks (240–278 K) via the absorption equilibrium partitioning expressed as eqn (1):2
at temperatures measured in Fairbanks (240–278 K) via the absorption equilibrium partitioning expressed as eqn (1):2 | (1) |
 and C0i, an activity coefficient (γi) of 1 was assumed, considering that
and C0i, an activity coefficient (γi) of 1 was assumed, considering that  . Here, γi represents the non-ideal behaviour of a compound in an aerosol mixture and generally lies between 0.3 (readily partitions to the particle phase) and 3 (readily partitions to the gas phase) for ambient atmospheric aerosol.2 We used this method to estimate C0, mainly because it is commonly used to describe the gas–particle partitioning of organic compounds.
. Here, γi represents the non-ideal behaviour of a compound in an aerosol mixture and generally lies between 0.3 (readily partitions to the particle phase) and 3 (readily partitions to the gas phase) for ambient atmospheric aerosol.2 We used this method to estimate C0, mainly because it is commonly used to describe the gas–particle partitioning of organic compounds.
Since ions present in different concentrations have varying impacts on the overall partitioning behaviour of OA, hourly time series of observed and estimated parameters are presented as weighted averages (subscript of ‘wa’, such as those in Fig. 2). These were calculated using all ions i = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n as expressed in eqn (2), where Y is the parameter, n is the number of ions that were overlapped in the gas and particle phases, and I is the campaign-averaged ion concentration normalised to the maximum concentration.
:n as expressed in eqn (2), where Y is the parameter, n is the number of ions that were overlapped in the gas and particle phases, and I is the campaign-averaged ion concentration normalised to the maximum concentration.
 | (2) |
3. Results and discussion
3.1. Molecular properties of the detected organic compounds
Fig. 1 shows a general overview of the composition of organic compounds in terms of the number of C and O atoms in the species detected in Fairbanks during winter 2022. A total of 195 ions were commonly present in the gas and particle phases above the noise-filtering threshold. They are the focus of this study to investigate partitioning behaviours; these ions contributed 46 ± 7 and 82 ± 16% of the total mass in the gas and particle phases, respectively. The mass concentrations of these ions ranged from 2.2 to 111 μg m−3 in the gas phase, and below 1 to 21 μg m−3 in the particle phase.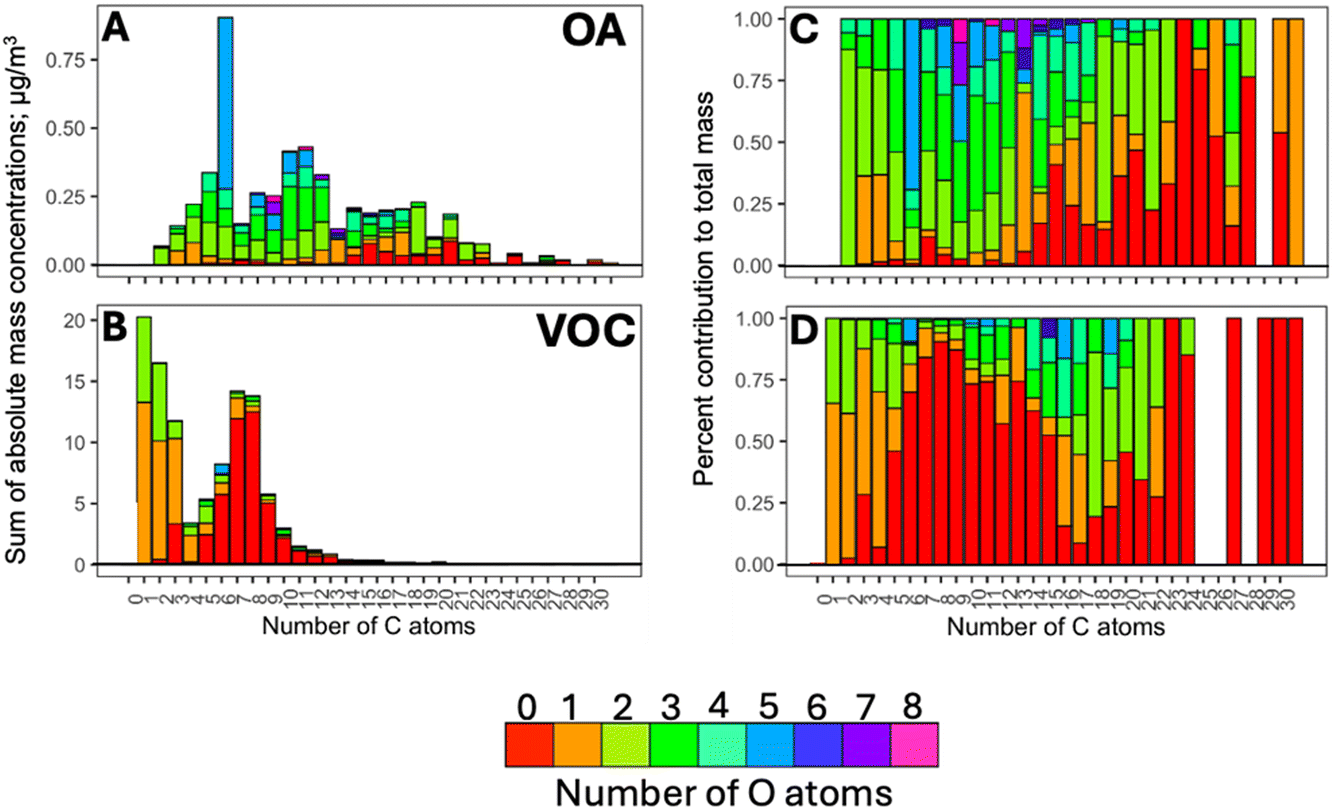 | ||
| Fig. 1 Overview of the mass distribution across the different chemical compositions of compounds detected with the PTR-ToF-MS, (A) in the particle phase, and (B) in the gas phase. Corresponding percent contributions are shown in (C) and (D). | ||
According to Fig. 1, the gas phase was dominated by compounds of generic composition, C6–9Hh (i.e., monoaromatics, particularly C7 and C8 aromatics, including m/z 107.09, xylene/ethyl benzene; and m/z 93.07, toluene), that largely originated from on-road transport in Fairbanks;44 such compounds contributed to 50% of the gas-phase signal compared to a mere 2% in the particle phase. Small, oxygenated VOCs were detected as well, such as m/z 33.03 (CH4O, methanol), m/z 31.03 (CH2O, formaldehyde), m/z 45.03 (C2H4O, acetaldehyde), m/z 47.01 (CH2O2, formic acid), m/z 59.05 (C3H6O, acetone), and m/z 61.03 (C2H4O2, acetic acid), etc., constituting ∼23% of the gas-phase mass concentration. However, they have not been included in the partitioning analysis in this study due to their exclusive association with the gas phase (or being below the detection limit in the particle phase).50 Larger oxygenated (i.e., C>3O>2Hh) and non-oxygenated species with more than 10 C atoms collectively accounted for only 5.2% of the gas-phase mass. These distributions are in line with previous studies that reported Fp values of 0.03–0.4 for less oxidised compounds with 2–3 C atoms, and higher Fp values of 0.3–0.9 for compounds with at least 4 carbons.24
