
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFurfural electrovalorisation to hydrofuroin with near-unity faradaic efficiency on a single-atom zinc catalyst†
Jiaxiang
Chen‡
 a,
Songbo
Ye‡
b,
Fangxin
She
a,
Xin
Yang
a,
Fangzhou
Liu
a,
Zixun
Yu
a,
Zhi
Zheng
a,
Ming
Hong
c,
Qiang
Wang
d,
Yuan
Chen
a,
Hao
Li
*b and
Li
Wei
*a
a,
Songbo
Ye‡
b,
Fangxin
She
a,
Xin
Yang
a,
Fangzhou
Liu
a,
Zixun
Yu
a,
Zhi
Zheng
a,
Ming
Hong
c,
Qiang
Wang
d,
Yuan
Chen
a,
Hao
Li
*b and
Li
Wei
*a
aSchool of Chemical and Biomolecular Engineering, The University of Sydney, Darlington, New South Wales 2006, Australia. E-mail: l.wei@sydney.edu.au
bAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, Sendai, 980-8577, Japan. E-mail: hao.li.b8@tohoku.ac.jp
cCentre for Future Materials and School of Engineering, University of Southern Queensland, Springfield Central, Queensland 4300, Australia
dState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, Shanxi 030001, China
First published on 23rd June 2025
Abstract
Electrochemical valorisation of biomass to value-added chemical feedstocks holds great potential to reduce the reliance on fossil fuels and accelerate the realisation of a sustainable future. In this work, we show that hydrofuroin, an important feedstock for sustainable aviation fuels, can be selectively produced on a zinc (Zn) single-atom catalyst via the electrochemical furfural reduction reaction (FRR). Initial theoretical results show that the weak binding capability of a zinc (Zn) single-atom active center effectively suppresses the parasitic hydrogen evolution reaction (HER) while enabling fast desorption and dimerization of furfural radicals towards hydrofuroin formation, which was proved by our experimental validation. The catalyst, obtained by depositing zinc phthalocyanine on purified multi-walled carbon nanotubes, exhibits near-unity faradaic efficiency for hydrofuroin production in a wide potential window, e.g., −0.5 to −0.8 VRHE. The kinetic study further provides mechanistic insights into hydrofuroin formation on the single-atom site. This catalyst can be integrated into a flow cell electrolyser to achieve highly efficient furfural conversion to sustainable fuel precursors, which is beneficial for biomass electrovalorisation to value-added green products and chemicals.
Broader contextElectrochemical valorisation can transform biomass-derived small molecules into value-added chemical feedstocks under mild conditions by utilizing electricity generated from renewable sources, providing a promising approach towards a sustainable future chemical industry. Among various biomass-derived small molecules, furfural is an important building block that could be transformed into precursors for polymer production (e.g., furfural alcohol and 2-methylfuran) and, more importantly, hydrofuroin for sustainable aviation fuel production. In comparison to thermochemical methods used for furfural conversion, herein, we combined theoretical prediction and experimental validation. We demonstrated that a single-atom site with weak binding capability could be beneficial for the formation of hydrofuroin. This single-atom site could significantly suppress the parasitic hydrogen evolution reaction and the further reduction of furfural to other by-products and, consequently, realize highly efficient hydrofuroin production with nearly 100% faradaic efficiency. The mechanistic insights into the molecule conversion over such single-atom active sites could provide valuable guidance for catalyst design to further improve the electrovalorisation efficiency and accelerate the transformation towards sustainability. |
Introduction
Green chemical production from abundant renewable biomass could play a critical role in addressing climate change and realizing a sustainable future.1 Hemicellulose derived from lignocellulosic biomass is a promising precursor, and it can be converted to multiple platform chemicals, such as furfural and 5-hydroxymethyl furfural (HMF).2 The U.S. Department of Energy has identified furfural and HMF as leading biomass-derived platform chemicals that can be valorised to valuable products, including polymer precursors, biofuel and biochemicals.3,4 State-of-the-art furfural upgrading mainly produces furfuryl alcohol (FAL), other 5-membered oxygen-containing heterocycles (e.g., furan) and other fuel supplements following the conventional thermochemical routes.5,6 However, chromium (Cr) in copper chromite catalysts,7 along with high-pressure hydrogen (H2) generated by parasitic side-reactions under high-temperature conditions, causes environmental and safety concerns.8 This energy- and emission-intensive process also consumes and discharges large amounts of waste solvents during product separation and purification, highlighting the need for greener biomass valorization processes.9Electrochemical hydrogenation (ECH) has recently emerged as a promising and sustainable alternative approach for furfural electrovalorisation via the furfural reduction reaction (FRR).10 Compared to the conventional hydrolysis method, FRR can be powered by renewable electricity, thereby avoiding fossil fuel consumption, and utilize water as the proton source instead of hydrogen gas (H2). As shown in Scheme 1, FRR affords value-added products, including furfural alcohol (FA),11 2-methylfuran (2-MF),12 and hydrofuroin (HF).13–15 Among these products, hydrofuroin, which has a higher carbon number as a C10 dimer, has drawn intense interest. It can be further hydrogenated to alkanes, forming crucial components in sustainable aviation fuels.16,17
 | ||
| Scheme 1 (a) Furfural electroreduction reaction (FRR) to various products and the corresponding standard potentials. (b) Selective formation of hydrofuroin on a weak binding metal centre. | ||
Achieving highly selective hydrofuroin production requires efficient catalysts and optimized electrolysis conditions, e.g., electrolytic pH, ion concentration, and operation potential.11 Hydrofuroin is formed via the dimerization of a protonated furfural radical (FCHOH˙, where F stands for the furan ring in the furfural molecule) generated during FRR. Currently, noble metal (palladium, Pd) or copper (Cu) and lead (Pb)-based bulk or nanosized materials are the focus of research. For example, a polycrystalline palladium (Pd) catalyst could reach an HF yield of up to 94.5% in organic electrolytes, but its wide application is limited by the scarcity of the noble metal.18 A cheap Pb metal surface could achieve a faradaic efficiency of around 30% in an organic-aqueous mixed environment,19 yet Pb is a heavy metal and poses environmental issues. As alternatives, a Cu2O-derived nanowire is reported to produce HF with 83.5% selectivity in a 1 M KOH electrolyte.15 More recently, nitrogen-coordinated transition metal single-atom catalysts (M–N–C) have emerged as promising candidates for HF electrosynthesis with high atomic efficiency.20 By combining density functional theory (DFT) calculations with experiments, cobalt phthalocyanine (CoPc) and copper phthalocyanine (CuPc) molecules were adsorbed on the carbon nanotube (CNT) surface, affording a family of heterogeneous molecular catalysts (HMCs) that could exhibit good furfural electrovalorisation performance, opening a new avenue to enable efficient furfural upgrading to hydrofuroin.13 Their well-defined atomic structure is also beneficial for mechanistic investigation. Despite their good FEHF of over 60%, the parasitic hydrogen evolution reaction (HER)21 and the formation of by-products (e.g., FA) demand high-performance catalysts with improved selectivity for HF production.
The formation of HF on single-atom catalysts proceeds through the dimerization of radicals, which are derived from surface-adsorbed furfural molecules by accepting an electron and a proton, and the subsequent desorption.13 Therefore, a metal center with weak binding capability could be favorable for HF production. It facilitates fast furfural radical formation while suppressing proton adsorption to form H* (* stands for the metal active site), thereby minimizing the parasitic HER. The weak adsorption of the furfural radical could also inhibit the further reduction to FA and 2-MF by sequential electron and proton transfer steps. Based on this assumption, we show that molecularly dispersed zinc phthalocyanine (ZnPc) on a CNT substrate can provide a high-performance catalyst (denoted as ZnPc/CNT) for selective electrovalorisation of furfural to HF (Scheme 1b). Initial DFT calculation and microkinetic modelling results show that the fully filled Zn 3d orbitals facilitate the expected weaker binding capability and suppress HER activity compared to Co and Cu single-atom sites reported earlier. Subsequent experiments further validated the theoretical results, showing an FEHF of over 95% with significantly suppressed formation of H2 and by-products (i.e., FA and MF) in a wide potential window. Moreover, this catalyst can be integrated into a two-electrode flow cell, enabling direct HF production and recovery from the aqueous electrolyte.
