
Combating eukaryotic and prokaryotic harmful algal blooms with visible-light driven BiOBrxI1−x/MFe2O4/g-C3N4 (M = Co & Ni) recyclable photocatalysts†
Anjitha
A
 a,
Shijina
K
a,
Ajayan
K. V.
b,
Sindhu
Swaminathan
a,
Irene M. C.
Lo
cd and
Kishore
Sridharan
*a
a,
Shijina
K
a,
Ajayan
K. V.
b,
Sindhu
Swaminathan
a,
Irene M. C.
Lo
cd and
Kishore
Sridharan
*a
aDepartment of Nanoscience and Technology, University of Calicut, P.O. Calicut University, Thenhipalam 673635, India. E-mail: sridharankishore@uoc.ac.in
bDepartment of Botany, University of Calicut, P.O.Calicut University, Thenhipalam 673635, India
cDepartment of Civil and Environmental Engineering, The Hong Kong University of Science and Technology, Hong Kong, China
dInstitute of Advanced Study, The Hong Kong University of Science and Technology, Hong Kong, China
First published on 6th November 2024
Abstract
Photocatalysis offers a promising avenue for completely eliminating harmful algal blooms (HABs), a significant threat to global freshwater reserves. In this study, a series of BiOBrxI1−x photocatalysts were synthesized and the most optimal catalyst was integrated with pristine g-C3N4 and pre-synthesized CoFe2O4/g-C3N4 and NiFe2O4/g-C3N4 to form binary and ternary composite heterojunction photocatalysts (BiOBr0.95I0.05/g-C3N4 – BG, CoFe2O4/BiOBr0.95I0.05/g-C3N4 – CBG, and NiFe2O4/BiOBr0.95I0.05/g-C3N4 – NBG). The synthesized photocatalysts were thoroughly characterized and their performance was evaluated through the visible light driven photocatalytic degradation of both Microcystis aeruginosa (prokaryotic) and Scenedesmus acuminatus (eukaryotic) algal cells sourced directly from ponds. The exceptional photocatalytic efficiency of CBG evidenced through the variation in chlorophyll-a content, malondialdehyde, and electrolytic leakage confirmed the complete rupture of the algal cells after 3 h of light exposure. This was further reconfirmed through fluorescence microscopy analysis and interestingly, both HABs failed to re-grow even after 10 days. The enhanced performance of CBG was attributed to the boosted generation of charge carriers facilitated by its extended visible light absorption, which in turn produced reactive oxygen species (˙O2− and ˙OH radicals) that caused irreparable oxidative damage to algal cells, while effectively suppressing the exciton pair recombination supported by its double Z-scheme heterojunction. Furthermore, the magnetic recyclability feature of CBG facilitated its easy removal from treated water for avoiding secondary pollution. The design of magnetically recyclable photocatalysts for degrading both prokaryotic and eukaryotic HABs demonstrated here is anticipated to inspire the development of efficient photocatalysts and design of cost-effective solutions required for treating ponds and lakes infected with HABs.
Environmental significanceHarmful algal blooms (HABs) significantly disrupt freshwater ecosystems, degrade water quality, and pose health risks through toxin release. Current mitigation strategies are often unsustainable or cause secondary pollution. This study evaluates magnetically recyclable double Z-scheme heterojunction photocatalysts (CBG and NBG) for the complete photodegradation of both prokaryotic and eukaryotic HABs, directly sourced from ponds. Photocatalytic treatment resulted in complete algae cell rupture, monitored via fluorescence microscopy, and by measuring chlorophyll-a, electrolytic leakage, and malondialdehyde concentration. Interestingly, both the algae failed to re-grow post-treatment, offering a long-term, sustainable approach for HAB management. The magnetic recyclability feature of the photocatalysts reduces secondary pollution risks, making this method suitable for large-scale water treatment and essential for protecting global freshwater resources. |
1. Introduction
Water is of paramount significance for all known forms of life. Unfortunately, the majority of water on our planet exists as salty seawater, and consumable freshwater constitutes only 3% of the total water resource. Rapid urbanization, population growth, and industrialization have directly or indirectly polluted our freshwater resources. The global availability of clean water is becoming a severe concern for our future generations. One of the serious threats to our freshwater resources worldwide is the proliferation of harmful algal blooms (HABs). This phenomenon arises due to industrial sewage, agricultural and aquaculture runoff and their discharge into eutrophic water bodies that typically contain excessive nutrient elements such as nitrogen and phosphorus. Favorable environmental conditions including temperature, light irradiance, pH and water currents promote the excessive growth of HABs. Furthermore, global warming and the subsequent climate change worldwide are reported to provide conditions that accelerate the growth of HABs.1 The adverse effects of HABs extend beyond their impact on water quality. The excessive growth of HABs drastically reduces the oxygen content in the water body and hinders the passage of light, thereby impeding the growth of other aquatic organisms and disrupting the biodiversity of the aquatic ecosystem. HABs, particularly cyanobacteria, can produce and release secondary metabolites, including bioactive compounds and potent biotoxins such as hepatotoxins (e.g., microcystins, nodularins, and cylindrospermopsins) and alkaloid neurotoxins (e.g., anatoxin-a, anatoxin-a (S), and saxitoxins). These toxins not only harm aquatic life but can also prove fatal to humans through ingestion (consumption of contaminated food or water), skin contact (e.g., fishing and swimming), and inhalation (via aerosols formed during wave breaking). Extensive research on microcystins, the most diverse and toxic class of cyanotoxins with over 100 different congeners, has revealed their potential to inhibit the activity of protein phosphatase types 1 and 2A, possibly promoting tumor progression.2,3 As a precaution, the World Health Organization has recommended a drinking water guideline of 1.0 ppb for microcystin-LR.Various approaches, including physical techniques (such as ultrasound, adsorption, UV irradiation, and membrane separation), chemical methods (including chemical algaecides, coagulation, and flocculation), and microbial technologies (utilizing algicidal bacteria, fungi, and protozoa), have been developed to combat HABs in natural water systems. While these methods have proven effective in removing HABs, they encounter challenges such as high operational costs, secondary contamination, and low removal rates.4 In comparison, advanced oxidation processes (AOPs) have gained considerable attention due to their high efficiency, lower costs, and environmental friendliness.5 Among AOPs, photocatalysis has emerged as a promising and widely employed method in environmental remediation. By harnessing UV and visible light, photocatalysis generates reactive oxygen species (ROS) that can effectively destroy algal cells, toxins, and other metabolites in water bodies.6 Studies have demonstrated the efficacy of certain photocatalysts, such as TiO2 and AgBiO3, in rupturing harmful algae and degrading the toxins under light irradiation.7,8 However, inherent shortcomings, such as rapid electron–hole pair recombination and the requirement for UV light activation, constrain the practical use of pristine single-component photocatalysts.9,10 Consequently, enhancing the photocatalytic activity of these materials has become a prominent focus in recent times. With appropriately chosen catalysts, photocatalysis can not only obliterate algal cells but also break down algal metabolites by generating reactive oxygen species (ROS).11,12
