
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmmonia synthesis from nitrate reduction by the modulation of a built-in electric field and external stimuli
Shaoce
Zhang
a,
Rong
Zhang
a,
Ying
Guo
b and
Chunyi
Zhi
 *acde
*acde
aDepartment of Materials Science and Engineering, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong 999077, China. E-mail: cy.zhi@cityu.edu.hk
bSchool of Interdisciplinary Studies, Lau Chun Him Building, Lingnan University, 8 Castle Peak Road, Tuen Mun, New Territories, Hong Kong, China
cHong Kong Center for Cerebro-Cardiovascular Health Engineering (COCHE), Shatin, NT, HKSAR, China
dCentre for Functional Photonics, City University of Hong Kong, Kowloon, Hong Kong, China
eHong Kong Institute for Clean Energy, City University of Hong Kong, Kowloon 999077, Hong Kong, China
First published on 7th January 2025
Abstract
Ammonia (NH3) is a vital chemical feedstock and a carbon-free energy source. The reduction of nitrate (NO3−) from environmental pollutants is a sustainable method for NH3 production compared with the industrially intensive Haber–Bosch method, which can mitigate energy and environmental concerns. However, due to the involvement of multi-electron transfer-proton coupling processes, the NO3− reduction reaction (NO3RR) exhibits sluggish kinetics and significant side reactions. This review provides a comprehensive summary of recent research progress in facilitating NO3RRs using a built-in electric field and external stimuli. The paper commences by introducing the mechanisms and challenges of the NO3RR, subsequently focusing on strategies for built-in electric field/external stimuli-assisted catalytic reactions. The internal electric field can be triggered by constructing a Mott–Schottky heterojunction and a semiconductor–semiconductor heterojunction, adjusting the coordination environment of active sites, and regulating the electrical double layer, while the external stimuli include optical, stress, and thermal stimuli. This review focuses on the activation and adsorption processes of reactants and intermediates by a built-in electric field/external stimuli, and their influence on the thermodynamics and kinetics of reactions. Finally, we summarize the strategies for built-in electric field/external stimuli-assisted NO3RRs, highlight the challenges of achieving high activity and selectivity in NH3 production, and provide clear guidance for future research.
Broader contextAmmonia (NH3) plays an essential role in agriculture, industry, and pharmaceuticals, with its global demand greatly increasing. However, traditional NH3 production relies on the energy-intensive Haber–Bosch process, which involves nitrogen and hydrogen as raw materials under high-temperature, high-pressure conditions, resulting in significant CO2 emissions. The aqueous-based reduction of NO3− utilizes water and the pollutant NO3− as raw materials, converting them into NH3 through various forms of energy input such as electrocatalysis and photocatalysis. This process concurrently addresses both energy crises and environmental pollution issues. However, the NO3− reduction reaction (NO3RR) to generate NH3 involves a nine-proton and eight-electron transfer process, leading to sluggish kinetics and severely competitive hydrogen evolution. Therefore, multiple strategies have been proposed to enhance the performance of NO3− reduction. Among them, introducing a built-in electric field/external stimuli to modulate the activation and adsorption behavior of reactants and intermediates, thereby promoting reaction kinetics and thermodynamics, becomes a promising strategy. Based on this, this review provides a comprehensive summary of the latest research progress in built-in electric field/external stimuli-assisted NO3RRs, including material analysis, mechanistic elucidation, problem summarization, and future prospects. This review aims to facilitate NO3RR performance and provide guidance for NO3RR processes in future industrial implementations. |
1. Introduction
Ammonia (NH3) is indispensable in fertilizer manufacturing and the pharmaceutical industry, and it is a promising energy carrier because of its high hydrogen content.1–4 However, the production of NH3 currently relies heavily on the industrially intensive Haber–Bosch process, which requires high pressure and high temperatures, leading to significant greenhouse gas emissions.5–7 Therefore, aqueous-based NH3 synthesis, utilizing water molecules as proton sources and nitrogen gas (N2)/nitrate ions (NO3−) as nitrogen sources, has garnered widespread attention.8–10 In comparison to the high dissociation energy of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond in nitrogen molecules (941 kJ mol−1), the dissociation energy of the N
N bond in nitrogen molecules (941 kJ mol−1), the dissociation energy of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is only 204 kJ mol−1,11 which suggests that the NO3− reduction reaction (NO3RR) has more potential in NH3 synthesis. Furthermore, NO3− exhibits higher solubility in aqueous solutions than N2, providing the foundation for scalable and efficient production of NH3.12,13 Simultaneously, the extensive application of nitrate fertilizers to boost crop growth, along with animal manure, particularly from intensive livestock farming, excessive irrigation on agricultural lands, decomposition of crop residues in soil, and activities like mining, explosives manufacturing, and fertilizer industries, lead to the accumulation of nitrates in water bodies, posing a significant threat to human health.14–18 In this context, the NO3RR not only produces high-value, zero-carbon hydrogen-rich fuel but also effectively removes NO3− pollutants from water bodies, which mitigates energy crises and environmental pollution, offering broad and crucial prospects for application.
O bond is only 204 kJ mol−1,11 which suggests that the NO3− reduction reaction (NO3RR) has more potential in NH3 synthesis. Furthermore, NO3− exhibits higher solubility in aqueous solutions than N2, providing the foundation for scalable and efficient production of NH3.12,13 Simultaneously, the extensive application of nitrate fertilizers to boost crop growth, along with animal manure, particularly from intensive livestock farming, excessive irrigation on agricultural lands, decomposition of crop residues in soil, and activities like mining, explosives manufacturing, and fertilizer industries, lead to the accumulation of nitrates in water bodies, posing a significant threat to human health.14–18 In this context, the NO3RR not only produces high-value, zero-carbon hydrogen-rich fuel but also effectively removes NO3− pollutants from water bodies, which mitigates energy crises and environmental pollution, offering broad and crucial prospects for application.
However, the NO3RR is a complex reaction involving proton-coupled electron transfer (PCET).19,20 A series of essential steps are interconnected in a sequential manner:21 (a) the NO3− migrates to the catalyst surface. (b) The electrons successively transfer to the active sites to participate in the catalytic process. (c) Active sites facilitate the adsorption and activation of NO3−, the intermediate conversion, and NH3 desorption. The primary challenge in achieving high-performance catalysis lies in the sluggish reaction kinetics induced by restricted mass transfer, complex multi-electron and proton transfer processes, and inappropriate adsorption/desorption of NO3− and intermediates, leading to poor activity for NH3 production and severe competing hydrogen evolution reactions.22–24 Based on this, various strategies have been proposed, including controlling the nanostructure of the catalyst,25,26 tandem catalysis,27,28 introducing defects,29,30 designing single/dual atoms,31,32 and forming alloys.33,34 Besides, constructing a built-in electric field is vital in optimizing the adsorption and desorption processes of reactants and intermediates.21,35 A composite catalyst leads to the spontaneous electron transfer resulting from the various work functions of different components, thereby establishing a built-in electric field.36 This strategy regulates the electronic distribution on the catalyst surface, thereby enhancing its intrinsic activity. Additionally, adjusting the coordination environment of active sites and modulating the catalyst-electrolyte electrical double layer can effectively introduce an electric field.37,38 Besides constructing a built-in electric field, introducing external stimuli can effectively regulate the thermodynamic and kinetic performance of the NO3RR. For instance, renewable solar energy can stimulate catalysts possessing localized surface plasmon resonance (LSPR) effects to generate high-energy hot electrons and an intense electric field, thereby promoting the NO3RR.39 The introduction of a built-in electric field and external stimuli has been proven to facilitate the NO3RR to produce NH3. However, a comprehensive summary and in-depth understanding of strategies involving built-in electric field/external stimuli for the NO3RR are deficient.
In this review, we thoroughly summarized various strategies involving a built-in electric field/external stimuli in enhancing the NO3RR and analyzed their mechanisms (Fig. 1). The aim is to provide insights into such modulation strategies for NO3− reduction to produce NH3. This paper begins by introducing the fundamental principles and challenges of the NO3RR, then reviews strategies for introducing a built-in electric field through constructing Schottky heterojunctions, semiconductor–semiconductor heterojunctions, adjusting active site coordination environments, and modulating electrical double layers in promoting the NO3RR. Additionally, we summarize the effects of various external stimuli, including light, stress, and thermal, on NH3 production. Finally, we emphasize the challenges and perspectives of modulation strategies from the built-in electric field or external stimuli to enhance the NO3RR.
 | ||
| Fig. 1 Schematic of a built-in electric field/external stimuli-assisted NO3RR. | ||
2. Fundamentals of NH3 synthesis
2.1. Mechanism of the NO3RR
Diverse reaction intermediates and products can be obtained in the NO3RR involving various pathways because of the wide range of nitrogen's oxidation states (−3 to +5).40 The NO3− concentration and pH value of the electrolyte closely influence the mechanism in the NO3RR.41 The NO3RR can be broadly categorized into indirect and direct reduction processes. The indirect reduction process involves intermolecular self-catalysis to generate active species, typically occurring in high NO3− concentrations (>1 mol L−1) in acidic electrolytes. Current research mainly centers on the direct reduction process, which includes electron-mediated and hydrogen atom-mediated reductions (Fig. 2).42 | ||
| Fig. 2 The electron-mediated (blue arrow) and hydrogen atom-mediated (green arrow) pathways of the NO3RR. Reproduced with permission.42 Copyright 2021, Elsevier B.V. | ||
In electron transfer-mediated NO3RRs, various products can be generated through the reaction, including NO2, NO2−, NO, N2O, N2, NH2OH, and NH3. Notably, N2 and NH3 are primary products owing to their high thermodynamic stability.43 In this process, NO3− is firstly adsorbed onto the active sites of the catalyst (eqn (1.1)).44 The subsequent reduction of NO3− to NO2− is usually considered the rate-determining step (RDS) in the NO3RR (eqn (1.2)).45 Thus, the NO3RR performance is affected by the concentration of NO3−. In the electrolyte with low NO3− concentrations, the reaction process is influenced by co-adsorbed ions, whereas in cases of high NO3− concentrations, the active site density of the catalyst becomes the pivotal factor.41 The intermediate NO2− demonstrates obvious reactivity on the catalyst, leading to adsorbed NO (eqn (1.3)), significantly determining product distribution.46 NO(ads) can either undergo reduction to NH4+ (eqn (1.4)), serving as the final product or desorb from the active sites to produce NO in the electrolyte (eqn (1.5)).47 The NO dimers with weak adsorption are formed in the presence of NO(aq) in the electrolyte, serving as precursors for N2O (eqn (1.6)), which is subsequently reduced to N2 (eqn (1.7)).48–51
| NO3(aq)− ⇄ NO3(ads)− | (1.1) |
| NO3(ads)− + 2H+ + 2e− → NO2(ads)− + H2O | (1.2) |
| NO2(ads)− + 2H+ + e− → NO(ads) + H2O | (1.3) |
| NO(ads) + 6H+ + 5e− → NH4+ + H2O | (1.4) |
| NO(ads) → NO(aq) | (1.5) |
| NO(ads) + NO(aq) + 2H+ + 2e− → N2O(ads) + H2O | (1.6) |
| N2O(ads) + 2H+ + 2e− → N2 + H2O | (1.7) |
Meanwhile, NO3RRs can also occur following the hydrogen atom (H(ads))-mediated reduction process. The Volmer step of water dissociation provides H(ads) as shown in eqn (1.8).52,53 Atomic hydrogen (H) is a potent reducing agent (E°(H+/H) = −2.31 V vs. SHE),54 which can reduce NO3−, NO2−, and NO adsorbed on the active sites (eqn (1.9)–(1.11)).55 Kinetically, the formation of N–H bonds occurs more readily than the formation of N–N bonds, resulting in NH3 being the primary product of this pathway (eqn (1.12)–(1.14)).56,57
| H2O + e− → H(ads) + OH− (Volmer) | (1.8) |
| NO3(ads)− + 2H(ads) → NO2(ads)− + H2O | (1.9) |
| NO2(ads)− + H(ads) → NO(ads) + OH− | (1.10) |
| NO(ads) + 2H(ads) → N(ads) + H2O | (1.11) |
| N(ads) + H(ads) → NH(ads) | (1.12) |
| NH(ads) + H(ads) → NH2(ads) | (1.13) |
| NH2(ads) + H(ads) → NH3(ads) | (1.14) |
The H(ads)-mediated NO3RR generally occurs at low overpotentials, which is essential for inhibiting side reactions. This pathway is particularly advantageous in noble metal catalysts, owing to their strong affinity for hydrogen.58 Conversely, the electron transfer pathway is more probable on catalysts that exhibit a high adsorption capacity for NO3−.
