
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategic synergism in CO2 and biomass valorization into sustainable solar fuels via stable hybrid halide perovskites: unlocking untapped potential
Bhawna
Rawat
a,
Ankita
Kumari
b,
Manvi
Sachdeva
a,
Himanshu
Bhatt
a,
Dibyajyoti
Ghosh
b,
Hirendra N.
Ghosh
*c,
Rajenahally V.
Jagadeesh
 *de and
Kamalakannan
Kailasam
*a
*de and
Kamalakannan
Kailasam
*a
aInstitute of Nano Science and Technology (INST), Knowledge City, Sector 81, SAS Nagar, Manauli PO, 140306 Mohali, Punjab, India. E-mail: kamal@inst.ac.in
bDepartment of Chemistry, Indian Institute of Technology, Delhi, Hauz Khas, New Delhi 110016, India
cSchool of Chemical Sciences, National Institute of Science and Education Research (NISER), Bhubaneshwar, Odisha 752050, India
dLeibniz-Institut für Katalyse, Albert-Einstein-Straße 29a, 18059 Rostock, Germany
eNanotechnology Centre, Centre for Energy and Environmental Technologies, VŠB–Technical University of Ostrava, Ostrava-Poruba, Czech Republic
First published on 2nd September 2025
Abstract
Sunlight-driven integration of photocatalytic CO2 reduction with biomass feedstock valorization constitutes a highly efficient strategy for the synergistic production of multi-electron products and high-value fine chemicals, adhering to photo-chemical circular economy and sustainability. To date, no halide perovskite has been utilized for CO2 reduction coupled with biomass oxidation as the development of more stable, efficient, reusable, and non-toxic halide perovskites continues to be challenging. Herein, we report the room-temperature synthesis of methylammonium tin bromide (MA2SnBr6) quantum dots (QDs), a vacancy-ordered hybrid halide perovskite (HHP), without additional capping agents. These novel QDs maintain structural integrity in air, moisture, and polar solvents, addressing a significant issue associated with halide perovskites. Remarkably, the MA2SnBr6 QDs remain stable under ambient conditions even after 1 year, as confirmed by PXRD analysis. Interestingly, MA2SnBr6 achieved exceptionally high electron consumption rates (Re) of 5110 μmol g−1 h−1 and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 383 μmol g−1 h−1 for CO2 reduction under simulated and natural sunlight, respectively, outperforming previous systems. In situ transient studies demonstrate that the photogenerated electrons of MA2SnBr6 diffuse from the conduction band to trap states, reducing CO2, while synergistically photogenerated holes oxidize biomass-derived alcohols. Additionally, in situ EPR experiments were performed to unravel mechanistic insights. Computational studies identify the Br p-orbitals of MA2SnBr6 as the reaction centre for CO2 reduction. Consequently, this work introduces a lead-free, single-component material that operates without a co-catalyst, sacrificial agent or redox additive, offering a promising path towards achieving photoredox processes in a more sustainable and efficient manner.
383 μmol g−1 h−1 for CO2 reduction under simulated and natural sunlight, respectively, outperforming previous systems. In situ transient studies demonstrate that the photogenerated electrons of MA2SnBr6 diffuse from the conduction band to trap states, reducing CO2, while synergistically photogenerated holes oxidize biomass-derived alcohols. Additionally, in situ EPR experiments were performed to unravel mechanistic insights. Computational studies identify the Br p-orbitals of MA2SnBr6 as the reaction centre for CO2 reduction. Consequently, this work introduces a lead-free, single-component material that operates without a co-catalyst, sacrificial agent or redox additive, offering a promising path towards achieving photoredox processes in a more sustainable and efficient manner.
Broader contextThe most challenging aspect and bottleneck in photocatalysis is the effective employment of charge carriers in the most sustainable manner for synergistic reduction and oxidation processes while achieving a robust catalyst without the use of co-catalysts, sacrificial agents, or redox additives. Particularly, in tackling excessive CO2 emissions, solar energy provides a sustainable route to transform CO2 into fuels, achieving carbon neutrality. Photocatalytic CO2 reduction in synergism with biomass valorization under sunlight offers a highly effective approach towards “Photo-biorefinery.” This study also breaks the perception of the instability of halide perovskites for photocatalytic organic transformation and photoredox reactions. Halide perovskites are widely explored for CO2-to-CO conversion; however, deep-reduced fuels, such as CH4, still face a challenge with perovskites. This study not only presents the facile mechanochemical synthesis of novel lead-free hybrid halide perovskites in MA2SnBr6 QDs, but also addresses the limitation of conventional halide perovskites—particularly their instability, toxicity, low efficiency, and poor reusability. Eventually, the single-component perovskite achieved record-high production of CO and CH4 under natural sunlight, even without any co-catalyst or sacrificial agent—outperforming the existing halide perovskite photocatalysts. These findings uniquely couple the production of fine chemicals, such as vanillin, veratraldehyde, and 2-phenoxy-1-phenylethanone, with the generation of solar CO and CH4 fuels, thus achieving a “photo-chemical circular economy.” |
Introduction
Nature, in all its ingenuity, achieves extraordinary feats such as photosynthesis, an effortlessly executed complex process. However, the real challenge is in replicating these natural phenomena, a task that persistently advances the boundaries of scientific innovations. Mimicking natural photosynthesis, through the coupling of CO2 reduction with H2O oxidation to achieve carbon neutrality, represents an ideal green process in the development of artificial photosynthesis.1 CO2 itself plays a vital role in maintaining the ecological balance of the Earth. However, the increased use of non-renewable fossil-based feedstocks and rapid industrialization are leading to an increase in CO2 levels, which represents a bottleneck in sequestering CO2 in fuels to reduce our carbon footprints.2 The CO2 conversion to CO and CH4 has gained significant scientific attention3 with the assistance of various photocatalysts such as metal oxides, metal chalcogenides, metal complexes, organic semiconductors, and perovskites. However, the H2O oxidation follows slow kinetics that can be replaced by biomass oxidation to effectively utilize the photogenerated holes to drive the desired oxidation reaction. Integrating photocatalytic CO2 reduction with biomass valorization using sunlight presents a highly efficient strategy for the synergistic production of solar fuels and value-added fine chemicals that meet the criteria of chemical economy and sustainability.4 In this regard, to date, no halide perovskite has been employed for synergistic CO2 reduction with biomass valorization.Halide perovskites have emerged as promising advanced materials,5 which find increasing applications in catalysis.6 Their remarkable properties such as high absorption coefficient, exceptional charge carrier mobility, long diffusion length, highly crystalline structure and low binding energy of excitons, as well as tuning of band gap by changing the halide, established them as highly essential materials, opening new opportunities in catalysis.7 Despite significant achievements in halide perovskite-based materials, still advancements have to be made to maintain their stability and non-toxicity, thereby enhancing their broad applicability.7,8 To achieve this, a major focus is on replacing the A cation in the ABX3 structure of perovskites. The scientific focus was also shifted majorly towards the replacement of the B cation,9 which stands for Pb in many of the previously reported works as Pb-based perovskites are toxic for our precious nature. Therefore, the development of lead-free halide perovskites without compromising their properties and efficiency is of prime importance. In this regard, we turned our interest to focus on the preparation of lead-free perovskite materials and their applications.