In this context, polymeric metal-free graphitic carbon nitride (g-C3N4) stands out as a highly effective and extensively researched environmentally friendly photocatalyst, widely employed for various photocatalysis applications, including the degradation of HABs.13 Noteworthy attributes of g-C3N4 include excellent chemical stability in acid/base environments, thermal stability, and non-toxicity.14,15 Furthermore, its appeal as a photocatalyst stems from its two-dimensional (2D) sheet-like morphology and suitable bandgap of around 2.7 eV. The reduced bandgap is a consequence of its distinctive structural arrangement, involving π-conjugated graphitic planes formed through carbon and nitrogen sp2 hybridization. The simplicity of synthesizing g-C3N4 through the thermal polycondensation of nitrogen-rich precursors further enhances its attractiveness in the field of photocatalysis.16,17 However, the practical applicability is hindered by its relatively small surface area, high quantum efficiency, and rapid electron–hole (e−/h+) recombination.18,19 To address these limitations, various techniques such as metal/non-metal doping, decoration of noble metal nanoparticles and hybridization with other photocatalysts have been employed.20 Among these methods, the construction of a heterojunction by coupling g-C3N4 with another semiconductor photocatalyst is considered to be the most effective. This leads to the formation of a Z-scheme heterojunction resulting in a synergistic effect which greatly improves the separation efficiency of the e−/h+ pairs.21–24
Recently, there has been significant interest in a novel class of semiconductor materials known as bismuth oxyhalides (BiOX, X = Cl, Br, I) for their promising applications in photocatalysis, by virtue of their unique and intrinsic lamellar structure and appropriate bandgaps.25–27 A layered crystal structure of BiOX is formed by interlacing [Bi2O2] slabs with double halogen slabs, creating an internal static electric field (IEF) perpendicular to each layer. This self-generated IEF in bismuth oxyhalides facilitates the transport of photo-induced e− and h+ inside the crystal, leading to enhanced photocatalytic activity by increased separation of photogenerated charge carriers.28,29 Among the BiOX semiconductors, BiOCl can absorb only UV light owing to its wide bandgap energy (Eg = 3.5 eV),30,31 while BiOI has the smallest bandgap energy (Eg = 1.8 eV), enabling photon absorption across the entire visible light spectrum. Despite the narrow bandgap, the usage of BiOI is limited by its poor redox capabilities and fast recombination of the photogenerated charges.32,33 In contrast, BiOBr has attracted considerable attention due to its suitable band gap energy (Eg = 2.7 eV), structural stability, and extended visible-light responsive photocatalytic activity.34,35 Therefore, the formation of bismuth oxybromo-iodide (referred hereafter as BiOBrxI1−x) solid solutions has been identified as a viable strategy to enhance the visible light absorption of BiOBr.36 Integrating BiOBrxI1−x with g-C3N4 offers a promising strategy to overcome the limitations inherent to each material. Their 2D layered structures promote the formation of a heterojunction and the favorable band alignment between them enhances photocatalytic performance.37,38
Unfortunately, one of the crucial challenges in the development of photocatalysts towards wastewater treatment applications is the secondary pollution that may be caused by the incomplete removal of the nanoscale powdered photocatalysts. Therefore, integration of magnetic nanoparticles with photocatalysts for achieving complete removal of the photocatalysts from the treated wastewater while preserving their photocatalytic properties is of significant interest. Towards this end, spinel ferrites (AB2O4, A, B: transition metals, O: oxygen) are known for their excellent magnetic properties, low cost of synthesis and high stability.39 Spinel ferrites have been reported to be coupled with photocatalysts for achieving magnetic recyclability. Among the various spinel ferrites, NiFe2O4 and CoFe2O4, are attractive due to their narrow bandgap (1.56 eV and 2.0 eV, respectively), rendering them active under visible light.40,41
Our work details the synthesis of BiOBrxI1−x solid-solution photocatalysts, from which the best was identified through the degradation of methylene blue (MB). In addition, the best among BiOBrxI1−x was coupled with g-C3N4 to form a 2D–2D heterojunction photocatalyst and was also combined with pre-synthesized CoFe2O4/g-C3N4 and NiFe2O4/g-C3N4 for obtaining a ternary composite heterojunction, facilitating magnetic recyclability. The photocatalytic activities of the as-synthesized 2D–2D heterojunction and ternary composite photocatalysts were compared by studying the degradation of MB. Further, the synthesized catalysts were also employed towards the visible light driven photocatalytic degradation and complete removal of both Microcystis aeruginosa (prokaryotic) and Scenedesmus acuminatus (eukaryotic) algal blooms sourced directly from ponds. It is worth noting that existing research has targeted only the photodegradation of prokaryotic HABs, primarily Microcystis aeruginosa, and to the best of our knowledge there are no reports on the photocatalytic degradation of eukaryotic HABs. Also, no prior reports are available in the literature that have provided the fluorescent microscopic evidence of algal cell rupture following their photocatalytic degradation after monitoring the variation in the chlorophyll-a pigment concentration, lipid peroxidation, and electrolytic leakage. Therefore, this study was carefully planned considering these gaps in the literature. Interestingly, the ternary composite catalysts demonstrated excellent photocatalytic efficiency and achieved complete rupture of the algal cells, inhibiting their growth that was confirmed through various analytical techniques.
2. Experimental
2.1. Materials
Cobaltous nitrate (Co(NO3)2·6H2O), nickel nitrate (Ni(NO3)2·6H2O), bismuth nitrate pentahydrate (Bi(NO3)3·5H2O), ferric nitrate (Fe(NO3)3·9H2O), urea, potassium bromide (KBr), potassium iodide (Kl) and methanol were purchased from Merck. The MB dye used for the photocatalytic study was received from Sigma Aldrich. All the chemicals were used as such without any purification and deionized (DI) water was used throughout the experiments.2.2. Synthesis of photocatalysts
2.3. Characterization
The crystalline nature of the as-synthesized semiconductor photocatalysts was determined with an X-ray diffractometer (X'pert 3 powder, PANalytical) using Cu Kα radiation (λ = 1.5418 Å) in the 2θ range 10–80° at a scan rate of 5° min−1. Morphology analysis was conducted using a field emission scanning electron microscope (ZEISS Gemini SEM 300) equipped with a high brightness Schottky field emission electron gun. High-resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) were performed on a JEOL JEM 2100 HRTEM operating at 200 kV. The optical properties of the as-synthesized photocatalysts were studied by recording the UV visible diffuse reflectance spectra (DRS) on a spectrophotometer (JASCO, UV-Win) with BaSO4 as the reference and photoluminescence (PL) studies were carried out using a spectrofluorometer (Perkin Elmer, LS 55) at an excitation wavelength of 350 nm. The Brunauer–Emmett Teller (BET) surface area analysis was performed employing a BELCAT-M surface area analyzer. The computer-aided microscopy imaging system, Leica DM 6B, fitted with an optical microscope of 40× objective lens and an LED lamp with a 530 nm cutoff filter, was used for analyzing the chlorophyll fluorescence.2.4. Photocatalytic degradation of MB
The photocatalytic activity of the as-synthesized photocatalyst was assessed by monitoring the degradation of MB under visible light irradiation. A photocatalytic reactor with a 300 W Xe lamp (Luzchem Xenon photoreactor) was employed as a visible light source. In a typical experiment, a catalyst suspension was prepared by dispersing 1 g L−1 of photocatalyst in 10 ppm aqueous MB solution. The resulting suspension was placed inside the photoreactor and was stirred for 30 min under dark conditions prior to irradiation, to attain the adsorption–desorption equilibrium between the catalyst and MB. The suspension was then irradiated, and aliquots were withdrawn at regular time intervals for measuring the changes in the concentrations of MB using a UV-vis spectrophotometer. For consistency, all experiments were conducted in triplicate. The efficiency of the photocatalyst was calculated using the following equation.where C0 is the initial concentration of MB and C is the concentration of MB after light irradiation.

2.5. Photocatalytic degradation of HABs
where OD0 is the optical density before degradation and OD is the optical density after degradation.