Results and discussion
Theoretical FRR activity prediction
We first analyze the FRR activity of several MPc/CNT catalysts with different binding capabilities. Atomistic models were created by placing an MPc molecule (M = Co, Cu and Zn) on graphene nanosheets and allowing all atoms to relax for structural optimization (Fig. 1a; see details in the Experimental section). According to previous research, a flat adsorption configuration of furfural is more stable than upright adsorption configurations under hydrogenation reaction conditions on metals.7 Therefore, in this study, furfural was modelled with a flat orientation on the MPc/CNT catalyst models. Furthermore, seven potential adsorption sites on the furfural molecules (bottom of Fig. 1a) were assessed by considering their binding capability to the ZnPc model, and the O2 site has been identified as the most active (Fig. S1 and S2 in ESI†). The same sites were examined on CoPc and CuPc to evaluate their FRR and HER activity, with the geometry-optimized adsorption models compared in Fig. S3 in the ESI.† | ||
| Fig. 1 Theoretical analysis of the FRR performance on HMC models. (a) Optimized atomistic models of the ZnPc/CNT catalyst with different intermediates and the assignment of potential adsorption sites in the furfural molecule. (b) and (c) Calculated reaction free energy schemes along various pathways on different metal centers. (b) FRR and (c) HER pathways. (d) Calculated proton coverage and (e) and (f) microkinetic modelling results of (d) the HER and (e) FRR on different metal centers. | ||
Despite their similar atomic structures displayed in the models, these MPc molecules exhibit varied activity due to their varied d-electron fillings. The free energy diagrams for FRR and competing HER on the MPc/CNT models were first calculated. As shown in Fig. 1b, the rate-determining step (RDS) for Zn and Cu corresponds to the formation of FCHO* + H*, with energy barriers of 1.70 eV and 1.94 eV, respectively. In contrast, the stronger binding capability of Co made the desorption of FCHOH* as the RDS with a lower energy barrier of 1.34 eV. However, as compared in Fig. 1c, the adsorption energy of H* on Co is merely 0.06 eV, significantly lower than that of Zn (1.70 eV) and Cu (1.94 eV). This indicates an intrinsically high theoretical HER activity for Co, which has been frequently reported.22 The Heyrovsky step in the HER pathway is likely to occupy the limited metal active sites, thereby substantially suppressing the formation of the FCHOH radical and its dimerization to HF.
Subsequently, the microkinetic volcano plot for furfural hydrogenation activity was calculated and is displayed in Fig. 1d. By using the adsorption energy of *FCHO (EFCHO) as the descriptor, it can be found that ZnPc/CNT, which exhibits weak binding capability to conventional intermediates, could exhibit a much stronger binding capability, which leads to improved activity for the FCHOH radical formation for the subsequent dimerization towards HF formation. Meanwhile, the H* coverage on the single-atom active center between 0 and −1 V vs. a reversible hydrogen electrode (VRHE) was determined. As shown in Fig. 1e, in this potential window, where the FRR likely takes place, the Co metal sites are very likely to be fully occupied by H* via the Volmer step (H+ + e− + * → H*), resulting in significant HER activity, as reflected by the calculated exchange current density (j0) displayed in Fig. 1f, and suppressed FRR activity. On the contrary, the weak adsorption capability of Zn and Cu centers is beneficial for deactivating their HER activity, resulting in low calculated j0 values. Conclusively, these computation results indicate that ZnPc/CNT is the most promising catalyst due to its strong inhibition of the HER at the metal sites and high FRR activity towards the FCHOH radical formation.
Synthesis and characterization of the ZnPc/CNT catalyst
Based on the theoretical prediction, we prepared a Zn single-atom catalyst by depositing ZnPc on a carbon nanotube substrate, as schematically illustrated in Fig. 2a, affording a ZnPc/CNT catalyst that represents the atomic model used in our DFT calculation. The CNT substrate was purified using a multi-step method (see details in the Experimental section, Fig. S4–S6 in ESI†) to remove metal residues and surface oxygen-containing functional groups so as to avoid interference with the accurate assessment of FRR activity.23 Inductively coupled plasma atomic emission spectroscopy (ICP-AES) and inductively coupled plasma mass spectroscopy (ICP-MS) revealed undetectable iron residue (which is used as the catalyst for CNT synthesis) in the purified CNT substrate. X-ray photoelectron spectroscopy (XPS, Fig. S4 in ESI†) survey scan revealed a low oxygen content of about 0.2 at% after the purification. The high-resolution C 1s spectrum further showed the good quality of the nanotube (Fig. S5 in ESI†). No Fe feature can be found in the Fe 2p region. Moreover, the purified CNT substrate showed a clean surface under transmission electron microscope (TEM) observation (Fig. S6 in ESI†). | ||
| Fig. 2 Structural characterization of the ZnPc/CNT catalyst. (a) Synthesis of the ZnPc/CNT catalyst and a photograph of ∼2 g catalyst obtained in an upscale synthesis process. (b) A HAADF-STEM image and (c) EDX-STEM elemental mapping result. (d) High-resolution Zn 2p XPS spectra, (e) Zn K-edge XANES and (f) k3-weighted EXAFS spectra of ZnPc/CNT compared to reference samples. | ||
The highly dispersed ZnPc molecules on the CNT substrate exhibited minimal impact on the specific surface area of the catalyst, which is nearly identical to that of the CNT substrate (94 vs. 96 m2 g−1). These properties was obtained from their N2 physisorption isotherms (Fig. S7 in ESI†) using the Brunauer–Emmett–Teller (BET) method. A zinc loading of ca. 0.46 wt% was found by ICP-AES, suggesting that the ZnPc molecules are highly dispersed on the CNT surface. X-ray diffraction pattern (XRD) of the ZnPc/CNT catalyst is comparable to that of the CNT substrate, with no ZnPc features detected (Fig. S8 in ESI†). Compared to the clean surface of the CNT substrate, abundant bright (high-contrast) spots were found on ZnPc/CNT under high-angle annular dark-field scanning transmission electron microscopy (HADDF-STEM, Fig. 2b), which can be attributed to the individual Zn atoms in the highly dispersed ZnPc molecules. The corresponding energy dispersive X-ray (EDX, Fig. 2c) elemental mapping results confirm the uniform distribution of Zn and N elements in the catalyst.
XPS and X-ray absorption spectroscopy measurements revealed electronic interaction between the adsorbed ZnPc molecule and the CNT substrate. The high-resolution Zn 2p XPS spectra of bulk ZnPc and the ZnPc/CNT catalyst are compared in Fig. 2d. Both spectra exhibit spin–orbit split 2p3/2 and 2p1/2 peaks at ca. 1022 and 1045 eV, respectively. Negligible satellite peaks can be found due to the filled Zn 3d orbitals. A red-shifted binding energy of approximately 0.3 eV can be found after ZnPc adsorption on CNT, indicating the possible electron donation from the CNT substrate to ZnPc, consistent with previous reports.24 This charge transfer effect is considered in our theoretical analysis. On the contrary, the N 1s XPS spectra (Fig. S9 in ESI†) exhibited negligible changes before and after adsorption, exhibiting two features at 398.68 and 399.78 eV that can be assigned to Zn–N and imine-N (C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) in the phthalocyanine molecular skeleton, respectively.
C) in the phthalocyanine molecular skeleton, respectively.
