
Robust calix[4]arene-polyethyleneimine coated iron oxide nanoparticles for enhanced recovery of gold and platinum chloride complexes†
Carlos
Moya
 abc,
Natacha
Brion
d,
Ludovic
Troian-Gautier
efg,
Ivan
Jabin
g and
Gilles
Bruylants
*a
abc,
Natacha
Brion
d,
Ludovic
Troian-Gautier
efg,
Ivan
Jabin
g and
Gilles
Bruylants
*a
aEngineering of Molecular NanoSystems, Ecole polytechnique de Bruxelles, Université libre de Bruxelles, Brussels 1050, Belgium. E-mail: gilles.bruylants@ulb.be
bDepartament de Física de la Matèria Condensada, Universitat de Barcelona, Martí i Franquès 1, 08028 Barcelona, Spain
cInstitut de Nanociència i Nanotecnologia (IN2UB), Universitat de Barcelona, 08028 Barcelona, Spain
dAnalytical and Environmental Geochemistry (AMGC), Vrije Universiteit Brussel, Pleinlaan 2, 1050 Brussels, Belgium
eUniversité catholique de Louvain (UCLouvain), Institut de la Matière Condensée et des Nanosciences (IMCN), Molecular Chemistry, Materials and Catalysis (MOST), Place Louis Pasteur 1, bte L4.01.02, 1348 Louvain-la-Neuve, Belgium
fWel Research Institute, Avenue Pasteur 6, 1300 Wavre, Belgium
gLaboratoire de Chimie Organique, Université libre de Bruxelles, Avenue F.D. Roosevelt 50, B-1050 Brussels, Belgium
First published on 26th October 2024
Abstract
Magnetic removal and recovery of precious metals from wastewater and complex biological media pose significant challenges mostly due to the need for efficient, selective, and stable materials. This work reports a methodology that allows these challenges to be addressed by synthesizing iron oxide nanoparticles (IONPs) coated with a covalent layer of calix[4]arene-tetracarboxylate (X4C4) capable of binding polyethylenimine (PEI) functionalities via electrostatic interactions. In contrast to citrate coating, which was previously utilized as an attachment layer for PEI, the reductive grafting of X4C4-tetra-diazonium salts onto IONPs results in a considerably more stable coating that proves to be an excellent substrate for the adsorption of PEI. This efficiently results in a synergistic interaction that significantly improves the durability of the PEI coating and maintains the particles in a dispersed state. The stability of the resulting IONPs@X4C4@PEI particles is demonstrated by their ability to withstand both acidic and alkaline conditions without significant particle aggregation or loss of magnetic properties. Moreover, these particles exhibit exceptional magnetic reusability, retaining their selectivity and recovery efficiency over multiple separation cycles. The selective affinity of IONPs@X4C4@PEI particles for gold (Au) and platinum (Pt) stems from the specific binding interactions between the complexes formed by these metals in solution and the PEI coating, enabling efficient recovery of these precious metals. This work places these IONPs at the forefront in terms of stability, reusability, and selectivity, which will undoubtedly open new avenues for environmental remediation and purification applications.
Environmental significanceThis study presents a novel method for the synthesis and functionalization of IONPs aimed at the selective extraction of precious metals from complex aqueous solutions. By using a polyethylenimine coating adsorbed onto a layer of covalently bound calix[4]arenes, we achieved enhanced stability and magnetic reusability of the nanoparticles. Our approach shows exceptional efficacy in the recovery of gold and platinum complexes. This research aligns with the focus of Environmental Science: Nano, which provides innovative solutions to environmental challenges. It provides valuable insights into the development of nanomaterials for efficient and selective metal recovery, which is critical for pollution reduction and resource conservation. We therefore believe that our manuscript is well suited to the journal's readership, highlighting advances in environmental nanotechnology. |
Introduction
The removal of precious metals like gold (Au) and platinum (Pt) from wastewater presents a multifaceted challenge, demanding a range of treatment methods with distinct advantages and limitations.1–3 For example, the adsorption of these metals on surfaces stands out for its simplicity, cost-effectiveness, and efficiency but faces disposal and regeneration issues.4–6 Membrane methods excel in heavy metal extraction but suffer from fouling complexity and high costs.7 Electrochemical treatment, while offering rapid and efficient metal removal with minimal sludge and chemical use, is constrained by the high costs associated with electrodes and energy consumption.8 The choice of the most suitable method for removing these metals from wastewater hinges on various factors, including operation cost, initial metal ion concentration, environmental impact, pH, matrix composition, removal efficiency, and economic feasibility.The integration of iron oxide nanoparticles (IONPs) into these treatment methods, particularly the adsorption and membrane processes,9 presents a promising avenue for enhancing their overall collection efficiency.10–13 These systems, composed of magnetite (Fe3O4), maghemite (γ-Fe2O3), or their combinations are ideal for selectively extracting metals from water residues for several reasons.12,13 Their large surface area and magnetic properties allow for high metal ion binding, reduce waste, and simplify separation processes.14 IONPs can also efficiently be used to recover valuable metals from either organic and aqua, residues, and lixiviates, avoiding the use of hazardous chemicals and energy-intensive processes, thereby offering an eco-friendly alternative. However, IONPs face stability issues due to their susceptibility to aggregation and redox reactions.14 Their coating with organic molecules is also required to enhance their ability to selectively recover specific metals.
To address the above-mentioned challenges, various coatings have been developed,5,15 including thiol-modified silica,16 squaramide derivatives,17 and chitosan-based coatings,18 each offering distinct benefits. Among the various polymers employed to coat IONPs, polyethylenimine (PEI) stands out for its hydrophilicity and abundance of amino groups. PEI's structure, consisting of repeating units of amine and aliphatic carbons, facilitates electrostatic interactions with negatively charged IONPs, enabling their effective coating. Moreover, PEI's amino groups can form coordination bonds with anionic substances, including biomolecules and metal complexes, thereby enhancing selectivity, and promoting chelation effects.19–21 PEI-coated IONPs have demonstrated promising performance in recovering Au from thiosulfate and ammonia-thiosulfate leaching solutions.22,23 While modified PEI coated-IONPs exhibit excellent characteristics such as enhanced surface area and increased adsorption capacity, this strategy presents challenges in precisely controlling the particle coating, which can lead to aggregation, and compromises the reliability of the functionalized IONPs.
This issue can be solved by the addition of an intermediate coating that can enhance the interaction between the PEI layer and the IONP core, improving the overall stability and functionality. By strongly bridging the iron oxide core and the PEI coating, intermediate layers can effectively mitigate aggregation and enhance the practical application of PEI-coated IONPs, particularly in environmental and biomedical applications where precise and stable IONP performance is crucial.
In this context, calix[4]arene-tetradiazonium salts have proven to be particularly successful in the functionalization of metallic nanomaterials with dense and compact monolayers,24–27 providing extreme chemical and colloidal stabilities to the particles. Furthermore, these compounds allow the grafting of layers of tailored compositions28 and defined densities of appending functional groups,29 that enable the additional coupling to functional molecules. This makes calix[4]arene-coated nanomaterials highly versatile for diverse applications.29–31 The choice of calix[4]arene as a platform for surface modification is primarily driven by its structural properties. Unlike other diazonium salts,32 which often lead to multilayer formation, calix[4]arenes effectively prevent this issue by inhibiting polymerization at the surface. Calix[4]arenes also offer the flexibility to be functionalized with various organic groups, such as carboxyls,26 or PEG chains,28 at their smaller rim.33
This work reports on the development of a novel method for synthesizing IONPs with a robust PEI coating mediated by a grafted calixarene layer onto the IONP surface. To achieve this, we used a calix[4]arene-tetracarboxyltetradiazonium salt, namely X4C4-(N2+)4, that is functionalized with diazonium groups on its large rim for anchoring to the IONP surface and with carboxyl groups on its small rim. The reductive covalent grafting of calixarenes X4C4 on the IONPs led to an ultrastable organic layer able to establish strong electrostatic interactions with the PEI polymer, optimizing the final performance of the functionalized IONPs. The physicochemical features of these systems including particle size, core composition, and surface characteristics, were thoroughly characterized to assess the impact of the particle coating. Besides, the stability of the synthesized IONPs was evaluated in ionic, acidic, and basic solutions, as well as under magnetic separations. Finally, the collection efficiency and selectivity of Au and Pt chloride complexes were evaluated in solution as a function of pH, time, and particle concentration.
Experimental
Materials
Ammonia (NH3; 32%), hydrochloric acid (HCl; 35%), iron(II) chloride tetrahydrate (FeCl2·4H2O; 97%), and nitric acid (HNO3; 68%) were obtained from VWR Chemicals. Copper(II) chloride dihydrate (CuCl2·2H2O; 99%), iron(III) chloride hexahydrate (FeCl3·6H2O; 99%), iron(III) nitrate (Fe(NO3)3; 98%), gold(III) chloride hydrate (HAuCl4; 99%), platinum(IV) chloride (PtCl4; ≥99%), PEI solution (Mw ∼ 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 50% in water), thiourea (SC(NH2)2; 99%), trisodium citrate dihydrate (Na3Cit·2H2O; 99%), and zinc chloride (ZnCl2, 99%) were bought from Sigma-Aldrich. The X4C4-tetra-diazonium tetra-fluoroborate and the methoxycarbonylbenzene-diazonium (XC-N2+) fluoroborate were supplied by the X4C company.33 Milli-Q water (MQ-H2O) was employed in all the experiments. Magnetic separations were performed with a 45 mm cylinder magnet made of an alloy of Nd, Fe, and Be from speromagnets.
000, 50% in water), thiourea (SC(NH2)2; 99%), trisodium citrate dihydrate (Na3Cit·2H2O; 99%), and zinc chloride (ZnCl2, 99%) were bought from Sigma-Aldrich. The X4C4-tetra-diazonium tetra-fluoroborate and the methoxycarbonylbenzene-diazonium (XC-N2+) fluoroborate were supplied by the X4C company.33 Milli-Q water (MQ-H2O) was employed in all the experiments. Magnetic separations were performed with a 45 mm cylinder magnet made of an alloy of Nd, Fe, and Be from speromagnets.
Synthesis of IONPs
The IONPs selected for this work were synthesized following a two-step co-precipitation protocol described by M. P. Morales et al.34,35 During the first stage, 29.5 mL of a 32% NH3 solution was added with a 0.1–0.3 mL s−1 rate to 200 mL of a mixture of FeCl3·6H2O and FeCl2·4H2O with a molar ratio of 1:2 (0.594 M). The resulting mixture was left under magnetic stirring for 3 h at 90 °C. This stage produced Fe3O4 NPs that required 3 magnetic washing processes with MQ-H2O (see eqn (1)–(4)). The second stage was an acid treatment to convert Fe3O4 to γ-Fe2O3. For this, 150 mL of 2 M HNO3 was added to the pellet resulting from the last magnetic precipitation, and the mixture was stirred for 15 minutes at room temperature. The resulting particles were precipitated by magnetic separation and 150 mL of 1 M Fe(NO3)3 was added to enhance the particle crystallization. The solution was brought to boiling and stirred for 30 minutes then left to cool. The supernatant was removed by magnetic separation and a second addition of 150 mL of 2 M HNO3 was conducted for 15 minutes to completely oxidize any remaining Fe(II) (see eqn (5)). Finally, the whole was magnetically washed 3 times with MQ-H2O to obtain the final magnetic γ-Fe2O3 NPs.| FeCl3(aq) + 3OH−(aq) → Fe(OH)3(aq) + 3Cl−(aq) | (1) |
| Fe(OH)3(aq) → FeOOH(aq) + H2O(l) | (2) |
| FeCl2(aq) + 2OH−(aq) → Fe(OH)2(aq) + 2Cl−(aq) | (3) |
| 2FeOOH(aq) + Fe(OH)2(aq) → Fe3O4 (s) + 2H2O(l) | (4) |
| 2Fe3O4(s) + 2HNO3(aq) → 3γ-Fe2O3(s) + 2NO2(g) + H2O(aq) | (5) |
Functionalization of IONPs
Functionalization with X4C4-(N2+)4 and XC-N2+. Naked IONPs were functionalized with X4C4-(N2+)4 at 30 °C with an incubation time of 16 h in a thermomixer in two steps to give the coated IONPs@X4C4. First, the pH of 20 mL of naked IONPs with a [Fe] = 1 mg mL−1 (0.0179 M Fe) was adjusted to 12 by the addition of 2 mL of NaOH 0.1 M. Then, 5 mL of the X4C4-(N2+)4 (5.26 mM, 29.25 mg) was added to the solution and the pH was adjusted to 8 by the addition of 0.1 M HCl. The washing process consisted in 4 cycles of centrifugation at 10000 rcf for 15 minutes. From the second to the fourth cycles, the resulting pellet was redispersed in 20 mL of 0.5 mM NaOH by sonication of the solution for 5 minutes. Finally, the pellet corresponding to the last wash was redispersed in 20 mL of MQ-H2O, and the pH of the solution was adjusted to 8 by the addition of 0.1 M HCl. The functionalization of IONPs with the XC-N2+ salt involved a similar procedure to that used for X4C4-(N2+)4, but with an initial pH adjustment: the XC-N2+ solution was first brought to pH 6 before being combined with the IONPs. The mixture was then further adjusted to pH 8 and left under agitation for 16 hours.
Functionalization with citrate (Cit). 1 mL of IONPs (32 mg Fe mL−1, 0.5728 M Fe) was added into 20 mL of a Cit solution (0.052 M, 0.268 g).22 The pH of the dispersion was adjusted by the addition of 0.1 M HCl to a final value of 6. The whole was then placed in a thermomixer at 1000 rpm and 90 °C for 6 hours. The obtained particles were washed by centrifugation for 15 minutes at 15000 rcf and redispersed in MQ-H2O. This washing cycle was repeated 2 times, and the resulting pellet was finally redispersed in 20 mL of MQ-H2O.
Functionalization with PEI. This functionalization step was directly linked to the above-mentioned functionalization procedures. In a general way,22 1.12 mL of a 5% w/w PEI aqueous solution was added to 20 mL of the above-prepared IONPs (0.95–1 mg Fe mL−1, 0.017–0.0179 M Fe). The pH was then adjusted to 8 using 0.1 M HCl and the resultant mixtures were kept in a thermomixer at 50 °C for 16 h. After that, the mixtures were cleaned 3 times by centrifugation at 10000 rcf for 15 minutes. Finally, the pellets were redispersed in 20 mL of MQ-H2O.
Characterization techniques
Information on the particle size distribution, morphology, and crystal structure of the different samples was obtained using a ThermoFisher Tecnai T20 Transmission Electron Microscope (TEM) operating at 200 kV. TEM samples were prepared by placing a drop of a dilute water suspension of IONPs onto a carbon-coated Cu grid and letting it dry under a low vacuum.Histograms of the IONPs were determined by counting at least 300 particles with the ImageJ software and fitting the results to log-normal distributions following eqn (6).36
 | (6) |
| DTEM = D0eS2/2 | (7) |
 | (8) |
The information about the crystal structure of the samples was determined by the analysis of selected area electron diffraction (SAED) patterns employing ImageJ graphical analysis.37 The interplanar SAED distances were calculated by measuring the separation between the central spot and the diffraction spots and converting the reciprocal space distances into real space. These results were compared to the Fe3O4 (ICSD: 01-089-3854) and γ-Fe2O3 (ICSD: 00-039-1346) reference phases using the X'Pert High Score Plus program.38
The chemical composition of single and small agglomerates of particles was analyzed by Energy-Dispersive X-ray Spectroscopy (EDS) data, which was recorded with an Ultim-Max (Oxford Instruments) detector, coupled to a probe-corrected Titan (ThermoFisher Scientific) operated at 300 kV. The spatial resolution of the titan microscope is about 0.1 nm, but the lateral resolution of EDS is approximately 1 nm.
XRD spectra of samples were collected using a PANalytical X'Pert PRO MPD diffractometer with Fe-filtered Co Kα radiation (λKα = 1.7903 Å; λKα1 = 1.7890 Å, λKα2 = 1.7929 Å). The XRD data was collected within a range of 7 to 135° 2θ with a step size of 0.017° and a measuring time of 100 seconds per step, making 8 repeated scans. The crystal size (DXRD) was determined following the Rietveld–Debye–Scherrer eqn (9)
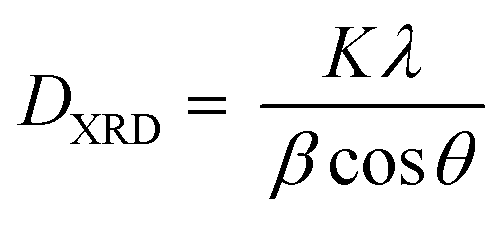 | (9) |
 | (10) |
Dynamic Light Scattering (DLS) was used to determine the hydrodynamic size (Dh) of samples using a Malvern Zetasizer Ultra ZSU5700 equipped with a He/Ne 633 nm laser. Results were obtained with DTS0012 disposable cuvettes by calculating the average of 3 measurements with 10 scans each. The settings chosen were Fe2O3 as the material with a refractive index of 2.36 and an absorption of 0.147. Zeta (ζ)-potential measurements were also conducted using the same instrument and DTS1070 cuvettes. For both types of analysis, solutions at 0.1 mg Fe mL−1 were employed.
Fourier-Transform Infrared Spectroscopy (FTIR) was performed using a Bruker Equinox 55 spectrophotometer at room temperature with a liquid nitrogen-cooled Hg–Cd–telluride detector. Samples were precipitated by centrifugation, deposited on Ge internal reflection pieces, and water removed by application of an N2 gas flow. For each sample, 128 scans were recorded with a 2 cm−1 resolution.
The organic fraction of the samples was evaluated by thermogravimetric analysis (TGA). Measurements were performed in a TGA-SDTA 851e/SF/1100 (Mettler Toledo) in a nitrogen atmosphere. Samples were kept for 2 h at 60 °C, followed by a linear ramp up to 700 °C at a rate of 10 °C min−1. The loss of organic material was obtained from the difference in weight at the plateau of the 60 °C isotherm and the remained mass at 700 °C.
The concentration of metals in synthetic metal solutions mimicked industrial wastewater metal concentrations.40 Those metal concentrations were determined by High-Resolution Inductively Coupled Plasma Mass Spectrometer measurements (HR-ICP-MS) with a Thermo Scientific Element II instrument (ThermoFisher Scientific) after aqua regia digestion and diluting controlled volumes respectively with HCl (2% w/w) and HNO3 (2% w/w) for Au and Pt, and Cu, Fe, and Ni.
UV-vis absorption spectra were recorded using a Shimadzu UV-3600 Spectrometer with a fast-scanning rate between 200–800 nm. 1 mL or 4 mL high-precision quartz cuvettes were used with a path length of 10 mm. The Fe concentration of the samples was determined using the protocol described by Torras et al.,41 which showed an absolute error of 4% with the ICP-MS measurements. A calibration curve was produced by measuring the absorbance of Fe standards with a concentration between 2.96 and 25.80 mg L−1versus a 5 M HCl blank (see Fig. S1 of the ESI†). The absorbance at a wavelength of 349 nm, which corresponds to the maximum of each curve, was picked to build the calibration curve using a linear regression of the absorbance obtaining the following expression:
 | (11) |
The magnetic properties of samples were measured with a Physical Properties Measurement System (PPMS) 9T magnetometer. Precipitated samples were dried with an airflow to remove a major part of the solvent. The resulting powders were kept in vacuum conditions for at least 48 h at room temperature to obtain compressed pellets. The magnetic measurements were then performed by encapsulating the pellets in a gelatine capsule. Magnetic features of the samples were evaluated first by measuring hysteresis loops M(H) recorded within ±50 kOe at 5 and 300 K. Saturation magnetization (Ms) was obtained by extrapolation of the high-field region of M(H) to zero field, assuming the high-field behavior,10,42
| M(H) = Ms + χpH | (12) |
Monitoring the robustness of the IONP particle coating in a 0.1 M NaCl solution
The stability of the naked, IONPs@X4C4, IONPs@XC, and IONPs@Cit samples was investigated in the presence of 0.1 M NaCl with a [Fe] = 0.1 mg mL−1 (0.0179 M Fe). The mixture was subjected to sonication for 15 minutes to ensure complete IONP redispersion, followed by mechanical stirring. The evolution of the Dh and ζ-potential were monitored over incubation periods ranging from 1 to 24 hours depending on the stability of the sample.Monitoring the robustness of the IONP coating in basic or acidic media
To elucidate the digestion kinetics of the iron oxide core under alkaline and acidic conditions, a combined approach of UV-vis absorption spectroscopy and DLS measurements was employed. First, the pH of 5 mL aliquots of IONPs@X4C4, IONPs@XC, and IONPs@Cit solutions containing 0.1 mg mL−1 iron (0.179 M Fe) was adjusted to 12 using 0.5 M NaOH and the suspensions were kept under mechanical shaking. The absorbance of these suspensions was monitored at wavelengths ranging from 200 to 600 nm over 120 minutes. Subsequently, the absorbance at 300 nm was chosen as the representative wavelength for tracking iron digestion. The same process was repeated with the three corresponding PEI-coated IONPs solutions but adjusting the pH to 1 using 0.5 M HCl. Their absorbance was continuously monitored over 2 hours to assess their stability under acidic conditions. Dh of the IONPs at both pHs was monitored using DLS.Magnetic reutilization of IONPs
The magnetic reutilization capability of three PEI-coated IONP batches was assessed through cycles of magnetic separation and redispersion. In each cycle, 2.5 mL of an initial IONP suspension at pH 8 was placed in a 4 mL flat-bottomed glass vial, which was then subjected to collection with a neodymium magnet for 30 minutes. After magnetic precipitation, the supernatant was carefully removed using a glass Pasteur pipette, ensuring the vial remained on the magnet to prevent nanoparticle loss. The IONPs remaining in the vial were re-suspended in 2.5 mL of MQ-H2O at pH 8, followed by a 15 minute sonication to ensure full redispersion. Subsequently, the Dh and ζ-potential of the IONPs were measured. This separation and redispersion process was repeated until the IONPs could no longer be effectively redispersed, indicating a loss of stability.Magnetic collection of precious metals
The extraction capacity of IONPs was studied in triplicates in synthetic solutions of Au(III) and Pt(IV) chlorides. This study monitored the influence of temperature, pH, particle concentration, and time on the extraction yield and selectivity in comparison to other metals, with concentrations of 76 μg L−1 (0.387–0.390 μM), replicating the metal levels present in industrial wastewater intended for treatment at certain urban sewage treatment facilities in Europe.1 In brief, a certain volume of particles was added to 1 mL of a synthetic solution containing NaCl 0.1 M and the metal salts at the desired concentrations. After 1 hour of incubation, particles were collected by magnetic separation with a neodymium magnet and separated from the supernatant. The supernatant was then acidified with either chloride or nitric acid solutions to determine the concentrations of Au, Pt or other metals, respectively, using ICP-MS. The content of the precipitate could be deduced from the analysis of the supernatant. For comparison purposes, control samples were prepared without the addition of particles.| [C]total = [C]supernatant + [C]precipitate | (13) |
Results and discussion
Synthesis and surface engineering of IONPs
IONPs synthesized in this work were produced by a two-step coprecipitation method, as described in the Experimental section.34,35 First, Fe3O4 NPs were prepared at 90 °C by mixing iron(II, III) chlorides with NH3 as a mild base to obtain particles around 16 nm. The resulting particles were oxidized to form γ-Fe2O3 which enhanced the particle stability while retaining approximately 80% of their initial magnetic properties.10,42Fig. 1 summarizes the overall structural properties of the synthesized IONPs. The TEM characterization of the as-prepared sample shows faceted spheroid particles with an average particle size of 16 ± 3 nm, which falls within the typical range of polydispersity of 20% for this synthesis method (see Fig. 1a and c). Besides, the interplanar distances determined by SAED were associated with the crystallographic planes of γ-Fe2O3, specifically (111), (220), (311), (400), (511), and (440) (Fig. 1b). The crystal quality of the sample was corroborated through High-Angle Annular Dark-Field (HAADF) imaging analysis, as depicted in Fig. 1d. This analysis shows particles with a good level of crystal quality throughout the entire particle ensemble, confirming single-crystal IONPs. Besides, The XRD analysis in Fig. S2,† comparing naked IONPs with reference patterns for γ-Fe2O3 and Fe3O4, reveals that the sample is mainly formed of the γ-Fe2O3 phase. This is evidenced by specific peak positions, the absence of the (222) plane indicative of Fe3O4, and a closer match of 2θ values to γ-Fe2O3 as detailed in Table S1.† The lattice parameter (a = 8.35 Å), as shown in Table S2,† further supports the γ-Fe2O3 structure. The observed DXRD of around 12 nm is slightly smaller than DTEM, likely due to particle size defects typically found in this synthesis method.10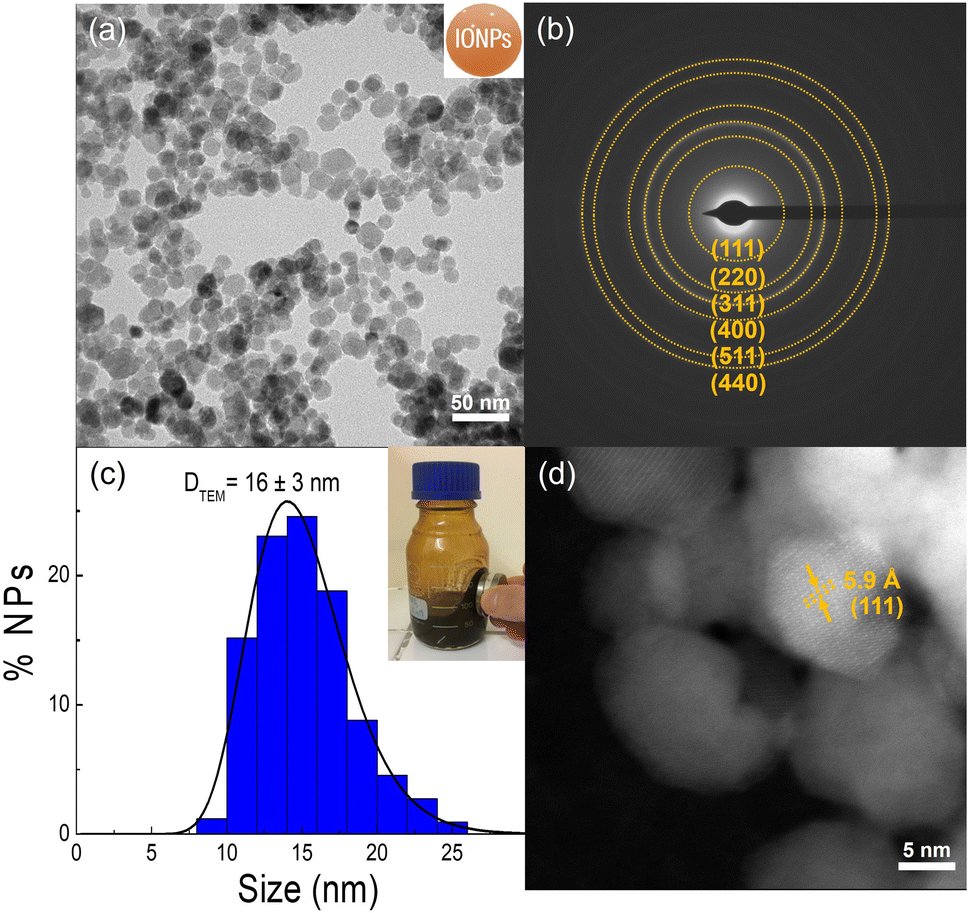 | ||
| Fig. 1 Structural TEM characterization of the synthesized IONPs. (a) Representative TEM image showing tens of IONPs. (b) SAED characterization shows the most intense pattern rings of the γ-Fe2O3 phase. (c) Particle size distribution obtained by fitting the histogram to a log-normal distribution function. The inset shows the magnetic response of the ferrofluid to an applied magnetic field from a neodymium magnet. (d) HAADF-STEM image of IONPs displaying an interplanar distance of 5.9 Å associated to the (111) plane of γ-Fe2O3 phase. | ||
The main objective of this work was to enhance the robustness of the PEI coating on IONPs beyond the current standards by generating an interface of calixarenes X4C4 (see Fig. S3†). A high robustness was anticipated as i) the reduction of the four aryldiazonium groups present on the calixarene large rim generates radicals that enable a strong binding of the macrocycles to the IONP surface,43 and ii) the large number of carboxylate groups on the calixarene small rim was expected to improve the electrostatic binding of PEI.24–27Fig. 2 summarizes the functionalization process performed for the IONPs@X4C4@PEI sample. The experimental details of this process can be found in the Experimental section. Briefly, X4C4-(N2+)4 was grafted onto the particle surface by the reduction of the diazonium group and release of nitrogen under slight basic conditions, allowing a slow grafting process onto the IONP surface. Second, the PEI functionalization was driven by electrostatic interactions at pH 8, which favors the interaction between deprotonated carboxylates and protonated amines.
 | ||
| Fig. 2 Two-step functionalization approach for the functionalization of IONPs with X4C4@PEI. The top left and right panels show the chemical structures of X4C4-(N2+)4 and PEI, respectively. | ||
The physicochemical features of the above-mentioned samples were compared to those made either with a classical aryl diazonium salt (i.e. XC-(N2+), see step 1 in the Fig. 2) or Cit, employing a similar functionalization methodology to X4C4 and a modified standard methodology, respectively (see Fig. S3†).22 Note that XC represents a subunit of X4C4 bearing only one diazonium group. Therefore, its grafting enables the study of the impact of forming multiple anchoring points on the coating's robustness. Moreover, XC is known to form multilayers at the surface through polymerization, while the methylene bridges of X4C4 prevent the polymerization of the macrocycle, resulting in ultra-thin organic layers.25 Cit is a common functionalization agent for IONPs since it provides stability and enables further modification of the particle surface.44–46 However, it shows the drawbacks of being sensitive to particle agglomeration, likely due to poor stability of the citrate layer to variations in pH, or the presence of other ions in the solution, which can disrupt the stabilizing effects of the citrate and lead to undesirable aggregation.47,48
The TEM characterization of the three series of functionalized IONPs before and after the addition of PEI are depicted in Fig. S4–S10.† They show particles with similar size (i.e. average diameter of 15–18 nm) and exhibiting only γ-Fe2O3 interplanar distances, regardless of the particle coating. This trend is also evident in the XRD spectra for the X4C4 series of samples, which consistently display the γ-Fe2O3 phase (see Fig. S2†).
These findings indicate that the functionalization processes have a low impact on both the size and the particle core composition. Therefore, these samples display analogous iron oxide cores, with only a slight increase in core size observed for the PEI-coated IONPs, probably due to the higher number of cleaning cycles achieved by precipitation. Remarkably, the TEM characterization of the IONP@X4C4@PEI sample reveals an extensive organic coating around the particles, suggesting a large presence of organic material on the particle surface (see Fig. S5†). Regarding the FTIR characterization, naked IONPs show bands typical of hydroxyl and nitrate groups adsorbed onto the particle surface, such as a broad peak centred at 3443 cm−1 attributed to O–H bond stretching, and a peak located at 1332 cm−1 corresponding to the asymmetric stretching of the N–O bond (Fig. 3a and S10, and Table S1†), due to the reagents employed in the synthesis method.48 Additionally, in the low-wavelength region, three bands characteristic of the γ-Fe2O3 phase are observed between 630 and 430 cm−1 (see Fig. S11 and Table S3†).
 | ||
| Fig. 3 Structural and magnetic characterization of naked IONPs (red line), IONPs@X4C4 (blue line), and IONPs@X4C4@PEI (orange line) samples. (a) FTIR spectra of the samples. The vibration modes of the most relevant functional groups are indicated for each spectrum. (b) ζ-Potential vs. pH for samples from pH 2 to 10 in MQ-H2O. (c) TGA thermograms of the samples from 60 to 700 °C. (d) Hysteresis loops of samples at room temperature with a H = ±50 kOe. The inset shows the low-field region of samples. | ||
In the case of the functionalized series of IONPs@X4C4, IONPs@Cit, and IONPs@XC samples, two characteristic bands appear around 1600–1400 cm−1, associated with the asymmetric and symmetric stretching vibrations of COOH groups present in X4C4, Cit, and XC molecules (Fig. 3a and S10, and Table S1†).49 Notably, the asymmetric stretching bands are shifted to higher energies suggesting the attachment of the organic molecules to the IONP surface.
The detection of PEI in the FTIR spectra was confirmed by the presence of the following prominent bands: PEI samples contain a typical doublet between 2900 cm−1 to 2800 cm−1, corresponding to the asymmetric and symmetric C–H stretching's vibrations, respectively (Fig. 3a and S12, and Table S4†). Besides, there are two peaks observed at 1600 cm−1 and 1400 cm−1 due to N–H and C–H bending vibrations, respectively; and a band at around 1000 cm−1 due to –CN– stretching of the primary amines.22 All the above-mentioned bands are shifted due to the interaction between the X4C4 and the PEI layers, as was confirmed by the presence of a band at 1300 cm−1 attributed to the symmetric COO stretching.49
Next, the particle stability of the three series of samples was monitored by DLS and ζ-potential in MQ-H2O solutions with pHs ranging from 2 to 10 (Fig. 3b, S13 and S14†). Naked IONPs show an isoelectric point (pI) of around 6.5 in good agreement with previously reported works on γ-Fe2O3.14,48,50 This means that IONPs are positively charged at pHs below 6.5 and negatively charged above this pH. IONPs@X4C4 have, nonetheless, a pI of ≈4 and more negative ζ-potential values at pH > 7. This suggests the presence of carboxylate groups exposed to the solvent, resulting from the successful attachment of X4C4. Consequently, this promotes the deagglomeration through a combination of electrostatic and steric interactions, in a similar manner to that found for AuNPs.27 This methodology was expanded for both IONPs@XC and IONPs@Cit samples (Fig. S14†). As expected, both samples show a similar trend to IONPs@X4C4 in both Dh and ζ-potential suggesting similar stabilization thanks to the carboxylates present on these organic molecules. In contrast, the PEI samples show positive ζ-potential values around 30 mV across the pH range from 2 to 9 in good alignment with previous works on PEI samples,22,23,51 indicating that the cationic charge density of PEI has shifted the highly negative surface potential to more positive ones (Fig. 3b and S14†). This fact can be attributed to the presence of the ammonium groups (–NH3+) of the polymer. It is worth mentioning that the dramatic decrease in potential at pH 10 can be explained by the partial deprotonation of the amines which have a pKa = 10–11.19,52
TGA analyses were conducted to assess the weight fraction of the organic coating on the iron oxide core. To achieve this, samples were heated first at 60 °C for 2 h and then heated to 700 °C at 10 °C min−1 under a nitrogen atmosphere. The results of the series of X4C4 samples are shown in Fig. 3c, while the corresponding XC and Cit samples are depicted in Fig. S15.† Additionally, the calculations derived from these findings are detailed in Table S5.† As a general trend, all samples show a slight diminution of mass of around 1–2% during the first stage of heating at 60 °C attributed to the removal of water present in the samples. Naked IONPs show the lowest organic fraction, with a mass loss of approximately 4% starting at around 200 °C and levelling off at around 600 °C. This mass loss can be attributed to the decomposition of some traces of reagents used for the synthesis.53,54 IONPs@X4C4 exhibit, on the other side, a progressive decrease in mass between 200 and 400 °C, which is associated with the removal of the X4C4 fraction, corresponding to 14%. This corresponds to 1900 ± 700 X4C4 per IONP (2.1 ± 0.3 X4C4 per nm2) assuming the TEM data. Note that these results resemble those obtained for spherical AuNPs of similar sizes suggesting an X4C4 monolayer at the IONP surface.26
IONPs@XC and IONPs@Cit samples show similar trends showing a single-step mass loss located between 200 and 400 °C, yielding a 6% organic loss. This organic loss corresponds to an average of 2700 ± 1300 XC per IONP, which translates to approximately 3.3 ± 0.9 XC per nm2; and 1800 ± 800 Cit per IONP, equating to roughly 2.5 ± 0.6 Cit per nm2. It is worth mentioning that the grafting density obtained for XC lies within the range of densities reported in the literature for the reductive grafting of aryldiazonium derivatives on different surfaces, which ranges from approximately 0.3 to 8 molecules per nm2.55 These compounds are known to lead to polyaryl film growth unless sterically hindered reagents are used.56 The Cit density found is also similar to that of other spherical IONPs of similar size.36
All the PEI functionalized samples show a two-step weight loss between 200 and 600 °C with different fractions of organic loss. The IONPs@X4C4@PEI sample shows the highest organic loss of approximately 23%, compared to the 11% in the other two PEI coated-IONPs in good agreement with the TEM findings described above.
This pattern suggests a denser and/or thicker coating (an increase of the organic weight fraction by 9% upon PEI addition on IONPs@X4C4, while it is only 5% for the two other sets of IONPs), which is probably due to the dense packing of carboxylates provided by the X4C4 layer that allows the interaction with a larger number of ammoniums groups of the PEI.
These results are linked to the magnetic response of the samples, which was studied by recording hysteresis loops with a H = ±50 kOe at 300 K (Fig. 3d and S16†). All samples exhibited zero Mr and Hc, consistent with a superparamagnetic behaviour and ruling out the presence of large IONPs or significant agglomerates. The measured Ms progressively decreased, in coherence with the increased amount of organic content present in samples. Besides, these values are in the same order of magnitude as the Ms of bulk γ-Fe2O3 (76 emu g−1) and consistent with those typically found in single-crystal IONPs.34–36
Table 1 recapitulates the main physicochemical features of the samples prepared for this work. As a general trend, all samples show similar sizes and crystallographic structures associated with γ-Fe2O3, regardless of the functionalization strategy, which suggests that all the samples have analogous iron oxide cores. Furthermore, the stability of the samples was evaluated by comparing the DLS and ζ-potential of aqueous solutions across a wide range of pH values. Consistent trends were observed for all samples in link to their organic coating. For example, DLS of IONPs@X4C4, IONPs@Cit, and IONPs@XC samples show a decreased Dh compared to the naked IONPs, which is due to the increased electrostatic repulsion between particles due to the presence of carboxylate groups at their surface. On the other side, PEI samples show a moderate increase of Dh due to the PEI's bulky polymer chains. The TGA results indicate a progressive increase in mass loss with increasing organic layer thickness. IONPs@X4C4@PEI have been shown to be the sample with the highest organic content. Finally, all samples show high Ms, typical of single nanocrystals, with a coherent decrease with the amount of organic fraction in the sample in good agreement with the TGA results. Stability experiments for all samples were assessed based on their organic content levels to highlight the significance of coating density in determining interaction and stability properties.
| Samples | D TEM ± σ (nm) | σ RSD | D h ± σ (nm) | PdI | ζ-Potential (mV) | M s (emu g−1) | % organic fraction |
|---|---|---|---|---|---|---|---|
| Naked IONPs | 16 ± 3 | 0.19 | 179 ± 1 | 0.2 | −28 ± 1 | 66 ± 1 | — |
| IONPs@X4C4 | 17 ± 3 | 0.18 | 116 ± 2 | 0.14 | −41 ± 1 | 60 ± 1 | 14 ± 1 |
| IONPs@XC | 16 ± 3 | 0.19 | 120 ± 2 | 0.2 | −36 ± 1 | 64 ± 1 | 6 ± 1 |
| IONPs@Cit | 15 ± 3 | 0.20 | 110 ± 4 | 0.2 | −38 ± 1 | 64 ± 1 | 6 ± 1 |
| IONPs@X4C4@PEI | 18 ± 3 | 0.17 | 188 ± 6 | 0.16 | 35 ± 1 | <56 | >23 |
| IONPs@XC@PEI | 17 ± 3 | 0.18 | 225 ± 5 | 0.18 | 35 ± 1 | 60 ± 1 | 11 ± 1 |
| IONPs@Cit@PEI | 18 ± 3 | 0.17 | 185 ± 5 | 0.19 | 32 ± 1 | 60 ± 1 | 11 ± 1 |
Monitoring the robustness of X4C4@PEI coatings of IONPs
A series of experiments were conducted to evaluate the stability of the different functionalized IONPs across various environments (Fig. 4 and S17–S19†). According to the Derjaguin–Landau–Verwey–Overbeek (DLVO) model,57 the colloidal stability of particles in suspension is the result of the balance between the attractive and repulsive forces between the charged particles, that are influenced by the ionic strength of the solution. By increasing the ionic strength, the counter ion cloud is compressed, which can induce a reduction of the repulsive forces between IONPs and lead to particle aggregation.57 Therefore, a simple manner to track the effect of the IONP coating on their colloidal stability is to monitor the Dh and ζ-potential of samples in an ionic 0.1 M NaCl solution at pH 7 as a function of time. As observed in Fig. 4a and S17,† the IONP@X4C4 sample shows the best stability behavior throughout the 24 hour incubation period, likely attributed to the protective effect of the compact and strongly grafted X4C4 layer, which effectively shields the metal oxide core from the destabilizing effects of the high ionic strength of the solution, even after 144 h of incubation (Fig. 4b). These findings indicate that the X4C4 coating remarkably enhances the colloidal stability of IONPs compared to XC and Cit. | ||
| Fig. 4 Comparative experiments of IONPs functionalized with various coatings in different aggressive media. (a) Time-resolved DLS measurements in 0.1 M NaCl of the naked IONPs, IONPs@X4C4, IONPs@XC, and IONPs@Cit samples. (b) Illustrative picture of the above-mentioned IONPs showed after 144 h of incubation. (c) Absorbance spectra in acidic conditions (pH 1) for the IONPs@X4C4@PEI particles with times ranging from 0 to 120 min. (d) Absorbance at 300 nm at pH 1 as a function of time for the samples IONPs@X4C4@PEI, IONPs@XC@PEI, and IONPs@Cit@PEI samples. | ||
Another common approach for assessing the durability of the IONP coating involves exposing the particles to either acidic or alkaline solutions. The iron oxide core can indeed react with both types of media, resulting in the formation of salts or hydroxides under acidic and basic conditions, respectively. So, it means that these reactions can be tracked using UV-vis spectroscopy, as the reactants and products exhibit distinct UV-vis signatures. Fig. S18 and S19† present the outcomes of UV-vis degradation kinetic experiments conducted on IONPs@X4C4 and IONPs@X4C4@PEI samples in basic and acidic conditions, respectively, and the comparison with the samples functionalized with XC and Cit. The UV-vis spectrum of the IONPs@X4C4 remains stable all through this experiment showing almost no modification for the duration of the experiment. This stability is further supported by DLS measurements, which indicated nearly identical hydrodynamic diameters for all samples throughout the experiment. On the contrary, the same experiments with IONP@XC and IONP@Cit samples show a clear decrease in the UV-vis signal over time, likely due to the digestion of the iron oxide core together with a broadening of the UV-vis spectra, certainly related to the particle agglomeration as confirmed by the DLS experiments (Fig. S18†).
Next, the robustness of the IONPs@X4C4@PEI coating was evaluated by tracking the UV-vis spectrum of the sample incubated at pH = 1 for times varying from 0 to 120 min (Fig. 4c and S19†). The obtained UV-vis spectra only show an insignificant decrease of the absorbance with time and no noticeable shifts in peak position or signs of broadening, indicating that the organic coating confers an efficient protective barrier, effectively safeguarding the IONP core against the aggressive acidic conditions. Additionally, the Dh remains stable and centred at 200 nm. In contrast, the IONPs@XC@PEI and IONPs@Cit@PEI samples demonstrated a faster digestion rate in the presence of HCl, showing a decrease of the absorbance ranging from 250 to 400 nm, likely due to the IONP digestion and/or agglomeration as was depicted by DLS. To recapitulate, X4C4-coated IONPs clearly exhibit the most robust coating in both ionic media and acidic/basic environments.
Magnetic reutilization of the IONPs
As described above, the IONPs prepared and functionalized in this work show a superparamagnetic behavior. Evaluating the limits of reusability of these systems could help to boost their applicability from an industrial point of view. An interesting manner to test the maximum magnetic stability of IONPs is by repeatedly precipitating them under a magnetic field and redispersing them by sonication. After several cycles, this can lead to the degradation of the IONP coating, which favours their irreversible aggregation.15Fig. 5 summarizes the experimental details carried out to determine the threshold in the reusability of the different functionalized PEI samples. The different batches of IONPs showed distinctive behaviours depending on the functionalization agent employed. IONPs@XC@PEI displayed early signs of irreversible aggregation after 7 magnetic cycles, accompanied by a dramatic 3200% increase in Dh. The IONPs@Cit@PEI sample exhibited a better stability, enduring 10 magnetic cycles before demonstrating a 200% Dh enhancement. Finally, the most resistant sample appeared to be the IONPs@X4C4@PEI batch, which supported 13 magnetic cycles before showing signs of aggregation with a 135% increase in its Dh. Notably, the ζ-potential of all samples transitioned from positive to negative values, showing the release of PEI from the particle surface. The stability of IONPs@X4C4@PEI is due to the dense packing of negatively charged X4C4 on the IONP surface, creating a strong electrostatic interaction with the dense PEI coating. This forms a protective barrier around the IONPs. This dense packing, confirmed by TGA results, favours a protective barrier around the IONPs, shielding them from the aggregation caused by magnetic forces. Additionally, the higher organic content in the IONPs@X4C4@PEI sample contributes to its stability, acting as a steric barrier, preventing aggregation, and providing extra protection against the deteriorating effects of the magnetic precipitation cycles.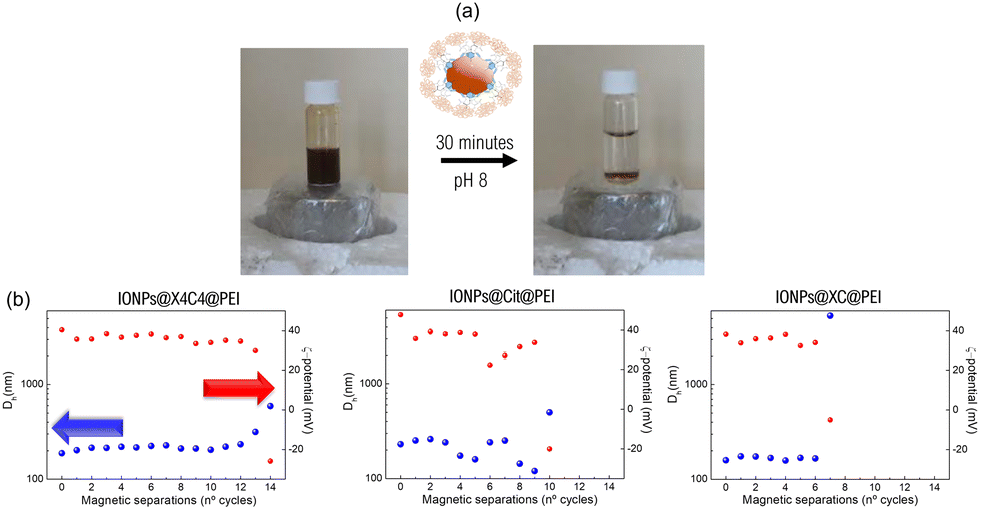 | ||
| Fig. 5 Monitoring of the particle reusability at pH 8 under magnetic separation of the PEI coated-IONPs. (a) Top side: Graphical view of the experimental procedure. (b) Dh and ζ-potential values along the different magnetic separations for the IONPs@X4C4@PEI, IONPs@XC@PEI, and IONPs@Cit@PEI samples. | ||
Magnetic extraction of Au and Pt chlorides
We investigated the efficiency of the most robust PEI coated-IONPs, i.e. IONPs@X4C4@PEI, in the collection efficiency of Au(III) and Pt(IV) solutions by monitoring the impact on the extraction yield of several variables including pH, particle concentration, extraction time, and the presence of other metals. These experiments were conducted with the same concentration of Au(III) and Pt(IV), i.e. 76 μg L−1, mimicking the metal concentrations found in industrial waters intended for treatment in some urban wastewater plants.1,40Fig. 6 shows the experimental design for the magnetic collection experiments. In brief, a certain number of particles was added to the Au(III) and Pt(IV) solutions in a NaCl 0.1 M buffer to prevent the precipitation of the metals.58,59 After 1 h of incubation, particles were precipitated by magnetic separation. Subsequently, the supernatants were treated with HCl or HNO3 and then subjected to ICP-MS for metal quantification. It is worth noting that this analysis indirectly revealed the composition of the precipitate, based on the principle that the total metal concentration equals the sum of the amounts in the supernatant and the precipitate. This method was validated by comparing the calculated and directly measured quantities of metals in the precipitate after dissolution across several samples, which showed consistent results. | ||
| Fig. 6 Experimental procedure to determine the extraction capacity of the IONPs@X4C4@PEI sample in synthetic Au or Pt solutions employing ICP-MS experiments. The concentration of [Au] and [Pt] selected for these experiments was the same for both metals and equal to 76 μg L−1. | ||
Fig. 7 shows the impact of pH, concentration of particles, and temperature on the collection of Au(III) and Pt(IV). As observed, pH is influencing differently the collection of these two precious metals, probably due to the tuning of the electrostatic interactions between the cationic PEI shell and the different metal complexes present in the solutions or favoring the diffusion of the different species present in low concentration. Particles display only a limited collection capacity for Au(III) at pH 2 and only at high particle concentrations, while the collection at pH 8 appears very efficient even at a low [γ-Fe2O3] of 5 mg L−1. This is because at pH 2, AuCl4− tends to hydrolyse to the neutral complex AuCl2(H2O)(OH) in the presence of NaCl, while the most abundant Au complex at pH 8 is the negatively charged Au(OH)4−.58,59 On the contrary, the collection of Pt follows a very similar trend at both pH values, showing the maximum collection of 80% at the concentration of 75 mg γ-Fe2O3 L−1 at 25 °C, probably because of the greater stability of the [PtCl6]2− complex, regardless of the pH. At the lowest IONP concentration, the collection capacity at 25 °C for Au and Pt is 12.6 mg Au g−1 γ-Fe2O3 and 12 mg Pt g−1 γ-Fe2O3, respectively. Besides, both solutions show a slight increase in metal collection at the highest temperature. It is worth mentioning that these values are one-fourth those published for PEI functionalized IONPs for Au(III) thiosulfate, most than probably because of the much higher metal concentration employed in these experiments (see Table S6†).22 Additionally, while our IONPs@X4C4@PEI show lower gold adsorption capacities compared to other systems, such as chitosan resin embedding IONPs (709.2 mg Au(III) g−1 IONP) and IONPs-thiourea (118.5 mg Au(III) g−1 IONP),18,60 they still achieve nearly 100% Au release under mild conditions.
 | ||
| Fig. 7 Percentage (%) of the initial Au and Pt content remaining in solution after extraction with IONPs@X4C4@PEI as a function of the [γ-Fe2O3] at 25 and 50 °C. Results for collection experiments at pH 2 and pH 8 at 25 and 50 °C are shown for Au(III) in (a) and (b) respectively, while those for Pt(IV) are shown in (c) and (d). | ||
To further explore the influence of the PEI layer on Au collection, naked IONPs, IONPs@X4C4, and IONPs@Cit@PEI were subjected to the same experimental conditions at pH 8. Interestingly, while the first two systems failed to demonstrate Au uptake (Fig. S20†), IONPs@Cit@PEI collected a comparable amount of Au(III) than IONPs@X4C4@PEI (Fig. S21†), suggesting a good efficiency of the PEI layer deposited on the Cit to collect Au(III) complexes in solution.
The reusability of a same batch of particles was tested for 5 successive cycles of immersion/magnetic separations in fresh Au(III) and Pt(IV) chloride solutions with metal concentrations equal to 76 μg L−1 at pH 8 (Fig. 8). For both precious metals, the particles seem capable of collecting large amounts of Au (95%) and Pt (from 90 to 70%) at each cycle even after 5 magnetic separations, which indicates that they were not saturated even after several magnetic collection cycles. Besides, the PEI shell likely maintains its integrity, as observed in prior analyses. Elucidating the minimum time required to collect most of the precious metals contained in the sample can save time and consequently increase the cost-effectiveness of the future industrial process. Additionally knowing the specific collection time for each metal could be an efficient manner to collect selectively the different metals, by playing on this parameter. For these reasons, the effect of the incubation times on Au(III) and Pt(IV) trapping in synthetic metal solutions was investigated at pH 8. Fig. 8c and d show the influence of the incubation times from 0 to 60 min at 25 °C. As a general trend, both metals show a large collection in the first incubation instants, which is more prominent in the case of Au, while the collection of Pt requires a longer incubation time of 60 minutes. The faster collection rate of Au by PEI compared to Pt might be attributed to the synergistic effects of electrostatic and steric interactions between PEI and the Au(OH)4− complex. This stabilization and strong electrostatic attraction might lead to efficient encapsulation of Au(OH)4− within the PEI matrix, contributing to the faster collection rate.
 | ||
| Fig. 8 The top panel shows the % of Au (a) and % of Pt (b) remaining in 5 magnetic separations employing a concentration of IONPs@X4C4@PEI of 25 mg mL−1 and 75 mg mL−1 in fresh synthetic Au(III) and Pt(IV) chloride solutions with equal Au and Pt concentrations of 76 μg L−1 at pH 8. The lower panel shows the effect of the incubation time on the collection of Au (c) and Pt (d), with times ranging from 0 to 60 minutes in synthetic Au and Pt chloride solutions at 25 °C. % Au and % Pt indicate the percentage of metal remaining in solution. | ||
To gain insights into the characteristics of the precious metal coating on the IONPs@X4C4@PEI surface after collecting Au or Pt, a comprehensive microscopic compositional analysis was performed. This analysis was conducted with the samples involving collection at pH = 8, 25 °C, and a γ-Fe2O3 concentration of 75 mg L−1. Fig. 9 displays the TEM study of the particles after the collection of Au(III), while Fig. S22† shows the particles after collection of Pt. For the Au case, the bright field TEM images reveal new dark features, indicating the formation of Au nanostructures at the IONP surface without the addition of any reducing agent. EDS analysis as shown in the HDDAF-STEM image corroborates this finding. This image exhibits bright spots, corresponding to a heavier element than the iron oxide cores. Moreover, EDX analysis in two distinct regions, labelled zone1 and zone 2 shown in Fig. 9d and e respectively, verifies the presence of Au. Fig. 9d shows a prominent Au peak, with a weight percent of 92.2%, along with O and Fe, confirming the growth of Au nanostructures at the surface of the IONPs. In contrast, Fig. 9 shows no Au signal, implying that large areas of the sample do not contain a measurable concentration of Au. The low amount of nanoscale Au is due to the low concentration used in these experiments. These results are likely caused by the amine groups in PEI acting as electron donors in good alignment with previous studies.61 In contrast, the experiments performed with Pt do not show the appearance of metallic nanostructures, suggesting that PEI does not reduce Pt(IV), which probably remains adsorbed on the IONP surface under the form of a complex (Fig. S22†). This finding aligns well with the typical larger redox potential of Pt complexes.
 | ||
| Fig. 9 Gallery of TEM images showing IONPs@X4C4@PEI particles after one collection process in synthetic Au(III) chloride solutions. (a) and (b) Show bright field TEM images at several magnifications of IONPs@X4C4@PEI containing large Au particles indicated with red arrows. (c) HAADF-STEM image highlighting bright spots indicative of heavy elements within a nanoparticle matrix, correlated with EDS spectra analysis for elemental composition determination. (d) Shows the spectrum 1 in zone 1 containing a high percentage of Au. (e) Depicts the spectrum 2 from zone 2 containing other elements. | ||
Next, the release of the two precious metals was studied under basic and acidic conditions, or with the addition of thiourea (SC(NH2)2) (Fig. S23†). Note that the latter stands out as an eco-conscious alternative to cyanide, enabling Au recovery and refinement from diverse sources, including electronic waste.2,62 The addition of SC(NH2)2 leads to the almost complete release of Au and Pt metals from the particles. As reported in previous works,2,62 the dissolution of Au in the presence of SC(NH2)2 is based on a redox reaction, in which Au is oxidized from Au(0) to Au(III) forming a stable gold thiocyanate complex, as shown in eqn (14).
| Au(s) + 2SC(NH2)2(aq) → Au[SC(NH2)2]+(aq) + e− | (14) |
| [PtCl6]2−(aq) + 2SC(NH2)2(aq) → Pt[SC(NH2)2]4+(aq) + 6Cl−(aq) | (15) |
In the final step of the study, we investigated the effectiveness of IONPs@X4C4@PEI in real processing solutions with water derived from the wet oxidation process of domestic sludge, referred to as WetOx solutions.40 Our experiments encompassed fresh WetOx solutions and thiosulfate enriched WetOx solutions, as depicted in Fig. S25.† Under these specific conditions, we observed that the IONPs@X4C4@PEI particles exhibited a lack of Au and Pt extraction ability, probably due to the saturation of the cationic layer by either negative organic substances present in the WetOx solutions or an excess of thiosulfate. Despite these challenges, IONPs@X4C4@PEI particles show promise as a viable material for Au recovery from inorganic matrices, such as those encountered in mine and electronic scrap leachates. These particles could also be envisaged for biotechnological applications such as nucleic acid purification.
Conclusions
In this work, we have developed a novel method for the synthesis of IONPs with a robust PEI coating mediated by the grafting of a X4C4 onto the IONP surface. This innovative approach results in magnetic particles that exhibit remarkable stability across a wide range of pH conditions, exceptional magnetic reusability, and outstanding selectivity in the recovery of Au and Pt from solutions containing competing metals.The developed methodology stands out from conventional PEI-coated IONPs due to its ability to effectively anchor the PEI layer onto the IONP surface, preventing its detachment and maintaining the particles in a dispersed state. This resilience may stem from the potent electrostatic attraction between the densely packed, negatively charged carboxylate groups of the X4C4 on the particles, and the positively charged ammonium groups of the PEI. Additionally, the enduring stability of the X4C4 coating is attributed to the formation of numerous covalent bonds with the IONP surface, which anchors the coating firmly in place and enhances the overall durability of the nanoparticle structure.
The exceptional stability of IONPs@X4C4@PEI particles is evident in their ability to withstand both acidic and alkaline conditions without significant particle aggregation or loss of magnetic properties. Moreover, these particles demonstrate exceptional magnetic reusability, maintaining their selectivity and recovery efficiency over multiple cycles of separation. The selectivity of IONPs@X4C4@PEI particles lies in their ability to preferentially bind anionic Au and Pt complexes over other neutral metal complexes or cations.
This work paves the way for the sustainable utilization of these particles across a diverse range of potential applications. In environmental remediation, these particles could selectively recover precious metals from wastewater, thereby reducing environmental pollution and fostering resource conservation. In addition to the promising results obtained for the recovery of Au and Pt, future work may explore the applicability of this chemical functionalization strategy to other hazardous metallic ions such as chromium, cobalt, and cadmium, thereby broadening the environmental relevance and impact of our approach. Additionally, these particles may find applications in the recovery of precious metals from leachates in mining or electronic scrap processes.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge INNOVIRIS (Brussels region) for its financial support (SUBLIMUS: becoming modern green urban alchemists: gold and silver mining from Brussels urban sewage, ref. 2019-RPF-2). CM acknowledges funding support from the University of Barcelona and the Spanish Ministry of Universities under the Maria Zambrano Program, funded by the European Union Next Generation-EU/PRTR, as well as the Beatriu de Pinós fellowship program (ref. 2022 BP 00243). L. T.-G. is a Chercheur Qualifié of the Fonds de la Recherche Scientifique – FNRS. The authors also acknowledge the use of the Servicio General de Apoyo a la Investigación-SAI at the University of Zaragoza, and the Scientific and Technical Services at the University of Barcelona for the TEM, TGA, and XRD characterization measurements, respectively.Notes and references
- N. A. A. Qasem, R. H. Mohammed and D. U. Lawal, npj Clean Water, 2021, 4, 1–14 CrossRef.
- Z. Wei Liu, X. Yi Guo, Q. Hua Tian and L. Zhang, J. Hazard. Mater., 2022, 440, 129778 CrossRef PubMed.
- A. O. Adeeyo, O. S. Bello, O. S. Agboola, R. O. Adeeyo, J. A. Oyetade, M. A. Alabi, J. N. Edokpayi and R. Makungo, Water Reuse, 2023, 13(2), 134–161 CrossRef CAS.
- U. Upadhyay, I. Sreedhar, S. A. Singh, C. M. Patel and K. L. Anitha, Carbohydr. Polym., 2021, 251, 117000 CrossRef CAS.
- P. Makvandi, S. Iftekhar, F. Pizzetti, A. Zarepour, E. N. Zare, M. Ashrafizadeh, T. Agarwal, V. V. T. Padil, R. Mohammadinejad, M. Sillanpaa, T. K. Maiti, G. Perale, A. Zarrabi and F. Rossi, Environ. Chem. Lett., 2021, 19(1), 583–611 CrossRef CAS.
- J. Dobrzynska, M. Dabrowska, R. Olchowski, E. Zieba and R. Dobrowolski, J. Environ. Chem. Eng., 2021, 9(4), 105302 CrossRef CAS.
- Z. Hubicki, M. Wawrzkiewicz, G. Wójcik, D. Kolodynska and A. Wolowicz, Ion Exch.: Stud. Appl., 2015, 3–36 CAS.
- S. Lei and Y. Song, Front. Chem., 2021, 9, 1–11 Search PubMed.
- M. I. A. Abdel Maksoud, A. M. Elgarahy, C. Farrell, A. H. Al-Muhtaseb, D. W. Rooney and A. I. Osman, Coord. Chem. Rev., 2020, 403, 213096 CrossRef.
- X. Batlle, C. Moya, M. Escoda-Torroella, Ò. Iglesias, A. Fraile Rodríguez and A. Labarta, J. Magn. Magn. Mater., 2022, 543, 168594 CrossRef CAS.
- N. Ajinkya, X. Yu, P. Kaithal, H. Luo, P. Somani and S. Ramakrishna, Materials, 2020, 13(20), 1–35 CrossRef PubMed.
- P. Saharan, G. R. Chaudhary, S. K. Mehta and A. Umar, J. Nanosci. Nanotechnol., 2014, 14(1), 627–643 CrossRef CAS PubMed.
- R. D. Alorro, N. Hiroyoshi, H. Kijitani, M. Ito and M. Tsunekawa, Miner. Process. Extr. Metall. Rev., 2010, 31(4), 201–213 CrossRef CAS.
- N. Zhu, H. Ji, P. Yu, J. Niu, M. U. Farooq, M. W. Akram, I. O. Udego, H. Li and X. Niu, Nanomaterials, 2018, 8(10), 1–27 CrossRef.
- V. C. Tai, H. X. Che, X. Y. Kong, K. C. Ho and W. M. Ng, J. Ind. Eng. Chem., 2023, 127(25), 82–100 CrossRef CAS.
- S. Zhang, Y. Zhang, J. Liu, Q. Xu, H. Xiao, X. Wang, H. Xu and J. Zhou, Chem. Eng. J., 2013, 226, 30–38 CrossRef CAS.
- P. Duel, M. S. Gutiérrez, P. Rodríguez, A. León, K. A. López, J. Morey and M. N. Piña, RSC Adv., 2018, 8(63), 36123–36132 RSC.
- A. M. Donia, A. A. Atia and K. Z. Elwakeel, Hydrometallurgy, 2007, 87(3–4), 197–206 CrossRef CAS.
- Z. Chen, Z. Lv, Y. Sun, Z. Chi and G. Qing, J. Mater. Chem. B, 2020, 8(15), 2951–2973 RSC.
- A. N. Danthanarayana, D. C. Manatunga, R. M. De Silva, N. V. Chandrasekharan and K. M. N. De Silva, R. Soc. Open Sci., 2018, 5(12), 181369 CrossRef CAS.
- D. D. Al-Araji, F. H. Al-Ani and Q. F. Alsalhy, Int. J. Environ. Anal. Chem., 2023, 103(16), 4752–4776 CrossRef CAS.
- N. D. Ilankoon, C. Aldrich, E. A. Oraby and J. J. Eksteen, Hydrometallurgy, 2020, 195, 105375 CrossRef CAS.
- J. C. Betancur, P. M. Montoya and J. A. Calderón, Hydrometallurgy, 2019, 189, 105128 CrossRef CAS.
- L. Troian-Gautier, A. Mattiuzzi, O. Reinaud, C. Lagrost and I. Jabin, Org. Biomol. Chem., 2020, 18(19), 3624–3637 RSC.
- A. Mattiuzzi, I. Jabin, C. Mangeney, C. Roux, O. Reinaud, L. Santos, J. F. Bergamini, P. Hapiot and C. Lagrost, Nat. Commun., 2012, 3(1), 1–8 Search PubMed.
- L. Troian-Gautier, H. Valkenier, A. Mattiuzzi, I. Jabin, N. Van Den Brande, B. Van Mele, J. Hubert, F. Reniers, G. Bruylants, C. Lagrost and Y. Leroux, Chem. Commun., 2016, 52(69), 10493–10496 RSC.
- L. Troian-Gautier, A. Mattiuzzi, P. Blond, M. Retout, G. Bruylants, O. Reinaud, C. Lagrost and I. Jabin, in Aryl Diazonium Salts and Related Compounds, Springer, Cham, 2022, pp. 247–262 Search PubMed.
- M. Retout, P. Blond, I. Jabin and G. Bruylants, Bioconjugate Chem., 2021, 32(2), 290–300 CrossRef CAS PubMed.
- B. Gosselin, M. Retout, R. Dutour, L. Troian-Gautier, R. Bevernaegie, S. Herens, P. Lefèvre, O. Denis, G. Bruylants and I. Jabin, Anal. Chem., 2022, 94(20), 7383–7390 CrossRef CAS PubMed.
- M. Retout, B. Gosselin, A. Adrović, P. Blond, I. Jabin and G. Bruylants, Nanoscale, 2023, 15, 11981–11989 RSC.
- B. Gosselin, M. Retout, I. Jabin and G. Bruylants, Sens. Diagn., 2024, 2, 248–255 RSC.
- D. Hetemi, V. Noël and J. Pinson, Biosensors, 2020, 10(1), 4 CrossRef CAS PubMed.
- https://X4c.Eu/ .
- M. P. Morales, S. Veintemillas-Verdaguer, M. I. Montero, C. J. Serna, A. Roig, L. I. Casas, B. Martínez and F. Sandiumenge, Chem. Mater., 1999, 11(11), 3058–3064 CrossRef CAS.
- P. De La Presa, Y. Luengo, V. Velasco, M. P. Morales, M. Iglesias, S. Veintemillas-Verdaguer, P. Crespo and A. Hernando, J. Phys. Chem. C, 2015, 119(20), 11022–11030 CrossRef CAS.
- M. Escoda-Torroella, C. Moya, A. F. Rodríguez, X. Batlle and A. Labarta, Langmuir, 2021, 37(1), 35–45 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9(7), 671–675 CrossRef CAS.
- X'Pert High Score Plus program, PANalytical B.V., Almelo, The Netherlands, 2006 Search PubMed.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS.
- M. Elskens and P. Claeys, BRIDGE 2019: SUBLIMUS – Becoming Modern Green Urban Alchemists: Gold and Silver Mining from Brussels Urban Sewage.
- M. Torras, C. Moya, G. A. Pasquevich and A. Roig, Microchim. Acta, 2020, 187(9), 488 CrossRef CAS.
- B. D. Cullity and C. D. Graham, Introduction to Magnetic Materials, 2011 Search PubMed.
- K. Brymora, J. Fouineau, A. Eddarir, F. Chau, N. Yaacoub, J. M. Grenèche, J. Pinson, S. Ammar and F. Calvayrac, J. Nanopart. Res., 2015, 17(11), 1–9 CrossRef CAS.
- C. Moya, R. Escudero, D. C. Malaspina, M. De La Mata, J. Hernández-Saz, J. Faraudo and A. Roig, ACS Appl. Bio Mater., 2019, 2(7), 3084–3094 CrossRef CAS PubMed.
- M. Vassallo, D. Martella, G. Barrera, F. Celegato, M. Coïsson, R. Ferrero, E. S. Olivetti, A. Troia, H. Sözeri, C. Parmeggiani, D. S. Wiersma, P. Tiberto and A. Manzin, ACS Omega, 2023, 8(2), 2143–2154 CrossRef CAS PubMed.
- T. Granath, K. Mandel and P. Löbmann, Part. Part. Syst. Charact., 2022, 39, 2100279 CrossRef CAS.
- A. Atrei, F. F. Mahdizadeh, M. C. Baratto and A. Scala, Appl. Sci., 2021, 11(15), 6974 CrossRef CAS.
- A. Walter, A. Garofalo, A. Parat, H. Martinez, D. Felder-Flesch and S. Begin-Colin, Nanotechnol. Rev., 2015, 4(6), 581–593 CAS.
- S. Yu and C. M. Chow, J. Mater. Chem., 2004, 14(18), 2781–2786 RSC.
- I. T. Lucas, S. Durand-Vidal, E. Dubois, J. Chevalet and P. Turq, J. Phys. Chem. C, 2007, 111(50), 18568–18576 CrossRef CAS.
- N. D. Ilankoon, C. Aldrich, E. A. Oraby and J. J. Eksteen, Miner. Process. Extr. Metall. Rev., 2020, 41(5), 311–322 CrossRef CAS.
- A. N. Solodov, J. R. Shayimova, E. A. Burilova and R. R. Amirov, Colloid Polym. Sci., 2018, 296(12), 1983–1993 CrossRef CAS.
- P. Liu, C. H. He, F. Liu, L. Xu, Y. Wan and J. H. He, Therm. Sci., 2016, 20(3), 967–972 CrossRef.
- P. Melnikov, V. A. Nascimento, I. V. Arkhangelsky and L. Z. Zanoni Consolo, J. Therm. Anal. Calorim., 2014, 115(1), 145–151 CrossRef CAS.
- N. Griffete, F. Herbst, J. Pinson, S. Ammar and C. Mangeney, J. Am. Chem. Soc., 2011, 133(6), 1646–1649 CrossRef CAS PubMed.
- J. Greenwood, T. H. Phan, Y. Fujita, Z. Li, O. Ivasenko, W. Vanderlinden, H. Van Gorp, W. Frederickx, G. Lu, K. Tahara, Y. Tobe, H. Uji-I, S. F. L. Mertens and S. De Feyter, ACS Nano, 2015, 9(5), 5520–5535 CrossRef CAS PubMed.
- L. Saunders, J. Pharm. Pharmacol., 1951, 3(1), 865–882 CrossRef CAS.
- I. Mironov, Russ. J. Inorg. Chem., 2000, 45(4), 633–637 Search PubMed.
- M. D. Urović, R. Puchta, Ž. D. Bugarčić and R. Van Eldik, Dalton Trans., 2014, 43(23), 8620–8632 RSC.
- T.-L. Lin and H.-L. Lien, Int. J. Mol. Sci., 2013, 14, 9834–9847 CrossRef PubMed.
- K. Bali, M. Bak, K. Szarka, G. Juhász, G. Sáfrán, B. Pécz, J. Mihály and R. Mészáros, J. Mol. Liq., 2021, 322, 114559 CrossRef CAS.
- D. A. Ray, M. Baniasadi, J. E. Graves, A. Greenwood and S. Farnaud, J. Sustain. Metall., 2022, 8(2), 597–612 CrossRef.
- M. H. Morcali, B. Zeytuncu, S. Akman and O. Yucel, Desalin. Water Treat., 2016, 57(14), 6582–6593 CrossRef CAS.
- J. D. Cuppett, S. E. Duncan and A. M. Dietrich, Chem. Senses, 2006, 31(7), 689–697 CrossRef CAS.
- K. J. Powell, P. L. Brown, R. H. Byrne, T. Gajda, G. Hefter, A.-K. Leuz, S. Sjöberg and H. Wanner, Chem. Int., 2015, 37(1), 15–19 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4en00408f |
| This journal is © The Royal Society of Chemistry 2025 |