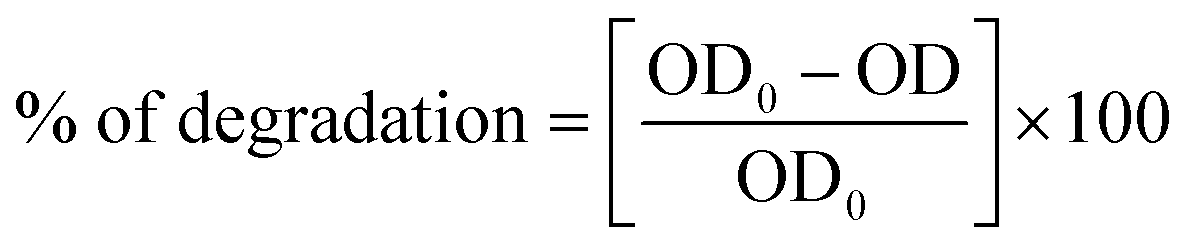
2.5.4.1. Pigment analysis. The degradation efficiency of the algal cells over the photocatalyst was evaluated by examining the chlorophyll-a content. This indicator serves for both quantitative and qualitative measurements to analyze the removal of algal cells. Qualitatively, it can be evaluated by measuring the absorbance value at 680 nm wavelength using a UV-vis spectrophotometer against algal blooms without a catalyst (control). The degradation efficiency of algal cells can be calculated using the following equation:
where C0 represents the initial chlorophyll-a concentration and C represents chlorophyll-a concentration at time t. Quantitatively, the degradation efficiency of the algal cells over the photocatalyst was examined by extracting the chlorophyll-a pigment using Mackinney's method.43 In brief, the samples filtered through the membrane filtration unit and biomass were washed with DI water, suspended in 4 mL of 80% methanol, and well homogenized. Subsequently, the solution was centrifuged at 12000 rpm for 10 min. The supernatant was saved, and absorbance was measured at 663 nm using a UV-vis spectrophotometer against methanol as the blank sample as per the equation:


2.5.4.2. Evaluation of oxidative damage. Lipid peroxidation was measured as the quantity of malondialdehyde (MDA). During photocatalytic degradation, the membrane lipid peroxidation can lead to an increase in MDA content and cell destruction. MDA was measured based on the thiobarbituric acid method and measurements were made using a UV-vis spectrophotometer at 532 nm,44 with values expressed in nmol MDA mL− 1. The electrolytic leakage (EL) of the control and treated solutions of algal samples was measured following the method of Chen et al.45 The calculation was performed using the formula mentioned in the equation:
where E1 and E2 are the electrical conductivities of the solution.

2.5.4.3. Chlorophyll fluorescence analysis. Fluorescence microscopy is one of the most powerful tools in the visualization of chlorophyll fluorescence in microalgal cells.46 The morphology of the HABs (both M. aeruginosa and S. acuminatus) before and after degradation was examined microscopically and confirmed by recording both bright field and dark field (fluorescence) micrographs employing a Leica DM6 B microscope at an incident wavelength of 530 nm.
2.5.4.4. Monitoring algal cell re-growth after photocatalytic treatment. After the 3 h photocatalytic degradation of HABs with CBG as the photocatalyst, both algae (M. aeruginosa and S. acuminatus) were carefully collected from the reaction slurry by magnetic separation (the CBG photocatalyst) for studying their re-growth. The degraded algae were added to BG-11 growth medium in an experimental vessel after which they were transferred to aerated conical flasks. Both M. aeruginosa and S. acuminatus were kept under laboratory conditions at 26 °C, illuminated with a constant light flux with 12 h/12 h light/dark cycles at pH 7 for a period of 10 days.47,48
2.5.4.5. Detection of reactive species. To identify the dominant reactive species responsible for the degradation of M. aeruginosa and S. acuminatus, scavenger studies were performed by adding potassium dichromate (K2Cr2O7) (as e− scavenger), ammonium oxalate (AO) (as h+ scavenger), isopropanol (IPA) (as ˙OH scavenger), and benzoquinone (BQ) (as ˙O2− scavenger). These scavengers were individually added to the algae water before the addition of the photocatalyst. Photocatalytic experiments were performed, and the reactive species were determined by assessing the changes in chlorophyll-a content before and after the photodegradation experiments in both algal cells.
3. Results and discussion
3.1. Structure, morphology and surface area analysis
The crystal phase of the as-prepared samples was evaluated by XRD. In Fig. 1a and b, the XRD pattern of the pure g-C3N4 sample revealed a broad peak at 13.2° and 27.5° corresponding to the (100) and (002) planes, respectively (JCPDS 87-1526). The weak peak at 13.2° signifies the in-plane structural arrangement of tri-s-triazine units and the high intensity peak at 27.5° is related to the interplanar separation between the conjugated aromatic systems. | ||
| Fig. 1 XRD patterns of (a) g-C3N4, BBI0.05, BG, CG and CBG, and (b) g-C3N4, BBI0.05, BG, NG and NBG. | ||
The XRD peaks of BBI0.05 were matched with the tetragonal phase of BiOBr (JCPDS 09-0393). Notably, a slight shift is observed in the most prominent (102) peak towards the lower angle (see Fig. S1†), which could be attributed to the larger ion radius of I− (2.02 Å) in comparison to Br− (1.82 Å).49 This shift is indicative of iodine doping,50 and the presence of additional peaks corresponding to BiOI (JCPDS 10-0445) further confirmed the formation of the BBI0.05 solid-solution photocatalyst. The diffraction pattern of BG exhibited consistency with that of BBI0.05, with no discernible peak for g-C3N4, attributable to its polymeric nature. For CG and NG samples, the XRD analysis revealed cubic phases corresponding to CoFe2O4 and NiFe2O4, respectively (JCPDS 22-1086 and 10-0325), without any observable peaks for g-C3N4 owing to its polymeric nature.51 In the case of the ternary composite CBG, the diffraction peaks were indexed to cubic CoFe2O4, tetragonal BiOBr, and BiOI. Similarly, for NBG, the peaks were indexed to cubic NiFe2O4, tetragonal BiOBr, and BiOI. Notably, the presence of g-C3N4 was not detected in CBG and NBG as well, which could be attributed to its polymeric nature. Other parameters such as crystallite size (D) and structural defects such as dislocation density (ρ) and microstrain (ε) for the synthesized photocatalysts calculated from the XRD data (see Fig. S1†) are presented in Table S1 (ESI†) and are discussed in detail in section 3.4, for correlating them with the performance.
The morphology of the synthesized photocatalysts was examined through FESEM. The low magnification FESEM micrograph presented in Fig. 2a depicts the overall morphology of CBG consisting of BBI0.05 nanoplates and CoFe2O4 nanoparticles that appear to be embedded on g-C3N4 nanosheets. The magnified micrograph presented in Fig. 2b clearly depicts the 2D nanoplate-like morphology of BBI0.05 with an average thickness of ∼15 nm. Further, CoFe2O4 nanoparticles are also observed to be intimately attached to BBI0.05 nanoplates. Similarly, in the case of NBG, the overall morphology presented in Fig. 2c is a mixture of BBI0.05 nanoplates and NiFe2O4 nanoparticles that seem to be embedded on g-C3N4 nanosheets. The magnified micrograph presented in Fig. 2d clearly depicts the 2D nanoplates of BBI0.05 with an average thickness of ∼15 nm, which are intimately attached to NiFe2O4 nanoparticles. Although the presence of g-C3N4 is not explicitly observed from the micrographs, all the particles seem to have grown on top of g-C3N4. This observation strongly suggests the successful formation of the ternary heterostructure.
 | ||
| Fig. 2 FESEM micrographs of (a and b) CBG and (c and d) NBG at two different magnifications. Nanoparticles of CoFe2O4/NiFe2O4 embedded on g-C3N4 and the stacked growth of BiOBr0.95I0.05 nanoplates can be observed from micrographs. | ||
The growth of CG and NG could follow the mechanism reported previously.51 During pyrolysis inside the sealed crucible, at ∼170 °C, the urea releases ammonia and isocyanic acid, while metal nitrates (Co2+/Ni2+ and Fe3+) decompose into metal oxides. At this point, ammonia and isocyanic acid react with the metal oxides and hydroxyl groups to form metal-oligomers and dicyanamide. When the temperature reaches ∼240–300 °C, the metal-oligomers get converted into cyanuric acid and ammelide. Next, at temperatures higher than 400 °C, two simultaneous reactions may lead to the formation of CG and NG: (a) self-combustion of metal cations in the organic matrix of cyanuric acid/ammelide by the release of H2 and (b) metal cations bound with amino groups (–NH2) in dicyanamide act as Lewis acid sites to facilitate the polymerization of g-C3N4. The theoretical growth mechanism is validated through TEM analysis.