The molecular species in the particle phase showed a very broad carbon distribution ranging from C2 to C20 (C2–20HhO2–6), where hydroxy/cyclic sugars, carbonyls, carboxyls, multifunctional oxygenated compounds, and methoxy phenols, accounted for ∼6, 6, and 7, 12, 13% of the total particulate mass (Fig. 1A).44 The strongest signal in the OA arose from m/z 163.06 (i.e., C6H10O5 assigned to levoglucosan and isomers) and several methoxy phenols, including m/z 125.06 (C7H8O2, guaiacol), m/z 139.08 (C8H10O2, creosol), m/z 155.07 (C8H10O3, syringol), etc. In Fairbanks, these compounds were emitted from residential wood burning44 in agreement with previous studies.51–54 Approximately, 40% of the particle mass was made up of oxygenated aromatics (i.e., C≥6HhO≥1) and 12% came from large (m/z > 200) unoxygenated polyaromatic or cyclic hydrocarbons. These included m/z 203.09 (C16H10, pyrene), m/z 229.10 (C18H12, chrysene), m/z 241.1 (C19H12, benzo[cd]pyrene), etc. mostly originating from residential use of heating fuel oil.44 Other large molecules, including dehydroabietic acid (m/z 301.21, C20H28O2) and fatty acids (e.g., m/z 255.23, palmitoleic acid; m/z 281.24, linoleic acid; m/z 283.26, oleic acid) originated from wood combustion and residential/commercial cooking, respectively.44
3.2. General trends of OA phase partitioning
Fig. 2A depicts the partitioning behaviour of organics in Fairbanks in terms of the observed Fp,wa as a function of recorded temperature at the CTC site and OA mass concentrations from the AMS. The Fp is a simple metric to describe the gas–particle partitioning of any compound, but it relies on the total mass of the organics9,24,55 as expressed in eqn (1). Accordingly, the highest average Fp,wa values, reaching 0.8, were observed at the highest OA loadings, which coincided with the lowest temperatures during the ALPACA campaign (Fig. 2A). Effective saturation mass concentrations (log10C*) were also calculated from the observed Fp using eqn (2) and are represented as a function of recorded temperatures in Fig. 2B. These results are in line with previous reports of biomass burning populating the SVOC bin.56 To tentatively distinguish the relationship of Fp from OA and temperature, the dataset is clustered in three regimes based on OA concentrations, i.e., low (5–8 μg m−3), medium (8–13 μg m−3), and high (>13 μg m−3) OA. An overall increase of 0.5 in the 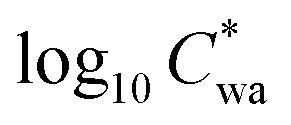 value is observed for increasing OA concentrations, which means a small change of ∼5 μg m−3 in the C* itself, and thus, the organics remain in the SVOC bin. Furthermore, as expected, within each of the three OA regimes, decreasing temperatures enhanced partitioning to the particle phase.
value is observed for increasing OA concentrations, which means a small change of ∼5 μg m−3 in the C* itself, and thus, the organics remain in the SVOC bin. Furthermore, as expected, within each of the three OA regimes, decreasing temperatures enhanced partitioning to the particle phase.
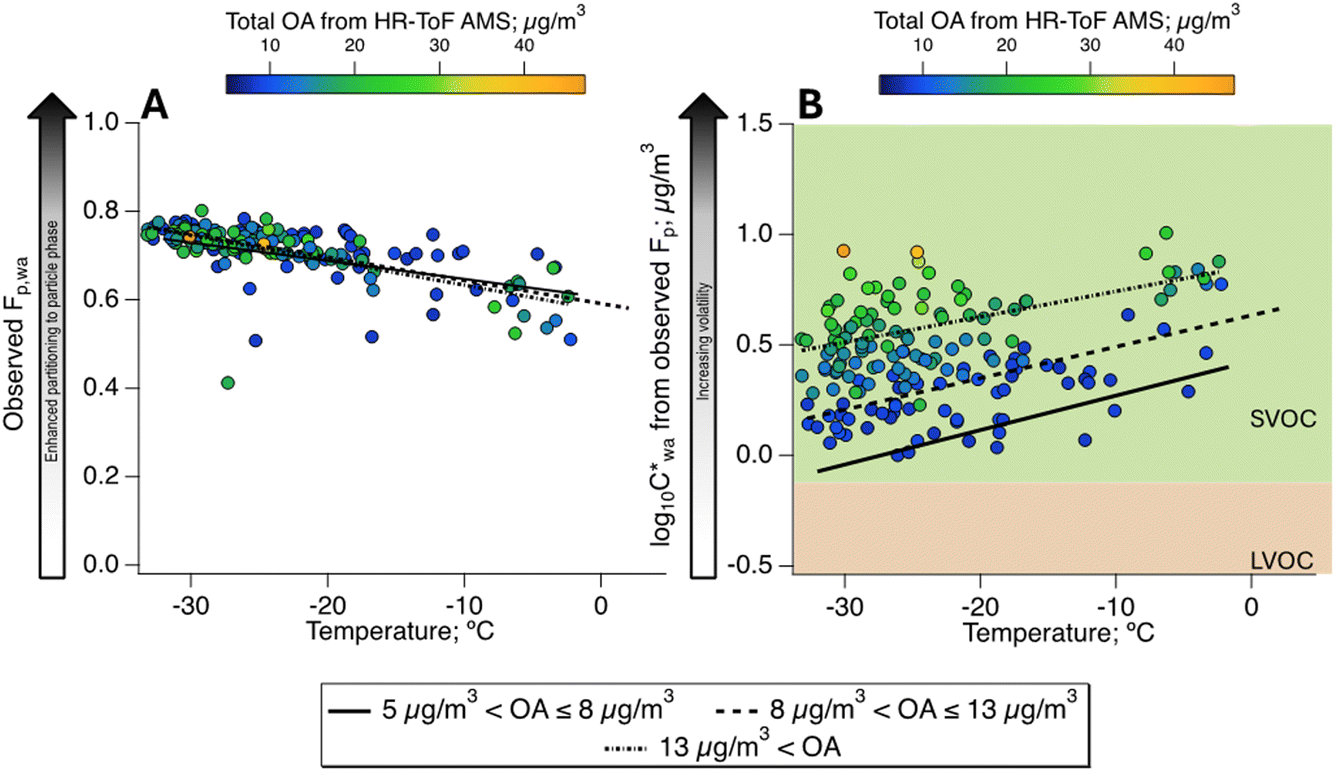 | ||
Fig. 2 Partitioning behaviour of total OA as a function of ambient temperature and OA concentrations using the (A) particle-phase fraction (Fp) and (B) effective saturation mass concentrations (C*). Concentration-weighted hourly averages, i.e., of Fp,wa and  , are shown to give appropriate weighting to the influence of ions according to their concentrations [in the OA]. The background of (B) delineates data points into volatility classes based on the volatility-basis set.4,9 , are shown to give appropriate weighting to the influence of ions according to their concentrations [in the OA]. The background of (B) delineates data points into volatility classes based on the volatility-basis set.4,9 | ||
Our observations align with a recent laboratory chamber study of SVOC, where the ratio of concentrations in the particle to gas phases (Ci,particle/Ci,gas) of individual compounds varied with temperature, RH and OA concentrations.24 SVOCs distributed increasingly more into the particle phase going from 20 to 0 °C, which was attributed to reduced vapour pressure of the compounds.24 In line with our analysis, the authors observed smaller Ci,particle/Ci,gas values (i.e., lower volatilities) at low OA concentrations. The only exception was observed for low-volatility compounds associated with long equilibration times, causing high Ci,particle/Ci,gas, due to their stronger propensity for the particle phase.24