In our current work, we aimed to address four major challenges of the perovskite's domain, namely stability, reusability, toxicity, and efficiency of hybrid halide perovskites (HHPs).10 To tackle these issues, we synthesized a vacancy-ordered HHP, specifically MA2SnBr6 QDs, which had not been explicitly synthesized so far. MA2SnBr6 was synthesized via a greener approach, a simple yet effective solvent-free mechanochemical synthesis at room temperature without the use of additional capping agents. Interestingly, the synthesized MA2SnBr6 QDs maintained structural integrity in air and moisture, addressing a common issue with HHPs. To our surprise, these QDs are also stable in polar solvents such as isopropanol, ethanol, and acetonitrile, overcoming a major challenge typically encountered with halide perovskites. Remarkably, the significance of QDs is now widely recognized, as emphasized by the 2023 Nobel Prize in Chemistry awarded for the discovery and synthesis of QDs, highlighting their wide-ranging applications. Inspired by this notable achievement, the potential use of the synthesized air and moisture-resistant MA2SnBr6 QDs has been investigated for the photocatalytic conversion of renewable resources, including carbon dioxide and biomass-derived compounds, under sunlight, mimicking the process of artificial photosynthesis.
Therefore, for performing artificial photosynthesis, MA2SnBr6 QDs were applied for the simultaneous photocatalytic reduction of CO2 into CO and CH4 and oxidation of biomass-based alcohols and lignin model compounds to carbonyl compounds under natural sunlight. In this context, halide perovskites have been considerably explored for photocatalytic CO2 reduction;11 however, the majority of these relied on Pb-based perovskites,12–36 which are not compatible with the sustainability goals of carbon capture, utilization, and fixation.11 Further, previous studies employed co-catalyst-loaded perovskites,12,13,19,29 ternary systems,23,25,32,37 and complex heterojunctions37–43 to increase the photocatalytic activity, yet not succeeded in a promising manner. Moreover, the complex synthesis procedure of these photocatalytic systems makes them economically non-viable, thereby limiting their potential for large-scale process. Additionally, the full potential of halide perovskites has not been realized because they were not employed for the simultaneous photo-reduction of CO2 and photo-oxidation of biomass-based feedstocks. Thus, during the photo-reduction of CO2, the unutilized photogenerated holes are employed for oxidation reactions. In response, MA2SnBr6 QDs serves as Pb-free, single-component systems offering a synergistic way for utilizing photogenerated electrons for CO2 reduction to CO and CH4, while performing the conversion of biomass-derived compounds using photogenerated holes. Thus a sustainable pathway has been adapted without loading any co-catalyst and even without any sacrificial agent.
Results and discussion
Synthesis and characterization of MA2SnBr6 QDs
The MA2SnBr6 QDs were synthesized for the first time via a simple mechanochemical route and employed for photocatalytic CO2 reduction in synergism with the oxidation of biomass-derived compounds (Scheme 1a and b). The initial characterization was conducted by powder X-ray diffraction (PXRD) of the samples, as shown in Fig. S1. The PXRD pattern of MA2SnBr6 showed no peak corresponding to the reactants, MABr or SnBr4, confirming the successful synthesis of pure MA2SnBr6 perovskites.44 | ||
| Scheme 1 Schematic of (a) the synthesis route followed for MA2SnBr6 QDs, and (b) MA2SnBr6 perovskite QDs applied for photocatalytic CO2 reduction coupled with oxidation of biomass-derived compounds. | ||
The MA2SnBr6 QDs were kept under ambient conditions and PXRD was conducted after one year, where no change was observed in the PXRD pattern of MA2SnBr6, demonstrating the retention of the MA2SnBr6 crystalline structure even after one year (Fig. 1a). The diffuse reflectance UV-vis (DR UV-vis) spectrum of MA2SnBr6 is presented in Fig. 1b, revealing an absorbance onset at 462 nm, and the corresponding Tauc plot indicates a band gap of 2.80 eV (Fig. 1c). To investigate the surface characteristics, initially, a survey scan of MA2SnBr6 was recorded by X-ray photoelectron spectroscopy (XPS). The survey scan showed the presence of C, N, Sn, and Br elements in the perovskite sample with elemental atomic compositions of 25.9%, 13.7%, 11.4%, and 49.0%, respectively (Fig. S2a), confirming the Br-enriched surface in the synthesized MA2SnBr6 material. The high-resolution XPS (HR-XPS) of Sn 3d was recorded, which showed two peaks at binding energies of 487.3 and 495.8 eV corresponding to Sn 3d5/2 and Sn 3d3/2, respectively (Fig. 1d). These peaks confirmed the presence of Sn in the +4 oxidation state. The HR-XPS of Br 3d was deconvoluted into two peaks centered at 69.0 eV and 70.2 eV attributed to binding energies of Br 3d5/2 and Br 3d3/2, respectively (Fig. S2b). Further, the HR-XPS of C 1s was deconvoluted into two peaks centered at 284.7 eV and 286.2 eV corresponding to the C–C/C–H and C–N binding energies, indicating the presence of methyl and methyl ammonium groups, respectively, in MA2SnBr6 (Fig. S2c). The C–N peak also appeared in the N 1s HR-XPS at 402.0 eV, further confirming the presence of methyl ammonium groups (Fig. S2d).
 | ||
| Fig. 1 (a) PXRD patterns of MA2SnBr6 after one year; (b) powder DR UV/Vis spectra of MA2SnBr6; (c) Tauc plot of MA2SnBr6; (d) Sn 3d HR-XPS of MA2SnBr6; and MAS solid-state NMR spectra of (e) 119Sn, and (f) 79Br of MA2SnBr6. | ||
The chemical structure of MA2SnBr6 was also scrutinized by CP/MAS solid-state NMR spectroscopy, where the 119Sn NMR spectra showed a peak with a chemical shift at −1970 ppm (Fig. 1e), indicating the presence of single sites of the Sn atom. Then, 79Br NMR was also recorded to see the bromine environment in MA2SnBr6. Notably, a single peak was observed at 66 ppm (Fig. 1f), stipulating the presence of one type of Br atoms, which are bonded to Sn in the perovskite structure. Next, the 13C NMR spectra showed a peak at a chemical shift of 33.2 ppm (Fig. S3a) which corresponded to the –CH3 group present in the MA2SnBr6 perovskite. In the 15N NMR spectra, the peak observed at 31.7 ppm (Fig. S3b) was attributed to the ammonium group present in MA2SnBr6. Subsequently, the chemical structure of MA2SnBr6 was characterized by ATR-FTIR spectroscopy (Fig. S4), where the stretching frequency at 900 cm−1 was attributed to the rocking vibration of CH3NH3+, the stretching at 1473 cm−1 corresponded to C–H bending, the peak at 1574 cm−1 was assigned to the N–H bending vibration, the peak at 3180 cm−1 was assigned to the N–H asymmetric stretching vibration, and the peak at 543 cm−1 was assigned to the Sn–Br stretching vibration. The Raman spectral analysis was performed to study the vibrational properties of MA2SnBr6, where the Br–Sn–Br asymmetric bending deformation was observed at δ(F2g) = 119.5 cm−1, Sn–Br asymmetric stretching was observed at v(Eg) = 140.7 cm−1 and Sn–Br symmetric stretching was noticed at 185.8 cm−1 (Fig. S5).