Fig. 2e compares the normalized Zn K-edge X-ray absorption near-edge spectra (XANES) of ZnPc/CNT to those of bulk ZnPc and Zn foil references. ZnPc/CNT and ZnPc both exhibited a pre-edge peak at about 9666 eV due to the D4h symmetry of the Zn atom in ZnPc molecules. Upon adsorption, the slightly decreased pre-edge peak intensity can be attributed to the reduced symmetry level caused by the strong interaction between the adsorbed ZnPc and the CNT substrate.25 Meanwhile, the slightly reduced Zn white line intensity and red-shifted edge energy of the ZnPc/CNT further confirmed the electron donation from CNT to the surface-adsorbed ZnPc molecules, showing good agreement with the XPS results.26 The Fourier-transformed extended X-ray absorption fine structure (EXAFS) spectrum of the ZnPc/CNT is displayed in Fig. 2f, and the fitting results revealed a single Zn–N path in the first shell with a coordination number of 4 (see fitting results in Table S3 in ESI†). Compared to the bulk ZnPc, the Zn–N bond has slightly expanded by 0.026 Å due to the acceptance of electrons from the CNT substrate. Besides, no Zn–Zn feature can be identified, further confirming the molecular dispersion of ZnPc on the CNT substrate.
Electrochemical catalytic performance assessment
The FRR performance of ZnPc/CNT was examined in an H-cell separated by a pre-activated Nafion 117 membrane using a three-electrode configuration. ZnPc/CNT spray-coated on carbon cloth (1 × 1 cm2) at 0.1 mg cm−2 was used as the working electrode. An argon-purged 0.1 M potassium bicarbonate-carbonate buffer (pH = 10.2) with and without furfural was used as the electrolyte. HF standard sample was synthesized following earlier reports (see details in the Experimental section), and calibration curves of furfural, FA and HF (Fig. S10 in ESI†) were created using high-performance liquid chromatography (HPLC). The linear sweep voltammetry (LSV) curves of the catalyst and CNT substrate are compared in Fig. 3a. Without furfural addition, the ZnPc/CNT catalyst exhibited slightly improved HER performance compared to the CNT substrate (dashed lines), delivering a current density (j) of about −2.6 mA cm−2 at −0.8 V vs a reversible hydrogen electrode (VRHE). Upon adding 5 mM furfural, the j of ZnPc/CNT increased significantly to −18.3 mA cm−2 at −0.8 VRHE, which is over 2 times that of the CNT substrate, suggesting that the ZnPc is responsible for the observed FRR performance. Meanwhile, the onset potential of ZnPc/CNT (defined as the potential required to reach −0.5 mA cm−2) has also improved by ∼155 mV from −0.583 to −0.428 VRHE. These observations demonstrate that ZnPc/CNT exhibits more favourable reaction thermodynamics for furfural conversion compared to the HER. | ||
| Fig. 3 FRR performance in a three-electrode test. (a) LSV curves obtained in an electrolyte with (solid) and without (dashed) 5 mM furfural. (b) Representative discharging curves collected for 20 min. Calculated (c) furfural conversion, (d) faradaic efficiency (FE), and (e) carbon product selectivity at applied potentials from −0.5 to −0.8 VRHE over MPc/CNT (M = Co, Cu and Zn) catalysts. | ||
The FRR product selectivity of the ZnPc/CNT was further determined by quantifying the liquid and gaseous products after continuous discharging at various potentials for 20 min using HPLC and gas chromatography (GC). Representative current densities obtained at different operation potentials are compared in Fig. 3b. ZnPc/CNT could deliver stable current during the tests. The slightly decreased current density near the end of the test, which is more pronounced under low potential conditions (i.e., −0.8 VRHE), can be attributed to the continuously reduced furfural concentration in the 15 mL electrolyte during the reaction. Meanwhile, the FRR performances of the reference CoPc/CNT and CuPc/CNT catalysts are also recorded under identical conditions. An increasing furfural conversion from 18% to nearly 70% for the ZnPc/CNT catalyst was obtained upon increasing the operation potential from −0.5 to −0.8 VRHE (Fig. 3c). Similar furfural conversion trends were observed for the other two catalysts CoPc/CNT and CuPc/CNT. The liquid and gaseous products obtained at different potentials were quantified using chromatography methods, and the results are compared in Fig. 3d and e, including the three HMCs. The production of HF exhibited an increasing trend in terms of faradaic efficiency and carbon product selectivity upon increasing the operation potential from −0.5 to −0.8 VRHE (Table S4 in ESI†). The FEHF quickly increased from 82% at −0.5 VRHE to >95% at −0.6 VRHE for ZnPc/CNT. Even at a very negative potential of −0.8 VRHE, an FEHF of up to 96% can be well maintained, while the H2 formation is suppressed continuously. Further increasing the operation potential to −0.85 VRHE would slightly reduce the FEHF to 89.5%. (Fig. S11 in ESI†). This observation further confirms that the weak-binding catalyst could effectively suppress the parasitic HER while affording high selectivity towards HF formation (Fig. 3d). Despite CoPc/CNT exhibiting an earlier onset potential and higher current density (Fig. S12 in ESI†), the optimal ZnPc/CNT catalyst continuously exhibited higher selectivity towards HF formation (Table S4 in ESI†), in good agreement with our DFT analysis.
We further calculated the carbon product selectivity from the HPLC analysis results, which revealed a similar trend towards HF formation to the FEHF. At −0.5 VRHE, there is a noticeable formation of FA and other products (similar to previous reports13) could account for ∼20% of total converted furfural. However, the selectivity towards HF quickly increased to over 95% with negligible by-product formation when the potential is below −0.6 VRHE. This can be attributed to the faster desorption and accumulation of furfural radicals at a more negative potential, which kinetically favours the radical dimerization to HF. We selected the potential range of −0.5 to −0.8 VRHE for its optimal selectivity and stable electrochemical activity. Moreover, we calculated the mass activity (jHF-mass) and site-specific turnover frequency (TOFHF) for HF formation on the ZnPc/CNT following a reported method.27 At −0.6 VRHE, the ZnPc/CNT catalyst could deliver a jHF-mass of 60.4 A gcat−1 and a TOFHF of 4.45 s−1, which further increased to 151.5 A gcat−1 and 11.15 s−1 at −0.8 VRHE, respectively. This performance is superior to those of recently reported catalysts for HF production in aqueous electrolytes, as detailed in Table S5 and Fig. S13 in ESI,† (data acquired from DigCat.org).11,13,14,19,28–33 To avoid the effect of low furfural concentration during long-term testing, we evaluated the stability of ZnPc/CNT in a three-electrode flow cell configuration. Under a continuous flow of 1.2 mL min−1 with 5 mM furfural, ZnPc/CNT exhibited a stable current density over a 4-hour period at −0.6 VRHE (Fig. S14 in ESI†). The LSV curves obtained before and after the test are almost identical, suggesting that the catalyst has good stability.