2.2. Challenges and dilemmas in the NO3RR
The NO3RR occurs at the solid–liquid interface, presenting a lower reaction barrier than the solid–gas–liquid interface associated with the NRR. However, thermodynamically, the overpotential for NH3 production is similar to that of several byproducts, such as N2O (1.12 V vs. RHE), NO (0.96 V vs. RHE), and NH2OH (0.73 V vs. RHE).59 Additionally, to achieve higher NH3 yield rates, it may be necessary to apply more negative electrode potentials, but this can simultaneously promote the competitive HER and reduce the faradaic efficiency of NH3 production, leading to unsatisfactory energy efficiency in the NO3RR.2,31,53,60,61 Furthermore, from a kinetic perspective, the NO3RR pathway for NH3 generation is complex, involving an eight-electron and nine-proton transfer process, resulting in sluggish kinetics.62 Consequently, developing catalysts for the NO3RR that achieve high NH3 yield rates and selectivity remains a significant challenge.Meanwhile, the necessary overpotential for the NO3RR and the source of protons depend on the pH of the electrolyte. Under acidic conditions, protons come from H+, leading to the following reaction equation:
Overall reaction:
| NO3− + 4e− + 5H+ → NH3 + O2 + H2O | (1.15) |
Cathode:
| NO3− + 8e− + 9H+ → NH3 + 3H2O (E0 = 0.88 V) | (1.16) |
Anode:
| 2H2O → O2 + 4H+ + 4e− | (1.17) |
Under neutral and alkaline conditions, H2O acts as the source of protons, resulting in the following reaction equation:
Overall reaction:
| NO3− + 2H2O → NH3 + 2O2 + OH− | (1.18) |
Cathode:
| NO3− + 8e− + 6H2O → NH3 + 9OH− (E0 = 0.69 V) | (1.19) |
Anode:
| 8OH− → 2O2 + 4H2O + 8e− | (1.20) |
The information above indicates that the reduction potential (E0) for the NO3RR is lower under acidic conditions (0.88 V) compared to alkaline electrolytes (0.69 V), resulting in more favorable thermodynamics.63,64 Nevertheless, under acidic conditions, the intense competition for hydrogen adsorption on the active sites impedes the NO3RR process. While NH3 synthesis costs in alkaline environments are already below the commercial price,2 the slow mass transfer requires higher voltages to achieve substantial yields, leading to increased energy consumption. This challenge is particularly pronounced in neutral media.
The activity and selectivity of the NO3RR are influenced by different factors, including the NO3− concentration, catalyst type, and diffusion dynamics. For a specific catalyst, enhancing the mass transfer can significantly improve the NO3RR. Increasing the concentration of NO3− enhances the accessibility of active sites to NO3−, thereby accelerating the reduction reaction. However, as the reaction progresses, the concentration of NO3− in the electrolyte diminishes, resulting in a gradual decrease in the NH3 yield rate. It is also crucial to understand whether the decrease in NH3 yield rate is related to catalyst deactivation or is simply due to a reduction in reactant concentration. Nevertheless, addressing mass transfer limitations in the NO3RR requires different strategies to those in CO/CO2 reduction reactions.65,66 To alleviate the limitations of mass diffusion issues in the CO/CO2 reduction reaction, gas diffusion electrodes can be utilized in flow-cell reactors to directly provide gaseous reactants to the electrolyte/electrode interface, which is not applicable to the NO3RR because NO3− is dissolved in the electrolyte.67 Consequently, developing methods to enhance the mass transport of NO3− under different conditions remains a significant challenge for future research.
Based on the mechanism and the challenges in the NO3RR, proposing a well-designed strategy to regulate the adsorption and activation of NO3− at active sites, control the transfer behavior of electrons and active hydrogen to suppress side reactions and facilitate the hydrogenation process, and manage mass diffusion to ensure a sufficient supply of reactants is essential for achieving high activity and selectivity for NH3 production via NO3RRs. The strategy not only enhances the efficiency of the reaction but also provides a pathway for the industrial application of the NO3RR.
3. NH3 synthesis enhanced by built-in electric fields
Enhancing NH3 synthesis through manipulating built-in electric fields represents a promising avenue in catalysis.68–72 These fields can profoundly impact the energies of key reaction steps, ultimately influencing the catalytic process. Researchers can fine-tune the adsorption energies of reactants and intermediates by designing these fields within catalysts, thereby steering the reaction towards more favorable pathways. This approach can boost the catalytic activity and facilitate the selectivity of the NH3 synthesis reaction, offering a pathway towards more sustainable and efficient NO3RRs.3.1. Mott–Schottky heterojunction
Utilizing the difference in work functions between metals and semiconductors to construct a Mott–Schottky heterojunction is an effective strategy for introducing a built-in electric field.73 When a metal and a semiconductor come into contact to form a rectifying junction, the energy bands of the semiconductor bend to match the work function (Φ) of the metal, triggering electrons to transfer from the component of lower work function to that with a higher work function, ultimately resulting in the alignment of the Fermi levels of both components.74 In detail, as shown in Fig. 3a, when the metal's work function is greater than that of the semiconductor, electrons irreversibly transfer towards the metal upon contact, leading to electron accumulation on the metal side and depletion on the semiconductor side at the interface, resulting in an upward bending of the semiconductor's conduction and valence bands at the interface, forming an intrinsic electric field.75 Conversely, when the metal's work function is lower than that of the semiconductor (Fig. 3b), electrons migrate irreversibly toward the semiconductor until the Fermi levels of both materials align. This process leads to electron accumulation on the semiconductor side and depletion on the metal side at the interface, resulting in a downward bending of the semiconductor's conduction and valence bands at the interface, thereby establishing a built-in electric field.73 Based on the mechanism mentioned above, the charge distribution state and the built-in electric field on the metal–semiconductor contact interface can be controlled by constructing different Mott–Schottky heterojunctions, potentially regulating the transfer of electrons in catalytic reaction processes and significantly altering the adsorption and desorption of specific reactants. Therefore, the development of Mott–Schottky heterojunction catalysts presents a strategy conducive to enhancing performance in the synthesis of NH3.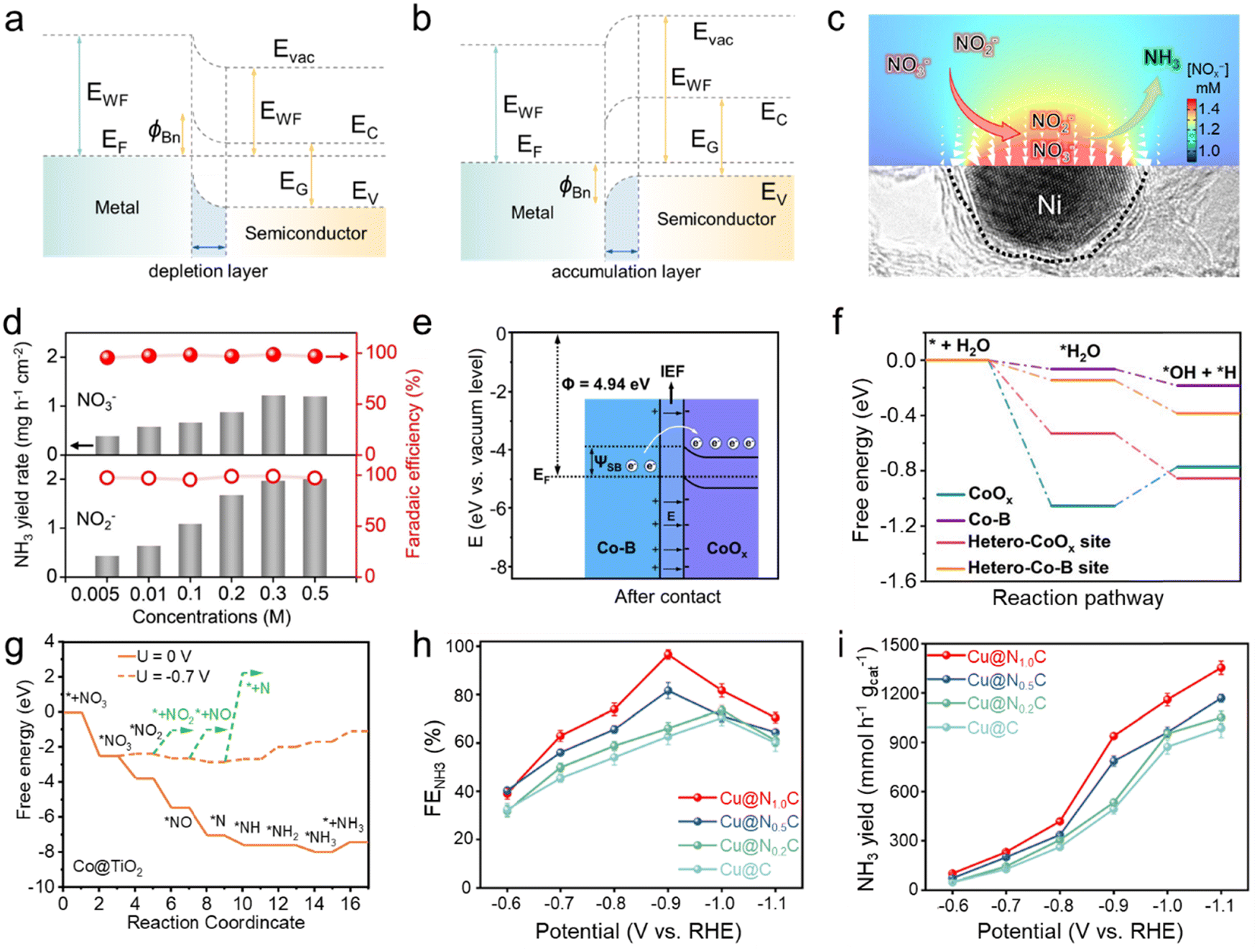 | ||
| Fig. 3 (a) and (b) Energy band diagrams. Evac, vacuum level; EG, energy gap; EWF, work function; Ec, conduction band edge; EV, valence band edge; EF, Fermi energy; ϕBn, Schottky barrier. (c) The distribution of NOx− anions on the surface of Nix/NC-sd. The size and orientation of each arrow indicate the density and location of NOx− anions. The lower part of the image shows a high-resolution transmission electron microscopy image of Nix/NC-sd. (d) NH3 yield rate and faradaic efficiency of Ni35/NC-sd in 0.5 M Na2SO4 electrolyte with various NOx− concentrations at −0.5 V vs. RHE. Reproduced with permission.76 Copyright 2021, Wiley-VCH. (e) Energy band diagrams of Co–B, and CoOx after contact. EF, Fermi energy; IEF, built-in electric field; ΨSB: Schottky barrier. (f) Free energy diagrams of H2O dissociation on different sites. Reproduced with permission.36 Copyright 2024, Royal Society of Chemistry. (g) Free energy diagram of Co@TiO2 adsorbing different intermediates in the NO3RR. Reproduced with permission.77 Copyright 2023, Wiley-VCH. (h) Faradaic efficiency and (i) yield rate of NH3 for Cu@NxC obtained in 0.5 M Na2SO4 and 0.1 M NaNO3. Reproduced with permission.78 Copyright 2024, Royal Society of Chemistry. | ||
To introduce the electric field induced by the Schottky barrier for promoting NH3 production, Li and colleagues76 fabricated electron-deficient Ni nanoparticles within nitrogen-rich carbon matrices (Nix/NC-sd). Computational simulations showed that the localized electric field at the charged electrodes can significantly enhance the density of NOx− ions (Fig. 3c), thereby accelerating the reduction process. This strategy facilitates the capture activation of NOx− and leads to high catalytic efficiencies for NH3 synthesis across a wide range of NOx− concentrations, maintaining a faradaic efficiency exceeding 95% in NH3 production and achieving a NH3 yield rate of 5.1 mg h−1 cm−2 from NO3− reduction and 25.1 mg h−1 cm−2 from NO2− reduction at −0.5 V vs. RHE (Fig. 3d). The remarkable NH3 production performance can be reproduced over 5 consecutive cycling tests, indicating its robust stability. The work by Wang et al.36 further evidenced the effectiveness of Mott–Schottky heterojunctions in enhancing NH3 production performance. Based on the work functions and band energies of metallic Co–B and p-type semiconductive CoOx, the formation of the Schottky contact in Co–B@CoOx results in charge redistribution and the establishment of a built-in electric field (Fig. 3e). This phenomenon regulates the adsorption behavior of reactants and intermediates during the NO3− reduction to NH3 process. Additionally, as protons derived from H2O serve as essential reactants in the NH3 production process, their research also demonstrates the influence of Schottky junction-induced electron redistribution on the behavior of H2O molecules. As illustrated in Fig. 3f, Co–B@CoOx exhibits enhanced capabilities in H2O molecule adsorption and dissociation compared to counterparts, thereby facilitating proton provision for participation in the hydrogenation process. Zheng and colleagues77 fabricated a Mott–Schottky heterojunction by loading Co nanoparticles onto TiO2 nanobelt arrays and demonstrated the promoting effect of the built-in electric field on the NO3RR by DFT calculations. The Schottky contact can mediate the electronic structure of Co@TiO2, increasing the electron density on the surface of Co, which facilitates the adsorption of NO3−, thereby enhancing the reduction process. The free energy diagram of Co@TiO2 adsorbing various species at different potentials in Fig. 3g exhibited the rate-determining step, *NH to *NH2, was only 0.69 eV. Meanwhile, the calculated free energy, which discourages the generation of NO2, NO, and N2, validates that the Schottky junction enhances the selectivity of the NH3 production catalytic process. In addition to directly utilizing the built-in electric field of Schottky junctions to regulate the adsorption of reactants and intermediates, Yu and his colleagues78 also employed Schottky junctions to promote the NO3RR by adjusting the state of active sites. They uniformly dispersed Cu nanoparticles on N-doped carbon substrates, where the N doping adjusted the carbon bandgap and regulated Mott–Schottky heterostructures. Due to the Schottky contact, electrons transferred irreversibly from Cu to the substrate, forming a built-in electric field and converting Cu sites into electron-deficient states, increasing the Cu+ concentration. This adjustment facilitated the activation and adsorption of NO3−, resulting in a faradaic efficiency for NH3 production of 96.2% at −0.9 V vs. RHE (Fig. 3h), along with an NH3 yield rate of 1353.1 mmol h−1 gcat−1 at −1.1 V vs. RHE (Fig. 3i).