In the microscopy study, the FESEM images were acquired to examine the morphology of MA2SnBr6, revealing a particle-like structure (Fig. 2a). To gain a clear insight, transmission electron microscopy (TEM) was performed, unveiling clusters of small particles for the MA2SnBr6 perovskite (Fig. 2b). The high-resolution TEM (HR-TEM) image provided a detailed picture, where it was found that the MA2SnBr6 perovskites were actually QDs. Further analysis of HR-TEM provided d-spacing values of 0.36 nm and 0.28 nm corresponding to the (311) and (040) planes, respectively (Fig. 2d). Additionally, the TEM analysis showed that these QDs have a size range of 3–4.6 nm with an average particle size of 3.8 ± 0.3 nm (Fig. 2f), and the inset of Fig. 2f presents the particle size distribution of MA2SnBr6 QDs. To assess the long-term stability of MA2SnBr6, TEM and HRTEM images of the same sample stored under ambient conditions for one year were recorded (Fig. 2c and 2e), and its morphology and dispersity with as-synthesized MA2SnBr6 were compared. The TEM image of MA2SnBr6 (after one year) shows clusters of small particles (Fig. 2c), and the HR-TEM images reveal agglomerated QDs for MA2SnBr6 (Fig. 2e) with an average particle size of 3.7 ± 0.3 nm comparable to that of the freshly synthesized material. Additionally, the measured lattice fringes exhibited a d-spacing value of 0.36 nm corresponding to the (311) plane and 0.28 nm representing the (040) plane (Fig. 2e). Hence, the morphology, dispersity, and crystallinity of MA2SnBr6 remained well-preserved even after one year of exposure to air and moisture. The observations, supported by PXRD, TEM, and HRTEM analyses, collectively demonstrated the excellent long-term stability of the MA2SnBr6 perovskite.
 | ||
| Fig. 2 (a) FESEM image; TEM image of (b) fresh MA2SnBr6 and (c) MA2SnBr6 after one year; HR-TEM image of (d) fresh MA2SnBr6 and (e) MA2SnBr6 after one year; and (f) TEM image showing the distribution of MA2SnBr6 QDs (inset shows the particle size distribution of QDs). | ||
Further, the thermogravimetric analysis (TGA) indicates that MA2SnBr6 exhibits good thermal stability up to 230 °C, after which decomposition begins, attributed to the degradation of the methylammonium group in MA2SnBr6 (Fig. S6a). Next, MA2SnBr6 was dissolved in different polar solvents such as ethanol, isopropanol, acetonitrile, and ethyl acetate, and the collected samples were subjected to PXRD analysis (Fig. S6b). No changes were observed in the PXRD patterns, demonstrating the excellent chemical resilience of the MA2SnBr6 perovskite in polar solvents. Further, the electronic band structure of MA2SnBr6 was deduced from ultraviolet photoelectron spectroscopy (UPS) measurements (Fig. S6c), and the corresponding schematic of the band energy diagram is presented in Fig. S6d.
Photocatalytic performance of MA2SnBr6 QDs
Showcasing their sufficient reduction potential, the MA2SnBr6 QDs were employed for photocatalytic CO2 reduction coupled with the simultaneous oxidation of biomass-derived alcohols and lignin model compounds under simulated light (Oriel AM 1.5 with 100 mW cm−2 intensity). The electrons from the MA2SnBr6 photocatalyst were utilized to carry out CO2 photoreduction producing CO and CH4 as reduced products, while the holes are synergistically employed for the selective oxidation of p-methoxybenzyl alcohol (pMBA) to p-methoxybenzaldehyde (pMBAL) as the model compound. The corresponding gas chromatography-mass spectrometry (GC-MS) chromatogram is presented in Fig. S7. Notably, the single-system MA2SnBr6 QDs achieved impressive production rates (Table 1) of 483 μmol g−1 h−1 for CO and 518 μmol g−1 h−1 for CH4, alongside a yield of 253 μmol g−1 h−1 of pMBAL with >99% of selectivity over 24 h (entry 1, Fig. 3a). In contrast, the optimized reaction performed without the catalyst resulted in no production of CO, CH4, or pMBAL. Similarly, when the reaction was conducted in the absence of light, only a yield of 13 μmol g−1 h−1 of pMBAL was observed, with no formation of CO and CH4 (Fig. 3a). These results highlighted the necessity of both the catalyst and light for the effective reduction of CO2 and oxidation of pMBA. To evaluate the practical applicability of MA2SnBr6 QDs for CO2 reduction with simultaneous pMBA oxidation, the photocatalytic reaction was carried out under natural sunlight. The reaction setup under sunlight is depicted in Fig. S8a, and the corresponding sunlight intensity and temperature are plotted in Fig. S8b. Under natural sunlight, the reaction offered impressive yields 1218.9 μmol g−1 h−1 of CO and 1243.2 μmol g−1 h−1 of CH4 with a yield of 167 μmol g−1 h−1 of pMBAL within 6 h. Further optimization extended the reaction time to 48 h during which a continuous increase in the yields of CO, CH4, and pMBAL was observed (Fig. 3b). Subsequently, the apparent quantum yield (AQY%) was measured under different monochromatic light irradiation. The AQY% was determined based on CH4 production under irradiation of monochromatic light of 400, 420, 450, and 500 nm and was compared with the DR UV-Vis absorption spectra of MA2SnBr6 (Fig. S9). Notably, the highest AQY of 2.75% was attained at 400 nm in comparison to 1.88%, 0.92%, and 0.19% at 420, 450, and 500 nm, respectively, demonstrating a conspicuous relation between the irradiated spectral profile and photocatalytic activity of MA2SnBr6. Moreover, applicability of MA2SnBr6 QDs was explored for the oxidation of lignin model compound and various biomass-derived alcohols (Table 1), with simultaneous CO2 reduction to CO and CH4 (entries 1–8). Interestingly, the alcoholic group in 2-phenoxy-1-phenylethanol, a lignin model compound, was selectively oxidized to a keto group without affecting other moieties or the core structure of the substrate. In addition, vanillyl alcohol, veratryl alcohol, furfuryl alcohol, benzyl alcohol, p-chlorobenzyl alcohol, and p-methylbenzyl alcohol were oxidized to their corresponding aldehydes with synergistic CO2 reduction to CO and CH4 (entries 3–8).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Oxidation product | Reduction product | ||
| Conversion (μmol g−1 h−1) | Selectivity (%) | CO (μmol g−1 h−1) | CH4 (μmol g−1 h−1) | |||
| a Reaction conditions: 5 mg photocatalyst, 5 mL ethyl acetate, 0.05 mmol substrate, CO2 atmosphere, simulated light: 100 mW cm−2, time = 24 h, and temperature = 25 °C; the yield of products was determined by GC and GC-MS analyses. | ||||||
| (1) |
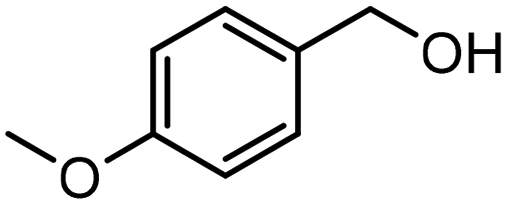
|

