DOI:
10.1039/D4EY00281D
(Paper)
EES Catal., 2025,
3, 701-711
Enabling the terpyridine ligand motif for Ir-based solid molecular catalysts†
Received
15th December 2024
, Accepted 11th February 2025
First published on 4th March 2025
Abstract
Terpyridine (tpy) and its derivatives are strongly coordinating ligands with a high degree of customizability. Due to their tendency to form stable bis(tpy) complexes with transition metals such as Ir and Ru, their application in thermal catalysis is limited, instead revolving mostly around electro-, photo- and supramolecular chemistry. Herein, it is demonstrated that immobilization of the tpy motif via incorporation into a polymer suppresses their formation in Ir-catalyzed formic acid dehydrogenation (FADH), highlighting a distinct advantage of solid molecular catalysts (SMCs). A catalytic activity of up to 175![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 was achieved at 160 °C and maintained at temperatures as low as 80 °C. Based on the results of a kinetic isotope effect (KIE) study, a catalytic cycle is proposed and the rate-determining step is identified. In a continuous setup, the most active SMC retained its activity over the course of 5 days, resulting in a TON upwards of 2800000. Through XPS, HAADF-STEM (-EDX) and EXAFS analyses, insights into the interaction between a metal precursor and poly-terpyridine are gained.
000 h−1 was achieved at 160 °C and maintained at temperatures as low as 80 °C. Based on the results of a kinetic isotope effect (KIE) study, a catalytic cycle is proposed and the rate-determining step is identified. In a continuous setup, the most active SMC retained its activity over the course of 5 days, resulting in a TON upwards of 2800000. Through XPS, HAADF-STEM (-EDX) and EXAFS analyses, insights into the interaction between a metal precursor and poly-terpyridine are gained.
Broader context
In heterogeneous catalysis, high metal dispersion presents a key factor in achieving sustainability goals. Ultimately, individual metal ions are stabilized on suitable support materials enabling full metal utilization. The control and tailoring of the nuclearity of metals and their anchoring on the support are often approached by trying to mimic homogeneous catalyst analogues as closely as possible. We herein present an SMC based on Ir and poly-terpyridines, which instead takes advantage of a distinct property of SMCs, the steric constraint of the polymer chain. Through immobilization the formation of inactive bis(tpy)complexes is suppressed, which is shown to take place for molecular tpy, and a highly active and selective catalyst is obtained for the exemplary dehydrogenation of formic acid. In addition, the interaction between a metal precursor and a polymer is illuminated by multiple state-of-the-art analytics, including an ex situ time-resolved XANES/EXAFS study showing the coordination of the metal to the macroligand and the changes occurring to it.
|
Introduction
Terpyridine (tpy) is a tri-dentate ligand with multiple possible modes of coordination. In its most simple form, it presents as an L3-ligand which coordinates using the free electron pairs of its three pyridinic nitrogens. Besides good σ-donor ability, the electron-deficiency of the pyridine rings also renders tpy a strong π-acceptor compared to other N-donor ligands.1 As a result, metal–tpy complexes exhibit an exceptional degree of stability. Functionalization of the individual pyridine rings enables adjustments to their steric and electronic properties. Hence, tpy recommends itself as a versatile ligand platform for high quality catalysis.2 As shown in recent reviews, homogeneous catalysts composed of first-row transition metals and the tpy ligand have been employed in a range of reactions, particularly with Cu and Ni.3–5 Examples include the Ni-catalyzed Claisen condensation6 or C–N bond cleavage of amides7 and the Cu-catalyzed aerobic oxidation of alcohols.8,9 For the rarer, albeit oftentimes more potent, second- and third-row transition metals, the nature of the available literature regarding tpy notably shifts. Looking at Ir or Ru, only a few examples of their use with tpy in thermal catalysis exist. These include the Ru-catalyzed transfer hydrogenation with unfunctionalized tpy by Beller et al.,10 which was later improved upon by Scymczak et al. by addition of hydrogen-bond directing groups at the outer pyridines.11 Instead, an abundance of research is available employing second- or third-row transition metals and tpy in the fields of electro-, photo-12–14 and supramolecular chemistry,15–17 very often in the shape of metal bis(tpy)-complexes.18–21 In these highly stable, octahedral bis(tpy)-complexes, all available coordination sites are occupied, which renders them thermocatalytically inactive. At the same time, Ru and Ir are two of the metals of choice in the fields of hydrogen storage and release via liquid organic hydrogen carriers (LOHCs) in the broader context of an energy transition, both homogeneously22–26 and heterogeneously.27–29 One such LOHC is formic acid (FA), the dehydrogenation (DH) of which is shown in Scheme 1 along with the unwanted dehydration side reaction. In the FADH, one class of highly active catalysts is composed of an appropriate precursor and the bipyridine ligand.30–32 This suggests that comparable tpy analogues would afford similar catalytic quality, if available. Herein, a solid molecular catalyst (SMC) based on molecular iridium and a tpy-containing macroligand is presented for the dehydrogenation of formic acid.33 In addition, the immobilization of the tpy motif through incorporation into a polymer network effectively suppresses the formation of inactive bis(tpy-complexes), which would take place for a homogeneous equivalent under identical conditions, showcasing a distinct advantage of SMCs. The results of the FADH using these SMCs are shown, before the interactions between the macroligand and the metal are closely investigated.