3.2. Semiconductor–semiconductor heterojunction
A semiconductor heterojunction is a structure composed of different types of semiconductor materials, mainly categorized as p–n and non-p–n junctions. Taking the p–n junction as an example (Fig. 4a), when p-type and n-type semiconductors come into contact, a built-in electric field is formed at the interface due to the diffusion of electrons and holes. This electric field effectively separates electrons and holes and inhibits their recombination, prolonging the lifetime of charges. Additionally, this unique structure allows for spatial separation of oxidation–reduction sites. To utilize the distinctive properties of semiconductor heterojunctions for NO3RR, Lu and colleagues35 developed a novel electrocatalyst by layering CuCl (111) and rutile TiO2 (110) with distinct work functions. The electron transfer from TiO2 to CuCl leads to a built-in electric field (BEF), and is hence named CuCl_BEF. They utilized molecular dynamics simulation in the electrolyte containing 100 mg L−1 NO3− and concluded that after 10 ns, the peak concentration of NO3− positioned on the CuCl_BEF side reached 12.3 ions per nm3, surpassing the concentration observed on the pure CuCl surface (6.3 ions per nm3, as shown in Fig. 4b). Consequently, NO3− with ultra-low concentrations (100 mg L−1) was converted to NH3 at a remarkable production rate of 1.82 mg h−1 cm−2 (Fig. 4c). In contrast, CuCl + MXene without a built-in electric field exhibited a lower rate. The CuCl_BEF catalyst displayed negligible fluctuations in selectivity during consecutive recycling tests, indicating its superior stability (Fig. 4d). The theoretical calculation shows that the electric field facilitated the accumulation of NO3− ions on the catalyst surface, enhancing the reaction rate and lowering the energy barrier of the RDS for the key reaction intermediate *NO (Fig. 4e). The presence of the electric field in this semiconductor heterojunction can enhance ion adsorption, thereby boosting catalytic efficiency. This strategy suggests new avenues for designing innovative catalysts tailored for reactions involving ions as reactants. | ||
| Fig. 4 (a) Schematic demonstrating the band structure and built-in electric field formation mechanisms in the p–n heterojunction. Reproduced with permission.79 Copyright 2024, Royal Society of Chemistry. (b) Spatial distribution of NO3− on the catalyst surface obtained from molecular dynamics simulation. CuCl_BEF represents a heterojunction composed of CuCl and TiO2 with an induced built-in electric field. (c) Average NH3 yield rate over 6 h for various electrocatalysts. (d) Consecutive recycling tests at −1.0 V vs. RHE for CuCl_BEF. (e) Free energy diagram of reaction pathways for reducing NO3− to NH3 on the CuCl_BEF. Reproduced with permission. Copyright 2021, Wiley-VCH. (f) Schematic presenting the band structures and built-in electric field formation of CuO/MnO2. Reproduced with permission.80 Copyright 2022, Royal Society of Chemistry. (g) Band structures of CuO and NiO pre-contact (left) and post-contact (right). Reproduced with permission.69 Copyright 2024, Elsevier B.V. | ||
Meanwhile, Wang and colleagues80 developed a novel hierarchical structure of CuO@MnO2 1D core–2D shell nanoarrays supported on a Cu foam substrate (CuO@MnO2/CF). Integrating MnO2 nanosheets onto the CuO nanowire arrays offers a high density of active sites and enhances mass transfer. Notably, the CuO/MnO2 heterojunction establishes an built-in electric field at the interface, promoting the accumulation of NO3− and expediting NO3− reduction kinetics through optimized adsorption processes (Fig. 4f). As a result, the CuO@MnO2/CF composite demonstrates an exceptional NH3 faradaic efficiency of 94.92%, remarkable NH3 selectivity of 96.67%, and superior NO3− conversion of 99.38% in the NO3RR. In another study, benefiting from the superior H2O-to-H* conversion capabilities of nickel-based semiconductors,81,82 Zou and co-workers69 created a heterojunction by combining CuO and NiO due to their matched work functions. Through analyses using X-ray photoelectron spectroscopy (XPS) and ultraviolet photoemission spectroscopy (UPS), a semiconductor–semiconductor heterojunction between CuO and NiO was validated, which can trigger a built-in electric field. The catalytic performance of CuO/NiO in the NO3RR under low NO3− concentrations including NO3− conversion rates, product selectivity (NH4+, NO2−, and N2), and faradaic efficiency, was assessed by comparing with the performance of pure CuO and NiO. The influence of applied potentials, pH of the electrolyte, and NO3− concentrations on the catalytic performance of CuO/NiO in the NO3RR was also investigated. Density functional theory (DFT) calculations revealed that the CuO/NiO system, with its built-in electric field, is conducive to enhancing reactant mass transfer and H* provision. Based on the experimental and theoretical results, they summarized the origination of the promotion of CuO/NiO in the NO3RR. As illustrated in Fig. 4g, upon the contact between CuO and NiO, the spontaneous electron flow leads to band bending, creating a built-in electric field that accelerates charge transfer and the rate of the NO3RR.
3.3. Coordination-environment modulation
In addition to heterojunction interfaces, introducing an electric field by adjusting the coordination environment of catalysts can also enhance catalytic processes. The coordination structure near active sites can effectively influence electron distribution and induce built-in electric fields, thereby modulating the adsorption behavior of reactants and intermediates. Moreover, it has been demonstrated that modulating the built-in electric field between dual-layer single-atom catalysts can also effectively adjust polarization, suppressing competitive reactions in the NO3RR.37Frauenheim and colleagues37 proposed a universal strategy of heterogeneous bilayer single-atom catalysts with tunable surface chemistry to enhance the NO3RR. The asymmetric atomic design of these catalysts leads to a dipole moment that can strengthen built-in electric fields, regulating the activation of reactants and the adsorption of intermediates, thus modulating reaction pathways and enhancing NH3 selectivity and activity. They investigated a series of bilayer N-doped graphene (GN) supported single-atom catalysts. High-throughput calculations revealed that TiV-N4, NbV-N4, and GaV-N4-GN can all suppress competitive HERs with favorable limiting potentials of −0.32, −0.20, and −0.25 V, respectively. In the free energy diagrams of V2-N4-GN (Fig. 5a), apart from the *NO3− → *NO3H and *NO → *NOH steps showing an upward ΔG, the remaining hydrogenation processes exhibit a downward ΔG. NH3 desorbs from the adsorption site after overcoming a small final free energy barrier of 0.1 eV, indicating fast NH3 desorption from the catalyst surface. Therefore, the rate-determining step in this system is the *NO → *NOH transformation. The authors also summarized the limiting potential and RDS of the NO3RR on MV-N4-GN (Fig. 5b), indicating a strong dependence of the RDS and limiting potential on polarization. Compared to the upward ΔG for *NO3− → *NO3H in V2-N4-GN, the corresponding step in NbV-N4-GN shows a downward ΔG (Fig. 5c). Furthermore, in the subsequent hydrogenation steps, polarization did not alter the most stable NO adsorption mode and the most favorable adsorption pathway for the NO3RR. Instead, the RDS shifted to the desorption of NH3 from the surface.