The TEM micrographs of CBG and NBG are depicted in Fig. 3. The low magnification TEM micrograph presented in Fig. 3a shows the presence of CoFe2O4 nanoparticles embedded on g-C3N4 nanosheets, followed by the growth of BBI0.05 nanoplates. The dark area on the middle and top part of the TEM micrograph in Fig. 3a is attributed to the stacking of BBI0.05 nanoplates, while the bottom part clearly depicts the presence of CoFe2O4 nanoparticles embedded on g-C3N4 nanosheets. The rod-like features seen on the right edge of Fig. 3a represent the BBI0.05 nanoplates stacked in the perpendicular direction as observed through FESEM. Similarly, the magnified TEM in Fig. 3b also depicts the stacked BBI0.05 nanoplates (at the top left corner and bottom middle area of Fig. 3b) and CoFe2O4 nanoparticles embedded on g-C3N4 nanosheets, clearly indicating the formation of a ternary heterostructure. Despite the presence of crystalline CoFe2O4 nanoparticles and BBI0.05 nanoplates, the SAED pattern (inset of Fig. 3b) depicts a ring shape, which can be attributed to the presence of g-C3N4 nanosheets in the background. Further, the lattice spacing values of 2.76 Å and 2.53 Å corresponding to the prominent (110) plane of BiOBr and (311) plane of CoFe2O4, respectively, from the HRTEM micrograph shown in Fig. 3c, are consistent with the XRD results.
 | ||
| Fig. 3 TEM micrographs of (a and b) CBG and (d and e) NBG at two different magnifications. HRTEM micrographs of (c) CBG and (f) NBG. Selected area electron diffraction patterns of CBG and NBG are presented in the insets of (b) and (e), respectively. | ||
Similarly, the presence of NiFe2O4 nanoparticles embedded on the g-C3N4 nanosheets and the growth of BBI0.05 nanoplates can be observed from the TEM micrograph shown in Fig. 3d. The formation of the ternary heterostructure is confirmed from the magnified TEM micrograph in Fig. 3e, wherein the dark areas clearly indicate the BBI0.05 nanoplates, which are grown over the NiFe2O4 nanoparticles embedded on the g-C3N4 nanosheets. The ring shaped SAED pattern (inset of Fig. 3e) confirms the presence of g-C3N4. Further, as observed from the HRTEM micrograph in Fig. 3f, the lattice spacing values of 2.76 Å corresponding to the prominent (110) plane of BiOBr and 2.51 Å corresponding to the (311) plane of NiFe2O4 are consistent with the XRD results.
The BET surface area and pore characteristics of the prepared materials were analyzed using N2 adsorption–desorption isotherms, as shown in Fig. 4a. The adsorption isotherms exhibited typical type II b behavior, based on BDDT classification, suggesting adsorption in sheet-like structures. The observed hysteresis loops indicated the presence of mesopores. Both CG and NG exhibited relatively low BET surface areas (3 m2 g−1 and 2.5 m2 g−1, respectively), whereas the surface areas for CBG (14 m2 g−1) and NBG (15 m2 g−1) were 4.5 and 6 times higher, respectively. This indicates that the addition of BBI0.05 led to the increase in surface area for the CBG and NBG samples. The BJH pore size distribution curve, presented in Fig. 4b, revealed a broad range of pore sizes confirming that all the samples had pore sizes within the mesoporous range (<50 nm). The mesopore volumes for CBG and NBG were 0.08 cm3g−1 and 0.11 cm3g−1, respectively, which were higher compared to other samples, suggesting that CBG and NBG have a greater capacity to adsorb molecules on their surfaces. However, it is essential to emphasize that the BET surface area is not the sole factor influencing photocatalytic activity. While it aids in enhancing the adsorption of organic/inorganic pollutants on the catalyst surface, thereby improving performance, several other factors also contribute significantly to boosting photocatalytic efficiency. These include the formation of heterojunctions and the suppression of exciton pair recombination.
 | ||
| Fig. 4 (a) N2 adsorption–desorption isotherms and (b) BJH pore size distribution of BG, CG, NG, CBG and NBG. | ||
3.2. Optical properties
UV-vis DRS was employed to analyze the optical absorption properties of the photocatalyst, as depicted in Fig. 5a. As observed from the reflectance spectrum in Fig. 5a, the pristine g-C3N4 has an absorption edge around 435 nm (see Fig. S3b, ESI†), while it is placed at around 540 nm for pure BBI0.05. The improved visible light absorption of BBI0.05 in comparison to pristine BiOBr (which typically has an absorption edge at 440 nm) can be attributed to the replacement of bromine with 5% iodine atoms. Therefore, BG, the 2D/2D heterojunction formed by clubbing g-C3N4 with BBI0.05, also demonstrated good absorption in the visible light region and exhibited an absorption edge at 525 nm due to their synergy. As observed from Fig. 5a, the percentage of reflectance was the lowest for CG and NG, indicating their broadband absorption, which can be attributed to the embedment of magnetic CoFe2O4 and NiFe2O4 nanoparticles onto g-C3N4. Similarly, the broadband absorption in CBG and NBG was also evident as the percentage of reflectance was much lower in comparison to BG. The physical appearance of all the synthesized photocatalysts is shown in Fig. S3 (ESI†) and the broadband absorption of CG, NG, CBG, and NBG due to their dark coloration is evident from the absorbance spectra shown in Fig. S4a (ESI†). The bandgap energy was calculated through a combination of Tauc and Kubelka–Munk functions as reported in the literature.52 From the plot of [hνF(R)]1/2 against hν, the bandgap energy of the photocatalysts was estimated by extrapolating the linear portions of the x-axis to [hνF(R)]1/2 = 0. As observed from Fig. S4c (ESI†) and Fig. 5b, the bandgap energies of BG, CBG and NBG (2.55 eV, 2.5 eV and 2.4 eV, respectively) were found to be lower than that of pristine g-C3N4 (2.9 eV), indicating enhanced visible light absorption. | ||
| Fig. 5 (a) UV-vis diffuse reflectance spectra of the as-synthesized photocatalysts and (b) their corresponding Tauc plots. | ||
The separation efficiency of the photogenerated excitons (e−/h+ pairs) in the photocatalysts was assessed through PL spectroscopy. A higher emission intensity in the PL spectrum suggests a rapid recombination of the photogenerated excitons, whereas a lower emission intensity indicates the efficient separation of charge carriers.53 The photocatalysts were initially excited at a wavelength of 350 nm and the corresponding emission was recorded between 400 nm and 600 nm. Fig. 6a illustrates a notable emission peak at 437 nm for g-C3N4 characteristic of the band−band emission specifically the n–π* electronic transitions involving lone pairs of the nitrogen atom in g-C3N4. The intense emission peak can be associated with the high recombination rate of the photogenerated e−/h+ pairs. Comparatively, as observed from Fig. 6a, the emission intensity of the composites including BG was much lower than that of pure g-C3N4. The IEF generated within BBI0.05 and the formation of the heterojunction with g-C3N4 lead to the enhanced suppression of the recombination rate of the charge carriers in BG. Similarly, as observed from Fig. 6b, the emission intensity of CG and NG decreased significantly owing to the embedment of CoFe2O4 and NiFe2O4 in g-C3N4. Interestingly, the PL emission intensity of the ternary composites CBG and NBG was significantly lower in comparison to the rest of the samples and could be attributed to a combination of factors such as the IEF generated within BBI0.05 and the possible formation of a double Z-scheme heterojunction.