In the upcoming sections, we will use the molecular-level information from the PTR-ToF MS to further investigate the partitioning behaviour of individual compounds.
3.3. Partitioning of organic compounds at the molecular level
Fig. 3 depicts the oxidation state (OSc) of the individual organic components versus the average log10C* above −15 °C and below −20 °C over the three OA concentration regimes described earlier. Temperatures above 0 °C were rarely experienced during the ALPACA campaign (Fig. S1†), and thus, these temperature windows were chosen to ensure statistically significant data points for comparison. Additionally, there was a sharp increase in NR-PM1 around −18 °C (Fig. S1†), allowing us to treat, in an arbitrary way, these ranges as “cold” and “extremely cold” temperatures. Some of the compounds unequivocally identified as markers of important pollution sources in Fairbanks, such as wood combustion, traffic, or cooking,44 are shown in blue (below −20 °C) and red (above −15 °C). The remaining compounds are drawn in light and dark grey for the two temperature ranges, respectively.
 | ||
| Fig. 3 The oxidation state of carbon in the detected ions (OSc) versus the average effective saturation mass concentration (C*) as a function of OA concentrations (i.e., vertical panelling) and temperature (within each panel). The average log10C* is shown for three ranges of OA concentrations: >13 μg/m3 (A), 8–13 μg/m3 (B) and 5–8 μg/m3 (C). Compounds associated with known sources of OA emissions in Fairbanks are annotated and shown in blue (below −20 °C) and red (above −15 °C) colours. The value written under each ion is the Δlog10C*, i.e., the difference between the average log10C* of each ion at the two temperature ranges; a positive Δlog10C* indicates an increase in volatility above −15 °C. Data points are sized by abundance normalised to the concentration of levoglucosan. The background delineates data points into volatility classes based on the volatility-basis set.4,9 Error bars show one standard deviation. | ||
Methoxy phenols (or derivatives), such as m/z 125.06 (guaiacol), m/z 139.07 (creosol), m/z 153.06 (vanillin), m/z 179.08 (coniferaldehyde), and m/z 165.09 (eugenol), and other biomass-burning-related aromatics, such as m/z 111.04 (resorcinol), consistently populate the SVOC bin (Fig. 3). As shown in Fig. S4† these species may be present in both the gas and particulate phases even though they exhibited variable Fp values from 0.2 to 0.9. While remaining in the semi-volatile bin, they all exhibited positive Δlog10C* values (i.e., the difference between log10C* at high- and low-temperature windows) compared to the other OA identified ions. This shift means that their volatilities appreciably decrease below −20 °C, and the shifts seemed more important for OA concentrations above 8 μg m−3. Such compounds, whose temperature-induced a Δlog10C* increase by at least 0.5, made up 19–22% of the total OA mass.
Other compounds behaved quite differently and showed weak responses to temperature change (i.e., low Δlog10C* values). This is the case for small, volatile molecules, notably monoaromatic compounds (e.g., m/z 79.05 (benzene), m/z 93.07 (toluene)) that are mostly found in the gas phase (Fig. S4,†Fp < 0.1), but traces are also observed in the particle phase because of the extremely low ambient temperatures. Other heavier compounds, such as PAHs (e.g., m/z 229.10 (chrysene), m/z 203.09 (pyrene)), together with other tracers such as m/z 163.06 (levoglucosan), m/z 301.21 (dehydroabietic acid), and m/z 281.25 (linoleic acid) populated the lower end of the SVOC and part of the LVOC bins. They all showed small volatility changes at all OA concentrations and were mostly in the particle phase (Fig. S4†). These observations are in agreement with previous reports on biomass-burning organic aerosol (BBOA)20,57,58 that found levoglucosan and other BBOA tracers mostly in the particle phase.