HF formation mechanism on single-atom sites
By using DMPO as the capture agent, the six well-patterned resonance peaks found in the electron paramagnetic resonance (EPR, Fig. 4a) spectrum of the electrolyte after discharging at −0.6 VRHE show sextet hyperfine splitting (AN = 15.6 G and AβH = 22.3 G) that agree well with the DMPO–R˙ radical, which is also found in earlier.19 Meanwhile, the lack of the DMPO–H˙ features suggests that the FRR process on the single-atom Zn site does not involve the hydrogen-atom transfer (HAT) pathway, which has been observed on metal surfaces.34 This observation indicates that the FRR on the ZnPc/CNT catalyst may be initiated by proton and electron transfer to the furfural molecule adsorbed on the active site, followed by desorption and subsequent fast radical dimerization.35 | ||
| Fig. 4 FRR mechanisms on ZnPc/CNT. (a) EPR spectrum of the ZnPc/CNT collected after discharging at −0.6 VRHE in a 0.1 M K2CO3/KHCO3 electrolyte (pH = 10.2) for 10 min. (b) Tafel plots. (c) KIE test results. (d) Reaction order of HF formation with respect to cfurfural at different pH. (e) EPR spectrum of the catalyst collected after discharging at −0.6 VRHE in a 0.1 M KOH electrolyte (pH = 12.6) for 10 min. (f) N 1s XPS spectra of the catalyst collected before and after testing in the 0.1 M KOH electrolyte. | ||
We further calculated the Tafel slope from the plot derived from the HF partial current density (jHF) obtained from −0.45 to −0.75 VRHE. As displayed in Fig. 4b, a two-stage Tafel slope of 121 and 280 mV dec−1 in the high (−0.45 to −0.55 VRHE) and low (−0.55 to −0.75 VRHE) potential regions, respectively, was obtained. This observation suggests that in the high-potential region, the rate-determining step (RDS) is the electron transfer (ET) step before the formation of adsorbed furfural radicals (FCHOH˙),19 and the proton transfer is not involved in the RDS. It is only at very negative potential (less than −0.55 VRHE) that the slow proton generation, due to the sluggish water dissociation in the near neutral pH conditions or internal diffusion, limits the reaction rate and results in the larger Tafel slope. This two-stage kinetic performance is found to be universal for single-atom catalysts, e.g., CoPc or CuPc-based heterogeneous molecular catalysts, as shown in Fig. S15 in the ESI.† This assignment is further supported by the kinetic isotope effect (KIE) observed under deuterated conditions. Replacing H with D will not impact the product selectivity (Fig. S16 in ESI†). Therefore, the KIE factor for HF formation was calculated by comparing the HF partial current density obtained at different potentials. As shown in Fig. 4c, a small KIE factor of about 1.1–1.15 was found at −0.45 and −0.5 VRHE. It quickly increased to about 1.4–1.5 afterwards; the primary KIE effect indicates H/D involvement in the overall RDS of the reaction in the more negative potential region,36 showing good agreement with the Tafel analysis.
The FRR kinetics on the single-atom Zn active center is further assessed by examining its dependence on furfural concentration (cfurfural) and electrolyte pH (Fig. S17 in ESI†). The FRR order with respect to furfural is calculated from the slope between the cfurfural (4 to 315 mM) and HF formation rate obtained at −0.6 VRHE, which is calculated from the HF partial current density, jHF, obtained from the HF concentration determined by HPLC after 10 min of discharging in the electrolytes of different pH (7.5 to 13.6). As shown in Fig. 4d, in the low-concentration range (4 to 50 mM), the FRR to HF exhibits a positive reaction order of about 0.68 to 0.81. This observation further confirms that the dimerization should occur in the electrolyte instead of on the ZnPc active sites. Otherwise, a second reaction order should be obtained.7,13,19
Further increasing the furfural concentration above 50 mM will impact the FRR towards HF formation differently, which is pH-dependent. A negative reaction order of −0.15 is found under near-neutral pH (7.5) conditions. The reaction order gradually approaches zero with increasing pH, i.e., −0.05, 0.10 and 0.13 at a pH of 10.2, 12.6 and 13.6, respectively. The changes of reaction order, including its switch from positive to negative and varied dependence on pH, indicate different reaction mechanisms on the Zn single-atom site under different reaction conditions. Under near-neutral to weak alkaline conditions (pH = 7.5 and 10.2), the positive-negative switch of the reaction order with respect to furfural concentration suggests that furfural and protons compete for adsorption on the Zn active center, following a Langmuir–Hinshelwood (LH) mechanism at high furfural concentration. The adsorbed furfural forms the ketyl radical by accepting an electron and then a proton before desorption and the subsequent dimerization to form HF. This reaction pathway agrees with the recently proposed mechanism on single-atom active metal sites. It is different from the previously reported HF formation mechanism on the bulk metal electrode, e.g., Pb surface, where the ketyl radical is protonated in the outer sphere.37 Upon increasing the electrolyte pH from neutral to alkaline, the reaction switches from the HL to Eley–Rideal (ER) mechanism at high furfural concentration, evident from the positive reaction order, i.e., 0.18 and 0.24 obtained at pH = 12.6 and 13.6, respectively. This observation further suggests that internal diffusion (active site saturation by furfural) and external diffusion (proton transfer to the near-surface electrolyte) are the possible limiting factors. The change in proton dependence is further confirmed by the reaction order with respect to pH, which gradually increases from 0.14 to 0.28 at higher furfural concentrations (Fig. S18 and S19 in ESI†). The weak binding capability of the Zn single-atom site could enable fast desorption of the FCHO˙ radical and prevent its further reduction (hydrogenation) to form FA or 2-MF. Meanwhile, the increased local FCHO˙ radical concentration is kinetically beneficial for its dimerization to form HF. Moreover, the weak binding capability of Zn to H* could also suppress its HER activity, leading to high FE towards HF formation.
The formation of H intermediate in the ER mechanism under higher pH conditions was further confirmed by EPR measurement using DMPO as the capture agent at an identical furfural concentration (50 mM) and discharging potential and time (−0.6 VRHE and 10 min). As shown in Fig. 4e, a new nonet feature with hyperfine coupling constants of AN = 16.2 G and AβH = 22.2 G and an intensity ratio of roughly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2:1:2:1:2:1:1 has emerged as an addition to the sextet of DMPO–R˙. This new feature can be assigned to the formation of DMPO–H˙.34,38 This observation clearly indicates the existence of an additional adsorption site for the local generation and supply of H* to the ketyl radical under high pH and furfural concentration conditions. The EPR spectra collected in these two electrolytes at different potentials (i.e., form −0.5 to −0.9 VRHE) are further compared in Fig. S20 in the ESI.† While the intensity of the sextet corresponding to DMPO–R˙ increased gradually with the applied potential in the near neutral electrolyte (pH = 10.2), this increasing trend staggered under the alkaline condition (pH = 12.6). Meanwhile, the nonet feature of the DMPO–H˙ increased constantly, indicating the formation of more H˙, which resulted in the relatively lower FEHF obtained in the alkaline electrolyte at more negative potentials (e.g., 96% vs. 82% from electrolytes of pH = 10.2 and 12.6 at −0.8 VRHE, respectively). Moreover, computational mesokinetic analysis could be further included to gain a deeper understanding of the mechanistic observations.39,40
:1:2:1:2:1:2:1:1 has emerged as an addition to the sextet of DMPO–R˙. This new feature can be assigned to the formation of DMPO–H˙.34,38 This observation clearly indicates the existence of an additional adsorption site for the local generation and supply of H* to the ketyl radical under high pH and furfural concentration conditions. The EPR spectra collected in these two electrolytes at different potentials (i.e., form −0.5 to −0.9 VRHE) are further compared in Fig. S20 in the ESI.† While the intensity of the sextet corresponding to DMPO–R˙ increased gradually with the applied potential in the near neutral electrolyte (pH = 10.2), this increasing trend staggered under the alkaline condition (pH = 12.6). Meanwhile, the nonet feature of the DMPO–H˙ increased constantly, indicating the formation of more H˙, which resulted in the relatively lower FEHF obtained in the alkaline electrolyte at more negative potentials (e.g., 96% vs. 82% from electrolytes of pH = 10.2 and 12.6 at −0.8 VRHE, respectively). Moreover, computational mesokinetic analysis could be further included to gain a deeper understanding of the mechanistic observations.39,40
Post-electrolysis XPS measurement of the ZnPc/CNT catalyst suggests the formation of protonated N from the Zn–N bonding, as shown in Fig. 4f. This protonation is feasible in the negative potential window, covering the HER process. This observation agrees with our recent work that, in the late 3d metal phthalocyanine catalyst with weak adsorption capability (e.g., NiPc and CuPc), the nitrogen coordinating with the metal could be protonated and serve as the active center to provide H* intermediates.41 No Zn–Zn bond can be found from the Zn K-edge EXAFS spectrum of the ZnPc/CNT catalyst collected after the stability test (Fig. S21 in ESI†). Hence, the Zn single-atom sites can be well-stabilized by the coordinating nitrogen atoms.41
HF production in a two-electrode electrolyzer
Finally, we integrated this ZnPc/CNT catalyst into a two-electrode flow electrolyzer to assess its performance towards HF production. The electrolysis system is schematically illustrated in Fig. 5a, and a photograph of the cell is displayed in Fig. S22 (ESI†). This device has a 4 cm2 reaction area for the cathode and anode chambers, which are separated by a pre-activated Nafion 115 membrane. A piece of carbon cloth loaded with 0.5 mg cm−2 ZnPc/CNT was prepared by spray coating and was used as the cathode. The performance of this electrode assessed in the three-electrode configuration in a 0.1 M KOH electrolyte containing 0.1 M furfural is displayed in Fig. S23a in the ESI.† It could deliver a current density of 100 mA cm−2 at ca. −0.91 VRHE. Product quantification by HPLC and GC also showed that the ZnPc/CNT electrode could maintain high selectivity towards HF formation with an FEHF between 93% and 96% (Fig. S23b in ESI†). A piece of nickel foam coated with electrodeposited NiFe hydroxides was used as the anode (Fig. S24 and S25 in ESI†).42 The 0.1 M KOH containing 100 mM furfural electrolyte was circulated in the cathode chamber at a flow rate of 1.2 mL min−1, while a 0.1 M KOH solution was supplied to the anode chamber. | ||
| Fig. 5 HF production from a two-electrode electrolyzer. (a) Schematic of the two-electrode electrolyzer system. (b) LSV curve of the device operating with 100 mM furfural in 0.1 M KOH electrolyte. (c) Chronoamperometric test results obtained at Ecell = −1.8 and −2.0 V and (d) the corresponding product assessment results. (e) Stability performance of the device operating at −1.8 V for 12 hours. The inset is a photograph showing the segregated hydrofuroin in the aqueous electrolyte. | ||
The voltage-current response of the electrolyzer is displayed in Fig. 5b. The electrolyzer could deliver steady current output as the cell voltage increased, e.g., delivering −10 and −50 mA cm−2 at a cell voltage of −1.72 and −2.06 V (without iR compensation), respectively. The device could respond to applied potential stably, further delivering 100 mA cm−2 at about −2.32 V. The electrolyzer was further discharged at −1.8 and −2.0 V continuously for 30 min, and the current densities collected are displayed in Fig. 5c. It can be found that the ZnPc/CNT catalyst could operate stably. The liquid and gas products were assessed, and an FEHF of about 93–95% can be well maintained at different operation voltages, as compared in Fig. 5d. The analysis also confirmed that HF is the major carbon product.