 | ||
| Fig. 6 Photoluminescence spectra of (a) g-C3N4, BG, CG, NG, CBG and NBG photocatalysts; (b) magnified plot corresponding to the area marked in (a). | ||
3.3. Visible light driven photocatalytic degradation of MB
The photocatalytic efficiency of the samples was assessed by measuring the degradation of MB under visible light irradiation. Initially, the photodegradation efficiency of various solid-solution photocatalysts of BiOBrxI1−x was first assessed by the degradation of 5 ppm MB dye. As observed from the degradation plot (see Fig. S5, ESI†), BBI0.05 demonstrated the highest MB dye adsorption (after 30 min of stirring in the dark) and the highest degradation efficiency. Among the others, the photodegradation efficiency of BiOBr was comparable, while the catalysts with higher iodine content exhibited poor efficiency, which can be attributed to the higher recombination rate of the photogenerated electron–hole pairs.36The plot showing the photocatalytic degradation of 5 ppm MB in the presence of GCN, BBI0.05, BG, CBG, and NBG as photocatalysts with respect to irradiation time is presented in Fig. S6a (ESI†). As observed from the plots, the photodegradation of MB was negligible under visible light irradiation in the absence of a photocatalyst, indicating that no photolysis occurred. Interestingly, after the addition of the photocatalysts to MB under dark conditions, the adsorption over the surface of NBG was close to 90%, while for CBG and BG it was ∼76% and was only 5% for GCN. Next, under visible light irradiation, over 95% of MB was degraded within 30 min in the presence of NBG, which could be attributed to the enhanced e−/h+ pair separation and higher surface area. Both CBG and BG demonstrated similar photodegradation efficiencies and achieved over 90% MB degradation within the same time period of 30 min. Almost 77% MB degradation was achieved in the presence of BBI0.05, while pristine g-C3N4 exhibited the lowest efficiency (18%). The assessment of MB degradation was quantitatively conducted through pseudo-first-order reaction kinetics, employing the rate equation given by

UV-vis absorption spectra corresponding to the photocatalytic degradation of 10 ppm MB in the presence of CBG and NBG are presented in Fig. S7a and S7b (ESI†), respectively, wherein the consistent reduction in the intensity of the characteristic absorption peak of MB dye at λmax = 665 nm is indicative of the reduction in its concentration. As observed from the UV-vis absorption spectrum, under dark conditions, the absorption peak of MB decreased to 10% of its initial value in the presence of CBG (see Fig. S7a, ESI†), while for NBG it decreased by 60% (see Fig. S7b, ESI†), revealing the excellent adsorption of MB over the surface of NBG. As observed from Fig. S7c (ESI†), under light irradiation over 95% of MB was degraded within 90 min in the presence of NBG, while only 60% of MB was degraded with CBG during the same time period. Despite NBG demonstrating higher adsorption than CBG, both catalysts consistently reduced the MB concentration under visible light irradiation. The k values for CBG and NBG were 0.009 and 0.0215 min−1 respectively, indicating a more than a two-fold increase in the reaction rate for NBG compared to CBG (see Fig. S7d, ESI†).
3.4. Photodegradation of HABs
The best performing photocatalysts (BG, CBG and NBG) were employed for studying their capability towards the degradation of HABs. Each of the selected photocatalysts was dispersed in the water samples containing both M. aeruginosa (prokaryotic organism) and S. acuminatus (eukaryotic organism) algae cells for assessing their impact under visible light irradiation. A plot showing the time-dependent photocatalytic degradation of M. aeruginosa is presented in Fig. 7a. As observed from Fig. 7a, after dark adsorption for 30 min, there was almost negligible adsorption on the surface of BG, with the adsorption efficiency being close to 10% on both NBG and CBG. Next, under light irradiation, there was a consistent decrease in the optical density, indicating their degradation. After 60 min of light exposure, over 93% degradation was achieved in the presence of CBG, while NBG was able to degrade close to 84% during the same time period. On the other hand, BG was able to degrade only 68% of M. aeruginosa after 60 min of light exposure. Likewise, as observed from Fig. 7b, the trend was almost similar, wherein over 98% of S. acuminatus was degraded within 60 min in the presence of CBG, while only 50% degradation was achieved during the same time period with both NBG and BG. The enhanced photodegradation efficiency exhibited by CBG is in accordance with previous reports indicating that CoFe2O4 nanoparticles cause severe mechanical damage to eukaryotic algae.54 In general, photocatalysts with smaller crystallite sizes are reported to exhibit enhanced photocatalytic efficiency since the distance required for the charge carriers to travel to reach the surface is much lower in comparison to those with larger crystallite sizes.55 On the other hand, photocatalysts with smaller crystallites often result in more grain boundaries and defects, which can act as trapping sites for charge carriers – either promoting or hindering the photocatalytic activity. As shown in Table S1 (ESI†), GCN with the smallest crystallite size (3.57 nm) exhibited poor photocatalytic efficiency owing to the very high rate of exciton recombination as evidenced from the PL spectra (Fig. 6), while the optimum crystallite size, apt bandgap, and Z-scheme heterojunction in CBG suppressing the exciton recombination could be considered as the factors supporting its enhanced photocatalytic efficiency. | ||
| Fig. 7 Plot showing the time-dependent photocatalytic degradation of (a) Microcystis aeruginosa (prokaryotic organism) and (b) Scenedesmus acuminatus (eukaryotic organism) algal cells under visible light irradiation in the presence of BG, CBG and NBG as photocatalysts. Corresponding pseudo first order kinetic plots of (c) Microcystis aeruginosa and (d) Scenedesmus acuminatus. | ||
The photodegradation of HABs, primarily M. aeruginosa using various g-C3N4 based photocatalysts reported in the literature, is detailed in Table S1.† Interestingly, the performance of CBG was found to be superior in comparison to the rest. The kinetic plots in Fig. 7c and d reveal that the performance of BG and NBG in the degradation of S. acuminatus was poorer than that of M. aeruginosa. This discrepancy may be attributed to the robust cellulose cell wall of S. acuminatus, offering greater resistance to the photocatalytic degradation, in contrast to the simpler peptidoglycan cell wall of M. aeruginosa. Nevertheless, the overall results demonstrate a superior degradation rate by CBG compared to NBG. Therefore, further studies on algal degradation were conducted exclusively using CBG.