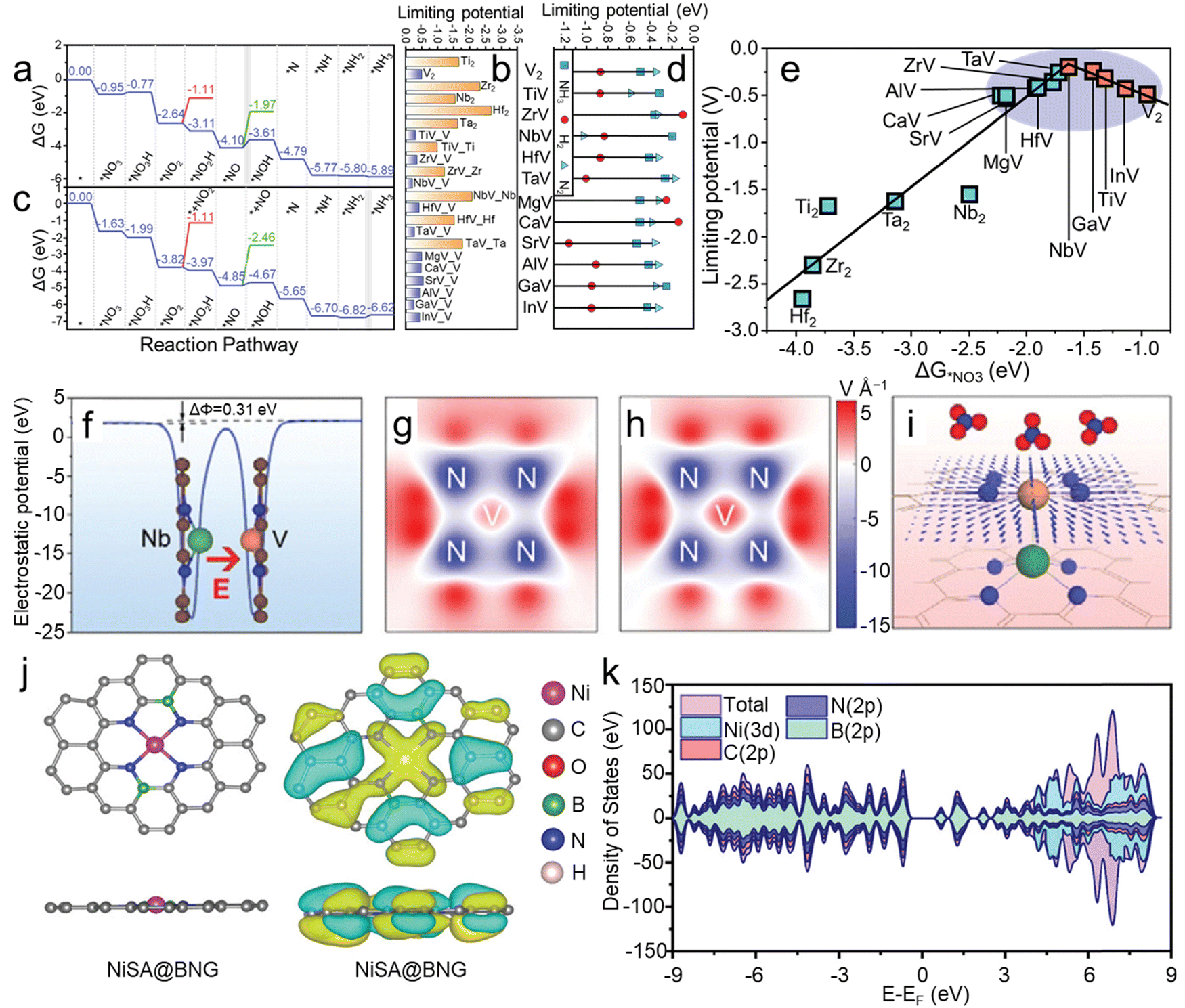 | ||
| Fig. 5 Free energy diagram for the reduction of NO3− to NH3 on (a) V2-N4-GN and (c) NbV-N4-GN. (b) Limiting potentials on M2-N4-GN and MV-N4-GN for the NO3RR. (d) Limiting potentials for generating various products during the NO3RR process. (e) Volcano-shaped relationship between UL and ΔG*NO3. (f) Plane-averaged potential of NbV-N4-GN. The simulated electric field on (g) V2-N4-GN and (h) NbV-N4-GN, in which the unit is V Å−1. (i) Schematic depicting the electric field from NbV-N4-GN, with each arrow's size and direction indicating the field's strength and orientation. Reproduced with permission.37 Copyright 2022, Wiley-VCH. (j) Optimized structures and charge distribution of NiSA@BNG. (k) The DOS for the NiSA@BNG catalyst. Reproduced with permission.83 Copyright 2024, Wiley-VCH. | ||
Polarization can adjust the strength of interactions between intermediates and catalysts, thereby influencing the selectivity of reactions. Consequently, the limiting potential of byproducts was calculated to investigate the ease of byproduct formation on NbV-N4-GN. For most MV-N4-GN catalysts (excluding ZrV, MgV, and CaV, as shown in Fig. 5d), the ΔG*H values are significantly higher than those of the NO3RR, indicating that MV-N4-GN retains its inhibitory effect on the HER. Therefore, the unique atomic structure and electronic properties of heterogeneous BSACs contribute to the polarization dependence and controllable selectivity of the NO3RR. Furthermore, the volcano-shaped relationship between the limiting potentials (UL) and adsorption free energy of NO3− (ΔG*NO3) indicates that heterogeneous BSACs exhibit a higher activity compared to their counterparts (Fig. 5e). The plane-average potential of NbV-N4-GN in Fig. 5f further confirms the existence of an electric field and that can be modulated through substituting other atoms. As shown in Fig. 5g and h, after Nb substitution, the color deepens around the V atom, indicating an enhanced electric field around V, further validating the feasibility of regulating the coordination environment to enhance the electric field. Therefore, the built-in electric field in BSACs can promote the accumulation of NO3− and mass transfer on the surface of the catalyst for an improved NO3RR (Fig. 5i). Meanwhile, Zhao and co-workers83 doped boron into carbon substrate to adjust the coordination structure of Ni SACs and construct B–N bonds to trigger the electric field that could facilitate the adsorption of NO3−. Compared to the catalyst without an electric field, that is, without doping boron, the faradaic efficiency of NiSA@BNG increased to ≈95% from 51% and the yield rate is 2 times higher. As shown in the density of states for NiSA@BNG, the B atoms significantly increase the density of states and redistribute the density of state (DOS; as shown in Fig. 5j and k), indicating the promoted conductivity of NiSA@BNG. Meanwhile, NiSA@BNG demonstrates superior stability, as evidenced by no significant decrease in faradaic efficiency and yield rate over 5 consecutive recycling tests. In conclusion, the introduced electric field declines the reaction energy barrier, promotes the adsorption of NO3− and enhances the stabilization of the catalyst.
3.4. Electrical double layer construction
Due to the electrode–electrolyte interface being the site of catalytic processes, research on the electrical double layer formed when the electrode contacts the electrolyte is crucial. As shown in Fig. 6a, the electrical double layer comprises the inner Helmholtz layer, outer Helmholtz layer, and diffusion layer.84 The type and concentration of solvated ions in the electrical double layer play a critical role in catalytic reactions, where cations in the electrolyte can stabilize negatively charged reaction intermediates through local electric field effects. In this context, Yu and colleagues38 studied the NO3RR performance by using bulk Cu as the cathode and adjusting the types and concentrations of alkali metal ions in the electrolyte. As shown in Fig. 6b, various alkali metal cations (Li+, Na+, K+, Cs+) were selected for COMSOL multi-physics simulations, and the resulting electric double layer (EDL) all exhibited negative potentials, which indicates the effectiveness of utilizing alkali metal cations in the electrolyte to modulate the electrolyte microenvironment within the EDL. This phenomenon can promote the dissociation of water molecules on the electrode surface and facilitate the rapid movement of generated OH− away from the electrode surface towards regions of positive electrostatic potential (ESP). During this process, OH− near alkali metal cations removed an H+ from adjacent water molecules, thus facilitating proton transfer. The authors further compared the NO3RR current obtained in electrolytes containing various alkali metal cations (Fig. 6c), with the current ordering as K+ > Na+ > Li+ ≈ Cs+, consistent with the results shown in Fig. 6b. This alignment demonstrates that modulating the EDL effectively promotes the NO3RR for NH3 production. Adding Li+, Na+, and K+ alkali metal cations had little effect on the potential-determining step, but introducing Cs+ increased the reaction barrier of the potential-determining step, thereby inhibiting the NO3RR (Fig. 6d). The evaluation through electrochemical testing of the NO3RR using Cu/Cu2O with different alkali metal cations added to the electrolyte also assessed the promoting effect of local electric field effects on the reaction. In the presence of NO3−, the addition of various cations showed significant NO3− catalytic activity (Fig. 6e and f). The charge accumulation at the catalytic interface shows the most significant decrease in Cdl after adding K+ (Fig. 6g). The conversion of NO3−, faradaic efficiency, selectivity for NH4+, and yield rate achieved 99%, 98%, 99.5%, and 980 μmol h−1 cm−2 in 1 M KOH, indicating the effectiveness of local electric field effects in promoting the reduction of NO3−. Combining the theoretical and experimental findings, a more comprehensive insight into the alkali metal cation-mediated strategy in the NO3RR process was proposed (Fig. 6h). Alkali metal cations near the OHP regulate proton migration and impact the interactions between protons and intermediates. Among these cations, K+ emerges as the most advantageous electrolyte component. It facilitates proton adsorption on the catalyst surface, promoting the hydrogenation process and enhancing the NO3RR. | ||
| Fig. 6 (a) Schematic of the EDL. Reproduced with permission.84 Copyright 2022, Elsevier B.V. (b) ESP distribution obtained from models with various alkali metal ions. (c) Simulated partial current densities of the NO3RR using electrolytes with various alkali metal cations. PZC represents the potential of zero charge. (d) Free energy diagram for reducing NO3− to NH3 with different alkali metal cations. (e) Linear sweep voltammetry (LSV) curves of Cu/Cu2O in the electrolyte containing 1 M MOH and 1 M MOH + 0.05 M MNO3. (f) NH4+ yield rate at different potentials in the electrolyte containing 1 M MOH and 0.05 M MNO3 at 0.4 V vs. RHE. (g) Double-layer capacitance (Cdl) values in 1 M MOH with 0.05 M MNO3. (h) Schematic diagram of the EDL-assisted NO3RR process. Reproduced with permission.38 Copyright 2024, Wiley-VCH. | ||
4. NH3 synthesis enhanced by external stimuli
The reasonable design of external stimuli-assisted catalysis, that is, efficiently harnessing energy abundant on earth, including solar energy, thermal energy, and mechanical energy, has significant potential for producing renewable fuels and removing environmental pollutants. Recent studies have indicated that certain external stimuli, such as light and stress, can trigger the built-in electric field within the catalyst, effectively achieving charge separation and impacting catalytic activity, reaction kinetics, and selectivity in reducing NO3− to NH3. Additionally, external stimuli can influence the catalytic reaction process through other pathways such as overcoming thermodynamic barriers in the NO3RR. However, there is a lack of comprehensive summaries and an in-depth understanding of the research progress on the NO3RR under different external stimuli.4.1. Optical stimuli
Photocatalysis is a process that utilizes light energy to drive chemical reactions and is widely applied in pollutant removal and energy conversion.85–87 It is primarily categorized into semiconductor photocatalysis and localized surface plasmon resonance (LSPR)-assisted photocatalysis. Semiconductor photocatalysis utilizes electron–hole pairs generated in semiconductor materials under light stimulation to catalyze reactions.88,89 On the other hand, LSPR-assisted catalysis relies on the localized electric field enhancement effect and high-energy hot electrons and holes produced by metal nanoparticles under specific wavelength light, enhancing the efficiency of photocatalytic reactions.90,91 | ||
| Fig. 7 (a) Schematic of semiconductor photocatalysis. Reproduced with permission.96 Copyright 2020, Elsevier Ltd. (b) EPR results of TNS before and after introducing oxygen vacancies. (c) The formation energy of BaONCs loading on pristine and defected TNS. (d) Time-resolved fluorescence emission decay spectra. The inset is UV-vis DRS. (e) Reaction rate of various products in the NO3RR. (f) Activation energy for NO3− reduction and H2O dissociation. (g) Stability test of BaONCs-TNS and the performance comparison with other works. Reproduced with permission.97 Copyright 2022, Springer Nature. (h) UV-Vis DRS and (i) photocurrent response of various samples. (j) Comparison of NH3 yield rate by different catalysts. Reproduced with permission.98 Copyright 2021, AAAS. | ||
Dong and co-workers97 synthesized subnanometric alkaline-earth oxide clusters (MONCs, M = Mg, Ca, Sr or Ba) through the operando construction for NH3 photosynthesis from NO3−. The electron paramagnetic resonance (EPR) measurements in Fig. 7b confirm the introduction of oxygen vacancies on TiO2 nanosheets (TNS). DFT calculations (Fig. 7c) demonstrated the feasibility of uniformly depositing MONCs on photoinduced vacancies on TNS. Time-resolved fluorescence emission decay spectra in Fig. 7d revealed a significant enhancement in the charge separation capability of the composite structure. They employed ethylene glycol (EG) as the hole sacrificial agent. The oxidation of EG and the reduction of nitrate ions (NO3−) occur simultaneously, where the oxidation of EG consumes the holes that, in turn, can accelerate the NO3RR, promoting NH3 synthesis. As an essential parameter for evaluating performance in the NO3RR, the selectivity of NH3 production on BaONCs-TNS was investigated. As shown in Fig. 7e, 29.26 mmol gcat−1 NH4+ was obtained after 3 h of photocatalysis. The total N species in the electrolyte during the catalyst remained stable, confirming the five-electron transfer process for N2 generation was suppressed. The resulting selectivity for NH3 production reached 97.67%. The activation energies for synthesizing NH4+ from NO3− and water splitting for H2 production were determined through calculations (Fig. 7f). A significant reduction of 1.42 eV in activation energy was observed for the NO3− reduction reaction compared to water splitting, indicating the potential for effectively suppressing electron consumption in side reactions. A long-term stability test of BaONCs-TNS was carried out as shown in Fig. 7g, in which 0.78 mmol NH4+ was obtained after 72 h of photocatalysis, demonstrating leading-edge performance in the NO3RR for NH3 generation. Meanwhile, Ni and colleagues98 loaded Ru on g-C3N4 in a photocatalyst for the NO3RR. As shown in the UV-vis diffuse reflection spectra (DRS) of various samples (Fig. 7h), a significant redshift and improved light absorption in the visible light region were observed after Ru decorating on 2D-C3N4. The introduction of Ru facilities the separation of photogenerated electrons and holes, leading to an increased photo response (Fig. 7i). Benefiting from this strategy, the optimal catalyst displays a superior NH3 yield rate of 2.627 mg h−1 gcat−1, 2.93 times higher than bulk g-C3N4 (Fig. 7j).