Finally, we performed long-term electrolysis at −2 V for 12 hours. About 200 mL of catholyte containing 100 mM furfural was circulated in the cathode chamber. The furfural concentration was periodically quantified and maintained by replenishing with a concentrated solution (0.4 M). As shown in Fig. 5e, the electrolyzer could maintain a stable current density of around −48 mA cm−2 throughout the test. At the same time, a high FEHF of about 95–96% can be well maintained during the operation (inset in Fig. 5e) and only trace amounts of FA and H2 were found as by-products. This device could deliver an HF productivity of 8.78 mol m−2 h−1 or 1.76 mol gcat−1 h−1. Additionally, the cost of the ZnPc/CNT is negligible compared to the costly precious metal-based catalysts. For example, at a low loading of ZnPc in the catalyst, the catalyst cost is about US$ 100 kg−1 based on the current cost of ZnPc (US$ 30 g−1) and MWCNT (US$ 100 kg−1). The synthesis process eliminated the need for high temperature thermal annealing and acid washing used in pyrolysis-based synthesis of SACs. The organic solvent used in this process can be fully recycled and reused, ensuring both environmental sustainability and cost efficiency. At a catalyst loading of 2 mg cm−2, 1 kg of catalyst is enough to prepare 50 m2 of electrode for the integrated electrolyzer, demonstrating excellent scalability and highlighting significant economic challenges for larger scale industrial applications.
Conclusion
In summary, starting with theoretical thermodynamic calculations and microkinetic modeling analysis, we demonstrate that a single-atom active site, i.e., Zn, with weak binding capability could afford good furfural hydrogenation while suppressing the parasitic hydrogen evolution reaction. Experimental results observed on a heterogeneous molecular catalyst constructed from depositing ZnPc on a purified CNT substrate showed good agreement with the DFT analysis. The Zn metal center could facilitate easy desorption of the intermediate radical for fast local dimerization towards hydrofuroin formation while almost eliminating the parasitic HER with a high HF faradaic efficiency of over 95% in a wide potential range. The kinetic analysis further revealed the important role of furfural and electrolyte concentrations in determining the HF formation rate, enabling the identification of an optimal reaction condition for the electrovalorisation of furfural to hydrofuroin. By further integrating the ZnPc/CNT catalyst in a two-electrode flow cell electrolyzer, a near-pure HF phase can be obtained during extended operation, showing great promise towards practical biomass electrovalorisation.Experimental methods
Calculation method
Spin-polarised density functional theory calculation was performed using the Vienna Ab initio Simulation Package (VASP) to determine the binding energies of the furfural hydrogenation reaction and hydrogen evolution reaction. The revised Perdew–Burke–Ernzerhof (RPBE) functional within the generalized gradient approximation was utilized to account for electronic exchange and correlation effects. Valence electrons were represented by Kohn–Sham wave functions, expanded in a plane-wave basis set with a cutoff of 520 eV. Core electrons were treated using a projector augmented-wave method. We ensured the convergence of electronic energy to within 10−5 eV and structural relaxation to 0.05 eV Å−1. The Brillouin zone was sampled using a Monkhorst–Pack grid, satisfying the criterion that the product of the number of k-points in any direction and the basis vector length in that direction (k × a) exceeds 15 Å. All calculations incorporated spin polarization. To maintain adequate spacing, a vacuum of at least 12 Å was added perpendicular to the surface. The adsorption energy of ketyl (ΔEFCHO) and proton intermediates (ΔEH) was calculated by| ΔEFCHO = Etotal − Eslab − Efurfural | (1) |
| ΔEH = Etotal − Eslab − 1/2EH2 | (2) |
The binding energies of both FCHO* and H* were used as descriptors, based on the scaling relationships established in our prior research, for the microkinetic modelling. The original model relied on calculated energetics for distinct fcc (111) and hcp (0001) surfaces. Given the established scaling relationship between the binding energies of FCHO* and H*, as demonstrated in our earlier study, we streamlined the activity volcano model to a one-dimensional framework by focusing solely on the binding energy of FCHO* as the descriptor. In the microkinetic model of the hydrogen evolution reaction, the Volmer–Heyrovsky mechanism was considered. The rate constant was calculated by
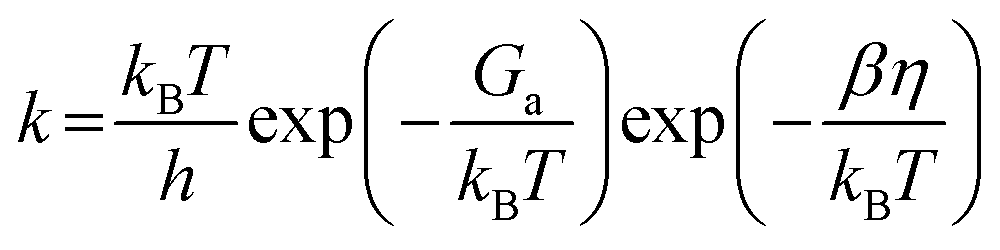 | (3) |
| j = ρneTOF | (4) |
The elementary steps for furfural hydrogenation towards FCHOH radicals are listed below:
| H2O + 2* ↔ H* | (5) |
| FCHO + * ↔ FCHO* | (6) |
| FCHO* + H* ↔ FCHOH* | (7) |
The considered elementary steps for the hydrogen evolution reaction are listed below:
| H+ + e− ↔ H* (Volmer) | (8) |
| H+ + e− + H* ↔ H2 (Heyrovsky) | (9) |
The energy barrier of the Heyrovsky step is derived from the scaling relationship between ΔGH* and Ga.