3.5. Chlorophyll-a reduction in algal cells
Chlorophyll is the building block of algal photosynthesis, absorbing and converting light. It is commonly employed as an indicator to assess the various growth phases of algae. The changes in chlorophyll-a per unit of water signifies the variations in overall photosynthetic activity, cell density, and biomass production.46 A decrease in chlorophyll-a level indicates lower photosynthetic activity and implies a stress response in microalgae.56 Therefore, the photodegradation of the algal cells under visible light can be elucidated by quantifying the chlorophyll-a content through optical density measurement as shown in Fig. 8. CBG exhibited a significant impact on the chlorophyll-a content in comparison to the control. Interestingly, after 180 min of light irradiation the chlorophyll-a content in M. aeruginosa was reduced from 11.038 μg ml−1 to 0.214 μg ml−1 (see Fig. 8a), achieving 98% degradation (see Fig. 8b). Likewise, the chlorophyll-a content in S. acuminatus was reduced from 5.2 μg ml−1 to 0.088 μg ml−1 (see Fig. 8a), achieving 100% degradation (see Fig. 8b) after 3 h of reaction. | ||
| Fig. 8 (a) Plot depicting the change in the chlorophyll-a content in M. aeruginosa and S. acuminatus treated with CBG under visible light irradiation. (b) Plot depicting the rate of photodegradation corresponding to the change in the chlorophyll-a content. | ||
3.6. Lipid peroxidation in algal cells
During the photocatalysis reaction, electrons and holes (exciton pair) are generated while light of energy greater than or equal to the bandgap of the photocatalyst is irradiated. These photogenerated excitons react with the hydroxyl ions and dissolved oxygen in water to form hydroxyl radicals (˙OH) and superoxide radicals (˙O2−), respectively as ROS (further explanation for the photocatalysis mechanism is discussed in section 3.10) The ROS primarily target the algal cell membrane, inducing oxidative damage through lipid peroxidation processes, leading to content leakage and eventual collapse of the cell membrane.57,58 MDA, a peroxidation product of membrane fatty acids, serves as an indicator of the lipid peroxidation degree and membrane system damage caused by environmental and abiotic stress.46,59 The key process in lipid peroxidation involves the removal of hydrogen from the unsaturated chains of phospholipids, leading to the conversion of phospholipids into lipid hydroperoxides, which further breaks down into MDA.11 Typically, during the photocatalytic treatment of HABs, the algal cell membrane collapses, leading to a surge in the MDA content with respect to the increase in irradiation time. Photocatalysis induced lipid peroxidation of the algal cells in the presence of CBG compared with the control samples shown in Fig. 9a. As observed, the MDA content in pristine algal cells of M. aeruginosa and S. acuminatus before the introduction of CBG was 0.96 nmol ml−1 and 1.17 nmol ml−1, respectively. Soon after the introduction of CBG (dark adsorption: 0 min), the MDA content in M. aeruginosa and S. acuminatus increased to 3.3 nmol ml−1 and 6.65 nmol ml−1, respectively. Interestingly, after light exposure for 3 h, the MDA content in M. aeruginosa and S. acuminatus increased to 7.68 nmol ml−1 and 56.76 nmol ml−1, respectively. Similar results were observed when a ternary composite photocatalyst of the form ZnFe2O4/Ag3PO4/g-C3N4 was subjected to photocatalytic treatment of M. aeruginosa under visible light, wherein the MDA content of algal cells increased from 0.292 nmol ml−1 to 0.876 nmol ml−1, indicating ROS-induced oxidative damage to the algae. The quantity and time-dependent rise in MDA serve as a biomarker of membrane lipid peroxidation in cyanobacteria and other photosynthetic organisms experiencing oxidative stress.57 Additionally, the increase in lipid peroxidation leads to growth inhibition, ion channel imbalance, modified permeability, and cell membrane damage. Therefore, the increase in MDA content observed herein in both M. aeruginosa and S. acuminatus indicates their complete degradation. | ||
| Fig. 9 Plot depicting (a) the photocatalysis induced lipid peroxidation in M. aeruginosa and S. acuminatus algal cells in the presence of CBG as the photocatalyst in comparison to the control, and (b) the electrolytic leakage in M. aeruginosa and S. acuminatus during their photocatalytic degradation in the presence of CBG as the photocatalyst. | ||
3.7. Electrolytic leakage
EL serves as a warning signal commonly used to measure stress-induced injury in plant cells and as a measure of cell stress tolerance. EL can occur across all species, tissues, and cell types and may be triggered by various stressors, irrespective of whether chemical, physical, or biological. Following the introduction of a stress factor, EL becomes noticeable almost immediately and can persist for a duration ranging from a few minutes to several hours.60 The amount of EL of both M. aeruginosa and S. acuminatus is shown in Fig. 9b. In comparison to the control (34.6% for M. aeruginosa and 70.57% for S. acuminatus), the percentages of EL after 3 h of photocatalytic treatment with CBG was 55.64% and 85.8%, respectively. Notably, the EL content steadily increased in both M. aeruginosa and S. acuminatus with respect to irradiation time in the presence of CBG, indicating their effective degradation.3.8. Microscopy of algal cells before and after photocatalysis
Bright (microscopic) and dark field (fluorescent) images of the algal cells before and after the photodegradation process in studying the reduction in the chlorophyll-a content serve as vital evidence of their effective degradation. As shown in Fig. 10, the reductions in chlorophyll-a content in both M. aeruginosa and S. acuminatus were observed microscopically, recorded at regular intervals of 60, 120 and 180 min of photocatalytic degradation in the presence of CBG as the photocatalyst. It should be noted that the dark field images indicating the bright red cells (Fig. 10b, f, d and h) are indicative of the live algal cells with good chlorophyll-a content, while the dead algal cells are indicated through their bright green color (Fig. 10n, p, r, t). The spherical and spindle shaped morphologies of the control samples (before the addition of the photocatalyst) of M. aeruginosa (Fig. 10a) and S. acuminatus (Fig. 10c), respectively, were confirmed from the bright field microscopy images, while the corresponding dark field images with high chlorophyll-a content can be seen in Fig. 10b and d. Even after the addition of the photocatalyst, there was no change in the morphology (see Fig. 10e and g) or the chlorophyll-a (see Fig. 10f and h) content of both the algal cells. However, after 60 min of light irradiation, the morphology (see Fig. 10i and k) of both the algal cells was found to be altered due to partial cell rupture and correspondingly the color of the dark field images turned yellow, revealing the decrease in the chlorophyll-a (see Fig. 10j and l) content. As the irradiation continued, the cell rupture progressed further as a result of effective photodegradation and after 180 min the morphology of the algal cells shrunk further (see Fig. 10q and s) and correspondingly the color of the dark field images turned to green (see Fig. 10r and t), revealing the complete removal of chlorophyll-a pigment. The microscopy data are consistent with the chlorophyll reduction studies discussed in section 3.4.1. | ||
| Fig. 10 Microscopic investigation of the algal cells before and after visible light driven photodegradation in the presence of CBG as the photocatalyst. Details of the microscopy images of M. aeruginosa: control (a and b), 0 min (e and f), 60 min (i and j), 120 min (m and n) and 180 min (q and r). Details of the microscopy images corresponding to S. acuminatus: control (c and d), 0 min (g and h), 60 min (k and l), 120 min (o and p), and 180 min (s and t). | ||
3.9. Monitoring algal cell regrowth
Following the 3 h photocatalytic degradation of both M. aeruginosa and S. acuminatus algal water samples with CBG as the photocatalyst were monitored to study the re-growth of the algal cells for a duration of 10 days. Owing to the presence of magnetic CoFe2O4 nanoparticles in CBG, they were efficiently separated from the algal water using a bar magnet (see Fig. S8, ESI†), which is a useful feature that helps in avoiding secondary pollution and for reusing the photocatalysts. Interestingly, the chlorophyll-a content remained at 0 mg ml−1 even after 10 days of incubation in the treated medium under ideal conditions, which confirmed the complete rupture of the algal cells treated with CBG under visible light, inhibiting their regrowth.3.10. Mechanism of the photocatalytic degradation of HABs, and role of ROS through scavenger studies
During photocatalysis, an electron (e−) is typically excited from the valence band of a semiconductor photocatalyst to the conduction band when it absorbs light energy equal to or greater than its bandgap, leaving behind a positively charged hole (h+). The photogenerated e− and h+ can then interact with dissolved oxygen (O2) and hydroxyl ions (OH−) in water, leading to the formation of ROS such as ˙O2− and ˙OH radicals. Additionally, ROS like hydrogen peroxide (H2O2) and singlet oxygen (1O2) may also be produced during the photocatalysis process.The mechanism of photodegradation of both HABs in the presence of CBG is proposed based on the experimental results discussed in the earlier sections of the manuscript and is illustrated in Scheme 1. The efficient adsorption of the prokaryotic and eukaryotic algal cell colonies onto the CBG surface is likely due to its large pore volume and optimal crystallite size. CBG's enhanced absorption in the visible light region allows it to generate e− and h+ upon visible light irradiation, which subsequently interact with water molecules and dissolved oxygen to form ROS. As depicted in Scheme 1, CBG, synthesized by coupling BiOBr0.95I0.05, g-C3N4 and CoFe2O4, forms a double Z-scheme heterojunction, attributed to the well-aligned valence and conduction band positions of the individual photocatalysts. During irradiation, the e− in the conduction bands of BiOBr0.95I0.05 and CoFe2O4 recombine with the h+ in the valence band of g-C3N4. Consequently, the e− in the conduction band of g-C3N4 and the h+ in the valence band of BiOBr0.95I0.05 and CoFe2O4 actively participate in algal cell photodegradation by producing ROS, which cause irreparable damage to the algal cell walls, leading to their rupture, as shown in Scheme 1.61–63 The cell rupture is evidenced by the loss of chlorophyll-a pigment, an increase in MDA due to lipid peroxidation, and elevated EL, indicating membrane damage. Additionally, the photocatalytic degradation of HABs was tracked via fluorescence microscopy, where the change in algal cell color in the dark field images from bright red to green confirmed cell rupture.