|
253 | >99 | 483 | 518 |
| (2) |

|

|
354 | >99 | 779 | 48 |
| (3) |

|

|
63 | >99 | 340 | 64 |
| (4) |

|

|
416 | >99 | 597 | 66 |
| (5) |

|

|
34 | >99 | 539 | 0 |
| (6) |

|

|
117 | >99 | 119 | 238 |
| (7) |

|

|
225 | >99 | 562 | 303 |
| (8) |

|

|
250 | >99 | 275 | 485 |

 | ||
| Fig. 3 (a) Photocatalytic reaction under different conditions, (b) time study, (c) PL spectra of MA2SnBr6 (PL excitation wavelength = 350 nm), and (d) time-resolved PL spectra of MA2SnBr6 (excitation wavelength 390 nm). | ||
Here, the synergistic CO2 reduction with biomass-derived alcohol oxidation shows marked differences in product selectivity. These differences can be explained by considering the structural and electronic properties of alcohol substrates, such as resonance (R) and inductive (I) effects. p-Methoxybenzyl alcohol (entry 1) shows nearly equal production of CO and CH4, attributed to the strong +R effect of the methoxy group, which increases the electron density on the benzylic carbon and promotes efficient oxidation, releasing sufficient electrons and protons for CO and CH4 production. A similar trend is observed in the case of p-methylbenzyl alcohol (entry 8), where the +I and hyperconjugative effects of the methyl group promote CH4 production. In contrast, significant CH4 production occurred in the case of p-chlorobenzyl alcohol (entry 7) despite the −I effect of the chloro group, possibly due to its moderate +R effect, which causes partial retention of electron density on the benzylic carbon. Further, benzyl alcohol (entry 6), where no substituent group is present, has higher CH4 formation than CO, indicating favorable proton and electron availability without any significant steric or electronic effect. Substrates such as 2-phenoxy-1-phenyl ethanol (entry 2) and veratryl alcohol (entry 4) exhibit higher CO selectivity, probably due to steric hindrance from bulky substituents and electron delocalization across the aromatic system, which can slow down the rate of oxidation and limit proton release, restricting CO2-to-CH4 conversion. Next, vanillyl alcohol (entry 3) possesses –OH and –OCH3 groups, which make the benzene ring electron-rich and stabilize the radical on the ring itself, diverting oxidation from benzylic position. Moreover, vanillyl alcohol undergoes intramolecular hydrogen bonding, which may hinder the substrate-catalyst interaction, thereby limiting oxidation and making fewer protons available for CH4 production. Notably, in the case of furfuryl alcohol (entry 5), only CO was produced, consistent with the electron-deficient furan ring, exhibiting −I and weak +R effects, which result in poor oxidation efficiency and minimal proton generation. Further, aliphatic alcohols, which are difficult to oxidize, provided corresponding aldehydes and esters in reasonably lower yields (entries 1 and 2, Table S1). In addition, the oxidation of benzylamine resulted in N-benzylidenebenzylamine in quantitative yields (entry 3, Table S1).
Electro- and photo-chemical studies of MA2SnBr6 QDs
Next, the photoelectrochemical measurements were carried out to inquest the charge transfer dynamics. First, the transient photocurrent response was conducted where MA2SnBr6 exhibited a swift response when intermittent light was irradiated at an interval of ∼30 s for 10 cycles (Fig. S10a). This proclaims the effective generation and separation of a significant number of charge carriers upon light illumination, emphasizing the critical role of light in generating photo-excited carriers.8 Subsequently, the electrochemical impedance spectroscopy (EIS) was conducted, where the Nyquist plot of MA2SnBr6 fitted to the Randles circuit model (Fig. S10b). It was found that the charge transfer resistance was significantly dropped after shining the light over the MA2SnBr6 electrode as compared to the dark state. This highlights the effective separation of charge carriers, facilitating enhanced charge migration across the surface of MA2SnBr6.45 The linear sweep voltammetry (LSV) was conducted to evaluate the CO2 reduction activity of MA2SnBr6 (Fig. S11). It was observed that the current density was increased when the light was irradiated showing the efficient charge separation and transfer over the surface of MA2SnBr6. Thereafter, the electrolyte was purged with CO2, and afterwards, LSV was performed. The increment in cathodic current accounts for the facile charge transfer of electrons from MA2SnBr6 to CO2, suggesting a good structure–activity relationship consistent with the photocatalytic experiments.Recycling and reusability of MA2SnBr6 QDs in the photocatalytic reaction
The chemical resilience of halide perovskites remains a challenge under photocatalytic reaction conditions. Notably, many previous studies have overlooked recyclability assessments post-photocatalysis. Therefore, the robustness of MA2SnBr6 was evaluated by employing it for five cycles of recyclability experiments (Fig. S12). The recyclability experiments revealed that the yields of pMBAL, CO, and CH4 were maintained without any significant decrease in photocatalytic activity, showcasing the excellent reusability of the MA2SnBr6 photocatalyst. After completing five cycles, the photocatalyst was examined by PXRD (Fig. S13a), DRUV-Vis spectroscopy (Fig. S13b), XPS (Fig. S14a–d), TEM (Fig. S15a), and HRTEM (Fig. S15b). The results indicated no change in the crystal structure, optical properties, surface characteristics, and morphology of MA2SnBr6, further proving its structural integrity even after repeated use.Reaction mechanism of the MA2SnBr6 QDs promoting the photocatalytic reaction
To investigate the underlying mechanism of the photoredox reaction of MA2SnBr6, illustrating the CO2 reduction coupled with biomass-derived alcohol oxidation, controlled experiments were conducted. First, the model reaction was carried out under an argon atmosphere, where no CO and CH4 were formed, showing that these products are exclusively formed through the photoreduction of supplied CO2 rather than from the degradation of the catalyst, solvent, or substrate (Fig. 3a). Additionally, 21 μmol g−1 h−1 of pMBAL was produced under an Argon atmosphere owing to the photogenerated hole-assisted oxidation of pMBA. Further, the isotopic labeling study was performed by supplying 13CO2 gas instead of 12CO2 to track the origin of formed CO and CH4 (Fig. S16). After the photocatalytic reaction, the gaseous samples were injected into GC-MS (Fig. S17), which detected the signal for 13CO and 13CH4 with m/z 29 and m/z 17, respectively (Fig. S18a and b), authenticating that CO2 is the only source to produce CO and CH4. Subsequently, a scavenger study was performed using CCl4 as an electron scavenger, where the production rate of CO and CH4 was significantly hampered, while the yield of pMBAL was comparatively increased. This result indicates that photogenerated electrons are responsible for CO2 reduction (Fig. 3a). When IPA was employed as a hole scavenger, a slight increase in CO production was observed with a noticeable decrease in CH4 yield compared to the optimized condition, whereas the pMBAL yield decreased significantly, which indicated the importance of holes in the oxidation of pMBA to pMBAL (Fig. 3a). The use of ethyl acetate as an aprotic solvent reduced the CH4 production rate, as it cannot supply protons necessary for CH4 formation, indicating that pMBA serves as the proton source in the reaction medium. The protons released during the oxidation of pMBA to pMBAL are essential for CH4 generation. However, the introduction of IPA suppresses this oxidation by scavenging photogenerated holes, thereby reducing proton availability in the reaction mixture. Consequently, the decreased proton concentration ultimately limits the formation of CH4. The in situ electron paramagnetic resonance experiments were conducted to trap the Cα radical of pMBA. The spin trap agent 5,5-dimethyl-1-pyrrolidine nitrogen oxide (DMPO) was used to trap the Cα