Recent works have emphasized that pre-existing OA compositions can be an important factor influencing the partitioning of intermediate-volatility organic compounds (IVOC)/SVOC species.20,59,60 During the FIREX-AQ-2018 campaign, an increasing BBOA fraction was related to enhanced Fp of polar compounds.20 Apparently wildfire organic aerosol enhanced the condensation of polar compounds into the particle phase, while this was not the case for nonpolar compounds, such as polycyclic aromatic hydrocarbons.20 In our case, due to the extremely low temperatures and the proximity to the sources, it is likely that the BBOA-rich and other oxygenated OA limited the evaporation of less oxygenated and lighter species, causing such small Δlog10C* values. The high presence of polyfunctional molecules containing carboxylic, hydroxyl, or carbonyl groups may induce reduced vapour pressure and increased viscosity.61
Fig. 4 shows the estimated phase state ratio associated with the field data using the DeRieux62 and Shiraiwa63 method. We clustered the dataset for two temperature regimes: between −15 °C and 5 °C and between −20 and −36 °C. According to Fig. 4, above −15 °C, approximately 60% of OA mass showed relatively medium range viscosity (PSR (phase state ratio) ∼0.7) and another 35% low viscosity (PSR <0.6), indicating that the majority of the OA is in a liquid state. Meanwhile, below −20 °C the PSR was substantially higher with more than 70% of the OA above ≥0.7 indicating the presence of a semi-solid phase associated with a slow or limited diffusive activity.
 | ||
| Fig. 4 The distribution of detected compounds across bins of saturation mass concentrations (C*) estimated from observed Fp in Fairbanks at cold (above −15 °C) and very cold (below −20 °C) temperatures. Mass fractions are coloured according to the phase state ratio measured using the method of (DeRieux et al., 2018)62 and (Shiraiwa et al., 2017).63 | ||
In addition to the temperature, atmospheric transport can also affect the aerosol phase state. A recent study on fresh BBOA showed a reduced intrinsic volatility when the BBOA mixture was diluted from 5000 μg m−3 to 5 μg m−3; this behaviour was attributed to enhanced evaporation of high-volatility species to the gas phase at higher concentration, leaving behind a diluted aerosol of higher viscosity (2–3 orders of magnitude higher than freshly emitted BBOA) substantially limiting multiphase chemistry.64 In our case pollution events were associated with stable conditions and strong temperature inversion;44,45 in addition the measurement site was in proximity to urban sources implicating the co-existence of high loading of fresh and moderately aged OA.
3.4. Performance of the absorptive-partitioning estimation approach under cold temperatures
Fig. 5A shows the distribution of organics according to the number of C and O atoms, where most compounds detected in Fairbanks would be expected to behave like SVOCs (0.3 < C0 < 300 μg m−3).8 Another substantive amount of the mass falls into the LVOC bin (3 × 10−4 < C0 < 0.3 μg m−3).8 The observed partitioning of ions in Fairbanks agrees with this classification, where ions with lower Fp populate the V/I/SVOC bins and ions of higher average Fp were E/LVOCs. This Fp can be used for a direct comparison between the observed and estimated volatilities viaeqn (1)20 if the C* estimated from the observed Fp is assumed to be equal to C0 (i.e., γ = 1).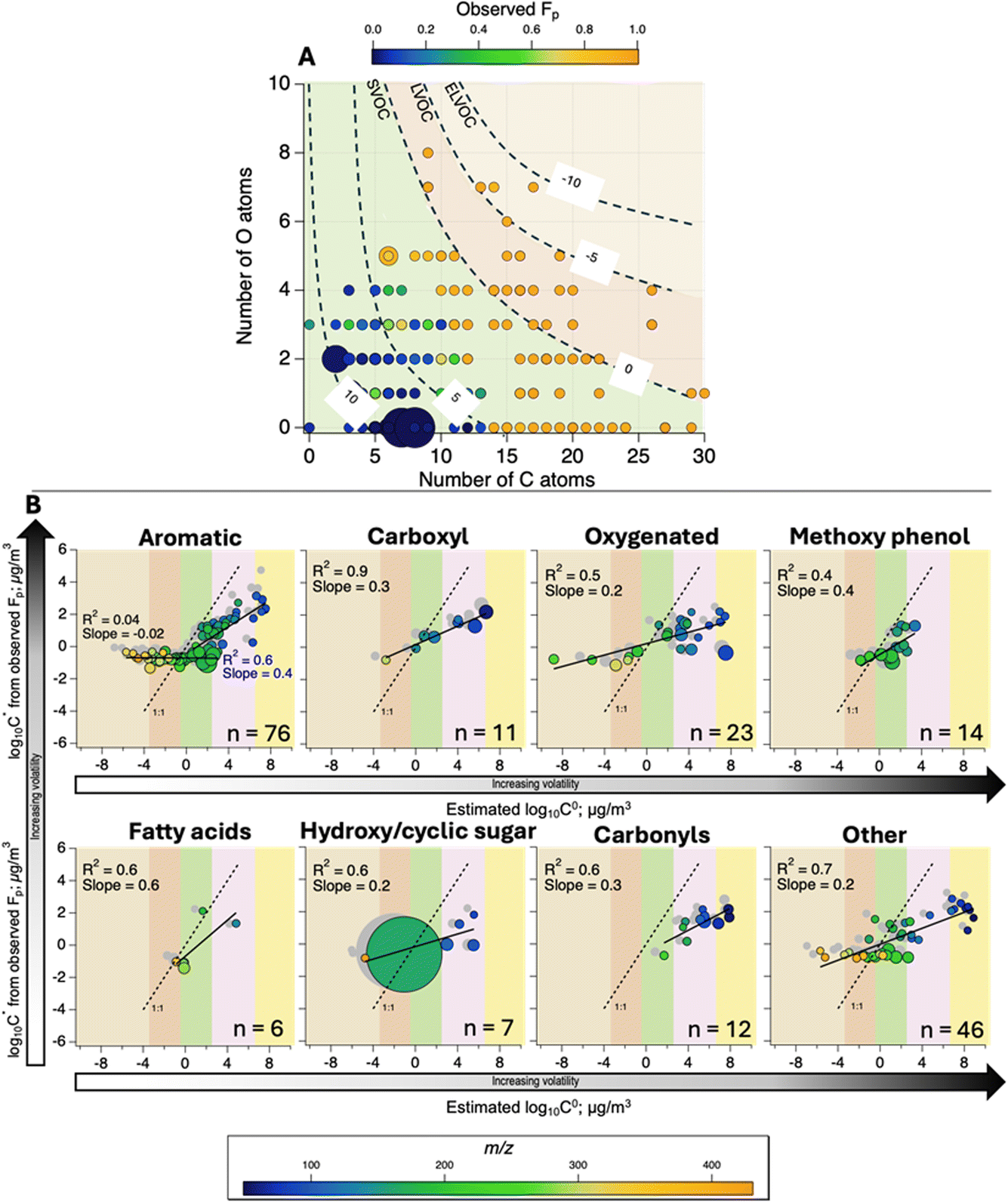 | ||
Fig. 5 Overview of the general partitioning behaviour (A) across the volatility-basis set (saturation mass concentrations from −10 to 10 μg m−3 are represented on the plot)5,9 in terms of the number of C and O atoms in the formulae tentatively assigned to ions. Data points are sized according to the total average concentration of ions in the gas and particle phases; they are coloured according to the observed average particle phase fraction of each ion (Fp); (B) estimated effective saturation mass concentration (C*) from observed Fp of individual ions with different functional groups and its agreement with estimated saturation mass concentration for pure compound (C0) above −15 °C (coloured data points) and below −20 °C (grey data points). Note that  . Data points are coloured according to the experimental mass of the ion and sized according to the abundance normalised to the concentration of levoglucosan. The background delineates the x-axis into volatility classes based on the volatility-basis set.4,9 . Data points are coloured according to the experimental mass of the ion and sized according to the abundance normalised to the concentration of levoglucosan. The background delineates the x-axis into volatility classes based on the volatility-basis set.4,9 | ||
We categorised our data by functional groups to assess the performance of the absorptive partitioning theory. According to Fig. 5B, the estimation approach showed a satisfactory linearity with observations (i.e., R2 > 0.4) but a clear disagreement with respect to the magnitude of volatilities (slopes from 0.02 to 0.5) similarly to what was observed for a wildfire study by Liang et al.20 A distinct dependence can be noticed between the molecular weight of ions and the performance of the estimation method. For compounds with m/z between 120 and 250, the estimated and measured values are in reasonable agreement, while the estimated C0 of compounds with m/z > 300 are significantly lower than the observed C*. This can be clearly seen for some polyaromatic hydrocarbons (in the plot of aromatic compounds), and some oxygenated compounds, in Fig. 5. On the other hand, for some very heavy and sticky molecules, we observed some instrumental memory effects (over-detection of very large compounds in the gas phase; Section S2†) that may induce overestimation of their volatility. Meanwhile, for smaller compounds with m/z < 120, the estimated C0 values indicate higher volatility with respect to the measured ones.