 | ||
| Fig. 8 (a) Schematic of LSPR. Reproduced with permission.107 Copyright 2021, Springer Nature. (b) NH4+ concentration, (c) NH4+ yield rate, (d) consumed charge, and (e) NH4+ faradaic efficiency obtained in the dark and under 532 nm light irradiation. (f) NH4+ yield rate at various ΔE under different conditions. (g) Scheme of LSPR-assisted NH4+ electrosynthesis. The working electrode in (b)–(g) is Au nanoparticles on a glassy carbon electrode. Reproduced with permission.39 Copyright 2022, American Chemical Society. (h) The finite-difference time-domain (FDTD) simulated electric-field distribution on Au NR@Cu2O–AuPd NPs. (i) NO3− conversion rate and (j) NH3 yield rate of different catalysts. Reproduced with permission.108 Copyright 2024, Elsevier B.V. | ||
The effect generated on the surface of metal nanostructures can decay through relaxation processes, generating numerous hot electron–hole pairs.109 Efficiently separating these hot electron–hole pairs shows promise for advancing catalytic processes like oxygen reduction, water splitting, methanol oxidation, and NH3 synthesis.110–113 Consequently, the effective separation and direct utilization of hot electron–hole pairs produced by the photoinduced metal LSPR effect, along with leveraging the improved catalytic activity and stability conferred by this effect, have captured considerable interest among researchers in the area of innovative sustainable and environmentally friendly energy research.
In LSPR-assisted NH3 electrosynthesis, Jain and colleagues39 synthesized Au nanoparticles with LSPR effects, serving as both photon-capturing materials and active sites for the NO3RR. Their electrosynthesis and overall electrochemical activities were enhanced by a factor of 15 compared to traditional electrocatalysis (Fig. 8b–d), while the faradaic efficiency for NH4+ production increased from 71% to 76% (Fig. 8e). Additionally, they proposed that the enhancement of kinetics due to the LSPR effect is most pronounced near the onset potential under applied bias. At ΔE = −10 mV, the LSPR effect boosted the NH4+ yield rate by 15 times, a level of activity that required an additional 140 mV of potential in the dark for the catalyst to achieve this (Fig. 8f). In conclusion, under light irradiation, the LSPR of Au nanoparticles is stimulated, generating electron–hole pairs through interband and intraband damping (Fig. 8g). High-energy electrons in the sp states drive the reduction of NO3− to NH4+, producing OH−, while the holes are consumed by electrons in the circuit. This leads to an alkaline oxygen evolution reaction at the counter electrode, with the OH− being oxidized.
Zhao and colleagues108 utilized the internal electromagnetic field generated by Au NRs to synthesize Au NR@Cu2O–AuPd NPs. The internal electromagnetic field (Fig. 8h) results from the distinct optical response properties of the bimetallic components and enhanced ability to adsorb NO3− and reaction intermediates, boosting the activity and selectivity in the NO3RR. This led to an NH3 yield rate of 4587.00 μg h−1 mgcat.−1 and a faradaic efficiency of 93.09% under neutral conditions (Fig. 8i and j).
4.2. Stress stimuli
Piezoelectricity is a fundamental material property that generates electric potential in response to stress stimuli. Typically, semiconductors exhibiting piezoelectric characteristics are polar materials possessing crystal structures that lack a center of symmetry. When subjected to stress, separating positive and negative charge centers within the lattice creates a piezoelectric potential on the material's surface. The most well-known piezoelectric materials, such as perovskite-structured Pb(Zr,Ti)O3 (PZT), zinc oxide (ZnO), gallium nitride (GaN), and zinc sulfide (ZnS), are extensively utilized in diverse fields like mechanical energy harvesting, active sensing, and self-powered systems. This generated potential can facilitate the transfer of electrons and holes towards the material's surface, leading to the development of catalytic research based on the mechanical-to-chemical energy conversion using piezoelectric materials.114The concept of piezoelectric catalysis is depicted in Fig. 9a. When subjected to external forces, piezoelectric materials generate an internal electric field because of spontaneous polarization. This electric field facilitates the separation of carriers towards the material's surface, thereby facilitating redox on the active sites. By adjusting external stress, such as controlling the frequency and power of the stress, both the orientation and strength of polarization can be manipulated. This dynamic regulation of surface properties has potential for the catalytic process. Research efforts have explored leveraging piezoelectric polarization to drive a series of catalytic processes, including water splitting, pollutant degradation, CO2 adsorption/reduction, and the selective transformation of organic compounds.115–118 In our previous work, we utilized the piezoelectric effect of the electron-rich BaTiO3 (BTO-OV1) to promote the catalytic performance of metal nanoparticles in the NO3RR.119 After introducing stress stimuli, the contact state between metal nanoparticles and the BTO-OV1 substrate was modulated. The I–V curves in Fig. 9b go from curves to straight lines as the pressure increases, which confirms the contact state was from Schottky contact to ohmic contact. The DFT calculations further demonstrated that the stress stimuli adjusts the local electron distribution and promotes the interaction between metal nanoparticles and BTO-OV1 (Fig. 9c). Based on the experimental and computational results, we proposed the mechanism of this strategy (Fig. 9d). After introducing stress stimuli, the contact state between metal nanoparticles and BTO-OV1 was modulated due to the piezoelectric effect. Because of the difference in the work function, a large number of the electrons in BTO-OV1 were transferred smoothly to the metal nanoparticles, which led to the generation of active hydrogen that participated in the hydrogeneration process in the NO3RR. The charge transfer also triggers the charge redistribution, suppressing the Volmer–Heyrovsky step of the competing HER. The optimized catalyst displays a superior faradaic efficiency of 95.3% and a yield rate of 6.87 mg h−1 mgcat.−1 for NH3 generation.
 | ||
| Fig. 9 (a) Schematic of the piezoelectric catalysis. Reproduced with permission.115 Copyright 2020, American Chemical Society. (b) I–V curves measured at different pressure using M/BTO-OV1 (M = Ru, Pd, and Pt). (c) Electron density difference mappings obtained from Ru/BTO-OV1 without (left) and with pressure (right). (d) Schematic diagram of piezoelectric-assisted electrochemical NO3RR. In (c) and (d), the red, gray, purple, and white balls represent O, Ti, Ru, and H atoms, respectively. Reproduced with permission.119 Copyright 2023, Elsevier Ltd. (e) Schematic of charge transfer of N-PDA under various conditions. (f) NH4+ yield rate measured under different conditions. Reproduced with permission.120 Copyright 2024, Elsevier B.V. | ||
The piezoelectric polarization electric field triggered by stress stimuli also has the potential to assist photocatalysis. Chen and colleagues120 synthesized a naphthalene-linked perylene diimide polymer (N-PDA) with rigidity and polarity for producing NH3 from NO3−. The stress stimuli triggers the piezoelectric polarization electric field of the catalyst, further facilitating the separation of photogenerated charge carriers and suppressing their recombination (Fig. 9e). The NH3 production rate through piezo-photocatalysis exceeded the sum of the yield rates obtained by piezoelectric catalysis and photocatalysis, further confirming the promotion of the stress stimuli in the NO3RR (Fig. 9f).
4.3. Thermal stimuli
Thermal catalysis primarily involves heating to provide the energy needed for catalytic reaction systems to overcome thermodynamic barriers, thus driving the conversion of reactants into products.121–124 In catalysis, electrons are typically driven and facilitated to transfer through electrical work under an applied voltage. Nevertheless, in steps except for electron transfer, the electric potential may not effectively influence the thermodynamics of the reaction. Introducing thermal stimuli offers a promising strategy for adjusting reaction thermodynamics, and the overpotential arising from kinetic barriers exceeding thermodynamic barriers can also be regulated through the thermal stimuli.125 In the NO3RR involving multi-PCET, electron transfer can be modulated through electrochemical methods. However, the proton transfer steps in the hydrogenation process cannot be controlled by the electric potential. Therefore, the introduction of thermal stimuli presents a promising approach for synergistically regulating both electron and proton transfer processes.Yu and colleagues125 dispersed Ni on a Cu substrate to prevent the accumulation of localized active hydrogen, synthesizing Ni-decorated Cu single-atom alloy oxide nanowire (Ni1Cu SAAO NW) to suppress the competing HER in NH3 production from NO3−. While inhibiting side reactions by enhancing the active hydrogen coverage, merely applying voltage alone cannot promote proton transfer in the hydrogenation process effectively. Therefore, they introduced thermal stimuli to facilitate the conversion of NO3− (Fig. 10a), enhance water molecule dissociation, and reduce the energy barrier in the reaction. Compared with Cu2O NWs under room temperature, Ni1Cu SAAO NWs exhibited significantly enhanced activity and selectivity in alkaline electrolytes at 60–80 °C (Fig. 10b and c). As shown in Fig. 10d, even at 0.1 V vs. RHE, the faradaic efficiency of NH3 production and temperature showed a positive correlation, increasing from 34.6% at 25 °C to 79.8% at 80 °C, with an NH3 yield rate of 9.7 mg h−1 cm−2. In contrast, with increasing temperature, Cu2O NWs showed only limited improvement in faradaic efficiency. Furthermore, Fig. 10e illustrates the performance of NO3− to NO2− conversion at different temperatures. With increasing temperature, Ni1Cu SAAO NWs demonstrated a gradual decrease in NO2− faradaic efficiency and yield rate, indicating that introducing thermal stimuli promoted the conversion of NO3− to NH3. Conversely, Cu2O exhibited a noticeable increase in NO2− yield with increasing temperature, maintaining a relatively high level of NO2− faradaic efficiency. This suggests that introducing thermal stimuli also promoted the reduction of NO3− to NO2− on Cu2O NWs, but the relatively weak hydrogen affinity of Cu2O NWs limited further performance enhancement.
 | ||
| Fig. 10 (a) Thermal-assisted electrocatalysis NO3RRs on Ni1Cu SAAO NWs. (b) Mechanism diagram of the NO3RR at different temperatures using Ni1Cu SAAO and Cu2O NWs. HT, RT, and FE are high temperature, room temperature, and faradaic efficiency, respectively. (c) The energy barrier overcome by thermally boosted electrocatalysis and surface-active hydrogen during NO3− reduction on Ni1Cu SAAO. Faradaic efficiency and yield rate for (d) NH3 and (e) NO2− production at different temperatures using Ni1Cu SAAO and Cu2O NWs at 0.1 V vs. RHE. Reproduced with permission.125 Copyright 2024, American Chemical Society. (f) The activity of various catalysts in thermocatalytic NO3RR at different H2 partial pressures in pH 7 solution with 0.1 M NaNO3. (g) Arrhenius plots of PtRu/C for thermocatalytic and electrocatalytic NO3RRs. (h) The cost per tonne of NH4NO3 in thermocatalytic and electrocatalytic NO3RRs vs. NH3 yield rate. Reproduced with permission.126 Copyright 2021, Royal Society of Chemistry. | ||
Singh and colleagues126 compared the thermal catalytic and electrocatalytic mechanisms of Pt/C, PtRu/C, and Pt75Ru25/C under different electrochemical reaction parameters, demonstrating the promoting effect of thermal stimuli in NH3 production from NO3RRs. They proposed that increasing the hydrogen (electro)chemical potential could enhance the conversion rate of NO3− (Fig. 10f). Furthermore, changes in pH significantly influenced the reaction performance of thermal catalysis and electrocatalysis in synthesizing NH3 (Fig. 10g). The NH3 yield rate of PtRu/C via thermal catalysis and electrocatalysis was also compared with industrial Haber–Bosch processes regarding rate and cost. As shown in Fig. 10h, the results indicated that the NH3 yield rate of PtRu/C under pH = 1 was comparable to the Haber–Bosch process, with lower standard costs per ton of NH4NO3 consumed.