radical of pMBA, and the resulting spectra (Fig. S19) presented a signal of six characteristic peaks, validating the formation of the adduct DMPO-Cα radical, verifying the intermediate α-hydroxy benzyl radical generation during photo-oxidation of pMBA to pMBAL.Next, the photoluminescence (PL) study of MA2SnBr6 was carried out at an excitation wavelength (λex) of 350 nm (Fig. 3c). A broad PL spectrum was observed, having a peak maximum of around 440 nm, which could be assigned to trap states. On subsequent addition of pMBA into MA2SnBr6, the PL emission intensity significantly decreased, which infers the participation of charge carriers in the oxidation of pMBA. Fig. 3d shows the time-resolved PL (TRPL) decay profile of MA2SnBr6, which reveals an average charge carrier lifetime of 4.5 ns. Upon the addition of the pMBA substrate, the decay dynamics significantly slowed down, resulting in an increased lifetime to 5.1 ns. The possible reason for this observation is the oxidation of pMBA, which is assisted by photogenerated holes, indicating that pMBA functions as an effective hole scavenger. The delayed dynamics suggest the active participation of photogenerated holes in the oxidation process, where they react with pMBA present in the system, facilitating its oxidation and leading to an increased average lifetime of charge carriers.
The excited-state dynamics of any catalyst significantly influences its photocatalytic activity.46,47 Hence, in order to delve deeper into the excited charge carrier dynamics of MA2SnBr6, we performed ultrafast transient absorption (TA) studies after 400 nm pump excitation. Fig. 4a shows the TA spectra of bare MA2SnBr6 upon 400 nm pump excitation, scanning in the spectral range from 420 nm to 650 nm at different pump–probe delay time scales. The pump fluence of 400 μJ cm−2 was used at which the possibility of auger recombination is negligible (as discussed in SI). The TA spectrum of MA2SnBr6 exhibits a positive absorption band spanning across the entire visible region, which peaked at 450 nm. This positive signal can be attributed to the excited state's absorption (ESA) of trapped charge carriers.48,49 Earlier, O. M. Bakr and co-workers similarly observed a positive band in the visible region upon 380 nm excitation for other lead-free perovskites, CsMnBr3.50 It is intriguing to see that no transient bleaching is observed here, which is possibly due to the presence of a large number of defects in MA2SnBr6.48 Keli Han and co-workers also observed a positive signal for the hydrothermally synthesized lead-free inorganic perovskite (Rb0.75Cs0.25)3InCl6 and attributed this signal to photoinduced absorption from trap states.51
 | ||
| Fig. 4 (a) Transient absorption spectra of bare MA2SnBr6, scanning range from 415 nm to 650 nm upon 400 nm pump excitation at various pump–probe delay time scales. Comparative dynamics between bare MA2SnBr6 and in the presence of (b) an electron scavenger, (c) hole scavenger, (d) CO2, and (e) pMBA upon 400 nm excitation. (f) Schematic of electron and hole pair generation in MA2SnBr6 QDs under 3.1 eV pump energy and their interaction with CO2 and pMBA. | ||
Further, we probed the TA kinetics of pristine MA2SnBr6 at a band maxima position of ∼450 nm (Fig. 4b) to investigate the charge carrier dynamics. The rise time of the signal was found to be 0.3 ps (Table S2), which suggests that there is almost no potential barrier for the trapping of the charge carriers.51 Further, the recovery dynamics were fitted with a bi-exponential function, obtaining two decay components (Table S2): an ultrafast time-scale component τ1 of 5.8 ps and a long lifetime component τ2 >1 ns. Here, the fast recovery component (τ2 = 5.8 ps) can be assigned to the surface defect trapping or cooling of hot self-trapped excitons in MA2SnBr6.48 The long-timescale component (τ2 >1 ns) is ascribed to the charge carrier recombination. Further, we conducted quenching experiments with electron and hole quenchers to determine whether the transient signal originated due to trapped holes or electrons. Fig. 4b and c show the comparative kinetics of bare perovskites before and after adding electron and hole quenchers, respectively. The signal intensity decreased (Fig. S20) in the presence of the electron scavenger, and faster decay dynamics were observed (Fig. 4b). The first recovery component τ1 decreased from 5.8 ps to 4.1 ps after adding the electron scavenger. Photogenerated electrons are scavenged by electron scavengers, leaving fewer electrons to recombine with the holes. Hence, the decrease in the transient signal intensity and faster recovery suggested that the signal corresponds to the trapped electrons. Thus, the transient signal in bare MA2SnBr6 can be easily assigned to ESA from trap states presented near the conduction band edge. However, in the presence of a hole-trapping agent, the decay dynamics became drastically slow (Fig. 4c), which validated the findings of the electron quenching experiment. The hole quencher scavenges the holes, leaving more electrons than holes. Hence, the slower decay observed further confirmed that electrons have a major contribution in the transient absorption signal of MA2SnBr6.
Next, we performed in situ transient studies in the presence of CO2 to examine its reactivity with photo-generated charge carriers in the bare MA2SnBr6 perovskite. Fig. 4d shows the comparative dynamics of bare MA2SnBr6 in the absence and presence of CO2. The decay dynamics becomes faster in the presence of CO2 than in the bare MA2SnBr6 perovskite. The first recovery component τ1 decreased from 5.8 ps in bare MA2SnBr6 to 4.9 ps in the presence of CO2. As explained earlier, the transient signal observed is due to the ESA of trapped electrons. Hence, the faster decay dynamics confirmed the active participation of MA2SnBr6 electrons in the CO2 reduction process,52 as schematically illustrated in Fig. 4f. In addition to that, we conducted ultrafast studies in the presence of pMBA to investigate the mechanism behind its oxidation. Fig. 4e represents the transient dynamics in the presence of pMBA. The slower dynamics was observed in the presence of alcohol substrates. The faster component (τ1) increased from 5.8 ps to 8.8 ps in the presence of pMBA. This delay in recovery suggests that holes are reacting with pMBA present in the system, eventually helping in the formation of pMBAL. Fig. 4f illustrates the photophysical mechanism responsible for the CO2 reduction and pMBA oxidation. The 3.1 eV pump generates the electrons and holes within the conduction and valence bands, respectively. The conduction band electrons diffuse to trap states, which react with CO2 molecules, producing CO and CH4. Conversely, the photogenerated holes react with biomass-derived pMBA to produce pMBAL.
The topographical image of MA2SnBr6 obtained via atomic force microscopy (AFM) is shown in Fig. S21, and its corresponding height profile reveals a thickness of 170 nm. Kelvin probe force microscopy (KPFM) was performed under dark and light conditions to analyze changes in the surface potential (Fig. 5a and c). In the dark, MA2SnBr6 delivers a contact potential difference (CPD) of 350 mV, which increased to 391 mV under illumination (Fig. 5b and d). The surface potential voltage (SPV) is defined as the change in the surface potential in the dark and under light irradiation (SPV = CPDlight – CPDdark), and is related to the photogenerated charge separation. The observed positive increment in CPD after irradiation of light (ΔCPD = 41 mV) suggests a more significant accumulation of photogenerated holes over the MA2SnBr6 surface than in the dark, enhancing exciton separation and improving the photocatalytic activity.53 To delve deeper into the mechanistic insights, the CO2 temperature-programmed desorption (CO2-TPD) analysis was performed to observe the adsorption behavior of CO2 over MA2SnBr6 (Fig. S22). The strong desorption peak observed between 180 and 190 °C indicates chemisorbed CO2 molecules over the surface of MA2SnBr6.