 | ||
| Scheme 1 Schematic representation of the double Z-scheme heterojunction formed in CBG and the pictorial representation of the mechanism describing the photodegradation of algal cells of both M. aeruginosa and S. acuminatus leading to their complete rupture. | ||
The relative role of different ROS generated during the photodegradation of the algae was investigated through a scavenger study. Typically, scavengers are added for suppressing the activity of specific reactive species during photocatalytic reactions to better understand the role of each ROS in the degradation process. As observed from the plots shown in Fig. 11, the photodegradation efficiency was significantly suppressed after the addition of various chemicals (BQ, AO, K2Cr2O7 and IPA) as scavengers for quenching specific reactive species in both M. aeruginosa and S. acuminatus. In the case of M. aeruginosa (see Fig. 11a), in comparison to pristine CBG, the addition of BQ significantly suppressed the degradation efficiency of CBG by 58%, indicating the dominant role of ˙O2−. Similarly, the addition of AO suppressed the degradation efficiency of CBG by ∼30%, indicating the contribution of h+. Further, the addition of K2Cr2O7 and IPA as e− and ˙OH suppressed the degradation efficiency of CBG to 20% and 18%, respectively. On the other hand, in the case of S. acuminatus (see Fig. 11b), all the chemical scavengers were observed to suppress the degradation efficiency of CBG, indicating the dominant role of all the reactive species. However, the order of suppression was ˙O2− > h+ > ˙OH > e−, wherein the degradation efficiency of CBG was suppressed by 46%, 44%, 39% and 36%, respectively. Therefore, it was conclusively evident that ˙O2− was the dominant ROS during the visible light driven photodegradation of both eukaryotic and prokaryotic HABs in the presence of CBG as the photocatalyst.
 | ||
| Fig. 11 Plot showing the time-dependent visible-light driven photocatalytic degradation of (a) M. aeruginosa and (b) S. acuminatus algae cells after the addition of various scavengers in the presence of CBG. | ||
4. Conclusion
In conclusion, novel magnetically recoverable BiOBr0.95I0.05/g-C3N4/MFe2O4 (M = Co, Ni) ternary composite photocatalysts were successfully synthesized through a combination of pyrolysis and hydrolysis techniques. Among a series of BiOBrxI1−x samples, BiOBr0.95I0.05 was found to be the most efficient photocatalyst and was chosen for the fabrication of the ternary heterojunction. Morphological analysis through FESEM and TEM revealed the 2D nanoplates of BiOBr0.95I0.05 stacked on CoFe2O4/g-C3N4, and their increased surface area and pore volume in comparison to the pristine counterparts through BET surface area analysis confirmed the formation of the ternary heterojunction. Further, UV-vis DRS revealed the apt bandgap of the ternary composites, and photoluminescence spectroscopy indicated the effective separation of the photogenerated charges, in comparison to the individual counterparts. Photocatalytic degradation studies of both Microcystis aeruginosa (prokaryotic organism) and Scenedesmus acuminatus (eukaryotic organism) algal cells were undertaken in the presence of the ternary composite photocatalysts under visible light. Interestingly, over 93% M. aeruginosa and S. acuminatus were degraded within 60 min of light exposure in the presence of CBG. Further, issues such as the decrease in chlorophyll-a content, increase in MDA due to lipid peroxidation, and increase in electrolytic leakage were studied for understanding the cell rupture through photocatalysis. Notably, the formation of the double Z-scheme heterojunction led to enhanced photodegradation of the algal cells facilitated by the effective separation of the photogenerated charges. Further, scavenger studies revealed the significant role of the superoxide radical species in the degradation of the algal cells, followed by holes, hydroxyl radicals and electrons. Furthermore, the attempt to regrow the degraded algal cells (recovered after magnetically separating the CBG photocatalyst) in a growth medium under ambient conditions exhibited no change in the chlorophyll-a content (0 mg ml−1) even after 10 days. Therefore, complete cell rupture was guaranteed through photocatalysis with CBG, while effectively mitigating the risk of secondary pollution associated with powdered photocatalysts through magnetic recovery.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Central Sophisticated Instrumentation Facility, University of Calicut for their support in FESEM and BET analysis.References
- M. L. Wells, V. L. Trainer, T. J. Smayda, B. S. O. Karlson, C. G. Trick, R. M. Kudela, A. Ishikawa, S. Bernard, A. Wulff, D. M. Anderson and W. P. Cochlan, Harmful Algae, 2015, 49, 68–93 CrossRef.
- J. M. O'Neil, T. W. Davis, M. A. Burford and C. J. Gobler, Harmful Algae, 2012, 14, 313–334 CrossRef.
- F. Imtiaz, J. Rashid, R. Kumar, J. O. Eniola, M. A. E. F. Barakat and M. Xu, Environ. Res., 2024, 248, 118251 CrossRef CAS.
- B. Balaji-Prasath, Y. Wang, Y. P. Su, D. P. Hamilton, H. Lin, L. Zheng and Y. Zhang, Environ. Chem. Lett., 2022, 20, 3133–3152 CrossRef CAS.
- B. Yu, Y. Zhang, H. Wu, W. Yan, Y. Meng, C. Hu, Z. Liu, J. Ding and H. Zhang, Sci. Total Environ., 2024, 906, 167650 CrossRef CAS PubMed.
- S. Sun, Q. Tang, H. Xu, Y. Gao, W. Zhang, L. Zhou, Y. Li, J. Wang and C. Song, Chemosphere, 2023, 312, 137239 CrossRef CAS PubMed.
- H. Mohan, S. Vadivel and S. Rajendran, Chemosphere, 2022, 302, 134827 CrossRef CAS.
- X. Yu, J. Zhou, Z. Wang and W. Cai, J. Photochem. Photobiol., B, 2010, 101, 265–270 CrossRef CAS.
- Y. Jin, S. Zhang, H. Xu, C. Ma, J. Sun, H. Li and H. Pei, Environ. Pollut., 2019, 245, 642–650 CrossRef CAS PubMed.
- M. Nemiwal, T. C. Zhang and D. Kumar, Sci. Total Environ., 2021, 767, 144896 CrossRef CAS PubMed.
- X. He, P. Wu, S. Wang, A. Wang, C. Wang and P. Ding, J. Cleaner Prod., 2021, 289, 125755 CrossRef CAS.
- G. Fan, L. Hong, J. Luo, Y. You, J. Zhang, P. Hua, B. Du, J. Zhan, R. Ning and M. Bao, Chem. Eng. J., 2020, 392, 123767 CrossRef CAS.
- D. Wang, J. Chen, X. Gao, Y. Ao and P. Wang, Chem. Eng. J., 2022, 431, 134105 CrossRef CAS.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- J. Wang and S. Wang, Coord. Chem. Rev., 2022, 453, 214338 CrossRef CAS.
- M. Reza Gholipour, C. T. Dinh, F. Béland and T. O. Do, Nanoscale, 2015, 7, 8187–8208 RSC.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- S. Shenoy, S. Ahmed, I. M. C. Lo, S. Singh and K. Sridharan, Mater. Res. Bull., 2021, 140, 111290 CrossRef CAS.
- P. Suyana, P. Ganguly, B. N. Nair, S. C. Pillai and U. S. Hareesh, Chem. Eng. J. Adv., 2021, 8, 100148 CrossRef CAS.