5. Conclusion and outlook
In this review, we comprehensively summarized strategies involving a built-in electric field and external stimuli to enhance the performance of the NO3RR for NH3 production. The approach of introducing built-in electric fields includes the construction of Mott–Schottky heterojunctions, semiconductor–semiconductor heterojunctions, modulation of coordination structures for active sites, and the construction of double layers. Additionally, the external stimuli modulation strategies such as introducing optical, stress, and thermal stimuli for enhancing NH3 production via NO3RR were also summarized. Thoughtful design of built-in electric fields and external stimuli can effectively boost both the activity and selectivity of NH3 production, with the potential to address energy crises and environmental pollution simultaneously. However, due to limited characterization techniques and design strategies, such modulation in the NO3RR still faces significant challenges.(1) Designing advanced characterization techniques to elucidate the specific roles of built-in electric field and external stimuli is crucial. For instance, introducing optical stimuli triggers the LSPR effect, generating high-energy hot electrons and holes and strong electric fields. However, distinguishing and quantifying the contributions of high-energy hot electrons and electric fields to catalytic reactions is challenging. Similarly, in semiconductor photocatalysis, light illumination induces the generation of carriers, accompanied by localized temperature changes on the electrode surface. Understanding the varying degrees of contribution from different enhancement mechanisms to catalysis facilitates the design of more efficient strategies to enhance catalytic efficiency.
(2) Developing in situ testing techniques is crucial for reactions involving built-in electric fields and external stimuli. Catalytic processes primarily occur on the catalyst surface, where such modulation can influence the adsorption and activation of reactants and intermediates, as well as control product distribution. In situ testing with built-in electric field/external stimuli participation can deepen the understanding of how they regulate catalytic processes. Catalytic reactions can be monitored in real-time, allowing observation of intermediate species behavior and changes in reaction kinetics. In short, developing in situ testing techniques will be pivotal in propelling the NO3RR, offering real-time insights into reaction mechanisms, catalyst performance, and reaction dynamics. This progress will aid in designing enhanced and environmentally friendly NH3 synthesis procedures.
(3) Optimize the methods of introducing external stimuli. For instance, stress stimuli are introduced primarily through ultrasonic waves. Nonetheless, it is vital to adjust catalyst structures and investigate approaches that make materials more responsive to external influences, allowing the piezoelectric effect to be activated by natural stimulation like water flow or wind pressure. Utilizing these natural disturbances augments the applicability of piezoelectric catalysis for enhancing NH3 production efficiency. Moreover, as a distinctive piezoelectric material, ferroelectric materials possess inherent polarization, creating an internal electric field that originates from the collective polarization effect formed by the spontaneous alignment of electric dipoles. Pre-treating ferroelectric materials and introducing electric fields to facilitate the NO3RR for NH3 synthesis could also be an effective approach.
(4) Designing cost-effective catalysts in NH3 production. For instance, using noble metals as the electrode material increases the cost of the NO3RR in LSPR-assisted NH3 production. Therefore, developing non-noble metal alternatives and designing effective strategies to amplify their LSPR effects and utilize high-energy hot electrons and strong electric fields to enhance NH3 synthesis is crucial for reducing the cost associated with this approach. One promising avenue is the exploration of plasmonic nanostructures composed of non-noble metals, such as Cu and Al. These materials also possess unique plasmonic properties that can be harnessed to enhance the NO3RR. By precisely tailoring the size, shape, and composition of these nanostructures, it is possible to tune their LSPR to the desired wavelength range for efficient NH3 synthesis. Additionally, synergistic coupling of LSPR with other catalytic mechanisms can further enhance the efficiency of NH3 synthesis. Integrating non-noble metal plasmonic nanostructures with suitable cocatalysts or semiconductors makes it feasible to harness the high-energy hot electrons generated during LSPR excitation and leverage the strong electric fields for selective and efficient NH3 production. Developing cost-effective strategies for LSPR-assisted NH3 synthesis using non-noble metals is essential for realizing sustainable and economically viable NH3 production processes. Continued research in this direction has great promise for reducing the overall cost and improving the scalability of LSPR-based NH3 synthesis technologies.
(5) In the research on regulating the NO3RR to produce NH3 through built-in electric field/external stimuli, exploring the coupling of multiple field/stimuli to enhance NH3 production performance has significant importance and promising prospects. A variety of built-in electric fields, produced by Motte–Schottky heterojunctions, semiconductor–semiconductor heterojunctions, coordinated active site environments, and electrical double-layer modulation, along with external stimuli, including optical, stress, and thermal stimuli, can synergistically enhance one another when introduced simultaneously. This synergy influences the reaction kinetics and site activity, ultimately enhancing the efficiency of NH3 production. Indeed, in the reaction of water splitting, studies have utilized the electric field generated by stress stimuli to further enhance the effective separation of photogenerated carriers, synergistically promoting reaction kinetics with the photogenerated electric field. In conclusion, by developing the combined effects of various field/stimuli, precise control over the catalytic process can be achieved, leading to improved performance in NH3 production.
(6) Developing theoretical computations is pivotal for designing rational strategies to modulate catalytic reactions using built-in electric field/external stimuli. Current theoretical models still exhibit significant discrepancies between simulating fields and actual systems, posing substantial challenges in designing built-in electric field/external stimuli-assisted reaction strategies. Therefore, optimizing theoretical methods to more precisely predict the atomic configurations and band structures of catalysts may obtain catalysts with higher performance. Meanwhile, tailored theoretical models can be explored for different fields to accurately assess the impact of built-in electric field/external stimuli on enhancing the performance of the NO3RR for NH3 synthesis from a theoretical perspective, elucidating their mechanisms. Machine learning can be employed to establish correlations between various built-in electric field/external stimuli and catalytic activity and selectivity, facilitating the identification of more suitable strategies.
(7) Developing stable catalysts. Stability is a crucial metric for evaluating catalyst performance. By designing catalysts with structural stability and employing methods to regulate the catalyst microenvironment for extending the catalyst's lifespan, there is potential to better adapt to external stimuli and to carry out NO3RRs for NH3 production under varying pH environments. Furthermore, common stability tests often consist of only a few dozen or even just a few cycles, which may not adequately reflect the catalyst's stability. Extending stability tests for longer durations until performance degradation occurs can help explore the catalyst's limit of stability, enabling rational design of the catalyst. Moreover, enhancing the stability of catalysts through strategic approaches can significantly increase their potential for industrial applications.
(8) A well-designed reactor effectively achieves high performance in reducing NO3− to produce NH3. One potential direction for future research is introducing a flow cell system into the NO3RR. The flow cells allow for continuous and controlled flow of reactants, enhancing mass transfer and improving reaction kinetics. This may lead to higher conversion rates and improved selectivity of electrochemical systems. Additionally, using flow cells enables better control of reaction conditions such as temperature, pressure, and flow rate, which is conducive to introducing external stimuli. Moreover, integrating flow cells can facilitate online monitoring and characterization techniques. This real-time monitoring capability can provide deeper insights into reaction kinetics, offering valuable insights for process optimization and catalyst design. Furthermore, flow cell systems can be easily scaled up for industrial applications, making it a promising approach for large-scale electrochemical NH3 synthesis. In conclusion, the application of flow cells in the NO3RR has the potential for improving reaction kinetics, better control of reaction conditions, enhanced online monitoring capabilities, and scalability for industrial implementation. Further research and development in this area will contribute to the advancement of efficient and practical NH3 production technologies.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts of interest to declare.References
- Y. Xiong, Y. Wang, J. Zhou, F. Liu, F. Hao and Z. Fan, Adv. Mater., 2024, 36, 2304021 CrossRef CAS PubMed.
- S. Han, H. Li, T. Li, F. Chen, R. Yang, Y. Yu and B. Zhang, Nat. Catal., 2023, 6, 402–414 CrossRef CAS.
- S. Zhang, H. Hong, R. Zhang, Z. Wei, Y. Wang, D. Chen, C. Li, P. Li, H. Cui, Y. Hou, S. Wang, J. C. Ho, Y. Guo, Z. Huang and C. Zhi, Angew. Chem., 2024, e202412830 Search PubMed.
- Z. Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F. Y. Chen, J. Y. T. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- Y. Guo, J. Gu, R. Zhang, S. Zhang, Z. Li, Y. Zhao, Z. Huang, J. Fan, Z. Chen and C. Zhi, Adv. Energy Mater., 2021, 11, 2101699 CrossRef CAS.
- H. Huang, K. Peramaiah and K.-W. Huang, Energy Environ. Sci., 2024, 17, 2682–2685 RSC.
- L. Li, C. Tang, X. Cui, Y. Zheng, X. Wang, H. Xu, S. Zhang, T. Shao, K. Davey and S. Z. Qiao, Angew. Chem., 2021, 133, 14250–14256 CrossRef.
- R. Zhang, H. Hong, X. Liu, S. Zhang, C. Li, H. Cui, Y. Wang, J. Liu, Y. Hou, P. Li, Z. Huang, Y. Guo and C. Zhi, Angew. Chem., Int. Ed., 2023, 62, e202309930 CrossRef CAS PubMed.
- Y. Kong, Y. Li, X. Sang, B. Yang, Z. Li, S. Zheng, Q. Zhang, S. Yao, X. Yang, L. Lei, S. Zhou, G. Wu and Y. Hou, Adv. Mater., 2022, 34, 2103548 CrossRef CAS PubMed.
- D. F. Abbott, Y. Z. Xu, D. A. Kuznetsov, P. Kumar, C. R. Müller, A. Fedorov and V. Mougel, Angew. Chem., 2023, 135, e202313746 CrossRef.
- R. Zhang, Y. Guo, S. Zhang, D. Chen, Y. Zhao, Z. Huang, L. Ma, P. Li, Q. Yang, G. Liang and C. Zhi, Adv. Energy Mater., 2022, 12, 2103872 CrossRef CAS.
- Q. Hu, Y. Qin, X. Wang, Z. Wang, X. Huang, H. Zheng, K. Gao, H. Yang, P. Zhang, M. Shao and C. He, Energy Environ. Sci., 2021, 14, 4989–4997 RSC.
- Y. Guo, R. Zhang, S. Zhang, Y. Zhao, Q. Yang, Z. Huang, B. Dong and C. Zhi, Energy Environ. Sci., 2021, 14, 3938–3944 RSC.