Materials
MWCNTs (∼20 nm diameter) were obtained from Nanografi and purified before use. Hydrochloric acid (HCl, 37%), sulfuric acid (H2SO4, 98%), hydrogen peroxide (H2O2, 30%), sodium hydroxide (NaOH, 98%), N,N′-dimethylformamide (DMF, 99%), isopropanol (IPA, HPLC grade), acetonitrile (CH3CN, HPLC grade), furfural (99%), furfuryl alcohol (FA, 98%), 5,5-dimethyl-1-pyrroline N-oxide (DMPO), magnesium (Mg) turnings (5–25 mm, 99.95%), sodium chloride (NaCl, ≥99.5%), sodium bicarbonate (NaHCO3, ≥99.7%), potassium bicarbonate (KHCO3, 99.7%), potassium carbonate (K2CO3, 99%), ammonium chloride (NH4Cl, ≥99.5%), magnesium sulfate (MgSO4, anhydrous, ≥99.5%), ethyl acetate (EtOAc, HPLC grade), n-hexane (LC grade), chloroform-d (CDCl3, 99.8 atom% D), deuterium oxide (D2O, 99.9 at% D), and Nafion 117 suspension (5% suspension) were obtained from Sigma-Aldrich and used without treatment. Zinc phthalocyanine (ZnPc) was obtained from Sigma-Aldrich and purified by triple-sublimation. The Nafion 115 membrane was obtained from fuel cell stores and pre-activated using H2SO4 and H2O2 treatment. The carbon cloth substrate was purchased from Sinerosz Technology (W1S1011). Deionized water (DI H2O) was prepared by using a MilliQ water system. Argon and hydrogen in argon (5% He/Ar) gas (Ar, 5.0) was obtained from BOC Australia.Carbon nanotube purification
The as-received CNTs were purified by first baking in air for 30 min at 300 °C, followed by bath sonication in 3 M HCl for 30 min and stirring in the acid for 6 h. The solid was collected by filtration, washed with DI H2O and dried in an oven before it was calcined at 1200 °C in a 100 sccm 5% H2/Ar flow for 2 hours. The purified CNTs are characterized to confirm their purity before being used for catalyst synthesis (Fig. S4–S6 in ESI†).Catalyst preparation
The ZnPc/CNT catalyst was prepared by dissolving 0.04 mM ZnPc in 50 mL DMF, followed by the addition of 50 mg purified CNT. The mixture was bath-sonicated for 30 minutes before being further stirred under ambient conditions for 24 hours. The catalyst was collected by filtration, washed with DMF and ethanol, and dried under vacuum. CoPc/CNT and CuPc/CNT catalysts were prepared following the same method.Synthesis of hydrofuroin (HF) standard
The HF standard is prepared following a reported method.43 Briefly, 200 mg furfural (1.89 mmol) was vigorously stirred with 1 g Mg turnings (41.67 mmol) in 10 mL 0.1 M NH4Cl aqueous solution at room temperature overnight. The mixture was then quenched with 3 M HCl before extraction with 20 mL EtOAc 3 times. The organic phase was collected, washed with a saturated NaHCO3 and NaCl solution, and dehydrated using anhydrous MgSO4. Afterwards, the organic phase was concentrated by rotary vaporisation to yield the crude product. Pure HF (36 mg) was then obtained by column chromatography using a gradient of n-hexane and EtOAc.Characterisation method
Powder X-ray diffraction (XRD) patterns were collected on a Stoe Stadi P diffractometer with a Cu-Kα source (λ = 1.5406 Å). Scanning electron microscope (SEM) images were obtained on a Zeiss Ultra Plus microscope. Spherical aberration-corrected high-angle annular dark field scanning transmission electron microscope (HAADF-STEM) images were recorded on a FEI Themis-Z microscope. The metal loading in different samples was determined on an inductively coupled plasma optical emission spectrometer (ICP-OES, Avio 500, PerkinElmer). X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Fisher Scientific K-Alpha+ spectrometer equipped with an Al-Kα (1486.3 eV) source. All spectra were collected using a pass energy of 20 eV and a spot size of 400 μm. X-ray absorption spectroscopy (XAS) measurements were acquired at the Australian Synchrotron. Data analysis and fitting were performed by using the Demeter Software package using the FEFF 9.0 code. Fourier-transformed extended X-ray absorption fine structure (EXAFS) analysis was performed with a K-range of 3 to about 12 Å−1 and an R-range of about 1 to 3 Å. Amplitude (S02), inner potential shift (ΔE0), shell radius correction (ΔR), and Debye–Waller factor (σ2) were used as fitting parameters.Electrochemical test
A catalyst ink was prepared by dispersing the catalyst powder in an IPA-H2O solution (9/1 = v/v) containing 0.05 wt% Nafion 117 ionomer at a concentration of 5 mg mL−1. The mixture was bath-sonicated for 30 minutes in an ice-water bath. The catalyst ink was spray-coated on a 1 × 1 cm2 carbon cloth electrode at 0.10 mg cm−2. The control electrode was prepared by loading only the carbon substrate at the same mass loading. Electrochemical tests were performed on a Metrohm Autolab PGSTAT302N potentiostat in a three-electrode configuration in a two-chamber H-shaped electrolyzer. The electrolysis temperature was maintained at 25 °C using water circulating through a jacket. The two chambers are separated by a pre-activated Nafion 117 membrane. The reference electrode was pre-calibrated and all potentials were reported to against the reversible hydrogen electrode (RHE, VRHE). A 0.1 M potassium carbonate-bicarbonate buffer (PCB, pH = 10) was used as the electrolyte. The electrolyte was pre-saturated by Ar bubbling for 30 min to remove dissolved oxygen and the catholyte was stirred using a magnetic stir bar at ∼600 rpm.Product quantification
About 0.5 mL catholyte was collected after electrolysis and diluted twice using CH3CN for product analysis on a Shimadzu LC-40XR high-performance liquid chromatography (HPLC) system equipped with a UV detector (220 nm) and a Shimadzu Shim-pack Scepter C18-120 column (3 μm, 4.6 × 50 mm). The sample was injected at 35 °C, and an H2O/CH3CN mixture (88.7/11.3 = v/v) was used as the mobile phase at a flow rate of 1 mL min−1. Calibration curves for furfural (F), furfuryl alcohol (FA) and hydrofuroin (HF) were established (Fig. S10 in ESI†). Furfural and FAL exhibit a retention time of 1.98 and 2.41 min, respectively. HF produced two peaks at 2.86 and 6.39 min due to two corresponding isomers.20 Hydrogen gas was quantified using a Shimadzu 2060 gas chromatography system with a thermal conductivity detector connected to the gaseous product outlet on the catholyte cell. By using the HPLC and GC results, the total furfural conversion rate (CF, %), product-specific selectivity (Yselectivityx, %, where x = F, FA, and HF) and faradaic efficiency (FEx, %, where x = F, FA, HF, and H2) were calculated using the equations shown below: | (10) |