 |
| | Scheme 1 Formic acid dehydrogenation (top) and dehydration (bottom). | |
Results and discussion
Macroligand synthesis and characterization
A synthetic route towards terpyridine polymers was developed based on the Suzuki–Miyaura-cross-coupling-reaction. To this end, di-brominated terpyridine monomers 3a–d were synthesized from 2-acetyl-6-bromopyridine 1 and aromatic aldehydes 2a–d according to a literature procedure, as exemplified in Fig. 1.34 Through the variation of the aromatic aldehyde 2 different backbone functionalizations were incorporated into the monomers. The polymerization of 3a–d was realized using the tetravalent boronic acid pinacol ester linker 4 in a two-phase system of toluene and 2 M K2CO3 solution in the presence of [Pd(PPh3)4] and trioctylmethylammonium chloride as phase transfer catalysts by refluxing for 24 h. Subsequently, terminal functional groups were end-capped by the addition of bromobenzene and phenylboronic acid pinacol ester and refluxing an additional 24 h, respectively.35 The resulting polymers P1–4 were filtered, washed in water, ethanol, and diethyl ether and stirred for 30 min in a solution of 6 wt% HCl/7 wt% H2O2 to remove residual Pd. Lastly, the polymers were treated with sodium carbonate solution to remove protonation from the previous step. Treatment, washing and filtration steps resulted in a negligible loss of polymers, and all polymers were received in quantitative yields. The proposed structures and thermal stability were verified using 13C solid state magic angle spinning NMR spectroscopy (SS-MAS-NMR), thermogravimetric analysis (TGA), N2 physisorption, and X-ray photoelectron spectroscopy (XPS). P1–4 showed little to no porosity in N2 physisorption experiments. TGA revealed a thermal stability of up to 200 °C. 13C-NMR spectroscopy results are in agreement with the proposed structures (see the ESI†). HAADF-STEM images of P2 revealed spherical particles of varying size, while XPS measurements of the polymer P1 were both in agreement with the polymer structure and showed the successful removal of Pd through the H2O2/HCl treatment (see the ESI†). Additional analytic data regarding the macroligands will be discussed later in the context of macroligand–metal interactions. The macroligands were designed to include specific motifs with respect to the structural limitations imposed by the monomer synthesis. P1 was designed as a benchmark for the terpyridine ligand. The additional phenyl ring in the para position of the central pyridine is supposed to have as little impact on the electronic properties of the tpy ligand as possible. P2 features a pyrrole ring in the para position of the central pyridine. Its added mesomeric effect is expected to have a considerable influence on the electronic properties of the tpy. Both P3 and P4 are functionalized with an aromatic tertiary amine. While in P3 it is di-methyl-substituted, P4 instead features di-ethanol substitutions. While the presence of amine sites on supports was shown to be beneficial in heterogeneous dehydrogenation catalysts,36 the presence of hydroxy groups has been shown to be beneficial in bipyridine-based homogeneous catalysts.37,38 For both P3 and P4, the amine functionalization is separated from the tpy ligand motif by an additional phenyl ring due to synthetic constraints of their respective monomers. This spatial separation hampers the ability of the functionalization to influence the electronic properties of the tpy ligand. Instead, the design idea behind P3 and P4 was to adapt the bulk properties of the macroligand (see Fig. 2). Possible ways that these could aid in the FADH include the improved wettability of the SMC, faster transport of the product gases away from the active site or faster hydrogen evolution through pre-coordination of H3O+. In all cases, catalysts were prepared from the macroligands by stirring in a solution of metal precursors in MeOH at 50 °C for 48 h, followed by filtration. A target metal loading of 1 wt% and IrCl3 as precursors was chosen as starting points for all catalysts of this study. Metal uptake was quantified through inductively coupled plasma optical emission spectroscopy (ICP-OES) of the filtrate. The metal uptake was near quantitative for all polymers except P1, where 79% of the metal loading was achieved (see the ESI†).
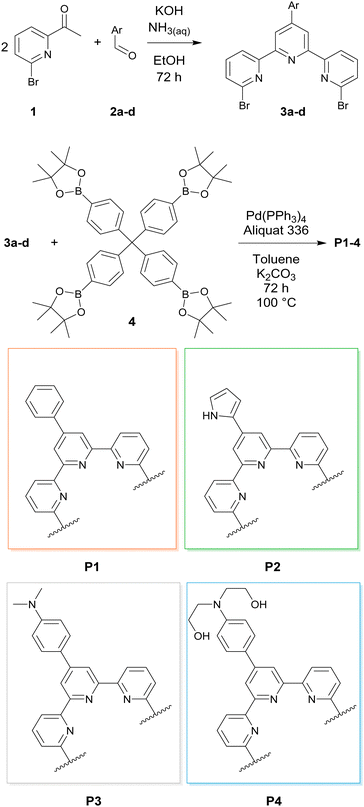 |
| | Fig. 1 Synthesis and structures of brominated terpyridine-containing monomers 3a–d and corresponding polymers P1–4. | |
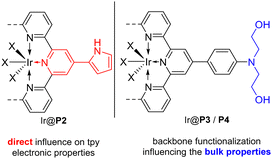 |
| | Fig. 2 Direct influence of para substitutions on the electronic properties of the central pyridine in P2 (left), and backbone functionalization of P3 and P4 (right). | |
Ir-catalyzed formic acid dehydrogenation
In an initial set of experiments, the FADH employing the SMC Ir@P1 was compared to its homogeneous counterpart to assess their behavior under reaction conditions. Either Ir@P1 or IrCl3 and molecular tpy were added to a 10 wt% aqueous FA solution and the FADH commenced in batch autoclaves at 160 °C. As can be seen in Table 1, entries 1 and 2, the benchmark SMC achieved a TOF of 11500 h−1 and a CO selectivity of 690 ppm, significantly outperforming the homogeneous analogue both in terms of activity and selectivity. Noteworthily, an observed maximum TOF of 3000 h−1 for the homogenous reaction was achieved in the initial stages of the reaction, after which it steadily declined without reaching full conversion. It can therefore be concluded that under these conditions, the molecular tpy formed inactive bis(tpy) complexes, while the initial catalytic activity stems from transient mono(tpy) complexes. A strong purple discoloration of the homogeneous reaction solution after the FADH is in agreement with this line of argument. In a second reaction (Table 1, entry 3), Ir@P1 and an additional equivalent of molecular tpy were employed to illuminate the behavior of the system in the presence of both immobilized and molecular tpy motifs. The results of these vary, but they all share incomplete conversion and worse catalytic quality compared to the SMC. As the respective reaction solutions also displayed a purple discoloration, a leaching of the Ir of the macroligand and subsequent inactivation in solution is likely. In summary, while the tpy-based SMC was capable of concluding the Ir-catalyzed FADH, the presence of the molecular ligand ultimately resulted in the formation of inactive bis(tpy) complexes. The suppression of this deactivation pathway through immobilization of the ligand motif represents a distinct advantage of solid molecular catalysts and has the potential to be transferred to other strongly coordinating ligand–metal combinations which would form inactive or otherwise unwanted di- or trimers. Experiments employing only precursors showed almost no activity, and with nanoparticular species showed significantly reduced activity (see the ESI†).