- H. Zhang, C. Wang, H. Luo, J. Chen, M. Kuang and J. Yang, Angew. Chem., Int. Ed., 2023, 62, e202217071 CrossRef CAS PubMed.
- F.-Y. Chen, Z.-Y. Wu, S. Gupta, D. J. Rivera, S. V. Lambeets, S. Pecaut, J. Y. T. Kim, P. Zhu, Y. Z. Finfrock, D. M. Meira, G. King, G. Gao, W. Xu, D. A. Cullen, H. Zhou, Y. Han, D. E. Perea, C. L. Muhich and H. Wang, Nat. Nanotechnol., 2022, 17, 759–767 CrossRef CAS PubMed.
- M. Cui, L. Zeng, W. Qin and J. Feng, Environ. Pollut., 2020, 263, 114553 CrossRef CAS PubMed.
- B. Chen, E. Liu, Q. Tian, C. Yan and Y. Zhang, Agron. Sustainable Dev., 2014, 34, 429–442 CrossRef CAS.
- M. J. Hendry, L. I. Wassenaar, S. L. Barbour, M. S. Schabert, T. K. Birkham, T. Fedec and E. E. Schmeling, Sci. Total Environ., 2018, 640, 127–137 CrossRef PubMed.
- D. Chen, S. Zhang, D. Yin, Q. Quan, Y. Zhang, W. Wang, Y. Meng, X. Liu, S. Yip, T. Yanagida, C. Zhi and J. C. Ho, Chem Catal., 2024, 4, 101024 Search PubMed.
- L. Bai, F. Franco, J. Timoshenko, C. Rettenmaier, F. Scholten, H. S. Jeon, A. Yoon, M. Rüscher, A. Herzog, F. T. Haase, S. Kühl, S. W. Chee, A. Bergmann and R. C. Beatriz, J. Am. Chem. Soc., 2024, 146, 9665–9678 CrossRef CAS PubMed.
- X. Zhao, M. Liu, Y. Wang, Y. Xiong, P. Yang, J. Qin, X. Xiong and Y. Lei, ACS Nano, 2022, 16, 19959–19979 CrossRef CAS PubMed.
- K. Kim, A. Zagalskaya, J. L. Ng, J. Hong, V. Alexandrov, T. A. Pham and X. Su, Nat. Commun., 2023, 14, 823 CrossRef CAS PubMed.
- R. Zhang, Y. Zhang, B. Xiao, S. Zhang, Y. Wang, H. Cui, C. Li, Y. Hou, Y. Guo, T. Yang, J. Fan and C. Zhi, Angew. Chem., Int. Ed., 2024, 63, e202407589 CrossRef CAS PubMed.
- K. Fan, W. Xie, J. Li, Y. Sun, P. Xu, Y. Tang, Z. Li and M. Shao, Nat. Commun., 2022, 13, 7958 CrossRef CAS PubMed.
- D. Chen, S. Zhang, X. Bu, R. Zhang, Q. Quan, Z. Lai, W. Wang, Y. Meng, D. Yin, S. Yip, C. Liu, C. Zhi and J. C. Ho, Nano Energy, 2022, 98, 107338 CrossRef CAS.
- J. Lim, C.-Y. Liu, J. Park, Y.-H. Liu, T. P. Senftle, S. W. Lee and M. C. Hatzell, ACS Catal., 2021, 11, 7568–7577 CrossRef CAS.
- G. Zhang, X. Li, K. Chen, Y. Guo, D. Ma and K. Chu, Angew. Chem., Int. Ed., 2023, 62, e202300054 CrossRef CAS PubMed.
- Y.-Y. Lou, Q.-Z. Zheng, S.-Y. Zhou, J.-Y. Fang, O. Akdim, X.-Y. Ding, R. Oh, G.-S. Park, X. Huang and S.-G. Sun, ACS Catal., 2024, 14, 5098–5108 CrossRef CAS.
- H. Du, H. Guo, K. Wang, X. Du, B. A. Beshiwork, S. Sun, Y. Luo, Q. Liu, T. Li and X. Sun, Angew. Chem., 2023, 135, e202215782 CrossRef.
- R. Daiyan, T. Tran-Phu, P. Kumar, K. Iputera, Z. Tong, J. Leverett, M. H. A. Khan, A. A. Esmailpour, A. Jalili, M. Lim, A. Tricoli, R.-S. Liu, X. Lu, E. Lovell and R. Amal, Energy Environ. Sci., 2021, 14, 3588–3598 RSC.
- H. Liu, X. Lang, C. Zhu, J. Timoshenko, M. Rüscher, L. Bai, N. Guijarro, H. Yin, Y. Peng, J. Li, Z. Liu, W. Wang, B. R. Cuenya and J. Luo, Angew. Chem., Int. Ed., 2022, 61, e202202556 CrossRef CAS PubMed.
- D. Chen, S. Zhang, D. Yin, W. Li, X. Bu, Q. Quan, Z. Lai, W. Wang, Y. Meng, C. Liu, S. Yip, F.-R. Chen, C. Zhi and J. C. Ho, Adv. Energy Mater., 2023, 13, 2203201 CrossRef CAS.
- L. Sun and B. Liu, Adv. Mater., 2023, 35, 2207305 CrossRef CAS PubMed.
- A. Paliwal, C. D. Bandas, E. S. Thornburg, R. T. Haasch and A. A. Gewirth, ACS Catal., 2023, 13, 6754–6762 CrossRef CAS.
- W. J. Sun, H. Q. Ji, L. X. Li, H. Y. Zhang, Z. K. Wang, J. H. He and J. M. Lu, Angew. Chem., Int. Ed., 2021, 60, 22933–22939 CrossRef CAS PubMed.
- X. Zhu, C. Ma, Y.-C. Wang, K. Qu, L. Song, J. Wang, Y. Gong, X. Liu, J. Zhang, Q. Lu and A.-L. Wang, Energy Environ. Sci., 2024, 17, 2908–2920 RSC.
- X. Lv, T. Mou, J. Li, L. Kou and T. Frauenheim, Adv. Funct. Mater., 2022, 32, 2201262 CrossRef CAS.
- W. Wen, S. Fang, Y. Zhou, Y. Zhao, P. Li and X. Y. Yu, Angew. Chem., Int. Ed., 2024, 63, e202408382 CrossRef CAS PubMed.
- E. Contreras, R. Nixon, C. Litts, W. Zhang, F. M. Alcorn and P. K. Jain, J. Am. Chem. Soc., 2022, 144, 10743–10751 CrossRef CAS PubMed.
- R. Zhang, C. Li, H. Cui, Y. Wang, S. Zhang, P. Li, Y. Hou, Y. Guo, G. Liang, Z. Huang, C. Peng and C. Zhi, Nat. Commun., 2023, 14, 8036 CrossRef CAS PubMed.
- M. T. de Groot and M. T. M. Koper, J. Electroanal. Chem., 2004, 562, 81–94 CrossRef CAS.
- X. Zhang, Y. Wang, C. Liu, Y. Yu, S. Lu and B. Zhang, Chem. Eng. J., 2021, 403, 126269 CrossRef CAS.
- S. Garcia-Segura, M. Lanzarini-Lopes, K. Hristovski and P. Westerhoff, Appl. Catal., B, 2018, 236, 546–568 CrossRef CAS.
- H. Luo, S. Li, Z. Wu, Y. Liu, W. Luo, W. Li, D. Zhang, J. Chen and J. Yang, Adv. Mater., 2023, 35, 2304695 CrossRef CAS PubMed.
- J. Zhou, M. Wen, R. Huang, Q. Wu, Y. Luo, Y. Tian, G. Wei and Y. Fu, Energy Environ. Sci., 2023, 16, 2611–2620 RSC.
- G. E. Dima, A. C. A. de Vooys and M. T. M. Koper, J. Electroanal. Chem., 2003, 554, 15–23 CrossRef.
- J. Long, S. Chen, Y. Zhang, C. Guo, X. Fu, D. Deng and J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 9711–9718 CrossRef CAS PubMed.
- A. C. A. de Vooys, M. T. M. Koper, R. A. Van Santen and J. A. R. Van Veen, J. Catal., 2001, 202, 387–394 CrossRef CAS.
- A. C. A. de Vooys, G. L. Beltramo, B. Van Riet, J. A. R. Van Veen and M. T. M. Koper, Electrochim. Acta, 2004, 49, 1307–1314 CrossRef CAS.
- J. Yang, M. Duca, K. J. P. Schouten and M. T. M. Koper, J. Electroanal. Chem., 2011, 662, 87–92 CrossRef CAS.
- R. Zhang, D. Shuai, K. A. Guy, J. R. Shapley, T. J. Strathmann and C. J. Werth, ChemCatChem, 2013, 5, 313–321 CrossRef CAS.
- S. Liang, X. Teng, H. Xu, L. Chen and J. Shi, Angew. Chem., 2024, 136, e202400206 CrossRef.
- O. Peng, Q. Hu, X. Zhou, R. Zhang, Y. Du, M. Li, L. Ma, S. Xi, W. Fu, Z.-X. Xu, C. Cheng, Z. Chen and K. P. Loh, ACS Catal., 2022, 12, 15045–15055 CrossRef CAS.
- A. C. A. de Vooys, M. T. M. Koper, R. A. Van Santen and J. A. R. Van Veen, J. Electroanal. Chem., 2001, 506, 127–137 CrossRef CAS.
- T. Hu, C. Wang, M. Wang, C. M. Li and C. Guo, ACS Catal., 2021, 11, 14417–14427 CrossRef CAS.
- H. Shin, S. Jung, S. Bae, W. Lee and H. Kim, Environ. Sci. Technol., 2014, 48, 12768–12774 CrossRef CAS PubMed.
- J. Gao, B. Jiang, C. Ni, Y. Qi and X. Bi, Chem. Eng. J., 2020, 382, 123034 CrossRef CAS.
- R. Liu, H. Zhao, X. Zhao, Z. He, Y. Lai, W. Shan, D. Bekana, G. Li and J. Liu, Environ. Sci. Technol., 2018, 52, 9992–10002 CrossRef CAS PubMed.
- J. M. McEnaney, S. J. Blair, A. C. Nielander, J. A. Schwalbe, D. M. Koshy, M. Cargnello and T. F. Jaramillo, ACS Sustainable Chem. Eng., 2020, 8, 2672–2681 CrossRef CAS.
- Y. Xiong, M. Sun, S. Wang, Y. Wang, J. Zhou, F. Hao, F. Liu, Y. Yan, X. Meng, L. Guo, Y. Liu, S. Chu, Q. Zhang, B. Huang and Z. Fan, Adv. Funct. Mater., 2024, 2420153 Search PubMed.
- Y. Xiong, Y. Wang, M. Sun, J. Chen, J. Zhou, F. Hao, F. Liu, P. Lu, X. Meng, L. Guo, Y. Liu, S. Xi, Q. Zhang, B. Huang and Z. Fan, Adv. Mater., 2024, 36, 2407889 CrossRef CAS PubMed.
- X. Deng, Y. Yang, L. Wang, X. Z. Fu and J. L. Luo, Adv. Sci., 2021, 8, 2004523 CrossRef CAS PubMed.
- Z. Fang, Z. Jin, S. Tang, P. Li, P. Wu and G. Yu, ACS Nano, 2022, 16, 1072–1081 CrossRef CAS PubMed.
- P. Mondol, D. Panthi, A. J. A. Ayala, S. O. Odoh and C. J. Barile, J. Mater. Chem. A, 2022, 10, 22428–22436 RSC.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- W. Luc, X. Fu, J. Shi, J.-J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu, J. Wu, Q. Yue, Y. Liu, F. Jiao and Y. Kang, Nat. Catal., 2019, 2, 423–430 CrossRef CAS.
- X. Fu, Chin. J. Catal., 2023, 53, 8–12 CrossRef CAS.