 | ||
| Fig. 5 KPFM images of MA2SnBr6 under (a) dark conditions and (b) its corresponding contact potential difference profile and (c) under light conditions and (d) its corresponding contact potential difference profile. | ||
Computational studies
Further, CO2 adsorption and activation on MA2SnBr6 were comprehensively studied by first-principles-based computational simulations, which elucidated the detailed mechanisms and pathways of the CO2 reduction reaction (Fig. 6a). Following the experimental results, two primary CO2 reduction pathways are considered: CO2 to CO and CO2 to CH4. Band-decomposed charge density analysis reveals that the localized charge around Br atoms originates from Br p-orbitals, indicating their critical role in CO2 reduction reaction (Fig. 6b and c). The Br active sites, enriched at the surface, play a pivotal role in CO2 activation and intermediate stabilization. As validated by XPS, a higher concentration of Br on the surface of MA2SnBr6 was observed (Fig. S2a), and computational results further highlight the localization of charge density around Br p-orbitals, making it a key catalytic site, thereby enhancing the catalytic efficiency of MA2SnBr6. | ||
| Fig. 6 (a) Optimized structure of MA2SnBr6. (b) Band decomposed charge density plot at the defect state. (c) 2D plot for localized charge density at the Br atom. (d) Reaction energy profile for CO2-to-CO and CO2-to-CH4 reduction. (e) Optimized structures of the catalyst and various intermediates involved in the reaction pathway (the number in each box represents the step number assigned in the reaction energy profile). (f) Schematic showing the mechanism for CO2 reduction over the surface of the MA2SnBr6 photocatalyst. Color keys: gray, tin; brown, bromine; blue, carbon; pink, nitrogen; and orange, hydrogen. | ||
Density functional theory calculations were conducted to optimize the photocatalyst design by simulating the reaction energetics of the CO2 reduction reaction, as shown in Fig. 6d. The optimized structures of the catalyst MA2SnBr6 and various intermediates involved in the reaction pathway are displayed in Fig. 6e, and the plausible mechanism for CO2 reduction on the MA2SnBr6 surface is presented in Fig. 6f. Typically, the photocatalytic CO2 to CO or CH4 reduction steps include the first proton-coupled electron transfer to generate a carboxyl intermediate (*COOH), and subsequently, the second charge transfer (one electron and one proton) for the formation of *CO intermediate as well as the desorption of CO for the final CO product (Fig. 6f, path-1). For CO2 to CH4, the reaction proceeds through path-2: CO2→ COOH*→ CO* → CHO* → CH2O* → CH3O* → CH4. Note that the adsorption energy value is scaled down to 0 eV here to track the energy changes in subsequent steps precisely. The formation of the COOH* intermediate is uphill by 0.18 eV. However, the transformation of COOH* to *CO is slightly downhill in energy by 0.13 eV. The formation of *COOH and its conversion to *CO are critical intermediates that determine the thermodynamics of the reaction pathway. At this stage, two competing possibilities arise: (1) desorption of CO as the final product or (2) the further reduction of *CO to form *CHO, leading to the formation of CH4. DFT calculations reveal that the desorption of CO from the catalytic surface requires an energy input of 0.49 eV, making this step thermodynamically feasible under appropriate reaction conditions. However, the formation of the *CHO intermediate is energetically downhill by −0.38 eV, favoring the progression towards CH4 formation. This downhill energy profile for *CHO formation indicates that, given sufficient energy, the catalyst can selectively drive the reduction pathway toward methane instead of terminating at carbon monoxide.
Despite the thermodynamic feasibility of both pathways, the rate-determining step (RDS) in the CO2 reduction process is the desorption of CO, which requires an energy input of 0.49 eV. This RDS acts as a bottleneck for CO formation, potentially limiting the efficiency of the CO2 reduction when CO is the desired product. However, the experimentally determined band gap of the photocatalyst is 2.8 eV, which is significantly higher than the energy required for CO desorption. Under light illumination, the catalyst generates sufficient photoexcitation energy to overcome the desorption barrier, ensuring that both CO desorption and subsequent *CHO formation become thermodynamically and kinetically favorable. This observation highlights the dual capability of the photocatalyst to selectively produce either CO or CH4, depending on the operating conditions.54 The computational analysis and comparison with experimental findings confirm that the photocatalyst can facilitate both CO and CH4 production.
These insights underscore the importance of catalyst design in tuning the product selectivity for the photocatalytic CO2 reduction reaction. The detailed mechanistic study highlights the dual functionality of the MA2SnBr6 surface in facilitating both CO2-to-CO and CO2-to-CH4 conversions. The localized charge density on Br p-orbitals enhances the catalytic efficiency, and the system's band gap provides sufficient driving force for CO2-to-CO and CO2-to-CH4 conversion. These findings underline the potential of MA2SnBr6 as an efficient photocatalyst for CO2 reduction.
Furthermore, the photocatalytic activity of MA2SnBr6 for CO2 reduction was compared with previously reported perovskites, as shown in Fig. 7 and Table S3. It was observed that most of the prior studies utilized lead-based perovskites, predominantly complex perovskite heterojunctions, and yet suffered a low production rate of CO. Moreover, even after significant attempts, the photoreduction of CO2 to CH4 was still not promising as it is an 8 e− process and demands high energy as compared to the 2 e− reduction process of CO2 to CO. Therefore, addressing the challenge, MA2SnBr6 displayed a remarkable electron consumption rate (Re) of 5110 μmol g−1 h−1 under simulated light and 12383 μmol g−1 h−1 under natural sunlight, significantly surpassing previously reported halide perovskite-based systems. Consequently, as a lead-free, single-component material, MA2SnBr6 not only achieves a superior CO2 reduction rate with a higher production rate of CO and CH4 but also simultaneously facilitates the photo-oxidation of biomass-derived compounds into value-added chemicals.
 | ||
| Fig. 7 Comparison of the electron consumption rate (Re) of previously reported halide perovskites. The referenced articles: ref. [S1–S44]. | ||
Conclusions
We successfully addressed the synergistic transformation of CO2 and biomass valorization to solar fuels and high-value fine chemicals utilizing the hybrid halide perovskite MA2SnBr6. Herein, the stability, reusability, toxicity, and efficiency issues of hybrid halide perovskites (HHPs) were solved. The air-stable, ambient, and polar solvent-friendly vacancy-ordered MA2SnBr6 QDs are disguised in solving one of the major challenges while dealing with the halide perovskites. For the first time, MA2SnBr6 QDs were employed in a photoredox process for CO2 reduction, and the icing on the cake is the simultaneous oxidation of biomass-derived alcohols to value-added products. MA2SnBr6 displayed an exceptionally high electron consumption rate (Re) of 5110 μmol g−1 h−1 under simulated light and 12383 μmol g−1 h−1 under natural sunlight, significantly surpassing previously reported halide perovskite-based systems. Based on in situ transient absorption, in situ EPR experiments, and computational studies, the detailed mechanism is unravelled for this photo-redox reaction to valorise CO2 and biomass-based feedstocks. Consequently, as a lead-free, single-component material, without any co-catalyst and sacrificial agent, MA2SnBr6 not only achieves significantly higher yields of CO and CH4 but also synergistically facilitates the photo-oxidation of biomass-derived alcohols into valuable chemicals, making a significant contribution towards sustainable solar fuel generation and fine chemical production.