Table 1 Comparison of catalytic quality between SMC Ir@P1 and homogeneous analogues
| # |
Catalyst |
Conv. (%) |
TOF (h−1) |
S
(CO) (ppm) |
(h) |
Ir leach (%) |
| Conditions: 6 ml of 10 wt% FA solution and 160 °C. 20 mg of the 1 wt% Ir-loaded polymer (SMC). Equivalent amounts of the molecular Ir precursor and the tpy ligand to 20 mg of the SMC. 20 mg of the 1 wt% Ir-loaded polymer (SMC) and an additional equivalent molecular tpy. |
| 1 |
Ir@P1a |
97.4 |
11500 |
687 |
2.8 |
0.2 |
| 2 |
IrCl3/tpyb |
71.6 |
3000 |
2800 |
4.2 |
— |
| 3 |
Ir@P1/tpyc |
73.5 |
6500 |
1428 |
3.6 |
n.d. |
SMCs Ir@P2–4 were then employed in the base-free FADH. Preliminary tests showed negligible thermal decomposition of FA in a blank experiment and minimal conversion of FA in the presence of just metal precursors (see the ESI†). In the base-free FADH at 160 °C, the results of which are summarized in Fig. 3, and all catalysts showed catalytic activity. Conversion of formic acid was calculated using HPLC and above 97% in all cases. Ir@P2, featuring a pyrrole functionalization at the center pyridine, exhibited the highest catalytic activity of the series with a TOF of 65500 h−1 and the lowest CO impurity of 192 ppm. Importantly, in the case of Ir@P2, the majority of the FA in the reaction solution was already decomposed by the time the reaction temperature reached 160 °C, resulting in an undervaluation of the catalytic activity. Ir@P3, the first of the two bulk-functionalized macroligands, performed similarly to the benchmark catalyst in terms of both activity and selectivity. In contrast, Ir@P4, which differs by the additional ethanol substituents at its functionalization, exhibited a significantly higher TOF of 49500 h−1 and improved selectivity at 254 ppm CO. Both the adaptation of the electronic properties of the tpy ligand (Ir@P2) and the bulk properties of the macroligand (Ir@P4) lead to improved activity in the FADH, highlighting the feasibility of both for future SMC designs. All SMCs exhibited the exceptional retention of Ir, with leaching levels around 0.2% Ir (see the ESI†), owing to the high stability of Ir(tpy) complexes. Since the FADH employing Ir@P2 likely concluded before 160 °C was reached, an additional experiment was conducted with a reduced catalyst amount of 7 mg. In this experiment, a TOF of 175000 h−1 was achieved, an almost five-time increase compared to a previous bipyridine SMC.35 A variation of the catalyst amount was also done to ensure that the change in activity does not stem from transport phenomena alone (see the ESI†), which it does not. The observed TOF can therefore be considered a good estimate of the true catalytic potential of Ir@P2. Still, this TOF exemplifies that changes in the electronic properties of the ligand motif result in a more pronounced impact on catalytic activity compared to changes to the bulk properties. Moving forward, additional catalytic experiments focused on SMC Ir@P2, since it exhibited both the highest activity and the lowest CO impurity.
 |
| | Fig. 3 Activity, selectivity and conversion in the base-free FADH. Conditions: 6 ml of 10 wt% FA solution, 20 mg of 1 wt% Ir-loaded polymer (SMC), and 160 °C. | |
Kinetic isotope effect study
In order to better understand the catalytic cycle and identify the rate-determining step for the SMC Ir@P2, a KIE study was carried out. A solvent and a substrate were replaced by deuterated versions and the influence on the catalytic activity quantified. As seen in Fig. 4, the use of D2O resulted in a limited impact on catalytic activity at a KIE of just 1.20. In contrast, the use of FA-d2 imposed a KIE of 10.6 (Fig. 4, entry 3). Using both in the deuterated form showed the expected compounding effect. These findings prove that the hydrogen of the FA is involved in the rate-determining step of the catalytic cycle, whereas the hydrogen of the solvent is not. Based on the available knowledge of the catalytic cycle of comparable systems,39 we propose the catalytic cycle depicted in Fig. 4. Initially, water ligand-bearing complex A exchanges its water ligand for formate (I). The resulting Ir–formate complex B undergoes a β-hydride elimination generating CO2 (II), to yield an Ir-hydride C. From the hydride species and H3O+, hydrogen is evolved (III) and the initial species A is recovered. Based on this catalytic cycle and the results of the KIE study, the β-hydride elimination step II can be identified as the rate-determining step. These results also imply that the availability of protons for the hydrogen evolution (III) is sufficient in the SMC Ir@P2 despite the absence of pendant bases.
 |
| | Fig. 4 KIE, the proposed catalytic cycle and the rate-determining step for the base-free FADH using Ir@P2. Conditions: 6 ml of 10 wt% FA solution, 7 mg of the SMC, and 160 °C. An octahedral Ir complex with the minimum required exchange of chloride ligands was assumed in the catalytic cycle. | |
Investigation of the viable temperature range and base addition
An important parameter towards the application of any catalyst is its lowest required reaction temperature. To identify it, the Ir@P2-catalyzed FADH was carried out in decreasing increments of 20 °C, the results of which are summarized in Table 2. To allow the reaction to conclude within a reasonable time frame, 20 mg of the SMC was used again. As expected, catalytic activity decreases with temperature (Table 2, entries 1–3). The lowest temperature at which the base-free FADH was successfully commenced was 100 °C with a remaining TOF of 960 h−1. The amount of CO impurity and Ir leaching each increased slightly at 140 and 120 °C, respectively, but a more significant decline in both metrics was observed at 100 °C, with close to 250 ppm CO and an Ir leach around 0.8%. This is surprising, as the main source of CO is expected to the thermal decomposition of FA, which should decline with temperature. At 80 °C, a reaction was observable. A known method to further improving the activity of comparable systems in the FADH is the addition of bases. While the best results are generally achieved by addition of amines, sodium hydroxide was chosen here in perspective to continuous applications where amine stripping would pose an issue. A set of experiments were run with an additional 30 mol% NaOH (respective to FA) added. At 160 °C, Ir@P2 achieved a TOF of 89000 h−1 in the presence of bases, a roughly 33% increase compared to the base-free FADH (Table 2). At the same time, catalytic quality deteriorated significantly in terms of CO selectivity at 680 ppm CO and 2.1% Ir leaching. Nonetheless, because of the observed increase in activity, it was attempted again to conduct the FADH at 80 °C in the presence of bases (Table 2, entry 5). The reaction went ahead with a TOF of 180 h−1 and at an improved selectivity of 380 ppm CO. However, catalytic activity ceased before full conversion and leaching increased significantly. Nonetheless, the step from 100 °C to 80 °C places the reaction firmly below the boiling point of the FA solution and marks an important achievement towards applicability of such SMCs, despite requiring future improvements. The increase in leaching could be attributed to the increased reaction time, although the Ir/tpy complex is highly stable. The higher CO impurity seen for both lower temperatures and base-addition defies expectations. Another possible explanation for both observations would be improper coordination of the metal to the macroligand in basic environments or under more ambient conditions.