- Y. Zou, Y. Yan, Q. Xue, C. Zhang, T. Bao, X. Zhang, L. Yuan, S. Qiao, L. Song, J. Zou, C. Yu and C. Liu, Angew. Chem., Int. Ed., 2024, 63, e202409799 CrossRef CAS PubMed.
- Y. Chen, X. Xia, L. Tian, M. Yin, L.-L. Zheng, Q. Fu, D. Wu and J.-P. Zou, Chin. Chem. Lett., 2024, 35, 109789 CrossRef CAS.
- M. Zhang, Z. Zhang, S. Zhang, Z. Zhuang, K. Song, K. Paramaiah, M. Yi, H. Huang and D. Wang, ACS Catal., 2024, 14, 10437–10446 CrossRef CAS.
- L. Min, S. Liping, H. Lihua and Z. Hui, Appl. Catal., A, 2024, 676, 119650 CrossRef CAS.
- X. Ji, C. Ma, F. Zhang, X. He, X. Fan, J. Li, Z. Li, L. Ouyang, L. Zhang, T. Li, D. Zhao, Y. Wang, J. Zhang, Z. Cai, S. Sun, A. A. Alshehri, Q. Lu and X. Sun, ACS Sustainable Chem. Eng., 2023, 11, 2686–2691 CrossRef CAS.
- D. Xu, S.-N. Zhang, J.-S. Chen and X.-H. Li, Chem. Rev., 2022, 123, 1–30 CrossRef PubMed.
- Z. Zhang and J. T. Yates Jr., Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- H. Zhang, R. Li, M. Humayun, Z. Huang, Y. Fu, Y. Cao, J. Duan, Y. A. Attia and C. Wang, Mater. Chem. Front., 2024, 8, 2811–2835 RSC.
- P. Gao, Z. H. Xue, S. N. Zhang, D. Xu, G. Y. Zhai, Q. Y. Li, J. S. Chen and X. H. Li, Angew. Chem., Int. Ed., 2021, 60, 20711–20716 CrossRef CAS PubMed.
- X. Fan, D. Zhao, Z. Deng, L. Zhang, J. Li, Z. Li, S. Sun, Y. Luo, D. Zheng, Y. Wang, B. Ying, J. Zhang, A. A. Alshehri, Y. Lin, C. Tang, X. Sun and Y. Zheng, Small, 2023, 19, 2208036 CrossRef CAS PubMed.
- Y. Zhou, W. Zhang, P. Guo, Y. Guo, J. Zhan, Y. Wang, B. Zhang, S. Zhang, L.-H. Zhang and F. Yu, Inorg. Chem. Front., 2024, 11, 3503–3510 Search PubMed.
- A. Balapure, J. R. Dutta and R. Ganesan, RSC Appl. Interfaces, 2024, 1, 43–69 RSC.
- Y. Xu, Y. Sheng, M. Wang, T. Ren, K. Shi, Z. Wang, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2022, 10, 16883–16890 RSC.
- F. Zhou and G. Tao, J. Phys. Chem. C, 2023, 127, 23180–23188 CrossRef CAS.
- T. Kou, M. Chen, F. Wu, T. J. Smart, S. Wang, Y. Wu, Y. Zhang, S. Li, S. Lall, Z. Zhang, Y.-S. Liu, J. Guo, G. Wang, Y. Ping and Y. Li, Nat. Commun., 2020, 11, 590 CrossRef CAS PubMed.
- S. Ajmal, A. Kumar, M. A. Mushtaq, M. Tabish, Y. Zhao, W. Zhang, A. S. Khan, A. Saad, G. Yasin and W. Zhao, Small, 2024, 20, 2310082 CrossRef CAS PubMed.
- B. Pan, Y. Wang and Y. Li, Chem Catal., 2022, 2, 1267–1276 CrossRef CAS.
- S. Zhang, Z. Liu, D. Chen, Z. Guo and M. Ruan, Chem. Eng. J., 2020, 395, 125101 CrossRef CAS.
- Y. Fan, W. Ma, D. Han, S. Gan, X. Dong and L. Niu, Adv. Mater., 2015, 27, 3767–3773 CrossRef CAS PubMed.
- Y. Guo, J. Sun, T. Guo, Y. Liu and Z. Yao, Angew. Chem., Int. Ed., 2024, 63, e202319664 CrossRef CAS PubMed.
- C. Zhang, Z. C. Shao, X. L. Zhang, G. Q. Liu, Y. Z. Zhang, L. Wu, C. Y. Liu, Y. Pan, F. H. Su, M. R. Gao, Y. Li and S. H. Yu, Angew. Chem., Int. Ed., 2023, 62, e202305571 CrossRef CAS PubMed.
- J. Xue, M. Fujitsuka, T. Tachikawa, J. Bao and T. Majima, J. Am. Chem. Soc., 2024, 146, 8787–8799 CrossRef CAS PubMed.
- S. Zhang, D. Chen, P. Chen, R. Zhang, Y. Hou, Y. Guo, P. Li, X. Liang, T. Xing, J. Chen, Y. Zhao, Z. Huang, D. Lei and C. Zhi, Adv. Mater., 2024, 36, 2310776 CrossRef CAS PubMed.
- J. Yang, L. Li, C. Xiao and Y. Xie, Angew. Chem., Int. Ed., 2023, 62, e202311911 CrossRef CAS PubMed.
- X. Sun, S. Jiang, H. Huang, H. Li, B. Jia and T. Ma, Angew. Chem., Int. Ed., 2022, 61, e202204880 CrossRef CAS PubMed.
- Y. Chen, C. Yan, J. Dong, W. Zhou, F. Rosei, Y. Feng and L. N. Wang, Adv. Funct. Mater., 2021, 31, 2104099 CrossRef CAS.
- K. Wenderich and G. Mul, Chem. Rev., 2016, 116, 14587–14619 CrossRef CAS PubMed.
- H. Li, Y. Zhou, W. Tu, J. Ye and Z. Zou, Adv. Funct. Mater., 2015, 25, 998–1013 CrossRef CAS.
- E. Sharma, V. Thakur, S. Sangar and K. Singh, Mater. Today: Proc., 2020, 32, 584–593 CAS.
- J. Li, R. Chen, J. Wang, Y. Zhou, G. Yang and F. Dong, Nat. Commun., 2022, 13, 1098 CrossRef CAS PubMed.
- D. Hao, J. Ren, Y. Wang, H. Arandiyan, M. Garbrecht, X. Bai, H. K. Shon, W. Wei and B.-J. Ni, Energy Mater. Adv., 2021, 2021, 9761263 Search PubMed.
- J. Homola, S. S. Yee and G. Gauglitz, Sens. Actuators, B, 1999, 54, 3–15 CrossRef CAS.
- E. K. Payne, K. L. Shuford, S. Park, G. C. Schatz and C. A. Mirkin, J. Phys. Chem. B, 2006, 110, 2150–2154 CrossRef CAS PubMed.
- C. L. Wong and M. Olivo, Plasmonics, 2014, 9, 809–824 CrossRef CAS.
- E. Hutter and J. H. Fendler, Adv. Mater., 2004, 16, 1685–1706 CrossRef CAS.
- H. Ahn, H. Song, J.-R. Choi and K. Kim, Sensors, 2018, 18, 98 CrossRef PubMed.
- A. Agrawal, S. H. Cho, O. Zandi, S. Ghosh, R. W. Johns and D. J. Milliron, Chem. Rev., 2018, 118, 3121–3207 CrossRef CAS PubMed.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS PubMed.
- T. Uwada, T. Asahi, H. Masuhara, D. Ibano, M. Fujishiro and T. Tominaga, Chem. Lett., 2007, 36, 318–319 CrossRef CAS.
- M. Ahlawat, D. Mittal and V. Govind Rao, Commun. Mater., 2021, 2, 114 CrossRef CAS.
- X. Yu, S. Du, Z. Xu, J. He, F. Liu, B. Wang, S. Sun, Y. Tang and K. Zhao, Chem. Eng. J., 2024, 480, 148152 CrossRef CAS.
- C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem Catal., 2022, 2, 52–83 CrossRef CAS.
- Z. Zheng, W. Xie, M. Li, Y. H. Ng, D.-W. Wang, Y. Dai, B. Huang and R. Amal, Nano Energy, 2017, 41, 233–242 CrossRef CAS.
- S. S. Rayalu, D. Jose, M. V. Joshi, P. A. Mangrulkar, K. Shrestha and K. Klabunde, Appl. Catal., B, 2013, 142, 684–693 CrossRef.
- X. Xie, J. Feng, X. Cui, J. Liu, L. Jiang and L. Dong, ACS Catal., 2021, 11, 13160–13168 CrossRef CAS.
- Y. Ma, X. Han, S. Xu, Z. Wang, W. Li, I. Da Silva, S. Chansai, D. Lee, Y. Zou, M. Nikiel, P. Manuel, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, Y. Cheng, S. Rudić, A. J. Ramirez-Cuesta, S. J. Haigh, C. Hardacre, M. Schröder and S. Yang, J. Am. Chem. Soc., 2021, 143, 10977–10985 CrossRef CAS PubMed.
- S. L. Wu and S. Yoo, Nat. Rev. Phys., 2022, 4, 143–144 CrossRef.
- S. Li, Z. Zhao, J. Zhao, Z. Zhang, X. Li and J. Zhang, ACS Appl. Nano Mater., 2020, 3, 1063–1079 CrossRef CAS.
- R. Su, H. A. Hsain, M. Wu, D. Zhang, X. Hu, Z. Wang, X. Wang, F. T. Li, X. Chen, L. Zhu, Y. Yang, Y. Yang, X. Lou and S. J. Pennycook, Angew. Chem., Int. Ed., 2019, 58, 15076–15081 CrossRef CAS PubMed.
- Y. Zhu, H. Chen, L. Wang, L. Ye, H. Zhou, Q. Peng, H. Zhu and Y. Huang, Chin. Chem. Lett., 2024, 35, 108884 CrossRef CAS.
- J. Ma, S. Jing, Y. Wang, X. Liu, L. Y. Gan, C. Wang, J. Y. Dai, X. Han and X. Zhou, Adv. Energy Mater., 2022, 12, 2200253 CrossRef CAS.
- S. Zhang, D. Chen, Y. Guo, R. Zhang, Y. Zhao, Z. Huang, J. Fan, J. C. Ho and C. Zhi, Mater. Today, 2023, 66, 17–25 CrossRef CAS.
- J. Zhang, K. Lv, J. Cheng, Y. Liu, Y. Wang, S.-F. Yin and P. Chen, Appl. Catal., B, 2025, 361, 124558 Search PubMed.
- S. Yuan, Y. Li, J. Peng, Y. M. Questell-Santiago, K. Akkiraju, L. Giordano, D. J. Zheng, S. Bagi, Y. Román-Leshkov and Y. Shao-Horn, Adv. Energy Mater., 2020, 10, 2002154 CrossRef CAS.
- Y. Zhao, W. Gao, S. Li, G. R. Williams, A. H. Mahadi and D. Ma, Joule, 2019, 3, 920–937 CrossRef CAS.
- J. Zhang, H. Chen, X. Duan, H. Sun and S. Wang, Mater. Today, 2023, 68, 234–253 CrossRef CAS.
- S. Zhang, D. Chen, Z. Liu, M. Ruan and Z. Guo, Appl. Catal., B, 2021, 284, 119686 CrossRef CAS.
- K. Liu, H. Li, M. Xie, P. Wang, Z. Jin, Y. Liu, M. Zhou, P. Li and G. Yu, J. Am. Chem. Soc., 2024, 146, 7779–7790 CrossRef CAS PubMed.
- Z. Wang, E. M. Ortiz, B. R. Goldsmith and N. Singh, Catal. Sci. Technol., 2021, 11, 7098–7109 RSC.
| This journal is © The Royal Society of Chemistry 2025 |