A recent study at two forested sites reports that the volatility estimated from SIMPOL-derived vapour pressure was much higher compared to that derived from gas–particle partitioning theory using the Fp measured with thermal desorption-AMS and FIGAERO-CIMS.22 In another work that compared multiple experimental and theoretical methods for volatility distributions, the elemental composition-based parameterisation8 also overestimated the volatility of organic compounds as compared to thermal-desorption methods and even the SIMPOL-driven estimations.65 Some discrepancies can be attributed to the effect of including the formulae of thermally decomposed ions, which cannot be excluded confidently in a complex ambient mixture.
In the CHARON PTR-ToF MS, although we used low voltages, we could observe some ion fragmentations for small hydrocarbons and levoglucosan (m/z 85.03, 127.04, and 145.05), which could also explain the general overestimation of the estimated C0 for all these ions in Fig. 5. There is, however, some evidence that vapor pressures of multifunctional compounds may not be very accurate when calculated using SIMPOL and other group contribution methods.66–68 According to Stark et al.,22 the partitioning method may not work well for very volatile or non-volatile compounds.
More generally, for a viscous compound emitted in particulate form, the estimated Fp from the absorptive partitioning theory is underestimated when an instantaneous equilibrium is considered. Thus, viscosity should be taken into consideration in the interpretation of discrepancies for the dataset investigated here, since the phase state of the aerosol affects the condensation and the evaporation of organic compounds on it.69
4. Conclusions and environmental implications
We investigated the gas–particle partitioning behaviour of organic compounds in Fairbanks, Alaska, during the wintertime. Temperature and OA loads were unequivocally critical factors driving the partitioning of organics. Major species associated with the combustion of softwood and heating fuel, and cooking, such as levoglucosan, dehydroabietic acid, PAHs, and fatty acids, remained almost exclusively in the particle phase, regardless of OA concentrations and temperature. However some semi-volatile compounds from hardwood burning, such as methoxy phenols, including guaiacol, resorcinol, etc., presented some temperature dependence in their partitioning.The partitioning behaviour of compounds from different emission sources has important implications for understanding local air quality and the frequent wintertime pollution episodes. For instance, the observed high Fp values for many organic compounds mean increased atmospheric lifetimes potentially aggravating pollution episodes in Fairbanks due to PM exceedances. Therefore, while attempting to control or investigate trends in emissions and pollution levels, the challenge is to appropriately distinguish the specific emission sectors, their chemical signature (PM) and the associated dynamics after release.
On the other end, the observed Fp fractions were often higher than estimated ones; mainly, the model underpredicted the particle phase fraction of OA, emphasising the need to examine shortcomings in partitioning-related parameterisations under extreme environmental conditions that are rarely simulated in laboratory studies. Overall, the uncertainties in the volatility parameterisations and current assumptions should be validated with further datasets from diverse sources, environmental conditions and also measurement techniques. Finally, we think that the present work may be useful to better understand pollution events in an extremely cold environment and could be used to improve volatility parameterisations and finally air quality models.
Data availability
Supporting material, including text, figures, and tables are available in the ESI.†Author contributions
AI and BD wrote the article. BT-R and BD set up, ran, and maintained the instrumentation during the campaign in Fairbanks. BT-R and AI processed the data. BD, AI, JK, and BT-R interpreted the data. WS and KSL coordinated the ALPACA and CASPA projects. WS and JM supported the fieldwork in Fairbanks. KL and BD contributed to funding acquisition for the CASPA project. All co-authors revised the article.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the researchers and other participants of the ALPACA campaign for providing the necessary logistical support to acquire the data presented in this study. This work was funded by the CASPA (Climate-relevant Aerosol Sources and Processes in the Arctic) project of the Agence Nationale de la Recherche (grant no. ANR-21-CE01-0017) Institut polaire français Paul-Émile Victor (IPEV) (grant no. 1215), and the CNRS-INSU programme. Les Enveloppes Fluides et l’Environnement (LEFE) ALPACA-France projects. We gratefully acknowledge the MASSALYA instrumental platform (Aix-Marseille Université, https://lce.univ-amu.fr/) for the analysis and measurements used in this publication. JM was supported by the National Science Foundation (NSF) Atmospheric Geoscience Program (grant no. AGS-2029747) and the NSF Navigating the New Arctic Program (grant no. NNA-1927750). The authors also thank J. Fochesatto (UAF) and other members of the ALPACA team for their assistance in the campaign planning, and deployment. AI is grateful to Subuktageen Qitta, Nastaran Mahmud, Laal Boo’on-Wali, and Minuit Mahmud for their immense intellectual input; without them, this report would not have been possible.Notes and references
- J. F. Pankow, Atmos. Environ., 1994, 28, 185–188 CrossRef CAS.