- H. Kato, Y. Sasaki, N. Shirakura and A. Kudo, J. Mater. Chem. A, 2013, 1, 12327–12333 RSC.
- K. Sridharan, E. Jang and T. J. Park, Appl. Catal., B, 2013, 142–143, 718–728 CrossRef CAS.
- Y. Ouyang, H. Jiang, H. Zhong, G. Li, X. Ji, Q. Jin, W. Xue, X. Hu and M. Zhu, Chem. Eng. J., 2023, 464, 142542 CrossRef CAS.
- Y. Xue, P. Wang, C. Wang and Y. Ao, Chemosphere, 2018, 203, 497–505 CrossRef CAS.
- S. Ding, D. Mao, S. Yang, F. Wang, L. Meng, M. Han, H. He, C. Sun and B. Xu, Appl. Catal., B, 2017, 210, 386–399 CrossRef CAS.
- K. Sridharan, S. Shenoy, S. Girish Kumar, C. Terashima, A. Fujishima and S. Pitchaimuthu, Catalysts, 2021, 11, 426 CrossRef CAS.
- J. Shang, T. Chen, X. Wang, L. Sun and Q. Su, Chem. Phys. Lett., 2018, 706, 483–487 CrossRef CAS.
- X. Xiao, C. Liu, R. Hu, X. Zuo, J. Nan, L. Li and L. Wang, J. Mater. Chem., 2012, 22, 22840–22843 RSC.
- J. Guo, X. Li, J. Liang, X. Yuan, L. Jiang, H. Yu, H. Sun, Z. Zhu, S. Ye, N. Tang and J. Zhang, Coord. Chem. Rev., 2021, 443, 214033 CrossRef CAS.
- L. Ye, Y. Su, X. Jin, H. Xie and C. Zhang, Environ. Sci.: Nano, 2014, 1, 90–112 RSC.
- L. S. Gómez-Velázquez, A. Hernández-GordilloCatedrático CONACYT, M. J. Robinson, V. J. Leppert, S. E. Rodil and M. Bizarro, Dalton Trans., 2018, 47, 12459–12467 RSC.
- A. M. Ganose, M. Cuff, K. T. Butler, A. Walsh and D. O. Scanlon, Chem. Mater., 2016, 28, 1980–1984 CrossRef CAS PubMed.
- L. Chen, R. Huang, M. Xiong, Q. Yuan, J. He, J. Jia, M. Y. Yao, S. L. Luo, C. T. Au and S. F. Yin, Inorg. Chem., 2013, 52, 11118–11125 CrossRef CAS.
- M. Zhang, C. Lai, B. Li, D. Huang, G. Zeng, P. Xu, L. Qin, S. Liu, X. Liu, H. Yi, M. Li, C. Chu and Z. Chen, J. Catal., 2019, 369, 469–481 CrossRef CAS.
- C. S. L. Fung, M. Khan, A. Kumar and I. M. C. Lo, Sep. Purif. Technol., 2019, 216, 102–114 CrossRef CAS.
- W. Yao, J. Zhang, Y. Wang and F. Ren, Appl. Surf. Sci., 2018, 435, 1351–1360 CrossRef CAS.
- S. Shi, M. A. Gondal, A. A. Al-Saadi, R. Fajgar, J. Kupcik, X. Chang, K. Shen, Q. Xu and Z. S. Seddigi, J. Colloid Interface Sci., 2014, 416, 212–219 CrossRef CAS.
- K. Sridharan, M. Agarwal, J. Philip, T. Endo and R. Philip, Trans. Mater. Res. Soc. Jpn., 2010, 35, 159–162 CrossRef CAS.
- C. Feng, Z. Lu, Y. Zhang, Q. Liang, M. Zhou, X. Li, C. Yao, Z. Li and S. Xu, Chem. Eng. J., 2022, 435, 134833 CrossRef CAS.
- U. Kumar, J. Kuntail, A. Kumar, R. Prakash, M. R. Pai and I. Sinha, Appl. Surf. Sci., 2022, 589, 153013 CrossRef CAS.
- S. Shenoy and K. Sridharan, Chem. Phys. Lett., 2020, 749, 137435 CrossRef CAS.
- G. Mackinney, J. Biol. Chem., 1941, 140, 315–322 CrossRef CAS.
- S. Zhou, H. Yin, S. Tang, H. Peng, D. Yin, Y. Yang, Z. Liu and Z. Dang, Ecotoxicol. Environ. Saf., 2016, 127, 214–221 CrossRef CAS PubMed.
- B. Chen, J. Huang, J. Wang and L. Huang, Colloids Surf., B, 2008, 61, 88–92 CrossRef CAS PubMed.
- K. V. Ajayan, P. J. Chaithra, K. Sridharan, P. Sruthi, E. Harikrishnan and C. C. Harilal, Environ. Res., 2023, 237, 116926 CrossRef CAS.
- H. Baniamerian, P. Tsapekos, M. Alvarado-Morales, S. Shokrollahzadeh, M. Safavi and I. Angelidaki, Chemosphere, 2020, 242, 125119 CrossRef CAS PubMed.
- G. Fan, M. Bao, X. Zheng, L. Hong, J. Zhan, Z. Chen and F. Qu, J. Hazard. Mater., 2019, 367, 529–538 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- M. Wang, R. Quesada-Cabrera, S. Sathasivam, M. O. Blunt, J. Borowiec and C. J. Carmalt, ACS Appl. Mater. Interfaces, 2023, 15, 49270–49280 CrossRef CAS.
- K. Sridharan, T. Kuriakose, R. Philip and T. J. Park, Appl. Surf. Sci., 2014, 308, 139–147 CrossRef CAS.
- K. Sridharan, E. Jang and T. J. Park, Appl. Catal., B, 2013, 142, 718–728 CrossRef.
- S. Shenoy, E. Jang, T. J. Park, C. S. Gopinath and K. Sridharan, Appl. Surf. Sci., 2019, 483, 696–705 CrossRef CAS.
- F. Ahmad, H. Yao, Y. Zhou and X. Liu, Chemosphere, 2015, 139, 479–485 CrossRef CAS PubMed.
- M. Abu Hanif, J. Akter, M. Akherul Islam, I. Lee, K. Prasad Sapkota, S. Shrestha, A. Pandey, N. Gyawali and J. Ryang Hahn, J. Photochem. Photobiol., A, 2022, 431, 114066 CrossRef CAS.
- A. Sharan and S. Nara, Aquat. Toxicol., 2020, 224, 105498 CrossRef CAS PubMed.
- G. Fan, X. Lin, Y. You, B. Du, X. Li and J. Luo, J. Hazard. Mater., 2022, 421, 304–3894 CrossRef PubMed.
- Y. Yang, H. Chen and J. Lu, Sci. Total Environ., 2023, 858, 159640 CrossRef CAS PubMed.
- M. S. Amritha, K. Sridharan, J. T. Puthur and O. P. Dhankher, J. Agric. Food Chem., 2021, 69, 10017–10035 CrossRef PubMed.
- V. Demidchik, D. Straltsova, S. S. Medvedev, G. A. Pozhvanov, A. Sokolik and V. Yurin, J. Exp. Bot., 2014, 65, 1259–1270 CrossRef CAS PubMed.
- M. Bhuvaneshwari, V. Iswarya, S. Archanaa, G. M. Madhu, G. K. S. Kumar, R. Nagarajan, N. Chandrasekaran and A. Mukherjee, Aquat. Toxicol., 2015, 162, 29–38 CrossRef CAS.
- P. Chen, B. A. Powell, M. Mortimer and P. C. Ke, Environ. Sci. Technol., 2012, 46, 12178–12185 CrossRef CAS.
- J. Ji, Z. Long and D. Lin, Chem. Eng. J., 2011, 170, 525–530 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4en00955j |
| This journal is © The Royal Society of Chemistry 2025 |