 | (11) |
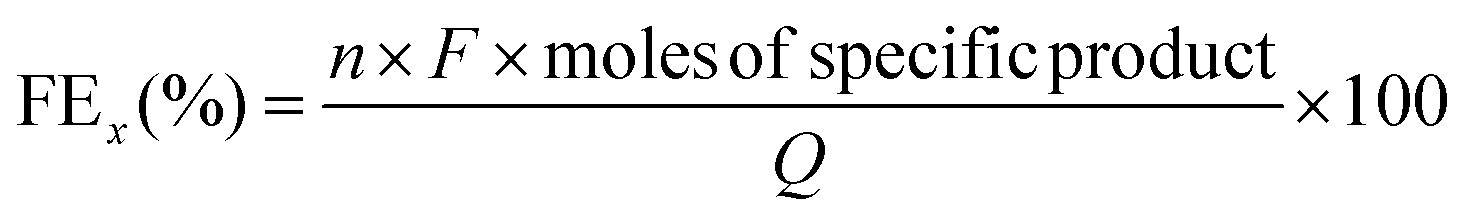 | (12) |
485 C mol−1), and Q is the total charge consumed (C).
Kinetic isotope effect (KIE) test
The KIE test was performed by replacing the DI H2O with D2O to prepare the electrolyte. 0.1 M K2CO3 was dissolved in DI H2O or D2O and saturated with Ar gas, before 5 mM furfural was added. LSV and chronoamperometric tests were performed in both electrolytes, and the product was quantified accordingly. Subsequently, the KIE factor was calculated as the ratio between the HF formation rates in the H2O and D2O electrolytes.Flow-cell assembly and test
A two-electrode flow cell was assembled and tested. The ZnPc/CNT catalyst loading on the 2 × 2 cm2 carbon cloth was increased to 0.5 mg cm−2 and served as the cathode. The catholyte was a 100 mM furfural in 0.1 M KOH solution circulated at 1.2 mL min−1 using a peristaltic pump for FRR. The anode is a nickel foam electrodeposited with NiFe hydroxides (NiFe/NiF), following a reported method.42 The cathode and anode are separated by a pre-activated Nafion 115 membrane. The catholyte is periodically sampled, and the furfural concertation is maintained by adding a concentrated furfural solution.Author contributions
J. X. designed and carried out the material synthesis and electrochemical test. S. Y. performed the DFT calculation with guidance from Q. W. and L. H. F. S. and Z. Y. helped with material characterization. X. Y. helped with EPR measurement. Z. Z. helped with XAS measurement. M. H. helped with electrolyzer design. Y. C., H. L. and L. W. formulated the idea and contributed to supervision and funding acquisition. J. X. and L. W. wrote the manuscript. All authors have contributed to the writing review and editing.Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was financially supported by the Australian Research Council under the Future Fellowship (FT210100218 and FT230100316) and Linkage Project (LP230200886). MH acknowledges support from iLAuNCH Trailblazer Programs. QW thanks the support from Science and Technology Cooperation and Communication Project of Shanxi Province (No. 202304041101016) and the Foundation of State Key Laboratory of Coal Conversion (Grant no. J24-25-610). The assistance and resources provided by Sydney Analytics, Sydney Nano, Sydney Microscopy & Microanalysis, and the Sydney Info Hub (SIH) at the University of Sydney are greatly appreciated. The authors are grateful for beamtime at the Australian Synchrotron, part of ANSTO, for XAS measurement under project ID 23466. The authors acknowledge the computational resources provided by the Hirose Foundation and Center for Computational Materials Science, Institute for Materials Research, Tohoku University (MASAMUNE-IMR, 202312-SCKXX-0203, 202312-SCKXX-0207, 202412-SCKXX-0209) and the National Computation Infrastructure (NCI, NCMAS-2024-59 and NCMAS-2025-116).References
- Y. Zhu, C. Romain and C. K. Williams, Sustainable polymers from renewable resources, Nature, 2016, 540(7633), 354–362, DOI:10.1038/nature21001.
- C. Tian, R. Dorakhan, J. Wicks, Z. Chen, K.-S. Choi, N. Singh, J. A. Schaidle, A. Holewinski, A. Vojvodic and D. G. Vlachos, et al., Progress and roadmap for electro-privileged transformations of bio-derived molecules, Nat. Catal., 2024, 7(4), 350–360, DOI:10.1038/s41929-024-01131-6.
- A. Jaswal, P. P. Singh and T. Mondal, Furfural – a versatile, biomass-derived platform chemical for the production of renewable chemicals, Green Chem., 2022, 24(2), 510–551, 10.1039/d1gc03278j.
- X. Li, P. Jia and T. Wang, Furfural: A Promising Platform Compound for Sustainable Production of C4 and C5 Chemicals, ACS Catal., 2016, 6(11), 7621–7640, DOI:10.1021/acscatal.6b01838.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Furfural: a renewable and versatile platform molecule for the synthesis of chemicals and fuels, Energy Environ. Sci., 2016, 9(4), 1144–1189, 10.1039/C5EE02666K.
- R. Bielski and G. Grynkiewicz, Furan platform chemicals beyond fuels and plastics, Green Chem., 2021, 23(19), 7458–7487, 10.1039/D1GC02402G.
- S. Liu, N. Govindarajan and K. Chan, Understanding Activity Trends in Furfural Hydrogenation on Transition Metal Surfaces, ACS Catal., 2022, 12(20), 12902–12910, DOI:10.1021/acscatal.2c03822.
- J.-P. Lange, E. van der Heide, J. van Buijtenen and R. Price, Furfural—A Promising Platform for Lignocellulosic Biofuels, ChemSusChem, 2012, 5(1), 150–166, DOI:10.1002/cssc.201100648.
- K. I. Galkin and V. P. Ananikov, When Will 5-Hydroxymethylfurfural, the “Sleeping Giant” of Sustainable Chemistry, Awaken?, ChemSusChem, 2019, 12(13), 2976–2982, DOI:10.1002/cssc.201900592.
- H. G. Cha and K.-S. Choi, Combined biomass valorization and hydrogen production in a photoelectrochemical cell, Nat. Chem., 2015, 7(4), 328–333, DOI:10.1038/nchem.2194.
- W. Xu, C. Yu, J. Chen and Z. Liu, Electrochemical hydrogenation of biomass-based furfural in aqueous media by Cu catalyst supported on N-doped hierarchically porous carbon, Appl. Catal., B, 2022, 305, 121062, DOI:10.1016/j.apcatb.2022.121062.
- S. Liu, Z. Mukadam, S. B. Scott, S. C. Sarma, M.-M. Titirici, K. Chan, N. Govindarajan, I. E. L. Stephens and G. Kastlunger, Unraveling the reaction mechanisms for furfural electroreduction on copper, EES Catal., 2023, 1(4), 539–551, 10.1039/D3EY00040K.
- Z. Mukadam, S. Liu, A. Pedersen, J. Barrio, S. Fearn, S. C. Sarma, M.-M. Titirici, S. B. Scott, I. E. L. Stephens and K. Chan, et al., Furfural electrovalorisation using single-atom molecular catalysts, Energy Environ. Sci., 2023, 16(7), 2934–2944, 10.1039/D3EE00551H.
- X. Shang, Y. Yang and Y. Sun, Electrohydrodimerization of biomass-derived furfural generates a jet fuel precursor, Green Chem., 2020, 22(16), 5395–5401, 10.1039/D0GC01720E.
- L. Ma, H. Liu and C. Wang, Switchable selectivity to electrocatalytic reduction of furfural over Cu2O-derived nanowire arrays, Dalton Trans., 2024, 53(24), 10338–10346, 10.1039/D4DT01078G.
- J. A. Muldoon and B. G. Harvey, Bio-Based Cycloalkanes: The Missing Link to High-Performance Sustainable Jet Fuels, ChemSusChem, 2020, 13(22), 5777–5807, DOI:10.1002/cssc.202001641.
- K. G. Ellis, D. W. Sopher, J. H. Utley, H. L. Chum and M. Ratcliff, Electro-Organic Reactions: XXX. Cleavage and Coupling of Some Biomass-Derived Organic Compounds, J. Electrochem. Soc., 1987, 134(12), 3058 CrossRef CAS.
- M. Temnikova, J. Medvedev, X. Medvedeva, N. H. Delva, E. Khairullina, E. Krivoshapkina and A. Klinkova, Electrochemical Hydrodimerization of Furfural in Organic Media as an Efficient Route to Jet Fuel Precursor, ChemElectroChem, 2023, 10(2), e202200865, DOI:10.1002/celc.202200865.
- H. Liu, D. M. Patel, Y. Chen, J. Lee, T.-H. Lee, S. D. Cady, E. W. Cochran, L. T. Roling and W. Li, Unraveling Electroreductive Mechanisms of Biomass-Derived Aldehydes via Tailoring Interfacial Environments, ACS Catal., 2022, 12(22), 14072–14085, DOI:10.1021/acscatal.2c03163.
- S. Mitchell and J. Pérez-Ramírez, Single atom catalysis: a decade of stunning progress and the promise for a bright future, Nat. Commun., 2020, 11(1), 4302, DOI:10.1038/s41467-020-18182-5.