- J. H. Seinfeld, Atmospheric Chemistry and Physics: from Air Pollution to Climate Change, John Wiley & Sons, Incorporated, Newark, 1st edn, 2016 Search PubMed.
- N. M. Donahue, A. L. Robinson, E. R. Trump, I. Riipinen and J. H. Kroll, in Atmospheric Aerosol Chemistry, 2014, pp. 97–143 Search PubMed.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- N. M. Donahue, J. Kroll, S. N. Pandis and A. L. Robinson, Atmos. Chem. Phys., 2012, 12, 615–634 Search PubMed.
- N. Donahue, W. Chuang, S. A. Epstein, J. Kroll, D. Worsnop, A. Robinson, P. Adams and S. Pandis, Environ. Chem., 2013, 10, 151–157 CrossRef CAS.
- N. M. Donahue, S. A. Epstein, S. N. Pandis and A. L. Robinson, Atmos. Chem. Phys., 2011, 11, 3303–3318 CrossRef CAS.
- Y. Li, U. Pöschl and M. Shiraiwa, Atmos. Chem. Phys., 2016, 16, 3327–3344 CrossRef CAS.
- N. M. Donahue, A. Robinson, C. Stanier and S. Pandis, Environ. Sci. Technol., 2006, 40, 2635–2643 Search PubMed.
- B.-H. Lee, E. Kostenidou, L. Hildebrandt, I. Riipinen, G. Engelhart, C. Mohr, P. DeCarlo, N. Mihalopoulos, A. Prevot and U. Baltensperger, Atmos. Chem. Phys., 2010, 10, 12149–12160 Search PubMed.
- C. Cappa and J. Jimenez, Atmos. Chem. Phys., 2010, 10, 5409–5424 Search PubMed.
- P. K. Saha, A. Khlystov, K. Yahya, Y. Zhang, L. Xu, N. L. Ng and A. P. Grieshop, Atmos. Chem. Phys., 2017, 17, 501–520 Search PubMed.
- K. Dzepina, R. M. Volkamer, S. Madronich, P. Tulet, I. M. Ulbrich, Q. Zhang, C. D. Cappa, P. J. Ziemann and J.L. Jimenez, Atmos. Chem. Phys., 2009, 9, 5681–5709 Search PubMed.
- P. L. Hayes, A. G. Carlton, K. R. Baker, R. Ahmadov, R. A. Washenfelder, S. Alvarez, B. Rappenglück, J. B. Gilman, W. Kuster and J. A. De Gouw, Atmos. Chem. Phys., 2015, 15, 5773–5801 Search PubMed.
- C. Mohr, J. A. Thornton, A. Heitto, F. D. Lopez-Hilfiker, A. Lutz, I. Riipinen, J. Hong, N. M. Donahue, M. Hallquist and T. Petäjä, Nat. Commun., 2019, 10, 4442 CrossRef CAS PubMed.
- O. Peräkylä, M. Riva, L. Heikkinen, L. Quéléver, P. Roldin and M. Ehn, Atmos. Chem. Phys., 2020, 20, 649–669 CrossRef.
- J. F. Pankow and W. E. Asher, Atmos. Chem. Phys., 2008, 8, 2773–2796 CrossRef CAS.
- S. Compernolle, K. Ceulemans and J.-F. Müller, Atmos. Chem. Phys., 2011, 11, 9431–9450 CrossRef CAS.
- G. Isaacman-VanWertz and B. Aumont, Atmos. Chem. Phys., 2021, 21, 6541–6563 CrossRef CAS.
- Y. Liang, R. A. Wernis, K. Kristensen, N. M. Kreisberg, P. L. Croteau, S. C. Herndon, A. W. H. Chan, N. L. Ng and A. H. Goldstein, Atmos. Chem. Phys., 2023, 23, 12441–12454 Search PubMed.
- R. Wu, L. Vereecken, E. Tsiligiannis, S. Kang, S. R. Albrecht, L. Hantschke, D. Zhao, A. Novelli, H. Fuchs and R. Tillmann, Atmos. Chem. Phys., 2021, 21, 10799–10824 CrossRef CAS.
- H. Stark, R. L. Yatavelli, S. L. Thompson, H. Kang, J. E. Krechmer, J. R. Kimmel, B. B. Palm, W. Hu, P. L. Hayes and D. A. Day, Environ. Sci. Technol., 2017, 51, 8491–8500 CrossRef CAS PubMed.
- A. Lutz, C. Mohr, M. Le Breton, F. D. Lopez-Hilfiker, M. Priestley, J. A. Thornton and M. Hallquist, ACS Earth Space Chem., 2019, 3, 1279–1287 CrossRef CAS.
- L. Li, D. Thomsen, C. Wu, M. Priestley, E. M. Iversen, J. Tygesen Skonager, Y. Luo, M. Ehn, P. Roldin and H. B. Pedersen, J. Phys. Chem. A, 2024, 128, 918–928 CrossRef CAS PubMed.
- S. A. Epstein, I. Riipinen and N. M. Donahue, Environ. Sci. Technol., 2010, 44, 743–748 CrossRef CAS PubMed.
- J. Ahn, G. Rao and E. Vejerano, Environ. Sci.:Processes Impacts, 2021, 23, 947–955 RSC.
- M. Bilde, K. Barsanti, M. Booth, C. D. Cappa, N. M. Donahue, E. U. Emanuelsson, G. McFiggans, U. K. Krieger, C. Marcolli and D. Topping, Chem. Rev., 2015, 115, 4115–4156 CrossRef CAS PubMed.