Table 2 Temperature, base and metal loading variations for the FADH employing Ir@P2
| # |
T (°C) |
mol% NaOH |
X (%) |
TOF (h−1) |
S
(CO) (ppm) |
(h) |
Leach (%) |
| Conditions: 6 ml of 10 wt% FA solution and 20 mg of the Ir-loaded polymer (SMC). Reaction was stopped on the 5th day due to time constraints. |
| 1 |
140 |
0 |
96.5 |
19750 |
143 |
2 |
0.2 |
| 2 |
120 |
0 |
98.9 |
4850 |
177 |
6 |
0.2 |
| 3 |
100 |
0 |
99.5 |
960 |
257 |
42 |
0.8 |
| 4 |
160 |
30 |
98.8 |
89000 |
682 |
1 |
2.1 |
| 5 |
80a |
30 |
62.6 |
180 |
380 |
110 |
4.0 |
Batch recycling and continuous experiments
To test the long-term stability of Ir@P2, batch recycling runs encompassing five consecutive FADHs were performed. A reaction temperature of 100 °C in the presence of bases was chosen as conditions for the recycling run to test the system at lower, i.e., closer to application temperatures while reaching full conversion in a reasonable time frame. After each run, the reaction vessel was cooled and an equivalent amount of pure FA was added to the autoclave via syringe and the next run started via heating. As can be seen in Fig. 5, Ir@P2 remained active over the course of the entire recycling. While conversion was not determined in between runs, analysis of the reaction solution after the recycling experiment showed full conversion. Interestingly, the catalytic activity increases from the first run to the second run and remains elevated until the last run. This observation suggests that the catalyst either needs to be formed in situ, or that a catalyst activation step such as the abstraction of a chloride ligand takes place. At the lower temperature at which the recycling was performed, it is plausible that either commences at a comparable rate to the reaction itself. In total, the catalyst maintained 90% of the initial activity by the end of the recycling. In terms of selectivity, an initial decrease in CO can be seen before CO impurities increase towards 580 ppm in the last run. This behavior is in line with an initial catalyst activation, followed by a slow catalyst deactivation in the later runs. Over the course of the recycling, a combined Ir leaching of 1.6% occurred. Based on the batch experiments, both the use of NaOH and lower reaction temperatures were shown to lead to a higher loss of metals. Hence, an increase in Ir leaching was expected for the recycling experiments. Instead, it represents a reduction compared to batch experiments under similar conditions (Table 2, entry 4). This suggests that the SMC was capable of retaining a higher amount of Ir over the course of multiple runs. Consequentially, the majority of the leaching does not scale with the reaction time, but instead occurs initially. The batch recycling resulted in an accumulated TON of 110000. A comparable solvent exchange recycling was also performed to rule out leached species as the main source of catalytic activity (see the ESI†).
 |
| | Fig. 5 Recycling experiments of Ir@P2 in the FADH reaction at 100 °C. Conditions: 6 ml of 10 wt% FA solution with 30 mol% NaOH and 20 mg of 1 wt% Ir@P2. FA was resupplied to the autoclave via syringe after the completion of each reaction cycle. | |
An additional experiment was carried out in a continuous stirred tank reactor (CSTR, see ESI†) to gauge the long-term stability of the SMCs. To this end, a lower metal content SMC 0.1 wt% Ir@P2 and 160 °C were chosen for the CSTR experiment to achieve high TONs faster. The time-resolved resupply of FA during the experiment is shown in Fig. 5. FA was rapidly replenished initially before FA decomposition linearized. Afterwards, FA resupply remained steady at small, frequent intervals, in line with the catalyst maintaining its activity over the entirety of the experiment. Longer intervals without FA resupply towards the end of the experiment suggest the onset of catalyst deactivation, and the reaction was stopped after 5 days. Over the course of the CSTR run, the catalyst reached a combined TON upwards of 2800000, highlighting the long-term activity of the tpy-based SMCs. In summary, SMCs based on Ir and the tpy-macroligand were shown to be highly active and selective in the FADH and showcased an exceptional level of Ir retention, particularly in the base-free FADH. While the addition of bases was shown to increase catalytic activity, an increase in metal leaching was also observed. Nonetheless, in the presence of NaOH, the SMC Ir@P2 was able to facilitate the FADH at temperatures as low as 80 °C. The long-term activity of the catalyst was highlighted in two types of experiments; a batch recycling where close to 90% of activity was maintained after 5 consecutive runs totaling a TON of 110000, and a continuous experiment in a CSTR where a TON of over 2800000 was achieved over the course of 5 days (Fig. 6).
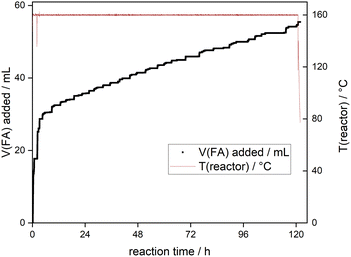 |
| | Fig. 6 Time-resolved, cumulative resupply of FA to the CSTR during continuous experiment. Conditions: 0.1 wt% Ir@P2, 160 °C, and 10 wt% aqueous FA solution was used to initially fill the reactor vessel. | |
Investigations into the metal–macroligand interaction
To better understand the nature of the metal–macroligand interactions, comparative analyses between the macroligand, fresh and spent SMC samples were performed. As shown in Fig. 7, XPS N 1s core-level analysis of macroligand P1 which only contains pyridinic nitrogen reveals a singular nitrogen species at 399.1 eV, thus attributed to the tpy motif. The singular species proves the indistinguishability of the three N of the terpyridine motif. To elucidate the interaction between the macroligand and the metal, an attempt was made to fully load P1 with iridium by stirring it in an excess of methanolic IrCl3 solution at 60 °C (equivalent to 20 wt% Ir). The XPS high-resolution spectra of Ir and N of the resulting sample are shown in Fig. 8. The high-resolution core-level Ir spectrum of the sample (Fig. 8, top) reveals two signals belonging to a singular Ir(III) species, in line with the metal precursor used. These XPS results show that the macroligand was successfully impregnated with the metal precursor in a molecular fashion, and that the oxidation state of the precursor was maintained. A total of three different nitrogen species are revealed for the high metal loading (Fig. 8, bottom). The first of the three species at 399.4 eV is again attributed to pyridinic nitrogen. The presence of unshifted pyridinic nitrogen reveals that not all tpy motifs contained in the macroligand coordinate Ir, despite sufficient metal being available during the impregnation step. This likely stems from the limited availability of these tpy motifs, either due to steric constraints of the polymer chain or due to limited diffusion to the inner lying parts of the polymer particles. A second species, at 400.7 eV, is attributed to metal-coordinating pyridinic nitrogen based on the extent of the shift from the previous species. The third, found at 402.2 eV, shows a relative shift of 2.8 eV compared to the pyridinic nitrogen. This shift is the characteristic for quaternary nitrogen species such as protonated nitrogen.40,41 Since no such species was found in the corresponding macroligand P1, this partial protonation likely occurred during the impregnation step with the acidic iridium chloride precursor.