Experimental section
Chemicals
Methylamine hydrobromide, ethyl acetate, and tetrabutylammonium hexafluorophosphate were purchased from TCI Chemicals, India. Isopropanol, acetonitrile, and tin(IV) bromide were purchased from Sigma-Aldrich, India. Ethanol was purchased from Merck, India.Characterization
A Bruker D8 Advanced diffractometer, which featured a scintillation counter detector and operated with Cu-Kα radiation (λ = 0.15418 nm, 2θ = 5–80°, scan rate = 4° min−1) source at 40 kV and 40 mA, was used to obtain powder X-ray diffraction (PXRD) patterns. The Raman spectrum was recorded using a WI Tech Raman Spectrometer with a 632 nm He–Ne laser. Diffuse reflectance spectra were recorded using an Agilent Cary 100 UV-vis spectrophotometer. ATR-FTIR spectroscopy was performed using a Bruker Vertex 70 spectrometer. X-ray photoelectron spectroscopy (XPS) and ultraviolet photoelectron spectroscopy (UPS) measurement of MA2SnBr6 was performed using an Al Kα X-ray source and a monochromator with ultra-high vacuum provided by Thermo Fisher Scientific. The He(I) (21.22 eV) gas discharge lamp was used as a source during UPS measurements. A Bruker Avance NEO 400 was employed for the measurement of solid-state MAS NMR spectra of 119Sn, 79Br, 13C, and 15N using a 5 mm FG NMR probe. Solid-state photoluminescence spectra (PL) were recorded using a Horiba Fluorolog instrument, and PL decay was studied using a TCSPC (time-correlated single-photon counting) assembly of the Horiba Fluorolog instrument. The SEM (scanning electron microscopy) images were collected using a JEOL instrument (JSM-7610FPlus). The morphology of MA2SnBr6 and the particle size of MA2SnBr6 were analyzed using a transmission electron microscope (TEM, JEOL) and a high-resolution TEM (HRTEM) at an accelerating voltage of 120 kV. EPR analysis was performed using a Bruker A300-9.5/12/S/W system. The measurement was performed at 25 °C using UV-Vis light procured from LOT-Quantum Design. For the experiment, 2 mg of MA2SnBr6 was dispersed in 1 mL of ethyl acetate followed by the addition of 10 μL of pMBA and spin-trap agent DMPO. The reaction mixture was purged with Ar to ensure an inert atmosphere. Initially, the EPR spectrum was recorded in the dark. Subsequently, the sample was illuminated for 30 min and the spectrum was recorded. The data acquisition setting involved a centre field of 3500 G, a sweep width of 300 G, and a static field of 3450 G with a modulation frequency of 100 kHz, a modulation amplitude of 4.91 G, and a sweep time of 81.92 s. The atomic force microscopy (AFM) and Kelvin probe force microscopy (KPFM) were performed using an Asylum Research MFP-3D AFM model with a Ti/Ir tip in the tapping mode. A Shimadzu GC-2014 instrument equipped with a mass detector was used to analyze the reaction products.Electrochemical measurement
Electrochemical analysis was performed in a standard three-electrode cell (a platinum wire as the counter electrode, a Ag/AgCl electrode as the reference electrode, and 0.1 M tetrabutyl ammonium hexafluorophosphate in dichloromethane as the electrolyte solution) using a Metrohm Autolab workstation. The ITO electrode coated with a catalyst was used as the working electrode and a 400 W xenon lamp was used as the light source. Prior to the use of the ITO substrate, it was washed by sonication in H2O, isopropanol, acetone, and again in H2O, and then the substrate was kept for drying in the oven. Then, 5 mg of MA2SnBr6 catalyst was dispersed in 50 μL of ethanol with 5 μL of Nafion. The prepared dispersion (10 μL) was drop-cast over the 0.25 cm2 area of the ITO electrode and allowed to dry at room temperature. The estimated amount of catalyst loading over the ITO electrode was about 0.8 mg cm−2. All the electrochemical measurements were carried out at 25 °C. Linear sweep voltammetry (LSV) run was carried out at 10 mV s−1 scan rate, and the potential value was reported in V vs. Ag/AgCl. CO2 was purged into the system for 30 min and LSV was recorded for the CO2-saturated electrolyte. To record the electrochemical impedance spectroscopy (EIS) spectra, a potential amplitude of 10 mV in the frequency range of 0.01 Hz to 100 kHz was employed. The Nova 2.1 FRA software was employed to analyse the impedance spectra.Transient absorption spectroscopy
Transient absorption spectroscopy was performed using a Ti:sapphire laser system with the regenerative 800 nm output pulsed at 1 kHz. The output was divided into two paths by a 70–30 beam splitter. The 30% of output of the regenerative pulse passed through the delay generator with a temporal resolution of 10 fs and focused on a sapphire crystal to produce a continuum probe pulse covering from 420 to 750 nm. The 70% percent of the output entered the spectrometer, where it passed through a BBO crystal to generate a 400 nm pump pulse. The pump pulse was chopped at 500 Hz and focused perpendicular to the sample. The acquired data were analysed using Surface Xplorer software. First, scattered light was subtracted from each scan prior to chirp correction. Later on, transient spectra were taken at specific time delay, and the kinetics was fitted at a particular probe wavelength. The samples were kept under continuous stirring to avoid the degradation of the sample. AgNO3 and KI were used as electron and hole scavengers, respectively, for transient studies.Computational details
The DFT calculations are performed using the Vienna Ab initio Simulation Package (VASP)55 with frozen-core projector augmented wave (PAW) pseudopotentials.56 The Perdew–Burke–Ernzerhof (PBE) functional of the generalized gradient approximation (GGA) was employed for the exchange–correlation potential. Long-range van der Waals (vdW) interactions were calculated using the empirical correction method PBE + D3.57 A Γ-centered Monkhorst–Pack mesh with k-points of 1 × 3 × 3 was used for the simulations, and the plane wave energy cutoff was set to 420 eV.58 The convergence criterion for electronic optimization was 10−6 eV, while atomic optimizations were deemed complete when the forces on atoms were below 0.02 eV Å−1. To simulate the MA2SnBr6 system, a vacuum of 40 Å was introduced along one direction. A surface was created, and the top layers are relaxed during optimization, while the remaining layers were fixed in their bulk geometry. The Fermi level was set to zero for the band structure and density of states (DOS) calculations, and Gaussian smearing was applied with a width of 0.02 eV. The VASPKIT tool was used for post-processing, including the calculation of Gibbs free energy.The adsorption-free energy (ΔG*) and adsorption energies of reactants and intermediates were calculated using the following equation:
| ΔG = ΔE + ΔEZPE − TΔS |
| ΔEabs = Eslab(/molecule) − (Eslab + Emolecule) |
Synthesis procedure
Vacancy-ordered methyl ammonium tin bromide (MA2SnBr6) perovskite QDs were synthesized for the first time via a straightforward mechanochemical route, where MABr (methylammonium bromide) and SnBr4 (tin(IV) bromide) were taken in 1:1 stoichiometric ratio as a starting material and ground in a mortar and pestle. A yellow powder was obtained via this solvent-free and eco-friendly process. The MA2SnBr6 powder was then recrystallized in ethanol to produce MA2SnBr6 crystals.
Photocatalytic experiment
The photocatalytic activity of MA2SnBr6 was tested under a solar simulator (Oriel AM 1.5 at 100 mW cm−2 intensity). In a typical reaction, 5 mL of ethyl acetate and 0.05 mmol of p-methoxybenzaldehyde were taken in a round-bottom flask containing a certain amount of catalyst. The reaction mixture was sonicated and purged with argon for 10 min. The reaction mixture was purged with a balloon filled with CO2 for 30 min and then the round-bottom flask was sealed. After the reaction, the gaseous sample was injected into a Shimadzu gas chromatograph (2014) equipped with an FID detector. After that, the liquid mixture was separated by centrifugation and filtration for quantitative and qualitative analyses. The liquid products were analyzed using a Shimadzu gas chromatograph-mass spectrometer (GC-MS).Author contributions