- A. P. Grieshop, M. A. Miracolo, N. M. Donahue and A. L. Robinson, Environ. Sci. Technol., 2009, 43, 4750–4756 CrossRef CAS PubMed.
- T. D. Vaden, D. Imre, J. Beránek, M. Shrivastava and A. Zelenyuk, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2190–2195 CrossRef CAS PubMed.
- Y. Chen, M. Takeuchi, T. Nah, L. Xu, M. R. Canagaratna, H. Stark, K. Baumann, F. Canonaco, A. S. H. Prévôt, L. G. Huey, R. J. Weber and N. L. Ng, Atmos. Chem. Phys., 2020, 20, 8421–8440 CrossRef CAS.
- F. D. Lopez-Hilfiker, C. Mohr, M. Ehn, F. Rubach, E. Kleist, J. Wildt, T. F. Mentel, A. Lutz, M. Hallquist, D. Worsnop and J. A. Thornton, Atmos. Meas. Tech., 2014, 7, 983–1001 CrossRef.
- J. A. Thornton, C. Mohr, S. Schobesberger, E. L. D’Ambro, B. H. Lee and F. D. Lopez-Hilfiker, Acc. Chem. Res., 2020, 53, 1415–1426 CrossRef CAS PubMed.
- F. D. Lopez-Hilfiker, C. Mohr, M. Ehn, F. Rubach, E. Kleist, J. Wildt, T. F. Mentel, A. J. Carrasquillo, K. E. Daumit, J. F. Hunter, J. H. Kroll, D. R. Worsnop and J. A. Thornton, Atmos. Chem. Phys., 2015, 15, 7765–7776 CrossRef CAS.
- A. Voliotis, M. Du, Y. Wang, Y. Shao, T. J. Bannan, M. Flynn, S. N. Pandis, C. J. Percival, M. R. Alfarra and G. McFiggans, Atmos. Chem. Phys., 2022, 22, 13677–13693 CrossRef CAS.
- A. Voliotis, Y. Wang, Y. Shao, M. Du, T. J. Bannan, C. J. Percival, S. N. Pandis, M. R. Alfarra and G. McFiggans, Atmos. Chem. Phys., 2021, 21, 14251–14273 CrossRef CAS.
- P. Eichler, M. Mueller, B. D’Anna and A. Wisthaler, Atmos. Meas. Tech., 2015, 8, 1353–1360 Search PubMed.
- M. Muller, P. Eicher, B. D’Anna, W. Tan and A. Wisthaler, Anal. Chem., 2017, 89, 10889–10897 CrossRef PubMed.
- J. Leglise, M. Muller, F. Piel, T. Otto and A. Wisthaler, Anal. Chem., 2019, 91, 12619–12624 Search PubMed.
- Y. Peng, H. Wang, Y. Gao, S. Jing, S. Zhu, D. Huang, P. Hao, S. Lou, T. Cheng and C. Huang, Atmos. Meas. Tech., 2023, 16, 15–28 Search PubMed.
- P. Eichler, M. Muller, C. Rohmann, B. Stengel, J. Orasche, R. Zimmermann and A. Wisthaler, Environ. Sci. Technol. Lett., 2017, 4, 54–58 Search PubMed.
- V. Lannuque, B. D’Anna, E. Kostenidou, F. Couvidat, A. Martinez-Valiente, P. Eichler, A. Wisthaler, M. Müller, B. Temime-Roussel, R. Valorso and K. Sartelet, Atmos. Chem. Phys., 2023, 23, 15537–15560 CrossRef CAS.
- F. Piel, M. Müller, K. Winkler, J. Skytte af Sätra and A. Wisthaler, Atmos. Meas. Tech., 2021, 14, 1355–1363 CrossRef CAS.
- G. I. Gkatzelis, T. Hohaus, R. Tillmann, I. Gensch, M. Müller, P. Eichler, K. M. Xu, P. Schlag, S. H. Schmitt, Z. Yu, R. Wegener, M. Kaminski, R. Holzinger, A. Wisthaler and A. Kiendler-Scharr, Atmos. Chem. Phys., 2018, 18, 12969–12989 CrossRef CAS.
- A. Ijaz, B. Temime-Roussel, B. Chazeau, S. Albertin, S. Arnold, B. Barret, S. Bekki, N. Brett, M. Cesler-Maloney, E. Dieudonné, K. K. Dingilian, G. J. Fochesatto, J. Mao, J. Savarino, W. Simpson, R. J. Weber, K. S. Law and B. D’Anna, Complementary aerosol mass spectrometry elucidates sources of wintertime sub-micron particle pollution in Fairbanks, Alaska, during ALPACA 2022, DOI:10.5194/egusphere-2024-3789.
- W. R. Simpson, J. Mao, G. J. Fochesatto, K. S. Law, P. F. DeCarlo, J. Schmale, K. A. Pratt, S. R. Arnold, J. Stutz, J. E. Dibb, J. M. Creamean, R. J. Weber, B. J. Williams, B. Alexander, L. Hu, R. J. Yokelson, M. Shiraiwa, S. Decesari, C. Anastasio, B. D’Anna, R. C. Gilliam, A. Nenes, J. M. S. Clair, B. Trost, J. H. Flynn, J. Savarino, L. D. Conner, N. Kettle, K. M. Heeringa, S. Albertin, A. Baccarini, B. Barret, M. A. Battaglia, S. Bekki, T. J. Brado, N. Brett, D. Brus, J. R. Campbell, M. Cesler-Maloney, S. Cooperdock, K. Cysneiros De Carvalho, H. Delbarre, P. J. DeMott, C. J. S. Dennehy, E. Dieudonné, K. K. Dingilian, A. Donateo, K. M. Doulgeris, K. C. Edwards, K. Fahey, T. Fang, F. Guo, L. M. D. Heinlein, A. L. Holen, D. Huff, A. Ijaz, S. Johnson, S. Kapur, D. T. Ketcherside, E. Levin, E. Lill, A. R. Moon, T. Onishi, G. Pappaccogli, R. Perkins, R. Pohorsky, J.-C. Raut, F. Ravetta, T. Roberts, E. S. Robinson, F. Scoto, V. Selimovic, M. O. Sunday, B. Temime-Roussel, X. Tian, J. Wu and Y. Yang, ACS ES&T Air, 2024, 1, 200–222 Search PubMed.
- P. F. DeCarlo, J. R. Kimmel, A. Trimborn, M. J. Northway, J. T. Jayne, A. C. Aiken, M. Gonin, K. Fuhrer, T. Horvath, K. S. Docherty, D. R. Worsnop and J. L. Jimenez, Anal. Chem., 2006, 78, 8281–8289 CrossRef CAS PubMed.
- M. Cesler-Maloney, W. R. Simpson, T. Miles, J. Mao, K. S. Law and T. J. Roberts, J. Geophys. Res.:Atmos., 2022, 127, e2021JD036215 Search PubMed.
- J. R. Campbell, M. Battaglia Jr, K. K. Dingilian, M. Cesler-Maloney, W. R. Simpson, E. S. Robinson, P. F. DeCarlo, B. Temime-Roussel, B. D’Anna and A. L. Holen, Sci. Adv., 2024, 10, eado4373 Search PubMed.
- L. K. Meredith, S. M. Ledford, K. Riemer, P. Geffre, K. Graves, L. K. Honeker, D. LeBauer, M. M. Tfaily and J. Krechmer, Front. Microbiol., 2023, 14, 1267234 CrossRef PubMed.
- B. Temime-Roussel, M. Cesler-Maloney, A. Ijaz, N. Brett, K. S. Law, S. Bekki, J. Mao, D. T. Ketcherside, V. Selimovic, L. Hu, W. R. Simpson and B. D’Anna, Concentrations and Sources of VOCs during wintertime urban pollution at Fairbanks, Alaska, Fall Meeting 2022, 2022 - AGU.
- L. T. Fleming, P. Lin, J. M. Roberts, V. Selimovic, R. Yokelson, J. Laskin, A. Laskin and S. A. Nizkorodov, Atmos. Chem. Phys., 2020, 20, 1105–1129 CrossRef CAS.
- A. Ijaz, W. Kew, Z. Cheng, S. Mathai, N. N. Lata, L. Kovarik, S. Schum, S. China and L. R. Mazzoleni, Environ. Sci.: Atmos., 2023, 3, 1552–1562 CAS.
- J. S. Smith, A. Laskin and J. Laskin, Anal. Chem., 2009, 81, 1512–1521 CrossRef CAS PubMed.
- J. Zhang, K. Li, T. Wang, E. Gammelsæter, R. K. Y. Cheung, M. Surdu, S. Bogler, D. Bhattu, D. S. Wang, T. Cui, L. Qi, H. Lamkaddam, I. El Haddad, J. G. Slowik, A. S. H. Prevot and D. M. Bell, Atmos. Chem. Phys., 2023, 23, 14561–14576 CrossRef CAS.
- S. L. Thompson, R. L. N. Yatavelli, H. Stark, J. R. Kimmel, J. E. Krechmer, D. A. Day, W. Hu, G. Isaacman-VanWertz, L. Yee, A. H. Goldstein, M. A. H. Khan, R. Holzinger, N. Kreisberg, F. D. Lopez-Hilfiker, C. Mohr, J. A. Thornton, J. T. Jayne, M. Canagaratna, D. R. Worsnop and J. L. Jimenez, Aerosol Sci. Technol., 2017, 51, 30–56 CrossRef CAS.
- A. A. May, A. A. Presto, C. J. Hennigan, N. T. Nguyen, T. D. Gordon and A. L. Robinson, Atmos. Environ., 2013, 77, 128–139 CrossRef CAS.
- V. O. Elias, B. R. T. Simoneit, R. C. Cordeiro and B. Turcq, Geochim. Cosmochim. Acta, 2001, 65, 267–272 CrossRef CAS.
- B. R. T. Simoneit, Appl. Geochem., 2002, 17, 129–162 CrossRef CAS.
- F. Mahrt, Y. Huang, J. Zaks, A. Devi, L. Peng, P. E. Ohno, Y. M. Qin, S. T. Martin, M. Ammann and A. K. Bertram, Environ. Sci. Technol., 2022, 56, 3960–3973 CrossRef PubMed.
- J. Ye, C. A. Gordon and A. W. H. Chan, Environ. Sci. Technol., 2016, 50, 3572–3579 Search PubMed.
- N. E. Rothfuss and M. D. Petters, Environ. Sci. Technol., 2017, 51, 271–279 Search PubMed.
- W.-S. W. DeRieux, Y. Li, P. Lin, J. Laskin, A. Laskin, A. K. Bertram, S. A. Nizkorodov and M. Shiraiwa, Atmos. Chem. Phys., 2018, 18, 6331–6351 CrossRef.
- M. Shiraiwa, Y. Li, A. P. Tsimpidi, V. A. Karydis, T. Berkemeier, S. N. Pandis, J. Lelieveld, T. Koop and U. Pöschl, Nat. Commun., 2017, 8, 15002 Search PubMed.
- K. Siemens, T. Paik, A. Li, F. Rivera-Adorno, J. Tomlin, Q. Xie, R. K. Chakrabarty and A. Laskin, ACS Earth Space Chem., 2024, 8, 1416–1428 CrossRef CAS.
- W. Chen, W. Hu, Z. Tao, Y. Cai, M. Cai, M. Zhu, Y. Ye, H. Zhou, H. Jiang, J. Li, W. Song, J. Zhou, S. Huang, B. Yuan, M. Shao, Q. Feng, Y. Li, G. Isaacman-VanWertz, H. Stark, D. A. Day, P. Campuzano-Jost, J. L. Jimenez and X. Wang, J. Geophys. Res.:Atmos., 2024, 129, e2023JD040284 Search PubMed.
- M. H. Barley and G. McFiggans, Atmos. Chem. Phys., 2010, 10, 749–767 CrossRef CAS.
- S. O’Meara, A. M. Booth, M. H. Barley, D. Topping and G. McFiggans, Phys. Chem. Chem. Phys., 2014, 16, 19453–19469 RSC.
- C. Dang, T. Bannan, P. Shelley, M. Priestley, S. D. Worrall, J. Waters, H. Coe, C. J. Percival and D. Topping, Aerosol Sci. Technol., 2019, 53, 1040–1055 CrossRef CAS.
- A. Zelenyuk, D. Imre, J. Beránek, E. Abramson, J. Wilson and M. Shrivastava, Environ. Sci. Technol., 2012, 46, 12459–12466 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00175c |
| ‡ Now at: Atmospheric, Climate, & Earth Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, USA |
| This journal is © The Royal Society of Chemistry 2025 |