 |
| | Fig. 7 XPS N 1s high-resolution core-level spectrum of polymer P1. | |
 |
| | Fig. 8 XPS Ir 4f high-resolution core-level spectrum (top) and N 1s (bottom) of Ir@P1 with a theoretical metal loading of 20 wt%. | |
As the most active SMC, fresh Ir@P2 was also analyzed accordingly via XPS (Fig. 9) Qualitatively, the N 1s high-resolution core level spectrum contains the same species as the previous sample, albeit at different ratios. A larger share of the nitrogen was pyridinic, and the second species at 400.5 eV, previously attributed to Ir-coordinating N, is a lot more pronounced. For Ir@P2, this stems from an overlap of the signal from the coordinating pyridinic N and the pyrrolic N entailed in the P2 macroligand. The third species at 402.2 eV, corresponding to protonated N, is less pronounced than in the previous sample. This is in agreement with the protonation occurring during the impregnation step. The Ir 4f spectra of fresh Ir@P2 revealed the same Ir(III) signals (see the ESI†).
 |
| | Fig. 9 XPS N 1s high-resolution core-level spectrum of fresh SMC Ir@P2. | |
HAADF-STEM and STEM-EDX images were also recorded for fresh 1 wt% Ir@P2. In these images, as shown in Fig. 10, Ir single sites can be seen finely dispersed across the particle surface. EDX imaging shows that the highly dispersed Ir is found almost exclusively on the organic, spherical polymer particles. This finding is in agreement with the proposed lower availability of the inner lying tpy motifs for coordination and the low porosity of the polymers. Combined with the findings of XPS analysis, it can be concluded that after impregnation, the Ir precursor is uniformly dispersed across the surface of the spherical polymer particles in a molecular fashion. At the same time, a significant share of the tpy nitrogen remains non-coordinating or protonated, and the latter of which likely occurs as a side-effect of the impregnation step. Subsequently, spent Ir@P2 retrieved from the batch recycling experiments was also analyzed to assess the changes occurring in the catalyst during the FADH. The Ir 4f and N 1s core-level spectra of spent Ir@P2 are shown in Fig. 11. In the Ir 4f core-level spectrum (Fig. 11, top), three additional signals can be seen. Besides the previously discussed Ir(III) species, the two signals at 61.2 and 64.2 eV, respectively, can be attributed to an Ir(0) species. This suggests a reductive deactivation of the catalyst, which usually takes shape in the form of nanoparticle formation. Additionally, a signal at 64.9 eV can be assigned to sodium, which was added to the reaction in the form of NaOH. While the three previously found N species are still present in the spent catalyst (Fig. 11, bottom), it can be seen that the share of protonated N significantly decreased, but the proportion of Ir-coordinating and pyridinic N increased.
 |
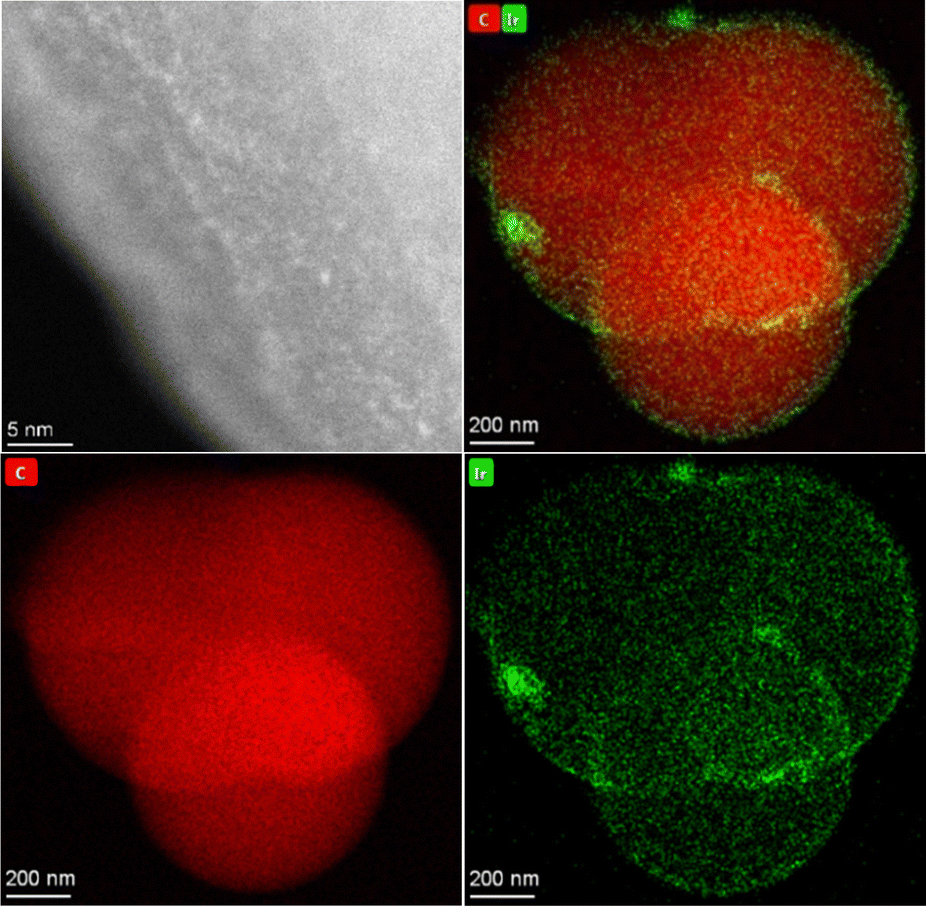
| | Fig. 10 HAADF-STEM image (top left) of the catalyst Ir@P2 showing highly dispersed metal species. HAADF-STEM-EDX image (rest) showing iridium (green) across the organic polymer particle (red). | |
 |
| | Fig. 11 XPS Ir 4f (top) and N 1s (bottom) high resolution core-level spectra of spent Ir@P2 with a metal loading of 1 wt%. The spent SMC was taken from the base-assisted FADH batch recycling at 100 °C. | |
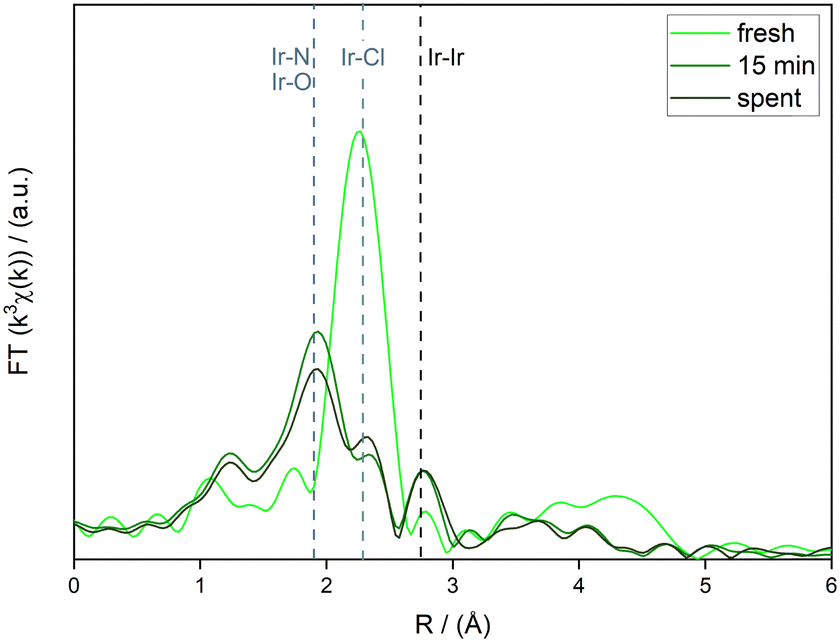
Since it was found that the recycling experiment reaches full conversion of FA after the fifth run, the observed deprotonation can be explained by the pH of the reaction solution. As the decomposition of the formic acid progresses and the pH of the reaction solution increases, the slightly acidic pyridinium ions (pKa = 5.18) deprotonate. The small amount of remaining protonated pyridine stems from the limited availability of the protonation site. The increase of pyridinic nitrogen is a direct consequence of the deprotonation. The elevated share of the Ir-coordinating N in the spent catalyst represents the strongest evidence so far that part of the coordination of Ir to the tpy motif of the macroligand occurred in reactio as opposed to during the impregnation step. This could either be caused by additional N becoming available as result of deprotonation, or because of the differing conditions in the FADH as compared to the impregnation. This would also explain the observed initial increase in catalytic activity during the batch recycling. HAADF-STEM and -EDX imagining of the spent SMC supports these findings. As can be seen in Fig. 12, HAADF-STEM imaging shows agglomerates on the particle surface of spent Ir@P2. EDX imaging reveals the agglomerates to consist of Ir. At the same time, it can be seen that highly dispersed iridium is still present in the spent catalyst and covers the polymer particle. The coexistence of molecular Ir single sites and Ir nanoparticles is congruent with the previous XPS findings. Lastly, an ex situ time-resolved resolved XANES and EXAFS study was carried out on Ir@P2. Comparisons were made between the metal precursor IrCl3, homogeneous Ir(tpy)Cl3, freshly prepared Ir@P2 and two Ir@P2 samples which were employed in the base-free FADH at 160 °C. The first was prepared by stopping the FADH after 15 minutes by rapidly cooling. At this time, conversion of FA was largely incomplete. This sample aimed at elucidating initial changes to the catalyst such as activation steps. The second sample is of a spent catalyst after one FADH cycle to allow a closer look at the resting state and possible deactivation mechanisms. Fig. 12 shows a comparison of the Ir–LIII XANES region of the five samples with the dotted lines representing the white lines (WLs) of the two homogeneous references. The WL for IrCl3 was found at 11215.0 eV, whereas that of Ir(tpy)Cl3 was found at 11215.9 eV. The difference between the WL position of these Ir(III) samples can be attributed to the different complex geometries and ligand environments (Fig. 13). The WL of the freshly prepared Ir@P2 was found at 11215.2 eV, placing it between those of the two homogeneous references, albeit very closely to the WL of IrCl3. This suggests that while an interaction between the metal and the macroligand likely exists after impregnation, it is not in the shape of the desired terpyridine coordination. In contrast, the WL for both Ir@P2 samples that underwent the FADH reaction (15 min, spent) are located at 11215.9 and 11216.0 eV, respectively. This is in excellent agreement with the WL of the homogeneous Ir(tpy)Cl3, and points towards a fast and complete terpyridine coordination under reaction conditions. The difference in the WL position between the fresh SMC and those that underwent FADH, the latter of which is in excellent agreement with molecular Ir(tpy)Cl3, suggests that the desired immobilized Ir(tpy) complex is not formed during impregnation. Rather, the formation takes place under reaction conditions, in a reliable and fast manner as evidenced by the extremely low leaching and the high catalytic activities recorded in the base-free FADH. To further underline these findings, EXAFS analysis was performed, and the results of which are shown in Fig. 14. Reference samples and literature values were used to determine the length of the most important bond types: Ir–N at 1.92 Å, Ir–Cl at 2.30 Å and Ir–Ir at 2.65 Å upwards (see the ESI†). For fresh Ir@P2, the dominant bond type remained Ir–Cl. Only a small shoulder around 1.92 Å indicates some degrees of the Ir–N interaction, in line with improper Ir coordination until under reaction conditions. A small Ir–Ir signal can also be seen, in disagreement with XPS and imaging analysis. Due to the small extent of the signal and the high likelihood of the Ir being improperly coordinated in the fresh sample, beam damage is a likely reason for this observation. In contrast, the 15 min sample exhibits Ir–N at around 1.90 Å as the main bond, accompanied by a strong reduction in the incidence of Ir–Cl. The emergence of Ir–N represents the coordination to the tpy macroligand motif, while the reduction of the Ir–Cl bond share can additionally be attributed to the abstraction of Cl ligands to allow the coordination of water or formate ligands. As Ir–O and Ir–N are not easily distinguishable by the bond length, both tpy and O-based ligands contribute to this main bond length. Additionally, a much stronger Ir–Ir signal emerges, corresponding to the formation of Ir nanoparticles. In the spent catalyst, the observed bond types are qualitatively identical to the sample at 15 minutes, albeit at slightly different ratios, suggesting that the majority of changes to the catalyst structure occur during the initial stages of the reaction. Ir–N/O is maintained as the main signal, highlighting the stability of the tpy coordination. An Ir–Cl shoulder also remains. The ratio between Ir–N/O and Ir–Cl shifts towards a higher Ir–Cl share. This can be explained by full conversion of the available formic acid and an exchange of formate ligands with chloride from the reaction solution. The Ir–Ir signal at around 2.7 Å remained identical to the one observed for the 15 min sample. Importantly, this implies that the formation of these agglomerates already takes place in the initial stages of the reaction, and indicates improperly coordinated Ir as its source, at least until much higher TONs. Another potential signal in the FADH samples was found at around 1.7 Å, at the lower end of Ir–O bonds.42 Such short Ir–O bonds are observed in small Ir–O clusters, and the existence of which is plausible in the context of the FADH, for example as a pathway towards the larger metal agglomerates. In retrospective, while leaching was near negligible for FADH reactions without bases and at high temperatures, it was shown to increase significantly in the presence of bases and lower temperatures. With the knowledge gained from XANES and EXAFS analysis, this likely stemmed from improperly coordinated iridium rather than extended reaction times. If both energy (e.g. elevated temperatures) and protons (e.g. low pH) are required for the coordination of Ir to the tpy macroligand, this would be severely hindered in base-assisted and low temperature reactions. This raises the question of the nature of the Ir after impregnation. Since imagining and XPS analysis of freshly impregnated SMCs showed no agglomerates and Ir(III) as the only species, the Ir sites on the macroligand prior to the FADH are likely a mixture of partially coordinated Ir (e.g. to a single or two of the pyridinic N of the tpy) and physisorbed IrCl3 di- or trimers. While metal retention in the base-free high temperature FADH was exceptional, it remained reasonable at lower temperatures and in the presence of bases. This consequentially includes physisorbed, molecular, and nanoparticular species and underscores the excellent ability of tpy-based SMCs to retain metals, a highly desirable quality for any heterogeneous catalyst.
 |
| | Fig. 12 HAADF-STEM image (left) and HAADF-STEM-EDX images (rest) of the spent catalyst Ir@P2 showing highly dispersed as well as agglomerated iridium (green) on the polymer particle surface (red). The spent SMC was taken from the base-assisted FADH batch recycling at 100 °C. | |
 |
| | Fig. 13 Iridium LIII-edge XANES region of IrCl3, Ir(tpy)Cl3 and ex situ time-resolved Ir@P2. Spectra are stacked for clarity, with dotted lines marking the WLs of the homogeneous samples. SMC samples taken from base-free FADH at 160 °C. | |
 |
| | Fig. 14 k3-weighted iridium LIII-edge FT-EXAFS of ex situ time-resolved Ir@P2 samples. Positions of the most important bond types were determined via referencing and marked with dotted lines. SMC samples taken from base-free FADH at 160 °C. | |
Conclusions and outlook
In this work, a series of customizable macroligands based on the tpy ligand motif were developed. SMCs for the Ir-catalyzed FADH were prepared from these by impregnation with IrCl3. It was possible to highlight a distinct advantage of SMCs in the suppression of the formation of inactive bis(tpy) complexes. Two avenues towards improving catalytic activity were explored via different functionalizations of the macroligands: the adaptation of the electronic properties of the entailed tpy ligand motif and the adaptation of the bulk properties of the macroligand. Both lead to improved catalytic activities, although the first approach resulted in an increased impact, yielding the most active SMC Ir@P2. In the FADH, Ir@P2 reached TOFs of up to 175000 h−1 at 160 °C without addition of bases and maintained catalytic activity at temperatures as low as 80 °C in the base-assisted FADH. As a result of the KIE study, a catalytic cycle was proposed and the rate-determining step was identified as β-hydride elimination. This finding also revealed that the evolution of hydrogen gas is not rate-determining, despite the absence of pendant bases or comparable structures in the macroligand design. The exceptional longevity of the Ir(tpy) system was highlighted both in batch recycling and in continuous operations, and in the latter of which a TON of 2800000 was reached over five days. XPS and HAADF-STEM (-EDX) analyses confirmed molecular, highly dispersed Ir single sites across the polymer. Analysis of the spent SMC via XPS and imaging revealed the emergence of Ir(0) in the shape of iridium nanoparticles, which co-existed with molecular single sites on the polymer surface. XANES and EXAFS analyses of an ex situ time resolved study revealed that the desired tpy coordination takes place under reaction conditions, not as intended during impregnation. It was possible to deduct from these findings that increased leaching and agglomeration for base-assisted and/or low-temperature FADHs can be traced back to a worse coordination of the metal by the tpy macroligand. Additional investigations into improved impregnation reactions and macroligand designs combining ligands and bulk improvements will be a part of future research in this context. All considered, the highly customizable tpy-macroligand recommends itself as the starting point for exceptionally stable and highly active and selective SMCs, not just in the Ir-catalyzed FADH but for the bountiful landscape of transition metal catalysis employing N-based aromatic ligands.
Author contributions
Keanu V. A. Birkelbach: writing – original draft, visualization, investigation, formal analysis, data curation, and conceptualization. Heinrich Hartmann: visualization, investigation, and formal analysis. Astrid Besmehn: visualization, investigation, and formal analysis. Alexander Meledin: visualization, investigation, and formal analysis. Isabella Kappel: visualization, investigation, and formal analysis. Peter J. C. Hausoul: writing – review and editing, supervision, and conceptualization. Regina Palkovits: writing – review and editing, supervision, resources, project administration, methodology, funding acquisition, and conceptualization.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy within the Exzellenzcluster 2186 “The Fuel Science Center” ID:390919832. The authors further thank the Diamond Light Source for providing beamtime on beamline B18 (SP32235). The authors acknowledge the funding by the German Federal Ministry of Education and Research (BMBF) and the Ministry of Economic Affairs, Industry, Climate Action and Energy of the State of North Rhine-Westphalia through the project HC-H2. We thank Dr Luke Keenan for his extensive help both during our measurement time at Diamond and thereafter while interpreting the XANES and EXAFS results. We would like to extend our thanks to Prof. Dr Claudia Weidenthaler for her help regarding XPS. We thank Frederic Thilmany for his support in the laboratory. We thank J. Heller, C. Franzen, H. Bergstein, H. Eschmann, H. Fickers-Boltz and N. Avraham-Radermacher for measuring parts of the analytics. Lastly, we thank S. Seidel and J. Baums for fruitful discussion. Open Access funding provided by the Max Planck Society.
Notes and references
- A. Czap, F. W. Heinemann and R. van Eldik, Inorg. Chem., 2004, 43, 7832–7843 CrossRef CAS PubMed.
- S. F. Kainat, M. B. Hawsawi, E. U. Mughal, N. Naeem, A. M. Almohyawi, H. M. Altass, E. M. Hussein, A. Sadiq, Z. Moussa, A. S. Abd-El-Aziz and S. A. Ahmed, RSC Adv., 2024, 14, 21464–21537 RSC.
- C. Wei, Y. He, X. Shi and Z. Song, Coord. Chem. Rev., 2019, 385, 1–19 CrossRef CAS PubMed.
- A. Winter, G. R. Newkome and U. S. Schubert, ChemCatChem, 2011, 3, 1384–1406 CrossRef CAS.
- A. Winter and U. S. Schubert, ChemCatChem, 2020, 12, 2890–2941 CrossRef CAS.
- J. Chen, M. Xu, S. Yu, Y. Xia and S. Lee, Org. Lett., 2020, 22, 2287–2292 CrossRef CAS PubMed.
- L. Hie, E. L. Baker, S. M. Anthony, J.-N. Desrosiers, C. Senanayake and N. K. Garg, Angew. Chem., Int. Ed., 2016, 55, 15129–15132 CrossRef CAS PubMed.
- G. Zhang, E. Liu, C. Yang, L. Li, J. A. Golen and A. L. Rheingold, Eur. J. Inorg. Chem., 2015, 939–947 CrossRef.
- Z. Ma, L. Wei, E. C. B. A. Alegria, L. M. D. R. S. Martins, M. F. C. Guedes Da Silva and A. J. L. Pombeiro, Dalton Trans., 2014, 43, 4048–4058 RSC.
- R. V. Jagadeesh, G. Wienhöfer, F. A. Westerhaus, A.-E. Surkus, H. Junge, K. Junge and M. Beller, Chem. – Eur. J., 2011, 17, 14375–14379 CrossRef CAS PubMed.
- C. M. Moore, B. Bark and N. K. Szymczak, ACS Catal., 2016, 6, 1981–1990 CrossRef CAS.
- A. Anthonysamy, S. Balasubramanian, V. Shanmugaiah and N. Mathivanan, Dalton Trans., 2008, 2136 RSC.
- J. Klein, A. Stuckmann, S. Sobottka, L. Suntrup, M. van der Meer, P. Hommes, H.-U. Reissig and B. Sarkar, Chem. – Eur. J., 2017, 23, 12314–12325 CrossRef CAS PubMed.
- L. Flamigni, B. Ventura, F. Barigelletti, E. Baranoff, J.-P. Collin and J.-P. Sauvage, Eur. J. Inorg. Chem., 2005, 1312–1318 CrossRef CAS.
- L. Flamigni, J.-P. Collin and J.-P. Sauvage, Acc. Chem. Res., 2008, 41, 857–871 CrossRef CAS PubMed.
- D. Rota Martir and E. Zysman-Colman, Coord. Chem. Rev., 2018, 364, 86–117 CrossRef CAS.
- M. Chiper, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2009, 30, 565–578 CrossRef CAS PubMed.
- J.-P. Collin, I. M. Dixon, J.-P. Sauvage, J. A. G. Williams, F. Barigelletti and L. Flamigni, J. Am. Chem. Soc., 1999, 121, 5009–5016 CrossRef CAS.
- E. Baranoff, J.-P. Collin, L. Flamigni and J.-P. Sauvage, Chem. Soc. Rev., 2004, 33, 147 RSC.
- R. R. Panicker and A. Sivaramakrishna, Coord. Chem. Rev., 2022, 459, 214426 CrossRef CAS.
- J. P. Sauvage, J. P. Collin, J. C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS.
- J. Campos, L. S. Sharninghausen, M. G. Manas and R. H. Crabtree, Inorg. Chem., 2015, 54, 5079–5084 CAS.
- F. J. Fernández-Alvarez, M. Iglesias, L. A. Oro and V. Polo, ChemCatChem, 2013, 5, 3481–3494 Search PubMed.
- R. Kanega, M. Z. Ertem, N. Onishi, D. J. Szalda, E. Fujita and Y. Himeda, Organometallics, 2020, 39, 1519–1531 CAS.
- N. Onishi, R. Kanega, H. Kawanami and Y. Himeda, Molecules, 2022, 27, 455 CrossRef CAS PubMed.
- C. Guan, Y. Pan, T. Zhang, M. J. Ajitha and K.-W. Huang, Chem. – Asian J., 2020, 15, 937–946 CrossRef CAS PubMed.
- Q. Wu, C. Shen, N. Rui, K. Sun and C. Liu, J. CO2 Util., 2021, 53, 101720 CrossRef CAS.
- C. Tébar-Soler, V. Martin-Diaconescu, L. Simonelli, A. Missyul, V. Perez-Dieste, I. J. Villar-García, J.-B. Brubach, P. Roy, M. L. Haro, J. J. Calvino, P. Concepción and A. Corma, Nat. Mater., 2023, 22, 762–768 CrossRef.
- A. Wang, P. He, J. Wu, N. Chen, C. Pan, E. Shi, H. Jia, T. Hu, K. He, Q. Cai and R. Shen, Energy Fuels, 2023, 37, 17075–17093 CrossRef CAS.
- Y. Himeda, S. Miyazawa and T. Hirose, ChemSusChem, 2011, 4, 487–493 CrossRef CAS PubMed.
- Y. Himeda, Green Chem., 2009, 11, 2018 RSC.
- H. Kawanami, M. Iguchi and Y. Himeda, Inorg. Chem., 2020, 59, 4191–4199 CrossRef CAS PubMed.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- J. Husson and L. Guyard, Heterocycl. Commun., 2015, 21, 199–202 CrossRef CAS.
- C. Broicher, S. R. Foit, M. Rose, P. J. C. Hausoul and R. Palkovits, ACS Catal., 2017, 7, 8413–8419 CrossRef CAS.
- S.-J. Li, Y.-T. Zhou, X. Kang, D.-X. Liu, L. Gu, Q.-H. Zhang, J.-M. Yan and Q. Jiang, Adv. Mater., 2019, 31, 1806781 CrossRef PubMed.
- M. Iguchi, H. Zhong, Y. Himeda and H. Kawanami, Chem. – Eur. J., 2017, 23, 17788–17793 CrossRef CAS PubMed.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- W.-H. Wang, S. Xu, Y. Manaka, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, ChemSusChem, 2014, 7, 1976–1983 CrossRef CAS PubMed.
- K. Artyushkova, J. Vac. Sci. Technol., A, 2020, 38, 031002 CrossRef CAS.
- I. Matanovic, K. Artyushkova, M. B. Strand, M. J. Dzara, S. Pylypenko and P. Atanassov, J. Phys. Chem. C, 2016, 120, 29225–29232 CrossRef CAS.
- S. Lee, Y.-J. Lee, G. Lee and A. Soon, Nat. Commun., 2022, 13, 3171 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Regina
Palkovits
*a and
Regina
Palkovits