Bhawna Rawat: conceptualization, methodology, investigation, writing – original draft, data curation, and visualization; Ankita Kumari: computational studies, writing; Manvi Sachdeva: performed transient absorption experiments and writing; Himanshu Bhatt: transient absorption methodology, data analysis; Dibyajyoti Ghosh: computational studies, writing – review and editing; Hirendra N. Ghosh: writing – review and editing; Rajenahally V. Jagadeesh: writing – review and editing; Kamalakannan Kailasam: conceptualization, supervision, writing – review and editing, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI. See DOI: https://doi.org/10.1039/d5ey00169b.Acknowledgements
B. Rawat thanks UGC New Delhi for fellowship under Grant 191620176340/(CSIR UGC NET) and INST Mohali for instrumental facilities. Prof. K. Kailasam expresses thanks to Anusandhan National Research Foundation (ANRF) for the Core Research Grant (CRG), sanction order No. CRG/2023/001875. Rajenahally V. Jagadeesh thanks the support from the European Union under the REFRESH-Research Excellence for Region Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Programme Just Transition.References
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966–977 CrossRef CAS PubMed
.
- S. M. Han, M. Park, S. Kim, C. Jeong, J. Kim and D. Lee, EES Catal., 2025, 3, 723–732 RSC
- Y. Li, H. Liu, J. Raj, M. Pishnamazi and J. Wu, EES Catal., 2025, 3, 843–855 RSC
- A. Jaryal, A. K. Singh, S. Dhingra, H. Bhatt, M. Sachdeva, H. N. Ghosh, A. Indra and K. Kailasam, EES Catal., 2024, 2, 1019–1026 RSC
- H. Li, D. Regaldo, C.-S. J. Wu, M. Prato, A. Treglia, H. Wang, W. Hempel, M. Sessolo, Y. Zhou, A. Olivati and A. Petrozza, Energy Environ. Sci., 2025, 18, 6618–6627 RSC
- W. Zhang, G. E. Eperon and H. J. Snaith, Nat. Energy, 2016, 1, 16048 CrossRef CAS
- J. Sheng, Y. He, J. Li, C. Yuan, H. Huang, S. Wang, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 13103–13114 CrossRef CAS PubMed
- N. Li, X. Chen, J. Wang, X. Liang, L. Ma, X. Jing, D. L. Chen and Z. Li, ACS Nano, 2022, 16, 3332–3340 CrossRef CAS PubMed
- S. Li, H. Lin, C. Chu, C. Martin, W. MacSwain, R. W. Meulenberg, J. M. Franck, A. Chakraborty and W. Zheng, ACS Nano, 2023, 17, 22467–22477 CrossRef CAS PubMed
- H. Huang, H. Yuan, K. P. F. Janssen, G. Solís-Fernández, Y. Wang, C. Y. X. Tan, D. Jonckheere, E. Debroye, J. Long, J. Hendrix, J. Hofkens, J. A. Steele and M. B. J. Roeffaers, ACS Energy Lett., 2018, 3, 755–759 CrossRef CAS
- J. Lee, S. P. Chai and L. L. Tan, ACS Energy Lett., 2024, 9, 1932–1975 CrossRef CAS
- Z. Zhang, M. Shu, Y. Jiang and J. Xu, Chem. Eng. J., 2021, 414, 128889 CrossRef CAS
- Y. F. Xu, M. Z. Yang, H. Y. Chen, J. F. Liao, X. D. Wang and D. Bin Kuang, ACS Appl. Energy Mater., 2018, 1, 5083–5089 CrossRef CAS
- M. Shu, Z. Zhang, Z. Dong and J. Xu, Carbon, 2021, 182, 454–462 CrossRef CAS
- N. Li, J. Wang, G. Zhao, J. Du, Y. Li, Y. Bai, Z. Li and Y. Xiong, ACS Mater. Lett., 2024, 6, 999–1006 CrossRef CAS
- A. Pan, X. Ma, S. Huang, Y. Wu, M. Jia, Y. Shi, Y. Liu, P. Wangyang, L. He and Y. Liu, J. Phys. Chem. Lett., 2019, 10, 6590–6597 CrossRef CAS PubMed
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., 2018, 130, 13758–13762 CrossRef
- Z. Mo, X. Zhu, Z. Jiang, Y. Song, D. Liu and H. Li, Appl. Catal., B, 2019, 256, 117854 CrossRef CAS
- J. F. Liao, Y. T. Cai, J. Y. Li, Y. Jiang, X. D. Wang, H. Y. Chen and D. Bin Kuang, J. Energy Chem., 2020, 53, 309–315 CrossRef
- L. Li, Z. Zhang, C. Ding and J. Xu, Chem. Eng. J., 2021, 419, 129543 CrossRef CAS
- Z. C. Kong, H. H. Zhang, J. F. Liao, Y. J. Dong, Y. Jiang, H. Y. Chen and D. Bin Kuang, Sol. RRL, 2020, 4, 1–7 Search PubMed
- Z. C. Kong, J. F. Liao, Y. J. Dong, Y. F. Xu, H. Y. Chen, D. Bin Kuang and C. Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS
- Y. Jiang, J. F. Liao, Y. F. Xu, H. Y. Chen, X. D. Wang and D. Bin Kuang, J. Mater. Chem. A, 2019, 7, 13762–13769 RSC
- Y. F. Xu, M. Z. Yang, B. X. Chen, X. D. Wang, H. Y. Chen, D. Bin Kuang and C. Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed
- Y. Jiang, J. F. Liao, H. Y. Chen, H. H. Zhang, J. Y. Li, X. D. Wang and D. Bin Kuang, Chem, 2020, 6, 766–780 CAS
- Y. Jiang, H. Y. Chen, J. Y. Li, J. F. Liao, H. H. Zhang, X. D. Wang and D. Bin Kuang, Adv. Funct. Mater., 2020, 30, 2–9 Search PubMed
- J. Hou, S. Cao, Y. Wu, Z. Gao, F. Liang, Y. Sun, Z. Lin and L. Sun, Chem. – Eur. J., 2017, 23, 9481–9485 CrossRef CAS PubMed
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS
- S. Bera, A. Patra, S. Shyamal, D. Nasipuri and N. Pradhan, ACS Energy Lett., 2022, 7, 3015–3023 CrossRef CAS
- Y. F. Xu, X. D. Wang, J. F. Liao, B. X. Chen, H. Y. Chen and D. Bin Kuang, Adv. Mater. Interfaces, 2018, 5, 1–8 CAS
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed
- L. Y. Wu, Y. F. Mu, X. X. Guo, W. Zhang, Z. M. Zhang, M. Zhang and T. B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed
- X. Wang, J. He, L. Mao, X. Cai, C. Sun and M. Zhu, Chem. Eng. J., 2021, 416, 2–10 Search PubMed
- X. Wang, J. He, J. Li, G. Lu, F. Dong, T. Majima and M. Zhu, Appl. Catal., B, 2020, 277, 119230 CrossRef CAS
- J. Wang, J. Wang, N. Li, X. Du, J. Ma, C. He and Z. Li, ACS Appl. Mater. Interfaces, 2020, 12, 31477–31485 CrossRef CAS PubMed
- S. Wan, M. Ou, Q. Zhong and X. Wang, Chem. Eng. J., 2019, 358, 1287–1295 CrossRef CAS
- S. Kumar, I. Hassan, M. Regue, S. Gonzalez-Carrero, E. Rattner, M. A. Isaacs and S. Eslava, J. Mater. Chem. A, 2021, 9, 12179–12187 RSC
- Z. Zhang, D. Li, Y. Chu, L. Chang and J. Xu, J. Phys. Chem. Lett., 2023, 14, 5249–5259 CrossRef CAS PubMed
- Z. Zhang, Y. Jiang, Z. Dong, Y. Chu and J. Xu, Inorg. Chem., 2022, 61, 16028–16037 CrossRef CAS PubMed
- X. D. Wang, Y. H. Huang, J. F. Liao, Y. Jiang, L. Zhou, X. Y. Zhang, H. Y. Chen and D. Bin Kuang, J. Am. Chem. Soc., 2019, 141, 13434–13441 CrossRef CAS PubMed
- Q. M. Sun, J. J. Xu, F. F. Tao, W. Ye, C. Zhou, J. H. He and J. M. Lu, Angew. Chem., Int. Ed., 2022, 61, e202200872 CrossRef CAS PubMed
- Y. Jiang, R. Zhou, Z. Zhang, Z. Dong and J. Xu, J. Mater. Chem. C, 2023, 11, 2540–2551 RSC
- Z. Zhang, D. Li, Z. Dong, Y. Jiang, X. Li, Y. Chu and J. Xu, Sol. RRL, 2023, 7, 1–10 Search PubMed
- B. Rawat, V. R. Battula, P. K. Nayak, D. Ghosh and K. Kailasam, ACS Appl. Mater. Interfaces, 2023, 15, 53604–53613 CrossRef CAS PubMed
- B. Rawat, V. R. Battula, A. Kumari, D. Ghosh and K. Kailasam, ACS Sustainable Chem. Eng., 2024, 12, 13124–13136 CrossRef CAS
- A. M. El-Zohry, B. Turedi, A. Alsalloum, P. Maity, O. M. Bakr, B. S. Ooi and O. F. Mohammed, Commun. Chem., 2022, 5, 1–7 CrossRef PubMed
- N. K. Tailor, S. Singh, S. K. Saini, Kaivalya, M. A. Afroz, M. Kumar, S. C. Peter, K. K. Pant and S. Satapathi, Adv. Funct. Mater., 2024, 2402894, 2–10 Search PubMed
- D. Sebastian, A. Pallikkara, H. Bhatt, H. N. Ghosh and K. Ramakrishnan, J. Phys. Chem. C, 2022, 126, 11182–11192 CrossRef CAS
- P. Singh, G. Kaur, N. Ghorai, T. Goswami, A. Thakur and H. N. Ghosh, Phys. Rev. Appl., 2020, 14, 14087 CrossRef CAS
- J. Almutlaq, W. J. Mir, L. Gutiérrez-Arzaluz, J. Yin, S. Vasylevskyi, P. Maity, J. Liu, R. Naphade, O. F. Mohammed and O. M. Bakr, ACS Mater. Lett., 2021, 3, 290–297 CrossRef CAS
- P. Han, W. Zhou, D. Zheng, X. Zhang, C. Li, Q. Kong, S. Yang, R. Lu and K. Han, Adv. Opt. Mater., 2022, 10, 2101344 CrossRef CAS
- X. Deng, J. Zhang, K. Qi, G. Liang, F. Xu and J. Yu, Nat. Commun., 2024, 15, 1–12 Search PubMed
- R. Chen, S. Pang, H. An, J. Zhu, S. Ye, Y. Gao, F. Fan and C. Li, Nat. Energy, 2018, 3, 655–663 CrossRef CAS
- Y. Jing and T. Heine, J. Mater. Chem. A, 2018, 6, 23495–23501 RSC
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef
| This journal is © The Royal Society of Chemistry 2026 |