- H. M. Castro-Cruz and N. A. Macías-Ruvalcaba, Porphyrin-catalyzed electrochemical hydrogen evolution reaction. Metal-centered and ligand-centered mechanisms, Coord. Chem. Rev., 2022, 458, 214430, DOI:10.1016/j.ccr.2022.214430.
- M. D. Hossain, Z. Liu, M. Zhuang, X. Yan, G.-L. Xu, C. A. Gadre, A. Tyagi, I. H. Abidi, C.-J. Sun and H. Wong, et al., Rational Design of Graphene-Supported Single Atom Catalysts for Hydrogen Evolution Reaction, Adv. Energy Mater., 2019, 9(10), 1803689, DOI:10.1002/aenm.201803689.
- B.-H. Lee, H. Shin, A. S. Rasouli, H. Choubisa, P. Ou, R. Dorakhan, I. Grigioni, G. Lee, E. Shirzadi and R. K. Miao, et al., Supramolecular tuning of supported metal phthalocyanine catalysts for hydrogen peroxide electrosynthesis, Nat. Catal., 2023, 6(3), 234–243, DOI:10.1038/s41929-023-00924-5.
- E. Jung, H. Shin, B.-H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. Hooch Antink, S. Park and K.-S. Lee, et al., Atomic-level tuning of Co–N–C catalyst for high-performance electrochemical H2O2 production, Nat. Mater., 2020, 19(4), 436–442, DOI:10.1038/s41563-019-0571-5.
- J. Su, C. B. Musgrave, Y. Song, L. Huang, Y. Liu, G. Li, Y. Xin, P. Xiong, M. M.-J. Li and H. Wu, et al., Strain enhances the activity of molecular electrocatalysts via carbon nanotube supports, Nat. Catal., 2023, 6(9), 818–828, DOI:10.1038/s41929-023-01005-3.
- W. Wan, C. A. Triana, J. Lan, J. Li, C. S. Allen, Y. Zhao, M. Iannuzzi and G. R. Patzke, Bifunctional Single Atom Electrocatalysts: Coordination–Performance Correlations and Reaction Pathways, ACS Nano, 2020, 14(10), 13279–13293, DOI:10.1021/acsnano.0c05088.
- A. Mehmood, M. Gong, F. Jaouen, A. Roy, A. Zitolo, A. Khan, M.-T. Sougrati, M. Primbs, A. M. Bonastre and D. Fongalland, et al., High loading of single atomic iron sites in Fe–NC oxygen reduction catalysts for proton exchange membrane fuel cells, Nat. Catal., 2022, 5(4), 311–323, DOI:10.1038/s41929-022-00772-9.
- P. Nilges and U. Schröder, Electrochemistry for biofuel generation: production of furans by electrocatalytic hydrogenation of furfurals, Energy Environ. Sci., 2013, 6(10), 2925–2931, 10.1039/C3EE41857J.
- C. Liu, R. Li, W. Zhou, Y. Liang, Y. Shi, R.-L. Li, Y. Ling, Y. Yu, J. Li and B. Zhang, Selectivity Origin of Organic Electrosynthesis Controlled by Electrode Materials: A Case Study on Pinacols, ACS Catal., 2021, 11(14), 8958–8967, DOI:10.1021/acscatal.1c01382.
- R. J. Dixit, K. Bhattacharyya, V. K. Ramani and S. Basu, Electrocatalytic hydrogenation of furfural using non-noble-metal electrocatalysts in alkaline medium, Green Chem., 2021, 23(11), 4201–4212, 10.1039/D1GC00579K.
- X. Liu, Y. Sun, H. Ren, Y. Lin, M. Wu and Z. Li, Cu–Sn Bimetallic Activated Carbon–Carbon Coupling for Efficient Furfural Electroreduction, ACS Catal., 2024, 14(8), 5817–5826, DOI:10.1021/acscatal.3c06346.
- W. Geng, D. Zhang, N. Zhen, J. Du, J. Dong, C. Liu, S.-L. Chen, Y. Chi and C. Hu, Electroreductive C–C Coupling of Furfural to Jet Fuel Precursors in Neutral Media via Synergistic Catalysis of the Polyoxotungstate and Cu Complex, ACS Catal., 2024, 14(13), 10040–10052, DOI:10.1021/acscatal.4c02524.
- S. Huang, B. Gong, Y. Jin, P. H. L. Sit and J. C.-H. Lam, The Structural Phase Effect of MoS2 in Controlling the Reaction Selectivity between Electrocatalytic Hydrogenation and Dimerization of Furfural, ACS Catal., 2022, 12(18), 11340–11354, DOI:10.1021/acscatal.2c02137.
- Z.-C. Yao, J. Chai, T. Tang, L. Ding, Z. Jiang, J. Fu, X. Chang, B. Xu, L. Zhang and J.-S. Hu, et al., Manipulating hydrogenation pathways enables economically viable electrocatalytic aldehyde-to-alcohol valorization, Proc. Natl. Acad. Sci. U. S. A., 2025, 122(8), e2423542122, DOI:10.1073/pnas.2423542122.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J.-P. Tessonnier and W. Li, Mechanisms of Furfural Reduction on Metal Electrodes: Distinguishing Pathways for Selective Hydrogenation of Bioderived Oxygenates, J. Am. Chem. Soc., 2017, 139(40), 14120–14128, DOI:10.1021/jacs.7b06331.
- W. Deng, P. Zhang, Y. Qiao, G. Kastlunger, N. Govindarajan, A. Xu, I. Chorkendorff, B. Seger and J. Gong, Unraveling the rate-determining step of C2+ products during electrochemical CO reduction, Nat. Commun., 2024, 15(1), 892, DOI:10.1038/s41467-024-45230-1.
- H. Dong, R. Luo, G. Zhang, L. Li, C. Wang, G. Sun, H. Wang, J. Liu, T. Wang and Z.-J. Zhao, et al., Electrochemical epoxidation enhanced by C2H4 activation and hydroxyl generation at the Ag/SnO2 interface, Nat. Commun., 2025, 16(1), 1901, DOI:10.1038/s41467-025-57223-9.
- B. Zhou, Y. Tong, Y. Yao, W. Zhang, G. Zhan, Q. Zheng, W. Hou, X.-K. Gu and L. Zhang, Reversed I1Cu4 single-atom sites for superior neutral ammonia electrosynthesis with nitrate, Proc. Natl. Acad. Sci. U. S. A., 2024, 121(37), e2405236121, DOI:10.1073/pnas.2405236121.
- W. Chen, G. Qian, Y. Wan, D. Chen, X. Zhou, W. Yuan and X. Duan, Mesokinetics as a Tool Bridging the Microscopic-to-Macroscopic Transition to Rationalize Catalyst Design, Acc. Chem. Res., 2022, 55(22), 3230–3241, DOI:10.1021/acs.accounts.2c00483.
- W. Chen, W. Fu, X. Duan, B. Chen, G. Qian, R. Si, X. Zhou, W. Yuan and D. Chen, Taming Electrons in Pt/C Catalysts to Boost the Mesokinetics of Hydrogen Production, Engineering, 2022, 14, 124–133, DOI:10.1016/j.eng.2020.11.014.
- S. Ye, F. Liu, F. She, J. Chen, D. Zhang, A. Kumatani, H. Shiku, L. Wei and H. Li, Hydrogen Binding Energy Is Insufficient for Describing Hydrogen Evolution on Single-Atom Catalysts, Angew. Chem., Int. Ed., 2025, 64(23), e202425402, DOI:10.1002/anie.202425402.
- X. Lu and C. Zhao, Electrodeposition of hierarchically structured three-dimensional nickel-iron electrodes for efficient oxygen evolution at high current densities, Nat. Commun., 2015, 6, 6616, DOI:10.1038/ncomms7616.
- W.-C. Zhang and C.-J. Li, Magnesium in water: simple and effective for pinacol-coupling, J. Chem. Soc., Perkin Trans. 1, 1998,(19), 3131–3132, 10.1039/A806553E.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ey00113g |
| ‡ J. X. and S. Y. contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |