
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of organic ligands, phosphate and Ca on the structure and composition of Fe(III)-precipitates formed by Fe(II) oxidation at near-neutral pH†
Ville V.
Nenonen
 ab,
Ralf
Kaegi
a,
Stephan J.
Hug
a,
Jörg
Luster
c,
Jörg
Göttlicher
d,
Stefan
Mangold
d,
Lenny H. E.
Winkel
ab and
Andreas
Voegelin
*a
ab,
Ralf
Kaegi
a,
Stephan J.
Hug
a,
Jörg
Luster
c,
Jörg
Göttlicher
d,
Stefan
Mangold
d,
Lenny H. E.
Winkel
ab and
Andreas
Voegelin
*a
aEawag, Swiss Federal Institute of Aquatic Science and Technology, Überlandstrasse 133, CH-8600 Dübendorf, Switzerland. E-mail: andreas.voegelin@eawag.ch
bETH Zurich, Swiss Federal Institute of Technology, Institute of Biogeochemistry and Pollutant Dynamics, Universitätstrasse 16, CH-8092 Zürich, Switzerland
cWSL, Swiss Federal Institute for Forest, Snow and Landscape Research, Zürcherstrasse 111, CH-8903 Birmensdorf, Switzerland
dKarlsruhe Institute of Technology, Institute for Photon Science and Synchrotron Radiation, Hermann-von-Helmholtz Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
First published on 7th July 2025
Abstract
The oxidation of dissolved Fe(II) in near-neutral natural waters leads to the formation of Fe(III)-precipitates. Organic ligands, PO4, calcium (Ca) and other solutes affect the composition, bulk and nanoscale structure and colloidal properties of Fe(III)-precipitates and their impacts on co-precipitated compounds in interdependent ways. In this study, we quantified the effects of four low molecular weight organic acids (LMWOAs) with different Fe(III) complexation strengths (2,4-dihydroxybenzoic acid (2,4-DHB) ∼ galacturonic acid (Galact) ≪ 3,4-dihydroxybenzoic acid (3,4-DHB) < citric acid (Citr)) and of leonardite humic acid (LH) in combination with PO4 and Ca on Fe(III)-precipitate structure and composition in a multifactorial experiment. Precipitates were synthesized by oxidation of 0.5 mM Fe(II) in bicarbonate-buffered solutions at pH ∼ 7, at molar (OC/Fe) ratios of 0.1 to 9.6, at molar (PO4/Fe) ratios of 0.05 and 0.25, and without/with 4 mM Ca. OC-free controls consisted of amorphous (Ca)Fe(III)–phosphate and ferrihydrite (Fh) aggregated in core–shell nanoparticles with lepidocrocite (Lp) crystallites attached to their surface, reflecting their formation sequence. The LMWOAs promoted the formation of OC-loaded Fh at the expense of Lp and thereby led to more effective PO4 retention and lower residual dissolved PO4. This effect increased with ligand concentration and Fe(III) complexation strength. At a higher PO4 level, the co-precipitation of all LMWOAs was reduced, reflecting the stronger affinity of PO4 for Fe(III). At higher (OC/Fe), the strong ligands 3,4-DHB and Citr increasingly co-precipitated with PO4 into (Ca)–Fe(III)–PO4–OC nanoparticles and led to the formation of Fe(III)-colloids and Fe(III)–organic complexes that passed 0.2 μm filter membranes at the highest tested (OC/Fe) levels. Macromolecular LH (humic acid) had similar effects on Fe coordination and PO4 uptake to 2,4-DHB and Galact, but distinct effects on the nanoscale precipitate structure and colloidal properties. Ca enhanced the co-precipitation of PO4 and OC with Fe(III), the aggregation of Fe(III)-precipitate particles, and LH coagulation. Collectively, the insights presented in this study highlight the need to take the type of OC, its concentration relative to Fe, and interdependent effects of OC, PO4, SiO4, and Ca into account when assessing the formation of Fe(III)-precipitates by Fe(II) oxidation in near-neutral natural waters and their impacts on the cycling of PO4, OC and other co-precipitating compounds.
Environmental significanceIron (Fe) precipitates formed by Fe oxidation at anoxic/oxic interfaces in environmental systems critically affect the cycling of phosphorus (P), organic carbon (OC), and other compounds. Based on laboratory experiments in near-neutral bicarbonate-buffered solutions, this study shows how organic ligands with different Fe(III) complexation strengths, combined with phosphate (PO4) and calcium (Ca), determine the structure of freshly formed Fe(III)-precipitates, their ability to retain PO4 and OC, and their colloidal properties. The observed interdependent effects of different solutes must be considered when assessing the role of Fe oxidation products in environmental P, OC, and trace element cycling. |
1. Introduction
The oxidation of dissolved Fe(II) at natural redox boundaries leads to the formation of amorphous to poorly crystalline Fe(III)-precipitates. These nanoscale precipitates can act as immobilizing sorbents or colloidal carriers for a wide range of compounds and can thereby critically influence their biogeochemical cycles and environmental impacts as nutrients or contaminants.1 Phosphorus (P) is an essential nutrient, but excessive P inputs into surface waters lead to eutrophication and aquatic system degradation.2 Due to the high affinity of PO4 to Fe(III), its environmental cycling is closely linked to Fe, including its co-precipitation with Fe(III) at redox transitions. As “rusty carbon sinks”, Fe(III)-precipitates also play a crucial role in the stabilization of organic carbon (OC) in soils and sediments, in its transfer across redox interfaces, and thereby in the global C cycle.3–5In natural waters, the oxyanions phosphate (PO4), silicate (SiO4) and bicarbonate and the major cations calcium (Ca) and magnesium (Mg)6–9 interdependently affect the formation, structure and reactivity of Fe(III)-precipitates. The co-precipitation of PO4 with Fe(III) upon Fe(II) oxidation in near-neutral natural waters and its impact on Fe(III)-precipitate structure have been extensively studied. Phosphate limits Fe(III) polymerization and co-precipitates with Fe(III) as amorphous Fe(III)–(hydroxy)phosphate (FeP; basic Fe(III)–phosphate)7,8,10–12 or, in the presence of Ca, Ca–Fe(III)–(hydroxy)phosphate (CaFeP) with a higher degree of Fe(III) polymerization and PO4 uptake.8–10 At initial dissolved molar P/Fe ratios ((P/Fe)init) below the ratio of (Ca)FeP, initial (Ca)FeP formation leads to the depletion of dissolved PO4 and subsequent formation of different Fe(III)-precipitates.7,8 In PO4-depleted solutions without SiO4, the formation of (Ca)FeP is followed by the precipitation of lepidocrocite (Lp) and concomitant transformation of (Ca)FeP into PO4-loaded ferrihydrite (Fh).7,8 In PO4-free solutions containing SiO4, initially formed (Ca)FeP is preserved and continuing Fe(II) oxidation leads to the precipitation of SiO4-containing Fh.7,8,12 These sequential processes are reflected in the nanoscale structure and heterogeneity of fresh Fe(III)-precipitates.7,8,13
Dissolved organic matter (DOM) also affects the Fe(III)-precipitate structure and composition by inducing the formation of Fe(III)–OM-co-precipitates or mono- and oligomeric Fe(III)–OM complexes.14–21 Typically, the presence of DOM reduces the long- and short-range structural order in Fe(III)-precipitates.14,20,22 Previous studies on the effects of natural OM (NOM) on Fe(III)-precipitate formation and structure conducted with root exudates such as citrate,23,24 sugars,25 amino acids,26 mono- and polysaccharides,27–30 lignin and hydroxybenzoic acids,22,31 or humic and fulvic compounds17,20 indicated that effects of OM on Fe(III)-precipitate formation depend especially on the OC/Fe ratio, OM functional groups and their Fe(III) binding strength, and solution pH. Organic carbon can compete with PO4 for co-precipitation with Fe(III),32–34 but can also alter the structure of Fe(III)-precipitates and potentially cause an increase in PO4 sorption capacity.31,35,36 Positive correlations between OC content and PO4 sorption in soils have been attributed to an increase in the reactive surface area of Fe(III)-precipitates.31,35,37,38 In addition, Ca co-precipitated with Fe(III) can affect the reactivity of Fe(III)–OM co-precipitates,21,39,40 and enhance OC co-precipitation with Fe.41
In previous work, we studied the interdependent effects of PO4, SiO4, Mg and Ca on the structure and composition of fresh and aged Fe(II) oxidation products in laboratory experiments closely mimicking conditions in bicarbonate-buffered near-neutral natural waters.8,13,42 To date, analogous multifactorial laboratory studies on the interdependent effects of organic ligands are still scarce. In this study, we therefore examined how organic ligands with different structures and Fe(III) binding strengths in combination with PO4 and Ca influence the composition and structure of Fe(III)-precipitates formed by oxidation of 0.5 mM dissolved Fe(II) in bicarbonate-buffered aqueous solutions at near-neutral pH. In a multifactorial approach, we studied the effects of four low molecular weight organic acids (LMWOAs) and one macromolecular humic acid at initial molar OC/Fe ratios from 0.1 to 9.6 at two initial molar PO4/Fe ratios of 0.05 or 0.25 in treatments without and with Ca (4 mM). The organic ligands and OC/Fe ratios were selected to represent structural moieties and OC concentrations commonly found in soils, groundwater and surface waters.11,43 The selected (P/Fe)init ratios are known to induce initial (Ca)FeP precipitation followed by Lp precipitation and concomitant partial (P/Fe 0.25) or complete (P/Fe 0.05) (Ca)FeP transformation into Fh in OC-free controls.7,8 The composition and structure of the formed Fe(III)-precipitates were analyzed with a broad range of analytical techniques and used to explain interdependent variations in OC and PO4 sequestration from solution.
2. Materials and methods
2.1. Organic model compounds
Four low molecular weight organic acids (LMWOAs) were selected as model compounds: 2,4-dihydroxybenzoic acid (2,4-DHB; β-resorcyclic acid; pKa = 3.43, 9.16), galacturonic acid (Galact; pKa = 3.51), 3,4-dihydroxybenzoic acid (3,4-DHB; protocatechuic acid: pKa = 4.60, 9.27), and citric acid (Citr; pKa = 3.13, 4.76, 6.40) (structures shown in Fig. S1 in the ESI†). Dihydroxybenzoic acids such as 2,4-DHB and 3,4-DHB are found in forest soils and represent structural moieties found in humic substances.22 Galact is the monosaccharide unit of polygalacturonate, a degradation product of pectin and model for exopolymeric mucilage on plant root tips.27,44 Citr is an α-hydroxy acid with three carboxyl groups and plays an important role as a root exudate for nutrient acquisition in the rhizosphere.45All four LMWOAs carry carboxyl groups that are deprotonated at near-neutral pH and can interact with Fe(III); 2,4-DHB and 3,4-DHB also carry phenolic hydroxyl groups. Whereas 2,4-DHB can form a salicylate-type chelate complex with Fe(III) via its carboxyl and phenolic hydroxyl groups in the ortho position, 3,4-DHB can form a much stronger catecholate-type chelate complex via its two phenolic hydroxyl groups in meta and para positions.22 Galact mainly interacts with Fe(III) via its carboxyl group,44 but has been proposed to also form a chelate complex with Fe(III) involving its ring O-atom.46 Citr can bind Fe(III) very strongly via two carboxyl groups and one hydroxyl group.47 Thermodynamic calculations (ESI Section 1†) indicated that the extent of Fe(III) complexation by the LMWOA at a given molar (OC/Fe) ratio increases along the sequence 2,4-DHB ∼ Galact ≪ 3,4-DHB < Citr. For comparison with the LMWOA, experiments were also conducted with leonardite humic acid (LH) as a proxy of macromolecular NOM (International Humic Substances Society (IHSS); C-content: 64.0%; carboxyl groups: 7.46 mol kg−1; phenolic hydroxyl groups: 2.31 mol kg−1; pKa = 4.59, 9.72; ref. 48).
2.2. Synthesis of Fe(III)-precipitates
The synthesis of Fe(III)-precipitates by Fe(II) oxidation in aqueous solutions mimicking near-neutral natural waters followed a similar protocol to that in earlier work without organic ligands.8,13,42,49 Experiments were conducted at a total Fe concentration of 0.5 mM at two initial PO4/Fe ratios ((P/Fe)init) of 0.05 or 0.25 in 4 mM Ca or 8 mM Na background electrolytes at initial molar OC/Fe ratios ((OC/Fe)init) of 0.0, 0.1, 0.3, 0.6, 1.2, 2.4, 4.8, or 9.6. The factorial combinations are summarized in Table S3,† and analytical methods are given in Table S4.† In this manuscript, based on the background electrolyte cation (Ca or Na) and (P/Fe)init (0.05 or 0.25), the electrolytes are referred to as Ca 0.05, Ca 0.25, Na 0.05 and Na 0.25. Sample labels indicate the type of electrolyte, the organic ligand, and the molar (OC/Fe)init. For example, Ca 0.25 Citr 0.1 refers to experiments in Ca 0.25 electrolyte containing Citr at an (OC/Fe)init ratio of 0.1 and Ca 0.25 Ctrl refers to the corresponding control (Ctrl) treatment without organic ligands.Organic stock solutions were freshly prepared for each experiment. 2,4-Dihydroxybenzoic acid (Sigma Aldrich; C7H6O4, ≥97.0%), D-(+)-galacturonic acid monohydrate (Sigma Aldrich; C6H10O7·H2O, ≥97.0%), 3,4-dihydroxybenzoic acid (Sigma Aldrich; C7H6O4, ≥97.0%), citric acid monohydrate (Sigma Aldrich; C6H8O7·H2O, puriss. p.a.), or leonardite humic acid (IHSS; 1S104H) were added to 250 mL doubly deionized water (DDI; 18.2 MΩ cm−1, MilliQ® Element, Millipore) in a glass flask to achieve a nominal concentration of 30 mM (initial experiments) or 100 mM (later experiments) organic C. The solutions were equilibrated on a magnetic stirring plate (800 rpm) for 15 min. Subsequently, 1 M NaOH was manually titrated into the stirred stock solutions until pH 7.0 (±0.3) was reached. The LMWOA stock solutions were used without further treatment; the LH stock solution was stirred overnight and filtered (0.1 μm cellulose nitrate) before use.
The background electrolytes were prepared by dissolving either 4 mM CaCO3 or 8 mM NaHCO3 in DDI water purged with CO2 gas in a glass flask (Schott Duran), resulting in solutions with pH 5.5–5.6. After complete dissolution of CaCO3 or NaHCO3, the pH of the solutions was raised to 7.0 (±0.1) by passing pressurized air. This resulted in solutions buffered with ∼8 mM HCO3− and ∼16 mM H2CO3 in equilibrium with ∼50 mbar CO2, and a saturation index for calcite of ∼0.4 in Ca electrolytes. Subsequently, 2 μM of arsenate (AsO4) (4 mM Na2HAsO4·7H2O; neutral stock) and 2 μM Cd (4 mM CdCl2; acidic stock; 1 mM HCl) were added to each electrolyte at pH 7.0 to study the co-precipitation of two important toxic contaminants (results to be published separately). Considering the much higher initial concentrations of PO4 (25 or 125 μM), OC (50–4800 μM), bicarbonate (∼8000 μM), Fe (500 μM) and Na (8000 μM) or Ca (4000 μM) in our experiments, these trace levels of AsO4 and Cd were not expected to significantly affect Fe(III)-precipitate formation. Either 200 mL (initial experiments) or 600 mL (later experiments) of background electrolyte were transferred into polypropylene (PP) bottles. Subsequently, PO4 was added (50 mM NaH2PO4·H2O; neutral stock solution) to achieve initial molar P/Fe ratios ((P/Fe)init) of 0.05 (0.025 mM PO4) and 0.25 (0.125 mM PO4). To avoid photochemical reactions, spiking of the organic ligands and Fe(II) to the electrolytes, oxidation and precipitation of Fe, and initial and final samplings were performed in a fume hood covered with red plastic foil (primary red 106, LEE filters, UK). Aliquots of the organic ligand stock solutions were spiked to reach OC concentrations between 0.05 and 4.8 mM. For comparison, control treatments (Ctrl) without organic ligands were prepared.
To initiate Fe(II) oxidation, 0.5 mM Fe(II) was spiked into the solutions (using freshly prepared 50 mM FeSO4·7H2O stock solution acidified with 1 mM HCl). Immediately after each Fe(II) spike, the solution was rapidly agitated to ensure complete mixing, and an unfiltered 10 mL aliquot was collected in a polyethylene (PE) tube and acidified with 0.65% (v/v) HNO3 for analysis of total initial concentrations of Fe, P, Ca and Na by inductively coupled plasma mass-spectrometry (ICP-MS; Agilent 8900). Total initial concentrations measured by ICP-MS (in % of target values) equaled 95% ± 8% for Fe (percentage and standard deviation; n = 198), 102% ± 17% for P (n = 198) and 90% ± 7% for Ca (n = 104). Unfiltered 20 mL aliquots were collected in glass vials and acidified with 1.0% (v/v) HCl for analysis of total organic carbon (TOC). The oxidation and precipitation of Fe were allowed to proceed for 18 h, while the suspensions were agitated on a horizontal shaker (150 rpm). After this period, the suspension pH had increased to 7.3 (±0.2) due to some CO2 outgassing. To validate that Fe(II) oxidation was complete, selected samples were analyzed for residual Fe(II) using UV-Vis spectrophotometry (ESI Section 11†). Solution TOC was measured in initial solutions to confirm the spiked levels. TOC analyses of unfiltered final suspensions indicated that Fe-induced OC decomposition was negligible (Table S14†).
Aliquots of the precipitate suspensions were filtered (0.2 μm nylon filters, Whatman) for the analysis of dissolved element concentrations by ICP-MS (10 mL in PE tubes, acidified to 0.65% (v/v) HNO3) and of dissolved organic carbon (DOC) (20 mL in glass vials, acidified to 1.0% (v/v) HCl). Unfiltered aliquots (without acidification) were sampled for measurements of the zeta potential and particle size by electrophoretic and dynamic light scattering (ELS and DLS) and for the analysis of suspension turbidity changes over a 24-h settling time.
For analysis by scanning transmission electron microscopy (STEM), precipitates from the unfiltered suspensions were sampled on a laminar flow bench by placing 20 μL of suspension on a transmission electron microscopy (TEM) grid (Cu, holey carbon) and sucking the solution through the grid using a filter paper. The samples were washed by passing 40 μL of DDI water through the TEM grids and stored in a desiccator cabinet for later analysis.
The Fe(III)-precipitates were collected by passing the remaining suspensions through cellulose nitrate filters (0.1 μm; 47 mm diameter; Whatman). The precipitates were washed by passing 50 mL of DDI water through the filter membranes and dried overnight under a stream of air. The dried solids were stored in a desiccator for later analysis by X-ray absorption spectroscopy (XAS), X-ray diffraction (XRD), and Fourier-transform infrared spectroscopy (FTIR) and for acid digestion and TOC quantification.
2.3. Analysis of stock, initial unfiltered and final filtered solutions
Unfiltered and filtered acidified samples were diluted with 1.0% HNO3 solution for the analysis of Fe, P, Ca, Na and other elements by ICP-MS (Agilent 8900 triple-quadrupole, Agilent Technologies, Tokyo, Japan). From the concentrations of Fe, P, and Ca in the initial unfiltered and final filtered suspensions, their concentrations in suspended solids and the corresponding element ratios (e.g. the molar precipitate P/Fe ratio (P/Fe)ppt) were calculated.To measure total organic carbon in the initial unfiltered and dissolved organic carbon in the final filtered suspensions, a Shimadzu TOC-L analyzer was used. Final filtered suspensions from 12 control (Ctrl) treatments returned a TOC concentration of 0.8 ± 0.2 mg L−1 (average and standard deviation), due to OC release from the 0.2 μm nylon filters. This blank value was subtracted from DOC values measured in final filtered solutions. The TOC of the (diluted) organic stock solutions was measured to validate their OC concentrations.
2.4. Analysis of elemental composition of Fe(III)-precipitates
In addition to the calculation of the elemental composition of the Fe(III)-precipitates from total initial and final dissolved element concentrations, the elemental composition was also determined by acid digestion: in duplicate, 2.000 ± 0.200 mg of the precipitates were exactly weighed into 15 mL PE tubes and dissolved in 10 mL of HCl (1% (v/v) of 32% HCl) overnight on a tabletop shaker (300 rpm). The acid digests were diluted for analysis of Fe, P, and Ca by ICP-MS. From the results, the molar ratios of P/Fe ((P/Fe)ppt) and Ca/Fe ((Ca/Fe)ppt) were calculated. Co-precipitated OC was quantified using a TOC analyzer (Soli TOC® cube, Elementar Analysensysteme GmbH, Langenselbold, Germany). 10.00 mg (±1.00 mg) of the sample was exactly weighed into ceramic crucibles, reacted with 200 μL of 1 M HCl for 15 min to remove inorganic C and dried in a fume hood (1 week) prior to analysis. The molar OC/Fe ratios of the precipitates ((OC/Fe)ppt) were calculated from their OC and Fe contents.2.5. Fe K-edge extended X-ray absorption fine structure (EXAFS) spectroscopy
For analysis of the solids by Fe K-edge extended X-ray absorption fine structure (EXAFS) spectroscopy, ∼1.5 mg of the air-dried precipitates (∼1.0 mg Fe) were mixed with ∼50 mg of cellulose and pressed into 7 mm diameter pellets (absorption edge step close to unity). Most spectra were recorded at the SUL-X and the XAS beamlines at the KIT Light Source (Karlsruhe Institute of Technology, Germany), and some spectra were recorded at the SuperXAS beamline at the Swiss Light Source (SLS, Paul Scherrer Institute, Villigen, Switzerland). For the extraction of the EXAFS spectra and their evaluation by linear combination fitting (LCF), the software code Athena was used.50 Photon energy was calibrated by setting the first maximum of the first derivative of the Fe K-edge of metallic Fe foil to 7112 eV. The spectra were normalized by subtracting a first-order polynomial fit to the data from −100 to −30 eV before the edge and subsequently dividing by a second-order polynomial fit to the data from 50 to 400 eV above the edge (E0 set to 7128.5 eV). The EXAFS spectra were extracted using the Autobk algorithm (Rbkg = 0.85; k-weight = 3; spline k-range 0–11.6 Å−1). For visual examination, the EXAFS spectra were Fourier-transformed over the k-range 2.5–10.5 Å−1 using a Kaiser–Bessel apodization window (dk = 2 Å−1). The k3-weighted EXAFS spectra were analyzed by linear combination fitting (LCF) over the k-range 2–11 Å−1 using reference spectra of four types of Fe(III)-precipitates formed by Fe(II) oxidation in bicarbonate-buffered solutions (poorly crystalline lepidocrocite (Lp*), ferrihydrite (Fh*), amorphous Fe(III)–(hydroxy)phosphate (FeP*), and amorphous Ca–Fe(III)–(hydroxy)phosphate (CaFeP*))8,13 and of an Fe(III)–3,4-DHB co-precipitate22 (FeOrg*; spectrum provided by the authors).2.6. X-ray diffraction (XRD)
For X-ray diffraction (XRD) analysis, 10 mg of sample were suspended in ethanol, deposited onto a low-background Si slide, and allowed to dry. XRD patterns were recorded from 10° to 90° 2θ with a step size of 0.017° and a measurement time of 150 min per sample using Co Kα radiation (X'Pert Powder diffractometer with an X'Celerator detector, PANalytical, Almelo, The Netherlands). The diffractograms were evaluated using HighScore+ (Malvern PANalytical). From the full width at half maximum (FWHM) of the Lp peak at 15.97° 2θ (d020 6.26 Å), coherently scattering domain (CSD) sizes were calculated using the Scherrer equation.2.7. Fourier-transform infrared spectroscopy
The precipitates were analyzed by Fourier-transform infrared spectroscopy (FTIR) on a Biorad FTS 575 C instrument with a liquid N2 cooled MCT detector and an attenuated total reflectance (ATR) unit (Harrick Meridian diamond single reflection Split Pea ATR unit; 20 kHz speed with 5 kHz filter, UDR: 2, resolution: 2, aperture: 1 cm−1 and sensitivity: 8). Spectra were recorded over the wavenumber range 400–4500 cm−1. For baseline correction, a linear function was fit through the data in the wavenumber regions 1800–1850 cm−1 and 2400–2450 cm−1 (where no absorbance was expected) and subtracted from the spectra. The baseline-corrected spectra were scaled to 0.4 absorbance units in the wavenumber region 455–460 cm−1 (Fe–O vibrations51). Baseline correction and scaling were performed with Octave (GNU).2.8. Scanning transmission electron microscopy
Scanning transmission electron microscopy (STEM) coupled with secondary electron (SE) and high-angle annular dark field (HAADF) detection was used to visualize the precipitates and characterize their morphology (HD2700Cs, Hitachi, Japan). For image analysis, the software package DigitalMicrograph (V.1.82, Gatan Inc., CA, USA) was used. STEM coupled with HAADF and energy dispersive X-ray (EDX) detection was performed to determine the spatial distribution of P, Fe, C and Ca within the Fe(III)-precipitates (FEI Talos, F200X, Super-X EDS, four-detector configuration, 200 kV acceleration voltage; ThermoFischer Scientific, OH, USA). Data evaluation including the derivation of elemental distribution maps and molar element ratios was performed with the Velox software (ThermoFischer Scientific).2.9. Electrophoretic and dynamic light scattering and turbidity measurements
The electrophoretic mobility and the hydrodynamic diameter of particles in the final suspensions were derived from electrophoretic (ELS) and dynamic light scattering (DLS) measurements, respectively (Malvern Zetasizer, Malvern Panalytical, Worcestershire, UK). ELS measurements were performed in disposable folded polymethyl methacrylate (PMMA) capillary cells (3 measurements per sample, 10 scans per measurement, 10 seconds per scan). The zeta potentials of the particles were calculated from their electrophoretic mobilities using the Helmholtz–Smoluchowski equation (Henry function; f(Ka) = 1.5; dielectric constant of 78.5). DLS measurements were performed in a 10 mm quartz glass cuvette (3 measurements per sample, 12 scans per measurement, 30 seconds per scan). The autocorrelation function was used to derive the translational diffusion coefficient from which the hydrodynamic particle diameter was calculated. Particle settling in unfiltered suspensions was measured in 30 mL glass vials using a turbidimeter (TL2300, Hach Lange GmbH, Germany; nephelometric turbidity units (NTU)). Prior to the first measurement, the vials were turned twice upside-down to resuspend the precipitate. Particle settling was followed over 24 h; the vials were stored in the dark between measurements.3. Results
3.1. Elemental composition of Fe(III)-precipitates
For the Fe(III)-precipitates formed in the Ca electrolytes, the molar (P/Fe)ppt, (Ca/Fe)ppt and (OC/Fe)ppt ratios are shown in Fig. 1, and the corresponding data for all treatments are listed in Table S5.† In Ca electrolytes, no precipitates could be collected in treatments with Citr at (OC/Fe)init > 0.6, as most Fe passed the filter membranes. In the Na electrolytes, no precipitates could be collected in treatments with Citr at (OC/Fe)init > 0.1 (Na 0.25) or > 0.3 (Na 0.05) and 3,4-DHB at (OC/Fe)init > 0.6. Precipitates formed in Na electrolytes in the presense of LH were strongly attached to the filter membranes and could not be recovered. | ||
| Fig. 1 Molar (P/Fe)ppt, (Ca/Fe)ppt and (OC/Fe)ppt ratios of Fe(III)-precipitates formed in Ca electrolytes at (P/Fe)init 0.05 (a, c and e) and 0.25 (b, d and f), based on solid-phase Fe, P, Ca and OC quantification. Horizontal red dashed lines indicate ratios in Ctrl treatment. The results for all treatments are listed in Table S5.† | ||
The molar (OC/Fe)ppt of precipitates formed in Ca 0.05 electrolyte increased with increasing (OC/Fe)init and along the sequence 2,4-DHB < Galact ≪ 3,4-DHB < Citr (Fig. 1c and Table S5†). The same sequence but somewhat lower (OC/Fe)ppt were observed at the higher PO4 level in the Ca 0.25 electrolyte. In the Na electrolytes, OC uptake followed similar trends to those in the Ca electrolytes (2,4-DHB < Galact < 3,4-DHB; (P/Fe)init 0.25 < (P/Fe)init 0.05; Table S5†), but was overall lower than that in the Ca electrolytes.
In all electrolytes (Na and Ca, (P/Fe)init 0.05 and 0.25), the (P/Fe)ppt of the precipitates formed at the lowest initial (OC/Fe)init ratios tended to be higher than that in the OC-free controls (Fig. 1a and Table S5†) and stayed elevated at increasing (OC/Fe)init. Notably, for 3,4-DHB in the Ca 0.05 and Ca 0.25 electrolytes at (OC/Fe)init 9.6, the (P/Fe)ppt were considerably higher than (P/Fe)init, indicating that relatively less Fe than P had been retained on the filters. The amounts of co-precipitated Ca increased along the order 2,4-DHB < Galact < 3,4-DHB < Citr and with increasing (OC/Fe)init and were higher at (P/Fe)init 0.25 than at 0.05 (Fig. 1b and Table S5†). For LH in the Ca electrolytes, higher (OC/Fe)ppt than for the LMWOA were observed at a given (OC/Fe)init (Fig. 1), and also the co-precipitation of Ca was enhanced.
3.2. Fe K-edge EXAFS spectroscopy
To characterize the Fe(III)-precipitates in terms of local Fe coordination and structurally distinct Fe(III)-precipitate fractions, Fe K-edge EXAFS spectroscopy was used. The Fourier-transformed EXAFS spectra of precipitates formed in the Ca 0.05 electrolyte and reference spectra used for LCF analysis are shown in Fig. 2. For spectral interpretation, reference spectra of four types of Fe(III)-precipitates formed by Fe(II) oxidation in bicarbonate-buffered electrolytes13 and a spectrum of organically complexed Fe(III)22 were used (Fig. 2a): (1) poorly crystalline lepidocrocite formed in Na electrolyte in the absence of PO4 and SiO4 (Lp*). (2) Ferrihydrite formed in SiO4-containing Na electrolyte (Fh*) (lower degree of corner-sharing linkage of FeO6-octahedra than in 2-line ferrihydrite formed by forced Fe(III) hydrolysis).8 (3) Amorphous Fe(III)–(hydroxy)phosphate formed in Na electrolyte (FeP*), (4) amorphous Ca–Fe(III)–(hydroxy)phosphate formed in Ca electrolyte (CaFeP*; with structural Ca and higher (P/Fe)ppt and degree of Fe polymerization than FeP*).8 (5) Solid Fe(III)–3,4-DHB-coprecipitate with molar 3,4-DHB/Fe ratio ∼0.9; with monomeric Fe(III) in a catecholate-type complex (FeOrg*).22 Notably, the first-shell Fe–O peak of Fh* is markedly reduced compared to Lp*, FeP*, CaFeP*, and FeOrg*, due to strong distortion of FeO6 octahedra in Fh* and potential signal contributions from a minor amount of tetrahedral Fe.8,22 The spectrum of FeOrg*, on the other hand, had a similar first-shell Fe–O amplitude to the Lp*, FeP* and CaFeP* reference spectra, but was lacking a detectable second-shell signal. Comparison of the sample XANES spectra to the spectra of the Fe(III) reference compounds and an Fe(II) reference spectrum and the results from LCF fits including an Fe(II) reference spectrum (Fig. S2†) showed that Fe(II) was a negligible component in the studied samples (no Fe(II) remaining from the oxidation experiment and no Fe(II) formed during XAS data collection). | ||
| Fig. 2 Fourier-transformed k3-weighted Fe K-edge EXAFS spectra of reference compounds used for LCF analysis (a), precipitates formed at (P/Fe)init 0.05 in Ca electrolytes at (OC/Fe)init 0.6 (b) and precipitates formed at (P/Fe)init 0.05 in Ca electrolytes from (OC/Fe)init 0.1 to 9.6 (c). Reference spectra: Lp*: poorly crystalline lepidocrocite; Fh*: ferrihydrite; FeP*: amorphous Fe(III)–phosphate; CaFeP*: amorphous Ca–Fe(III)–phosphate; FeOrg*: Fe(III)-3,4-DHB co-precipitate. | ||
Spectra of precipitates formed in Ca 0.05 electrolyte at (OC/Fe)init of 0.6 are shown in Fig. 2b. The first- and second-shell peaks of the Fourier-transformed (FT) EXAFS spectrum of the OC-free control indicated that it mainly consisted of Lp. The largest decrease in the magnitude of the second-shell Fe–Fe peak at R ∼ 2.7 Å was observed for Citr, followed by 3,4-DHB, Galact, and 2,4-DHB. For a given organic ligand, the second-shell Fe–Fe peak magnitude decreased with increasing (OC/Fe)init (Fig. 2c), indicating decreasing Lp formation with increasing (OC/Fe)init. Concomitantly, the magnitude of the first-shell Fe–O peak at R ∼ 1.5 Å tended to decrease, indicative of the formation of an increasing amount of Fh.
For quantitative interpretation, the k3-weighted EXAFS spectra from all treatments were analyzed by LCF using the reference spectra shown in Fig. 2a. The LCF results for precipitates from the Ca and Na electrolytes at both (P/Fe)init are shown in Fig. 3, scaled using the fraction of Fe retained by filtration (0.2 μm) and together with the filter-passing Fe fraction. The LCF results are tabulated in Tables S6 and S7† and shown without scaling and filter-passing Fe in Fig. S4.† All EXAFS and reconstructed LCF spectra are shown in Fig. S5 and S6.†
 | ||
| Fig. 3 Results from LCF analysis of the Fe K-edge EXAFS spectra of the precipitates formed in Ca (a and c) and Na (b and d) electrolytes at (P/Fe)init 0.05 (a and b) and at (P/Fe)init 0.25 (c and d). The fit results for the precipitates at (P/Fe)init 0.05 are listed in Table S6† and for the precipitates at (P/Fe)init 0.25 in Table S7.† Reference spectra (shown in Fig. 2): Lp*: poorly crystalline lepidocrocite; Fh*: ferrihydrite; FeP*: amorphous Fe(III)–phosphate; CaFeP*: amorphous Ca–Fe(III)–phosphate. LCF fractions were scaled using the Fe fraction retained on 0.2 μm filter membranes; blue columns (FePass) indicate filter-passing Fe. | ||
For the Ca 0.05 electrolyte, the LCF results for the OC-free control sample showed that the precipitate consisted mainly of Lp and a minor fraction of Fh, as expected at (P/Fe)init < 0.1 in the absence of other strongly interfering ligands, where initial formation of (Ca)FeP is followed by (near-)complete (Ca)FeP transformation into Fh and precipitation of Lp.7,8,13 With increasing (OC/Fe)init, the fraction of Lp decreased and the fraction of Fh increased for all organic ligands, and the magnitude of this effect increased along the sequence 2,4-DHB < Galact < 3,4-DHB < Citr. For 3,4-DHB and to a lesser extent Galact and LH, the LCF-derived FeOrg* fractions pointed to increasing monomeric Fe(III) complexation by the organic ligands with increasing (OC/Fe)init. In the Na 0.05 electrolyte, the organic ligands had a similar effect on the Fe(III)-precipitate structure to that in the Ca 0.05 electrolyte (Fig. 3).
For the Ca 0.25 electrolyte, the LCF of the OC-free control indicated that the precipitate consisted of (Ca)FeP, Fh and Lp, in line with previous work.8,13 In the presence of organic ligands, as in the Ca 0.05 electrolyte, the fraction of Lp decreased and the fraction of Fh increased with increasing (OC/Fe)init, roughly along the sequence 2,4-DHB < Galact < 3,4-DHB < Citr. As in the Ca 0.05 electrolyte, LCF returned the largest fractions of FeOrg* for 3,4-DHB and minor fractions for Citr and Galact. For the OC-free control from the Na 0.25 electrolyte, LCF returned a higher (Ca)FeP fraction than that for the Ca 0.25 electrolyte, due to lower PO4 uptake by FeP than CaFeP and the need for a larger fraction of Fe for PO4 removal.8,13
For LH, the LCF results from all electrolytes suggested that its impact on local Fe coordination fell in between those of 2,4-DHB and Galact.
3.3. X-ray diffraction (XRD)
XRD served to gain insights into crystalline components in the precipitates. The XRD data for precipitates formed in the Ca electrolytes are presented in Fig. 4, and the data for the Na electrolytes are presented in Fig. S7.† The XRD pattern of the Ca 0.05 Ctrl exhibited peaks of calcite (Calc) that were mostly absent in treatments with OC and the control at (P/Fe)init 0.25, indicating that in addition to PO4, the organic ligands also hinder calcite precipitation. The XRD pattern of the Ca 0.05 Ctrl exhibited the most intense Lp peaks of all Ca electrolyte treatments. With increasing (OC/Fe)init, the heights of the Lp peaks decreased and their widths increased, indicating the formation of less and less crystalline Lp. For Citr at an (OC/Fe)init of 0.6 and for 3,4-DHB and a Galact at (OC/Fe)init of 2.4 and 9.6, only the broad peak of Fh remained. The Ca 0.25 Ctrl exhibited broader Lp peaks than the Ca 0.05 Ctrl13 and featured a broad peak indicative of (Ca)FeP and Fh. Effects of organic ligands on Lp crystallinity in the Ca 0.25 electrolyte were similar to those in the Ca 0.05 electrolyte. Patterns of precipitates from LH treatments most closely matched with those from 2,4-DHB treatments. XRD patterns of precipitates from Na electrolyte exhibited similar trends (Fig. S7†) to those observed in Ca electrolyte (Fig. 4). | ||
| Fig. 4 X-ray diffraction patterns of Fe(III)-precipitates from the Ca electrolyte with a (P/Fe)init of 0.05 (a) and 0.25 (b). Peak positions are indicated for lepidocrocite (Lp, orange), 2-line ferrihydrite (Fh, brown), amorphous Fe(III)–phosphate (FeP, olive) and calcite (Calc, green). For the Na 0.05 and Na 0.25 treatments, the XRD data are shown in Fig. S7.† | ||
Coherently scattering domain (CSD) sizes derived from the Lp (020) peak at 15.97° 2θ (Table S8†) indicate the extent of Lp sheet stacking. The largest CSD (1.55 nm) was observed for the precipitate Na 0.05 Ctrl. Considering a layer spacing of 0.6 nm along the unit-cell b-axis, this CSD size corresponds to 3–4 stacked sheets. Because PO4 interferes with Lp sheet stacking,8,13,52 Lp formed at (P/Fe)init 0.25 in general had a smaller CSD than Lp in analogous precipitates formed at (P/Fe)init 0.05.8,13,52 The lower CSD of Lp formed in OC-free controls in Ca than that in Na electrolytes was due to Ca enhancing the effect of PO4.13 Increasing organic ligand levels caused a decrease in the CSD, indicating that they also inhibited sheet stacking. At a given (OC/Fe)init, this effect increased along the sequence 2,4-DHB < Galact < 3,4-DHB < Citr. High levels of Citr, 3,4-DHB and Galact fully inhibited Lp formation. In contrast, even at the highest tested (OC/Fe)init of 9.6, 2,4-DHB did not fully inhibit Lp nucleation as indicated by weak and broad Lp peaks. However, the lack of the (020) peak showed that mostly unstacked Lp sheets had formed. The same was observed for precipitates formed in the presence of LH.
3.4. Fourier-transform infrared spectrometry (FTIR)
FTIR spectra were recorded to obtain information on organic ligand and oxyanion uptake and interactions with Fe(III). The FTIR spectra of precipitates formed in Ca electrolyte are presented in Fig. 5, and the spectra for the Na treatments are presented in Fig. S8.† | ||
| Fig. 5 Fourier-transform IR spectra of Fe(III)-precipitates from the Ca electrolyte with a (P/Fe)init of 0.05 (a) and 0.25 (b). The precipitates Ca 0.05 LH 2.4/9.6 and Ca 0.25 LH 9.6 could not be removed from the cellulose nitrate membranes. They were measured on the membrane, and the membrane spectrum was subsequently subtracted. The spectra of precipitates from the Na 0.05 and Na 0.25 treatments and of the cellulose nitrate membrane are shown in Fig. S8.† | ||
Considering precipitates formed in the Ca electrolyte at (P/Fe)init 0.05, the narrow bands at 740 and 1018 cm−1 are characteristic for Lp,51 and their presence and intensity were in line with the extent of Lp formation observed by XAS and XRD. Bands from 940 cm−1 to 1082 cm−1 represent co-precipitated PO4 (adsorbed or in (Ca)FeP).8,53 In the presence of the strongly interacting organic ligands, characteristic bands between 1100 and 1650 cm−1 increase with increasing ligand concentrations. Bands in the regions of 1360–1390 and 1560–1640 cm−1 represent carboxylate groups (symmetric and asymmetric stretching vibrations).54 As expected at pH 7.0, the marked C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of protonated carboxyl groups (∼1710 cm−1) was not observed.55 Fully protonated catechol would show peaks at 1250 and 1280 cm−1 arising from C–O vibrations, whereas these peaks merge into a single band when catechol forms a bidentate complex with Fe(III).56 The pronounced peak at ∼1271 cm−1 in the case of 3,4-DHB thus indicated catecholate-type Fe(III) binding.56,57 Similarly, for 2,4-DHB, the peak at 1253 cm−1 pointed to deprotonated phenolic hydroxyl groups in salicylate-type complexes with Fe(III).57 The vibrations at around 1480 cm−1 are attributed to the benzene ring.56 For precipitates formed in the presence of Citr, peaks at 1385 cm−1 and 1561 cm−1 can be attributed to carboxyl stretching vibrations.55,58 Precipitates formed in the presence of LH showed bands at 1570 and 1390 cm−1 due to carboxyl stretching vibrations and at 1280 cm−1 due to deprotonated phenolic hydroxyl groups, suggesting that LH interacted with Fe(III) via both carboxyl and phenolic hydroxyl groups, in line with previous work.15 In the case of weakly sorbing Galact and 2,4-DHB, the two broad bands in the wavenumber ranges 1370–1410 cm−1 and 1480–1500 cm−1 resulted from co-precipitated bicarbonate.59 The band at around 1635 cm−1 is due to adsorbed H2O.13
O stretching vibration of protonated carboxyl groups (∼1710 cm−1) was not observed.55 Fully protonated catechol would show peaks at 1250 and 1280 cm−1 arising from C–O vibrations, whereas these peaks merge into a single band when catechol forms a bidentate complex with Fe(III).56 The pronounced peak at ∼1271 cm−1 in the case of 3,4-DHB thus indicated catecholate-type Fe(III) binding.56,57 Similarly, for 2,4-DHB, the peak at 1253 cm−1 pointed to deprotonated phenolic hydroxyl groups in salicylate-type complexes with Fe(III).57 The vibrations at around 1480 cm−1 are attributed to the benzene ring.56 For precipitates formed in the presence of Citr, peaks at 1385 cm−1 and 1561 cm−1 can be attributed to carboxyl stretching vibrations.55,58 Precipitates formed in the presence of LH showed bands at 1570 and 1390 cm−1 due to carboxyl stretching vibrations and at 1280 cm−1 due to deprotonated phenolic hydroxyl groups, suggesting that LH interacted with Fe(III) via both carboxyl and phenolic hydroxyl groups, in line with previous work.15 In the case of weakly sorbing Galact and 2,4-DHB, the two broad bands in the wavenumber ranges 1370–1410 cm−1 and 1480–1500 cm−1 resulted from co-precipitated bicarbonate.59 The band at around 1635 cm−1 is due to adsorbed H2O.13
FTIR spectra of precipitates formed in Ca electrolyte at (P/Fe)init of 0.25 showed similar features to those observed for (P/Fe)init of 0.05 (Fig. 5). However, the intensity of the PO4 bands was higher and the spectral contributions of the organic ligands lower, showing that a higher PO4 level reduced organic ligand co-precipitation.15
FTIR spectra of precipitates formed in Na electrolyte (Fig. S8†) showed similar trends to those observed for Ca electrolyte. However, the intensities of the bands arising from the organic ligands were lower, in line with the lower OC contents of Fe(III)-precipitates formed in Na electrolytes than in Ca electrolytes (Table S5†).
3.5. Scanning transmission electron microscopy
The nanoscale morphology and elemental heterogeneity of the Fe(III)-precipitates were analyzed by STEM, including secondary electron (SE) and high-angle annular dark field (HAADF) imaging and energy-dispersive X-ray (EDX)-based element distribution mapping. SE images and EDX elemental distribution maps of P, Fe and Ca of precipitates formed in the Ca 0.05 and Ca 0.25 electrolytes at high organic ligand levels are shown in Fig. 6, and analogous data for low organic ligand levels are shown in Fig. 7. Additional STEM images (Fig. S9–S17†) and EDX-derived molar element ratios in morphologically distinct nanoscale entities (Tables S9 and S10†) are provided in the ESI.† Nanoscale precipitate characteristics offered insights into precipitate formation pathways and were, in general, in line with bulk Fe(III)-precipitate composition as inferred from the spectroscopic results. | ||
| Fig. 6 Secondary electron (SE) images of Fe(III)-precipitates in Ca electrolytes at a (P/Fe)init of 0.05 (a, e, i, m and q) or 0.25 (c, g, k, o and s), and energy-dispersive X-ray (EDX) elemental distribution maps of P, Fe and Ca from the respective treatments at a (P/Fe)init of 0.05 (b, f, j, n and r) or 0.25 (d, h, l, p and t). SE images and EDX maps of precipitates from the Ca electrolytes with LH, (OC/Fe)init at 0.6, and from the Na electrolytes are shown in Fig. S9–S11.† Corresponding HAADF images with elemental distribution maps of P, Fe and C are shown in Fig. S13–S16.† The SE/HAADF images and EDX elemental distribution maps for precipitates from the Ca and Na electrolytes with LH are shown in Fig. S11 and S17.† | ||
 | ||
| Fig. 7 Secondary electron (SE) images of Fe(III)-precipitates in Ca electrolytes at a (P/Fe)init of 0.05 (a, e, i, m and q) or 0.25 (c, g, k, o and s), and energy-dispersive X-ray (EDX) elemental distribution maps of P, Fe and Ca from the respective treatments at a (P/Fe)init of 0.05 (b, f, j, n and r) or 0.25 (d, h, l, p and t). SE images and EDX maps for precipitates from the Ca electrolytes with LH, (OC/Fe)init at 0.6, and from the Na electrolytes are shown in Fig. S8–S10.† Corresponding HAADF images with elemental distribution maps of P, Fe and C are shown in Fig. S13–S16.† The SE/HAADF images and EDX elemental distribution maps for precipitates from the Ca and Na electrolytes with LH are shown in Fig. S11 and S17.† | ||
For the Ca electrolyte at (P/Fe)init 0.05, SE images of the OC-free control combined with EDX maps showed PO4-loaded Fh particles with attached Lp platelets (Fig. 6a, b and S13a, b†), in line with previous work.13 The effects of the organic ligands increased in the order 2,4-DHB < Galact ≪ 3,4-DHB ∼ Citr: the precipitate Ca 0.05 2,4-DHB 9.6 consisted of spherical particles with a (Ca)FeP core and a rough surface characteristic of Fh,60 surrounded by Lp platelets (Fig. 6e, f and S9e, g†). The precipitate Ca 0.05 Galact 9.6 consisted of the same type of core–shell particles, but without attached Lp (Fig. 6i, j and S9i, j†). In strong contrast, the precipitates Ca 0.05 3,4-DHB 9.6 (Fig. 6m and n) and Ca 0.05 Citr 0.6 (Fig. 6q and r) featured spherical particles without core–shell structures. At lower (OC/Fe)init, effects of the ligands on particle structure were less pronounced (Fig. 7 and S10†). SE images showed Lp attached to P-rich Fh-aggregate cores in all treatments, with Lp abundance decreasing in the order 2,4-DHB ∼ Galact < 3,4-DHB < Citr.
In the Ca electrolyte at (P/Fe)init 0.25, the OC-free control consisted of core–shell (Ca)FeP–Fh particles covered with attached Lp platelets (supported P/Fe ratios from STEM-EDX, Table S9†), as described in previous work.7,13 In the precipitate Ca 0.25 2,4-DHB 9.6, some Lp platelets were still observed, whereas the precipitate Ca 0.25 Galact 9.6 consisted exclusively of (Ca)FeP–Fh core–shell particles. The precipitate Ca 0.25 3,4-DHB 9.6 featured spherical particles with a smooth surface (Fig. 6o and p) characteristic for (Ca)FeP,13 consistent with its high (P/Fe)ppt of 0.51 (Table S5†) and the XRD results. In contrast, the precipitate Ca 0.25 Citr 0.6 consisted of particles with a rough surface typical for Fh and Ca and P rich cores, consistent with core–shell (Ca)FeP–Fh particles (Fig. 6s and t).
Effects of the organic ligands on particle morphology were lower at lower (OC/Fe)init (Fig. 7), and Lp platelets attached to CaFeP–Fh core–shell particles were observed in all treatments.
Precipitates formed in Na electrolytes showed similar trends in particle morphology as a function of (P/Fe)init and ligand type and concentration (Fig. S9, S10 and Table S10†) to those described for the Ca electrolytes, but precipitate particles were in general smaller and less aggregated, as exemplified for Galact at a (OC/Fe)init of 9.6 in Fig. S12.†
In the case of LH at (OC/Fe)init 0.6, the precipitates consisted of Fh particles surrounded by Lp sheets (Fig. S11a–d†), as observed for 2,4-DHB. At (OC/Fe)init 9.6 in Ca electrolyte, the precipitate consisted of dispersed Fh and CaFeP nanoparticles embedded in an organic matrix (Fig. S11e–h†), indicating that Ca promoted the coagulation of LH61 and that LH inhibited the formation CaFeP–Fh core–shell particles. In Na electrolyte, in contrast, only few small Fe(III)-precipitate particles could be observed (Fig. S11m and n†).
3.6. Electrophoretic and dynamic light scattering and turbidity measurements
The results of zeta potential (ZP) measurements are shown in Fig. 8. In the Na electrolytes, precipitates formed in OC-free controls were characterized by negative ZP. This can be attributed to the co-precipitation of PO4, considering that the suspension pH (∼7.3) was in the range of the point of zero charge (PZC; 7–8) of pure Fe-oxides51 and consistent with the lower ZP in the Na 0.25 electrolyte than in the Na 0.05 electrolyte. In the Ca electrolytes, the ZPs of the OC-free controls were close to zero, due to charge compensation by co-precipitated Ca. In general, increasing (OC/Fe)init led to more negative ZP in both Na and Ca electrolytes. This effect increased in the order 2,4-DHB < Galact ≪ 3,4-DHB < Citr. In Na electrolytes, LH induced similar to or even more negative ZP than Galact, whereas in the Ca electrolytes, LH led to less negative ZP than Galact, indicating that Ca binding to LH reduced its negative charge. | ||
| Fig. 8 Zeta potential (ZP) of freshly suspended Fe(III)-particles in the Ca 0.05 (a), Na 0.05 (b), Ca 0.25 (c) and Na 0.25 (d) treatments at pH 7.3 (±0.2). For experiments conducted in duplicates, symbols and error bars indicate the average and the difference between the two measured values. | ||
Dynamic light scattering (DLS) offered insights into the size of suspended aggregates (Tables S11 and S12†), but was affected by uncertainties due to aggregate polydispersity and settling. Nevertheless, for increasing (OC/Fe)init, DLS indicated a shift towards smaller aggregate sizes. Opposite to this general trend, aggregates formed in the presence of 2,4-DHB exhibited sizes similar to or larger than those of the respective controls. Similarly, the size of aggregates formed in the presence of 3,4-DHB increased from 0.1 to 0.6, before it increased again towards higher (OC/Fe)init. In general, aggregates formed in Ca 0.05 electrolyte exhibited larger sizes compared to analogous precipitates in Na 0.05 or Ca 0.25 electrolyte, indicating that Ca enhanced and PO4 reduced particle aggregation via their effects on particle charge.
The turbidity of fresh suspensions was examined over a period of 24 h upon re-suspension (ESI Section S9†). The measurements revealed a noticeable impact of the organic ligands on the rate of turbidity reduction compared to the OC-free controls. For the Ca electrolyte, turbidity measurements indicated that increasing organic ligand concentrations decreased the rate of turbidity reduction, but that suspended particles were effectively removed by sedimentation within 24 h. In the Na electrolyte, the organic ligands significantly decreased the rate of turbidity removal, with near-complete inhibition in some treatments. At (P/Fe)init 0.25, turbidity removal was in general slower than that in analogous treatments at (P/Fe)init 0.05, pointing to the stabilization of suspended particles by PO4. As a notable exception, increasing levels of 2,4-DHB enhanced particle aggregation in the Ca 0.05, Na 0.05 and Na 0.25 electrolytes, in agreement with the effect of 2,4-DHB on aggregate sizes observed by DLS.
3.7. Filter-passing Fe, PO4 and OC
The freshly formed Fe(III)-precipitate suspensions were filtered through 0.2 μm filter membranes to analyze residual filter-passing Fe, PO4 and OC. The results for Fe and PO4 are shown in Fig. 9 and results for OC are shown in Fig. S23.† Since preliminary tests confirmed that residual concentrations of Fe(II) were negligible after the 18-h reaction period (Fig. S24†), filter-passing Fe was considered to be colloidal or organically complexed Fe(III). Accordingly, filter passing PO4 was considered to be bound to colloidal or organically complexed Fe(III) or dissolved. | ||
| Fig. 9 Residual filter-passing (0.2 μm) Fe (a–d) and PO4 (e–h) in the fresh suspensions from the Ca 0.05 (a and e), Na 0.05 (b and f), Ca 0.25 (c and g) and Na 0.25 (d and h) treatments. The error bars represent standard deviation from triplicate independent experiments. | ||
In the Ca electrolytes, concentrations of filter-passing Fe were very low in most treatments, indicating that readily filterable Fe(III)-precipitates had formed. Notable exceptions were the treatments with Citr and 3,4-DHB at an (OC/Fe)init of 2.4 and 9.6, where nearly all Fe passed the membranes in the presence of Citr and up to 55% Fe passed the membranes in the presence of 3,4-DHB. In the Na electrolytes, higher fractions of filter-passing Fe(III) already formed at lower levels of Citr or 3,4-DHB, and also high levels of Galact and LH led to filter-passing Fe(III).
Filter-passing fractions of PO4 as a function of filter-passing fractions of Fe(III) are shown in Fig. 10. Depending on the fraction of filter-passing Fe(III), distinct trends with increasing (OC/Fe)init and as a function of ligand type could be observed. At low filter-passing Fe fractions (<3%), much higher filter-passing PO4 than Fe(III) fractions were considered to represent truly dissolved PO4. Filter-passing PO4 for these treatments is shown in Fig. 11. In OC-free controls, residual dissolved PO4 fractions reflected the more effective PO4 uptake in Ca than in Na electrolytes8 and slightly more effective uptake at (P/Fe)init 0.25 than at 0.05, in line with previous work.8 In the presence of organic ligands, residual dissolved PO4 fractions decreased with increasing (OC/Fe)init, and the magnitude of this effect increased in the order 2,4-DHB < Galact < 3,4-DHB ∼ Citr.
 | ||
| Fig. 10 Filter-passing (0.2 μm) fractions of PO4 plotted against the corresponding filter-passing fractions of Fe (in % of the total concentration in initial solution). At filter-passing Fe < 3% (vertical red-dashed line), filter-passing PO4 is considered to represent dissolved PO4. | ||
 | ||
| Fig. 11 Dissolved PO4 in final suspensions from Ca 0.05 (a), Na 0.05 (b), Ca 0.25 (c), and Na 0.25 (d) electrolytes; i.e., filter-passing PO4 for all treatments with <3% filter-passing Fe (Fig. 10). Error bars represent the standard deviation of independent triplicate experiments, where available (not all treatments were done in replicates). | ||
In treatments where filter-passing Fe fractions exceeded 3%, nearly similar fractions of filter-passing PO4 and Fe(III) were in general observed (Fig. 9 and 10). For intermediate fractions of filter-passing Fe and PO4, this correlation points to filter-passing Fe(III)-precipitate colloids or Fe(III)–organic complexes with similar (P/Fe) to the Fe(III)-precipitate retained on the filter membrane. Notable outliers were the treatments with 3.4-DHB in Ca electrolytes at an (OC/Fe)init of 2.4 and 9.6. In these cases, and contrary to the respective treatments in Na electrolyte, filter-passing PO4 was low despite high fractions of filter-passing Fe(III), pointing to separation between the PO4-loaded Fe(III)-precipitate particles retained on the filter membranes and filter-passing PO4-free Fe(III)–organic complexes.
Filter-passing fractions of OC depended on ligand type (Fig. S23†). Citr and 3,4-DHB were effectively retained by the Fe(III)-precipitates in treatments with low filter-passing Fe(III) fractions (<3%), but followed filter-passing Fe(III) in treatments with higher filter-passing Fe(III), supporting the interpretation that filter-passing Fe(III) corresponded to OC-stabilized colloidal Fe(III) or Fe(III)–organic complexes. Galact and 2,4-DHB were weakly retained by the Fe(III)-precipitates (Fig. 1 and Table S5†), and due to the high fraction of the residual dissolved ligand, their filter-passing fractions were therefore high (>75%) in all treatments.
4. Discussion
Our results reveal how the tested organic ligands combined with PO4 and Ca profoundly and interdependently affect the bulk and nanoscale structure, elemental composition and colloidal properties of freshly formed Fe(III)-precipitates. In general, the magnitude of the effects of the organic ligands increased in the order 2,4-DHB < Galact ≪ 3,4-DHB < Citr, i.e., with increasing Fe(III) complexation strength (Table S2†), and with an increasing ligand concentration ((OC/Fe)init). In the following sections, the results are discussed with respect to (i) effects of the organic ligands, PO4 and Ca on Fe(III)-precipitate structure, (ii) interdependent co-precipitation of OC, PO4, and Ca and residual dissolved PO4, (iii) formation of Fe(III) colloids and Fe(III)–organic complexes, and (iv) effects of macromolecular humic acid (LH) relative to the studied LMWOA.4.1. Bulk and nanoscale structure of Fe(III)-precipitates
The extent to which the ligands affected the precipitate structure increased along the sequence 2,4-DHB < Galact ≪ 3,4-DHB < Citr and with increasing (OC/Fe)init. Their primary effect was to induce the formation of less and less crystalline Lp in favor of increasing fractions of Fh, similar to the effect of SiO4. The much weaker effects of 2,4-DHB and Galact than of 3,4-DHB and Citr can be attributed to the fact that both 2,4-DHB and Galact have only a single carboxyl group (and 2,4-DHB two separate phenolic hydroxyl groups), whereas 3,4-DHB can form catechol-type complexes with Fe(III) via its two neighboring phenolic hydroxyl groups22 and Citr can form chelate complexes involving two of its carboxyl and its hydroxyl group.47 Analogous differences in the effects of 3,4-DHB relative to 2,4-DHB and of Citr relative to Galact have been reported in studies on their effects on ferrihydrite-type Fe(III)-precipitates formed by forced Fe(III) hydrolysis.22,23,27,30 The LCF results sorted by increasing (ligand/Fe)ppt (Fig. 12) indicated that the effects of the ligands on Fe(III)-precipitate structure increased relativey consistently with increasing (ligand/Fe)ppt, irrespective of ligand type, and were mostly additive to the effects of PO4.
 | ||
| Fig. 12 Fe K-edge EXAFS LCF results for Ca (a and c) and Na (b and d) electrolytes at (P/Fe)init 0.05 (a and b) and 0.25 (c and d), sorted along increasing (ligand/Fe)ppt. The (ligand/Fe)ppt of each sample is indicated on the top of each column; the nominal (OC/Fe)init at which precipitates were formed is indicated in parentheses behind the ligand name. | ||
For 2,4-DHB and Galact, the combined STEM and XAS data show that sequential precipitation resulted in core–shell (Ca)FeP–Fh particles, with attached Lp platelets in treatments where Lp could still form. For 3,4-DHB, the results offered evidence for catechol-type Fe(III) complexation in the precipitates and for the formation of dissolved Fe(III)–catechol complexes in solution at high (OC/Fe)init. Sequential precipitate formation was still observed at lower (OC/Fe)init, whereas at high (OC/Fe)init, formation of dissolved Fe(III)–3,4-DHB complexes and CaFeP–3,4-DHB co-precipitates occurred in parallel. Citr had a stronger effect on Fe(III)-precipitate structure than 3,4-DHB, even in the Ca electrolytes where it also forms complexes with Ca. On the other hand, Citr at high (OC/Fe)init can promote the formation of oligomeric Fe(III)–Citr complexes that may bind PO4,23 whereas 3,4-DHB induces the formation of monomeric Fe(III)–3,4-DHB complexes that did not complex PO4 in our experiments.
Galact has only one carboxyl group like 2,4-DHB (Fig. S1†) and speciation calculations suggested slightly stronger Fe(III) binding by 2,4-DHB than by Galact (Table S2†). Therefore, the markedly stronger effect of Galact than 2,4-DHB on Fe(III)-precipitate structure at equal (OC/Fe)init was unexpected. One explanation could be that Galact forms a chelate complex with Fe(III) via its carboxyl group and ring O atom at near-neutral pH.46 Alternatively, by reaction with Fe(III), a minor amount of Galact could have become oxidized to compounds with two carboxyl groups46,67 that complex Fe(III) more strongly, an interpretation supported by the more negative zeta potentials of co-precipitates formed in the presence of Galact than in the presence of 2,4-DHB (see Section 4.3.1†).
At (OC/Fe)init 0.6 in Ca electrolyte, the precipitate formed at (P/Fe)init 0.25 consisted of core–shell CaFeP–Fh particles (Fig. 7). Because nearly all PO4 but only ∼63% of the 3,4-DHB became co-precipitated with Fe(III) (Table S5†), this suggested that, at an initial PO4 concentration (0.125 mM) exceeding the initial 3,4-DHB concentration (0.042 mM) by factor ∼3, CaFeP formed first, followed by Fh. At (P/Fe)init 0.05 and (OC/Fe)init 0.6, the precipitate consisted of Fh with attached Lp sheets (Fig. 7, S14 and S16†); indicating that strong binding of 3,4-DHB with Fe(III), similar to PO4, leads to effective 3,4-DHB uptake into the precipitate and thereby limits the ability of 3,4-DHB to prevent Lp nucleation.
At (OC/Fe)init 9.6 in Ca electrolytes, only ∼48% of the total Fe was retained by filtration, together with nearly all PO4 but only 21–28% of the total 3,4-DHB (Fig. 9 and Table S5†). At (P/Fe)init 0.05, XAS (Fig. 3) and XRD (Fig. 4) data and the rough particle surface observed by STEM (Fig. 6m) showed that the retained precipitate consisted mainly of Fh. At a (P/Fe)init of 0.25, on the other hand, XRD data (Fig. 4) and the smooth particle surface (Fig. 6o) suggested the formation of mainly CaFeP, whereas the LCF results pointed to a significant fraction of 3,4-DHB-complexed Fe(III). Based on its high 3,4-DHB content (Ca0.20FeP0.51(3,4-DHB)0.31; Table S5†) and lack of nanoscale heterogeneity (Fig. 6o and p), the precipitate Ca 0.25 3,4-DHB 9.6 could be referred to as Ca–PO4–Fe(III)–3,4-DHB co-precipitate. The filtered solutions from the experiments at (OC/Fe)init 9.6 in Ca 0.05 and Ca 0.25 electrolytes exhibited a distinct blue color indicative of dissolved Fe(III)–3,4-DHB complexes68 and contained 3,4-DHB and Fe at a molar ratio of ∼2.2, consistent with the expected formation of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex (Table S1†). As the blue color started to evolve right after Fe(II) addition, we postulate that, at an initial 3,4-DHB concentration (0.69 mM) exceeding initial PO4 concentration ∼6-fold (Ca 0.25) to ∼27-fold (Ca 0.05), co-precipitation of Ca, PO4 and 3,4-DHB into a filterable Fe(III)-precipitate and formation of PO4-free dissolved Fe(III)–3,4-DHB complexes proceeded in parallel.
:1 complex (Table S1†). As the blue color started to evolve right after Fe(II) addition, we postulate that, at an initial 3,4-DHB concentration (0.69 mM) exceeding initial PO4 concentration ∼6-fold (Ca 0.25) to ∼27-fold (Ca 0.05), co-precipitation of Ca, PO4 and 3,4-DHB into a filterable Fe(III)-precipitate and formation of PO4-free dissolved Fe(III)–3,4-DHB complexes proceeded in parallel.
4.2. Co-precipitation of PO4, OC and Ca with Fe(III) and residual dissolved PO4 and OC
In Fig. 13, the (OC/Fe)ppt values are plotted as a function of residual dissolved OC for all organic ligands in Ca and Na electrolytes at both (P/Fe)init. The data reflect the decreasing Fe(III) binding strength along the sequence Citr > 3,4-DHB ≫ Galact > 2,4-DHB. The data further show that the co-precipitation of all LMWOAs was lower at (P/Fe)init 0.25 than 0.05, in line with trends in the FTIR spectra (Fig. 5 and S8†), revealing a higher affinity of PO4 than the studied organic ligands for binding with Fe(III). This observation is in line with competitive adsorption studies that showed that adsorption of PO4 outcompetes Citr on goethite32 and that only stronger organic ligands such as mellitic acid can outcompete PO4.71,72
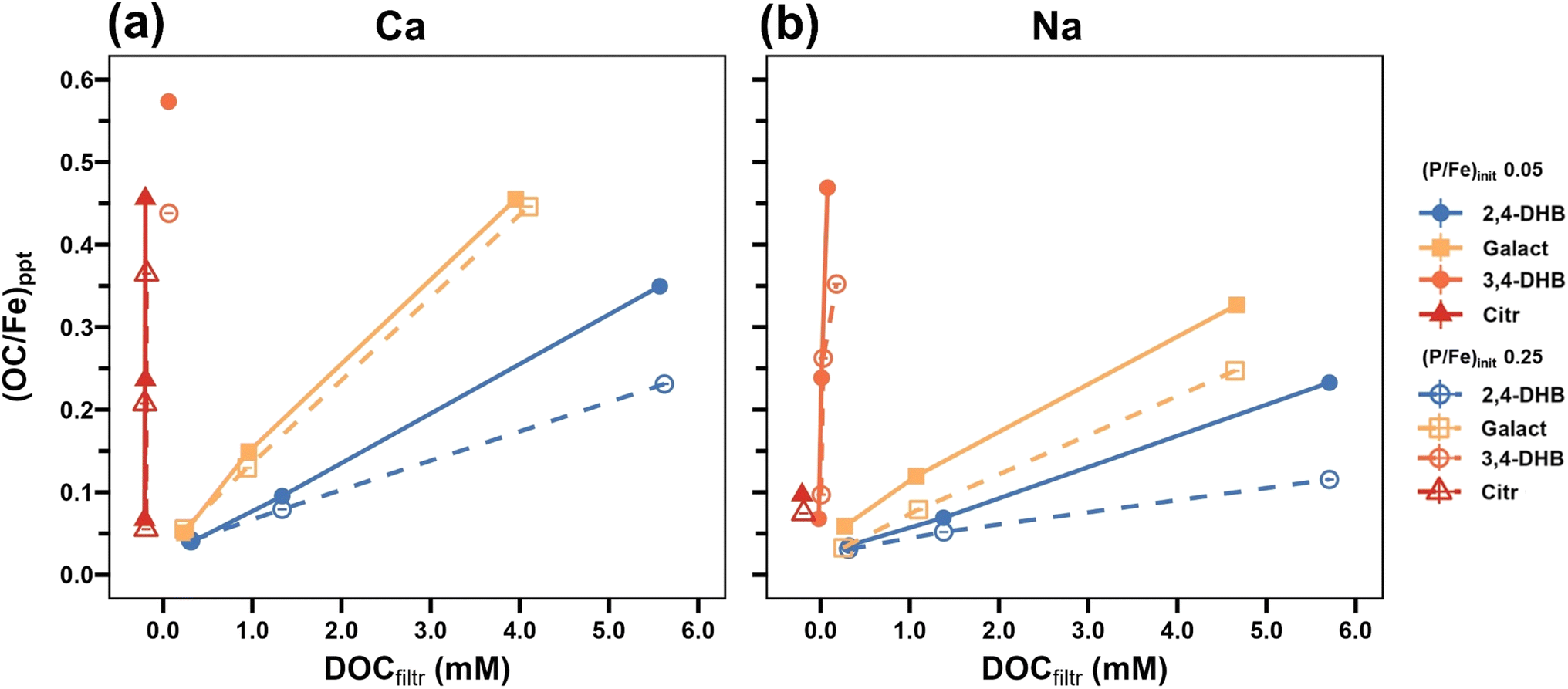 | ||
| Fig. 13 Molar precipitate OC/Fe ratio as a function of residual DOC in suspensions from experiments with 2,4-DHB, Galact, 3,4-DHB and Citr at a (P/Fe)init of 0.05 and 0.25 in (a) Ca- and (b) Na-electrolytes. The Citr results are shown with an x-axis offset of −0.2 for clarity. | ||
The presence of Ca also significantly enhanced LMWOA co-precipitation (Fig. 13), in line with higher organic ligand adsorption onto Fe(III)–(hydr)oxides in the presence of Ca.41,74 At the same time, the amounts of co-precipitated Ca (in general) increased with (OC/Fe)init (Fig. 1 and Table S5†). These synergistic effects can be explained by favorable electrostatic interactions between bivalent Ca and the organic ligands,40,75 direct Ca complexation by the organic ligands,76 and the formation of ternary Fe–OM–Ca complexes.77 Thereby, Ca may also indirectly promote PO4 co-precipitation, in analogy to enhanced AsO4 uptake by Fe(III)–OM co-precipitates in the presence of Ca.40
4.3. Particle zeta potential and aggregate size
With increasing (OC/Fe)init, both Citr and 3,4-DHB caused a substantial decrease in particle ZP, especially in the absence of Ca (Fig. 8), and thereby limited primary particle growth and aggregate formation (Tables S11 and S12†). Above a certain (OC/Fe)init, this led to a marked increase in the fractions of Fe(III) and PO4 that passed 0.2 μm filter membranes (Fig. 9 and 10).
4.4. Formation of OC-stabilized Fe(III)-colloids and dissolved Fe(III)–organic complexes
In experiments with Citr, hardly any Fe(III) could be retained by 0.2 μm filtration at (OC/Fe)init ≥ 2.4 in Ca 0.05 and 0.25 electrolytes and at (OC/Fe)init ≥ 0.3 in Na electrolytes (Fig. 9). Considering that (OC/Fe)init ratios of 0.3, 2.4 and 9.6 correspond to (Citr/Fe)init ratios of 0.05, 0.4 and 1.6 and assuming a molar Citr/Fe ratio of 1:1 for the dominant dissolved Fe(III)–Citr complexes (Fe2L2 or Fe3L3),47 it is evident that Citr-stabilized Fe(III)-colloids must have accounted for most of the filter-passing Fe(III) at an (OC/Fe)init of 0.3 and 2.4 and that dissolved Fe(III)–Citr complexes could only have dominated at (OC/Fe)init 9.6. Considering that AsO4 can bind to Fe(III)-trimers in ternary AsO4–Fe(III)–Citr complexes,23,70,81 we postulate that increasing (Citr/Fe)init led to a concomitant increase in the colloidal stability of the Fe(III)-precipitates, a decrease in primary Fe(III)-precipitate particle size towards Citr-stabilized Fe(III)-polymers, and an increasing amount of oligomeric Fe(III)–Citr complexes. The close correlation between filter-passing Fe(III) and PO4 pointed to a similar P/Fe in these different Fe(III) fractions.
In the case of 3,4-DHB, an (OC/Fe)init of 2.4 corresponded to a (3,4-DHB/Fe)init of 0.34, suggesting that filter-passing Fe(III) in the Na electrolytes could be mostly attributed to PO4-loaded Fe(III)-precipitate colloids (Fig. 9). For an (OC/Fe)init of 9.6 (i.e., (3,4-DHB/Fe)init of 1.4), on the other hand, UV-Vis spectra of 0.2 μm filtered suspensions from the Ca 0.05 3,4-DHB 9.6 and Na 0.05 3,4-DHB 9.6 treatments68,82 pointed to the formation of dissolved Fe(III)–catechol complexes68,82 (ESI Section 11†), in line with the deep blue color of these suspensions (Fig. S20†). In Na electrolytes, filter-passing PO4 closely followed Fe(III) (Fig. 9 and 10). In the Ca electrolytes at (OC/Fe)init 2.4 and 9.6, in contrast, PO4 retained in 0.2 μm filterable Fh (Ca 0.05 3,4-DHB 9.6) or CaFeP (Ca 0.25 3,4-DHB 9.6), whereas a significant portion of the Fe(III) passed the 0.2 μm membranes. Thus, Ca induced the formation of PO4-loaded Fe(III)-precipitate particles that could be separated from soluble Fe(III)–3,4-DHB complexes by filtration, whereas colloidal Fe(III)-particles formed in the Na electrolytes passed the filter membranes together with the Fe(III)–3,4-DHB complexes.
4.5. Effects of macromolecular LH in comparison to LMWOAs
Even at (OC/Fe)init 9.6, ∼15% of the Fe still formed Lp (Fig. 3), mostly as unstacked Lp nanosheets, as observed for 2,4-DHB (Fig. 3 and 4). Such nano-Lp has previously been identified in precipitates formed by the oxidation of anoxic DOM-containing soil solution86 and in Fe–organic flocs from a peatland87 and was also shown to form during Fe(II)-catalyzed transformation of Fh–OM co-precipitates.30,44 At (OC/Fe)init 0.6 in Ca electrolytes, LH promoted the formation of core–shell (Ca)FeP–Fh particles at (P/Fe)init 0.25 and PO4-rich Fh particles at (P/Fe)init 0.05 with attached Lp platelets (Fig. S11†), matching the morphology of precipitates formed in the presence of 2,4-DHB (Fig. 7). At (OC/Fe)init 9.6, however, the coagulation of LH by Ca61 resulted in LH flocs with diffusely embedded nanoscale Fe(III)-particles (Fig. S11†). Thus, coagulated LH effectively inhibited Fe(III)-precipitate growth, in line with recent work on the effects of Ca on Fe(III)–humic-acid co-precipitates.21,40
In the Ca electrolytes, co-precipitation of LH with Fe(III) at (P/Fe)init 0.25 was only marginally lower than that at (P/Fe)init 0.05, in contrast to the co-precipitation of the LMWOA (Fig. 1, 13 and Table S5†). This indicated that Ca-induced LH coagulation was more relevant for LH co-precipitation than LH competition with PO4 for binding to Fe(III), in line with enhanced Ca co-precipitation with LH (Fig. 1) and with a recent study on structural effects of Ca on Fe(III)–humic acid co-precipitates.21,40 In the presence of Ca and LH, enhanced PO4 co-precipitation could, to some extent, also have resulted from ternary PO4–Ca–LH complexation.90
In the Na electrolytes, on the other hand, filter-passing LH-stabilized Fe(III)-colloids formed at (OC/Fe)init above 0.6. For (OC/Fe)init 9.6, STEM-EDX and DLS indicated the formation of nano-sized Fe(III)-precipitates (Fig. S10m and n†) with diameters of ∼150 nm (Table S12†). Consistent with these results, a recent study showed that Fe(II) oxidation in the presence of a humic acid (1 mM NaCl, pH 6, 0.09 mM Fe(II) and ∼0.03 (P/Fe)) indicated a transition from unstable Fe(III)-colloids at (OC/Fe)init ∼ 1 to very small and stably suspended Fe(III)-colloids at (OC/Fe)init > 10.89
5. Environmental implications
This study aimed to assess how organic ligands in combination with PO4 and Ca impact the composition and structure of Fe(III)-precipitates formed by Fe(II) oxidation in near-neutral natural waters, for example at sites of anoxic soil or groundwater exfiltration into surface waters,11,12 in groundwater treatment for Fe removal, at redoxclines in lakes91,92 or in the rhizosphere of wetland plants.93 The concentrations of Fe(II) (28 mg L−1), PO4 (0.8–3.9 mg L−1 P), OC (0.6–59 mg L−1) and Ca (160 mg L−1), the molar ratios (OC/Fe)init and (P/Fe)init, and bicarbonate/CO2 as pH buffer were selected to mimic conditions in anoxic soil porewaters or groundwater upon oxygenation and to strengthen the transferability of the results to field systems. For 2,4-DHB and Citr as representative weakly and strongly Fe(III)-binding ligands, Fig. 14 schematically depicts their effects on the nanoscale structure of Fe(III)-precipitates as a function of (OC/Fe)init at (P/Fe)init of 0.05 and 0.25 in Ca and Na electrolytes. | ||
| Fig. 14 Effects of weakly Fe(III)-binding 2,4-DHB and strongly Fe(III)-binding Citr on nanoscale Fe(III)-precipitate structure as a function of (OC/Fe)init in Ca and Na electrolytes at (P/Fe)init 0.05 or 0.25. | ||
The results for 2,4-DHB can be considered representative for monocarboxylic acids and other weakly Fe(III)-binding ligands. They show that, upon initial (Ca)FeP formation, 2,4-DHB promotes the formation of Fh instead of Lp during continuing Fe(II) oxidation and thereby enhances the retention of co-precipitated PO4, in analogy to the mechanism by which SiO4 enhances PO4 retention.8,13 Even at high (OC/Fe)init, 2,4-DHB cannot compete with PO4 for binding with Fe(III) or infer a high negative charge to Fe(III)-precipitate particles and therefore cannot inhibit PO4 co-precipitation or induce the formation of mobile PO4-loaded Fe(III)-colloids. Relating the concentration-dependent effects of 2,4-DHB to reported effects of SiO4, and considering that concentrations of monocarboxylic acids in soil pore water may reach up to 1 mM (ref. 45 and 94) while SiO4 typically varies in the range 0.1–1 mM, effects of such weak organic ligands and SiO4 on Fe(III)-precipitate formation and PO4 sequestration are expected to be additive, their individual contributions depending on the specific conditions.
For Citr as a strongly Fe(III)-binding ligand, our results show similar effects on Fe(III)-precipitate structure and PO4 sequestration at low (OC/Fe)init to low-affinity ligands such as 2,4-DHB at high (OC/Fe)init. Thus, even if concentrations of di- and tricarboxylic acids in soil pore waters are often much lower (<0.05 mM) than concentrations of monocarboxylic LMWOAs (<1 mM),45,94 they may still add to the effects of monocarboxylic acids and SiO4 discussed before.
In the soil rhizosphere, plant roots exude ligands with high Fe(III) binding strength for nutrient acquisition, and Citr can reach high concentrations (up to 0.8 mM).95 In cases where Fe(II) proceeds in the presence of Citr at high (OC/Fe)init, Citr (or other strong ligands) form complexes with Fe(III) in parallel with PO4, resulting in mixed (Ca–)Fe(III)–PO4–OC co-precipitates in which individual particles represent aggregates of (sub)nanometric Fe(III)–OC and Fe(III)–PO4 polymers. Because Citr and 3,4-DHB infer a high negative particle charge and can form dissolved complexes with Fe(III) in oligomeric or monomeric form, they can induce the formation of mobile PO4- and OC-loaded Fe(III)-polymers or (PO4–)Fe(III)–ligand complexes that promote PO4 and OC mobility.
Humic acids are macromolecular compounds with a high number of rather weak Fe(III)-binding groups (e.g., isolated carboxyl groups) and lower numbers of high-affinity configurations of functional groups that can strongly bind Fe(III) in chelate complexes. At low (OC/Fe)init, we therefore observed effects of LH on Fe(III)-precipitate structure and PO4 uptake similar to the effects of 2,4-DHB. For typical concentrations of DOC (<0.5 mM) and SiO4 (0.1–1 mM) in anoxic groundwaters, our results suggest that SiO4 may often have a stronger impact on Fe(III)-precipitate structure and PO4 co-precipitation than DOC. In DOC-rich near-neutral environments such as fens4 or peak lake sediments,36 in contrast, effects of humic compounds may often dominate effects of SiO4. At representative molar OC/Fe ratios (∼10)4 in the presence of Ca, humic acids can stabilize individual (sub)nanometric PO4-loaded Fh or (Ca)FeP polymers in coagulated humic acid networks and prevent their aggregation into precipitate particles, as recently documented for Ca–Fe(III)–PO4–OC associations in the sediment of a peat lake.36 At even higher OC/Fe ratios (50 to 500; not studied here), humic acids increasingly form chelate complexes with monomeric or oligomeric Fe(III) via carboxyl or phenolic hydroxyl groups. Depending on the concentratino of Ca as coagulating cation, these Fe(III)-OC associations can occur in coagulated humic acid flocs or as mobile Fe(III)–PO4–OC colloids.
Co-precipitation of OC with Fe(III) during anoxic/oxic transitions plays an important role in OC storage in sediments and soils, OC transfer from soils and aquifers to surface waters, and hence in global C cycling.4,5 Carboxyl-rich OC has been shown to readily co-precipitate with Fe(III).96 Our results confirm the limited co-precipitation of monocarboxylic ligands such as 2,4-DHB and Galact with Fe(III). In addition, they demonstrate that not only ligands with several carboxyl groups such as Citr but also ligands that form catecholate-type complexes such as 3,4-DHB effectively co-precipitate with Fe(III). The formation of colloidally stable Fe(III)–OC co-precipitates or Fe(III)–OC complexes in the presence of strong ligands at high (OC/Fe)init, however, may limit OC retention at anoxic/oxic boundaries and enhance OC mobility in surface waters.43,97 The results of this study further emphasize that high (P/Fe)init ratios may effectively limit the co-precipitation of OC with Fe, whereas Ca can enhance the co-precipitation of OC via its effects on both OC and PO4 uptake and on particle charge.
In summary, this study shows that the type and concentration of OC and the interdependent effects of OC, PO4, SiO4 and Ca must be taken into account in studies on Fe(III)-precipitate formation in near-neutral bicarbonate-buffered waters and on the sequestration of PO4 and OC by Fe oxidation products. The compositional and (nanoscale) structural diversity of Fe(III)-precipitates formed in the presence of OC, PO4, Ca and other solutes must also be considered with respect to their transformation over time or the co-precipitation and cycling of other compounds, including essential or toxic trace elements.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Numa Pfenninger, Irene Brunner, Brian Sinnet, Anna Hasenfratz, Mischa Romer, and Jennifer Eberhard (Eawag, Switzerland) are acknowledged for assistance with laboratory experiments and analyses. The light source of the Karlsruhe Institute of Technology (KIT, Karlsruhe, Germany) and Swiss Light Source (SLS, Villigen, Switzerland) are acknowledged for providing synchrotron beamtime. Christian Mikutta (Leibnitz University Hannover, Germany) is acknowledged for sharing the XAS spectrum of an Fe(III)–3,4-DHB co-precipitate. The Scientific Center for Optical and Electron Microscopy (ScopeM) at ETH Zurich is acknowledged for granting access to electron microscopy. This project was funded via the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 813438 (P-TRAP).References
- A. Kappler, C. Bryce, M. Mansor, U. Lueder, J. M. Byrne and E. D. Swanner, An evolving view on biogeochemical cycling of iron, Nat. Rev. Microbiol., 2021, 19, 360–374 CrossRef CAS PubMed.
- V. H. Smith, Eutrophication of freshwater and coastal marine ecosystems a global problem, Environ. Sci. Pollut. Res., 2003, 10, 126–139 CrossRef CAS PubMed.
- K. Lalonde, A. Mucci, A. Ouellet and Y. Gélinas, Preservation of organic matter in sediments promoted by iron, Nature, 2012, 483, 198–200 CrossRef CAS PubMed.
- T. Riedel, D. Zak, H. Biester and T. Dittmar, Iron traps terrestrially derived dissolved organic matter at redox interfaces, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10101–10105 Search PubMed.
- J. D. Hemingway, D. H. Rothman, K. E. Grant, S. Z. Rosengard, T. I. Eglinton, L. A. Derry and V. V. Galy, Mineral protection regulates long-term global preservation of natural organic carbon, Nature, 2019, 570, 228–231 CrossRef CAS PubMed.
- D. T. Mayer and W. M. Jarrell, Phosphorus sorption during iron(II) oxidation in the presence of dissolved silica, Water Res., 2000, 34, 3949–3956 CrossRef.
- A. Voegelin, A.-C. Senn, R. Kaegi, S. J. Hug and S. Mangold, Dynamic Fe-precipitate formation induced by Fe(II) oxidation in aerated phosphate-containing water, Geochim. Cosmochim. Acta, 2013, 117, 216–231 CrossRef CAS.
- A.-C. Senn, R. Kaegi, S. J. Hug, J. G. Hering, S. Mangold and A. Voegelin, Composition and structure of Fe(III)-precipitates formed by Fe(II) oxidation in near-neutral water: interdependent effects of phosphate, silicate and Ca, Geochim. Cosmochim. Acta, 2015, 162, 220–246 CrossRef CAS.
- C. M. van Genuchten, A. J. Gadgil and J. Peña, Fe(III) nucleation in the presence of bivalent cations and oxyanions leads to subnanoscale 7 Å polymers, Environ. Sci. Technol., 2014, 48, 11828–11836 CrossRef CAS PubMed.
- A. Voegelin, R. Kaegi, J. Frommer, D. Vantelon and S. J. Hug, Effect of phosphate, silicate, and Ca on Fe(III)-precipitates formed in aerated Fe(II)- and As(III)-containing water studied by X-ray absorption spectroscopy, Geochim. Cosmochim. Acta, 2010, 74, 164–186 CrossRef CAS.
- S. Baken, C. Moens, B. van der Grift and E. Smolders, Phosphate binding by natural iron-rich colloids in streams, Water Res., 2016, 98, 326–333 CrossRef CAS PubMed.
- B. van der Grift, T. Behrends, L. A. Osté, P. P. Schot, M. J. Wassen and J. Griffioen, Fe hydroxyphosphate precipitation and Fe(II) oxidation kinetics upon aeration of Fe(II) and phosphate-containing synthetic and natural solutions, Geochim. Cosmochim. Acta, 2016, 186, 71–90 CrossRef CAS.
- V. V. Nenonen, R. Kaegi, S. J. Hug, J. Göttlicher, S. Mangold, L. H. Winkel and A. Voegelin, Formation and transformation of Fe(III)- and Ca-precipitates in aqueous solutions and effects on phosphate retention over time, Geochim. Cosmochim. Acta, 2023, 360, 207–230 CrossRef CAS.
- T. Karlsson and P. Persson, Coordination chemistry and hydrolysis of Fe(III) in a peat humic acid studied by X-ray absorption spectroscopy, Geochim. Cosmochim. Acta, 2010, 74, 30–40 CrossRef CAS.
- T. Karlsson and P. Persson, Complexes with aquatic organic matter suppress hydrolysis and precipitation of Fe(III), Chem. Geol., 2012, 322–323, 19–27 CrossRef CAS.
- D. Vantelon, M. Davranche, R. Marsac, C. La Fontaine, H. Guénet, J. Jestin, G. Campaore, A. Beauvois and V. Briois, Iron speciation in iron–organic matter nanoaggregates: a kinetic approach coupling Quick-EXAFS and MCR-ALS chemometrics, Environ. Sci.: Nano, 2019, 6, 2641–2651 RSC.
- H. Guénet, M. Davranche, D. Vantelon, J. Gigault, S. Prévost, O. Taché, S. Jaksch, M. Pédrot, V. Dorcet and A. Boutier, Characterization of iron–organic matter nano-aggregate networks through a combination of SAXS/SANS and XAS analyses: impact on As binding, Environ. Sci.: Nano, 2017, 4, 938–954 RSC.
- M. Sodano, C. Lerda, R. Nisticò, M. Martin, G. Magnacca, L. Celi and D. Said-Pullicino, Dissolved organic carbon retention by coprecipitation during the oxidation of ferrous iron, Geoderma, 2017, 307, 19–29 CrossRef CAS.
- C. Chen, J. J. Dynes, J. Wang and D. L. Sparks, Properties of Fe-organic matter associations via coprecipitation versus adsorption, Environ. Sci. Technol., 2014, 48, 13751–13759 CrossRef CAS PubMed.
- M. Kleber, K. Eusterhues, M. Keiluweit, C. Mikutta, R. Mikutta and P. S. Nico, Mineral–Organic Associations: Formation, Properties, and Relevance in Soil Environments, Adv. Agron., 2015, 130, 1–140 Search PubMed.
- A. Beauvois, D. Vantelon, J. Jestin, C. Rivard, M. Bouhnik-Le Coz, A. Dupont, V. Briois, T. Bizien, A. Sorrentino and B. Wu, How does calcium drive the structural organization of iron–organic matter aggregates? A multiscale investigation, Environ. Sci.: Nano, 2020, 7, 2833–2849 RSC.
- C. Mikutta, X-ray absorption spectroscopy study on the effect of hydroxybenzoic acids on the formation and structure of ferrihydrite, Geochim. Cosmochim. Acta, 2011, 75, 5122–5139 CrossRef CAS.
- C. Mikutta, J. Frommer, A. Voegelin, R. Kaegi and R. Kretzschmar, Effect of citrate on the local Fe coordination in ferrihydrite, arsenate binding, and ternary arsenate complex formation, Geochim. Cosmochim. Acta, 2010, 74, 5574–5592 CrossRef CAS.
- C. Liu and P. Huang, Atomic force microscopy and surface characteristics of iron oxides formed in citrate solutions, Soil Sci. Soc. Am. J., 1999, 63, 65–72 CrossRef CAS.
- R. Porras, C. H. Pries, M. Torn and P. Nico, Synthetic iron(hydr)oxide-glucose associations in subsurface soil: effects on decomposability of mineral associated carbon, Sci. Total Environ., 2018, 613, 342–351 CrossRef PubMed.
- B. Yang, N. Graham, P. Liu, M. Liu, J. Gregory and W. Yu, Atomic-Level Structural Differences between Fe(III) Coprecipitates Generated by the Addition of Fe(III) Coagulants and by the Oxidation of Fe(II) Coagulants Determine Their Coagulation Behavior in Phosphate and DOM Removal, Environ. Sci. Technol., 2023, 57, 12489–12500 CrossRef CAS PubMed.
- C. Mikutta, R. Mikutta, S. Bonneville, F. Wagner, A. Voegelin, I. Christl and R. Kretzschmar, Synthetic coprecipitates of exopolysaccharides and ferrihydrite. Part I: Characterization, Geochim. Cosmochim. Acta, 2008, 72, 1111–1127 CrossRef CAS.
- C. S. Chan, S. C. Fakra, D. C. Edwards, D. Emerson and J. F. Banfield, Iron oxyhydroxide mineralization on microbial extracellular polysaccharides, Geochim. Cosmochim. Acta, 2009, 73, 3807–3818 CrossRef CAS.
- T. D. Sowers, K. L. Holden, E. K. Coward and D. L. Sparks, Dissolved organic matter sorption and molecular fractionation by naturally occurring bacteriogenic iron(oxyhydr)oxides, Environ. Sci. Technol., 2019, 53, 4295–4304 CrossRef CAS PubMed.
- L. K. ThomasArrigo, R. Kaegi and R. Kretzschmar, Ferrihydrite growth and transformation in the presence of ferrous iron and model organic ligands, Environ. Sci. Technol., 2019, 53, 13636–13647 CrossRef CAS PubMed.
- K. Eusterhues, F. E. Wagner, W. Häusler, M. Hanzlik, H. Knicker, K. U. Totsche, I. Kögel-Knabner and U. Schwertmann, Characterization of ferrihydrite-soil organic matter coprecipitates by X-ray diffraction and Mossbauer spectroscopy, Environ. Sci. Technol., 2008, 42, 7891–7897 CrossRef CAS PubMed.
- J. S. Geelhoed, T. Hiemstra and W. H. Van Riemsdijk, Competitive interaction between phosphate and citrate on goethite, Environ. Sci. Technol., 1998, 32, 2119–2123 CrossRef CAS.
- Y. Mao, A. Ninh Pham, Y. Xin and T. David Waite, Effects of pH, floc age and organic compounds on the removal of phosphate by pre-polymerized hydrous ferric oxides, Sep. Purif. Technol., 2012, 91, 38–45 CrossRef CAS.
- M. Spohn, K. Diáková, F. Aburto, S. Doetterl and J. Borovec, Sorption and desorption of organic matter in soils as affected by phosphate, Geoderma, 2022, 405, 115377 CrossRef CAS.
- J. Gerke and R. Hermann, Adsorption of orthophosphate to humic-Fe-complexes and to amorphous Fe-oxide, Zeitschrift für Pflanzenernährung und Bodenkunde, 1992, 155, 233–236 CrossRef CAS.
- M. A. Münch, A. Voegelin, L. C. C. Hurtarte, J. Göttlicher and T. Behrends, Combining sequential extractions with bulk and micro X-ray spectroscopy to elucidate iron and phosphorus speciation in sediments of an iron-treated peat lake, Environ. Sci.: Processes Impacts, 2025, 27, 563–585 RSC.
- T. Hiemstra, J. Antelo, A. M. D. van Rotterdam and W. H. van Riemsdijk, Nanoparticles in natural systems II: the natural oxide fraction at interaction with natural organic matter and phosphate, Geochim. Cosmochim. Acta, 2010, 74, 59–69 CrossRef CAS.
- T. Hiemstra, J. Antelo, R. Rahnemaie and W. H. van Riemsdijk, Nanoparticles in natural systems I: the effective reactive surface area of the natural oxide fraction in field samples, Geochim. Cosmochim. Acta, 2010, 74, 41–58 CrossRef CAS.
- D. Adhikari, T. Sowers, J. W. Stuckey, X. Wang, D. L. Sparks and Y. Yang, Formation and redox reactivity of ferrihydrite-organic carbon-calcium co-precipitates, Geochim. Cosmochim. Acta, 2019, 244, 86–98 CrossRef CAS.
- A. Beauvois, D. Vantelon, J. Jestin, M. Bouhnik-Le Coz, C. Catrouillet, V. Briois, T. Bizien and M. Davranche, How crucial is the impact of calcium on the reactivity of iron-organic matter aggregates? Insights from arsenic, J. Hazard. Mater., 2021, 404, 124127 CrossRef CAS PubMed.
- T. D. Sowers, J. W. Stuckey and D. L. Sparks, The synergistic effect of calcium on organic carbon sequestration to ferrihydrite, Geochem. Trans., 2018, 19, 1–11 CrossRef PubMed.
- A.-C. Senn, R. Kaegi, S. J. Hug, J. G. Hering, S. Mangold and A. Voegelin, Effect of aging on the structure and phosphate retention of Fe(III)-precipitates formed by Fe(II) oxidation in water, Geochim. Cosmochim. Acta, 2017, 202, 341–360 CrossRef CAS.
- N. Gottselig, W. Amelung, J. W. Kirchner, R. Bol, W. Eugster, S. J. Granger, C. Hernández-Crespo, F. Herrmann, J. J. Keizer, M. Korkiakoski, H. Laudon, I. Lehner, S. Löfgren, A. Lohila, C. J. A. Macleod, M. Mölder, C. Müller, P. Nasta, V. Nischwitz, E. Paul-Limoges, M. C. Pierret, K. Pilegaard, N. Romano, M. T. Sebastià, M. Stähli, M. Voltz, H. Vereecken, J. Siemens and E. Klumpp, Elemental composition of natural nanoparticles and fine colloids in European forest stream waters and their role as phosphorus carriers, Global Biogeochem. Cycles, 2017, 31, 1592–1607 CrossRef CAS.
- L. K. ThomasArrigo, J. M. Byrne, A. Kappler and R. Kretzschmar, Impact of Organic Matter on Iron(II)-Catalyzed Mineral Transformations in Ferrihydrite-Organic Matter Coprecipitates, Environ. Sci. Technol., 2018, 52, 12316–12326 CrossRef CAS PubMed.
- D. L. Jones, Organic acids in the rhizosphere–a critical review, Plant Soil, 1998, 205, 25–44 CrossRef CAS.
- S. Deiana, C. Gessa, V. Solinas, P. Piu and R. Seeber, Analytical study of the interactions of D-galacturonic acid with iron(III) and iron(II) in solution and with iron(III)-bentonite, Anal. Chim. Acta, 1989, 222, 315–322 CrossRef.
- A. M. Silva, X. Kong, M. C. Parkin, R. Cammack and R. C. Hider, Iron(III) citrate speciation in aqueous solution, Dalton Trans., 2009, 8616–8625 RSC.
- J. D. Ritchie and E. M. Perdue, Proton-binding study of standard and reference fulvic acids, humic acids, and natural organic matter, Geochim. Cosmochim. Acta, 2003, 67, 85–96 CrossRef CAS.
- A. C. Senn, S. J. Hug, R. Kaegi, J. G. Hering and A. Voegelin, Arsenate co-precipitation with Fe(II) oxidation products and retention or release during precipitate aging, Water Res., 2018, 131, 334–345 CrossRef CAS PubMed.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, John Wiley & Sons, 2003 Search PubMed.
- J. Cumplido, V. Barrón and J. Torrent, Effect of phosphate on the formation of nanophase lepidocrocite from Fe(II) sulfate, Clays Clay Miner., 2000, 48, 503–510 CrossRef CAS.
- Y. Arai and D. L. Sparks, ATR–FTIR Spectroscopic Investigation on Phosphate Adsorption Mechanisms at the Ferrihydrite–Water Interface, J. Colloid Interface Sci., 2001, 241, 317–326 CrossRef CAS.
- P. Persson and K. Axe, Adsorption of oxalate and malonate at the water-goethite interface: molecular surface speciation from IR spectroscopy, Geochim. Cosmochim. Acta, 2005, 69, 541–552 CrossRef CAS.
- P. Borer, S. J. Hug, B. Sulzberger, S. M. Kraemer and R. Kretzschmar, Photolysis of citrate on the surface of lepidocrocite: an in situ attenuated total reflection infrared spectroscopy study, J. Phys. Chem. C, 2007, 111, 10560–10569 CrossRef CAS.
- H. Gulley-Stahl, P. A. Hogan, W. L. Schmidt, S. J. Wall, A. Buhrlage and H. A. Bullen, Surface complexation of catechol to metal oxides: an ATR-FTIR, adsorption, and dissolution study, Environ. Sci. Technol., 2010, 44, 4116–4121 CrossRef CAS PubMed.
- E. C. Yost, M. I. Tejedor-Tejedor and M. A. Anderson, In situ CIR-FTIR characterization of salicylate complexes at the goethite/aqueous solution interface, Environ. Sci. Technol., 1990, 24, 822–828 CrossRef CAS.
- M. S. Barreto, E. J. Elzinga and L. R. Alleoni, Attenuated total reflectance–Fourier transform infrared study of the effects of citrate on the adsorption of phosphate at the hematite surface, Soil Sci. Soc. Am. J., 2020, 84, 57–67 CrossRef CAS.
- M. Villalobos and J. O. Leckie, Surface complexation modeling and FTIR study of carbonate adsorption to goethite, J. Colloid Interface Sci., 2001, 235, 15–32 CrossRef CAS PubMed.
- R. Kaegi, A. Voegelin, D. Folini and S. J. Hug, Effect of phosphate, silicate, and Ca on the morphology, structure and elemental composition of Fe(III)-precipitates formed in aerated Fe(II) and As(III) containing water, Geochim. Cosmochim. Acta, 2010, 74, 5798–5816 CrossRef CAS.
- N. Kloster, M. Brigante, G. Zanini and M. Avena, Aggregation kinetics of humic acids in the presence of calcium ions, Colloids Surf., A, 2013, 427, 76–82 CrossRef CAS.
- E. Doelsch, J. Rose, A. Masion, J. Y. Bottero, D. Nahon and P. M. Bertsch, Speciation and crystal chemistry of iron(III) chloride hydrolyzed in the presence of SiO4 ligands. 1. An Fe K-edge EXAFS study, Langmuir, 2000, 16, 4726–4731 CrossRef CAS.
- J. Rose, A. Manceau, J.-Y. Bottero, A. Masion and F. Garcia, Nucleation and Growth Mechanisms of Fe Oxyhydroxide in the Presence of PO4 Ions. 1. Fe K-Edge EXAFS Study, Langmuir, 1996, 12, 6701–6707 CrossRef CAS.
- L. C. Roberts, S. J. Hug, T. Ruettimann, M. M. Billah, A. W. Khan and M. T. Rahman, Arsenic removal with iron(II) and iron(III) in waters with high silicate and phosphate concentrations, Environ. Sci. Technol., 2004, 38, 307–315 CrossRef CAS PubMed.
- M. A. Sabur, C. T. Parsons, T. Maavara and P. Van Cappellen, Effects of pH and dissolved silicate on phosphate mineral-water partitioning with goethite, ACS Earth Space Chem., 2021, 6, 34–43 CrossRef.
- U. Schwertmann and H. Thalmann, The influence of [Fe(II)], [Si], and pH on the formation of lepidocrocite and ferrihydrite during oxidation of aqueous FeCl2 solutions, Clay Miner., 1976, 11, 189–200 CrossRef CAS.
- S. Deiana, C. Gessa, V. Solinas, P. Piu and R. Seeber, Complexing and redox properties of the system D-galacturonic acid-iron(III), J. Inorg. Biochem., 1989, 35, 107–113 CrossRef CAS.
- M. Elhabiri, C. Carrër, F. Marmolle and H. Traboulsi, Complexation of iron(III) by catecholate-type polyphenols, Inorg. Chim. Acta, 2007, 360, 353–359 CrossRef CAS.
- M. Gracheva, Z. Klencsár, Z. Homonnay, Á. Solti, L. Péter, L. Machala, P. Novak and K. Kovács, Revealing the nuclearity of iron citrate complexes at biologically relevant conditions, BioMetals, 2023, 1–15 Search PubMed.
- P. Sharma, J. Ofner and A. Kappler, Formation of binary and ternary colloids and dissolved complexes of organic matter, Fe and As, Environ. Sci. Technol., 2010, 44, 4479–4485 CrossRef CAS PubMed.
- M. Lindegren and P. Persson, Competitive adsorption between phosphate and carboxylic acids: quantitative effects and molecular mechanisms, Eur. J. Soil Sci., 2009, 60, 982–993 CrossRef CAS.
- C. R. Evanko and D. A. Dzombak, Influence of structural features on sorption of NOM-analogue organic acids to goethite, Environ. Sci. Technol., 1998, 32, 2846–2855 CrossRef CAS.
- J. C. Mendez and T. Hiemstra, Ternary Complex Formation of Phosphate with Ca and Mg Ions Binding to Ferrihydrite: Experiments and Mechanisms, ACS Earth Space Chem., 2020, 4, 545–557 CrossRef CAS.
- M. S. C. Barreto, E. J. Elzinga, M. Ramlogan, A. A. Rouff and L. R. F. Alleoni, Calcium enhances adsorption and thermal stability of organic compounds on soil minerals, Chem. Geol., 2021, 559, 119804 CrossRef CAS.
- C. Moyne and T. Sterckeman, Effect of calcium and trace metals (Cd, Cu, Mn, Ni, Zn) on root iron uptake in relation to chemical properties of the root-excreted ligands, BioMetals, 2023, 36, 1013–1025 CrossRef CAS PubMed.
- B. Dinkelaker, V. Römheld and H. Marschner, Citric acid excretion and precipitation of calcium citrate in the rhizosphere of white lupin (Lupinus albus L.), Plant, Cell Environ., 1989, 12, 285–292 CrossRef CAS.
- M. A. Ali and D. A. Dzombak, Effects of simple organic acids on sorption of Cu2+ and Ca2+ on goethite, Geochim. Cosmochim. Acta, 1996, 60, 291–304 CrossRef CAS.
- P. Liao, W. Li, Y. Jiang, J. Wu, S. Yuan, J. D. Fortner and D. E. Giammar, Formation, aggregation, and deposition dynamics of NOM-iron colloids at anoxic–oxic interfaces, Environ. Sci. Technol., 2017, 51, 12235–12245 CrossRef CAS PubMed.
- A. Philippe and G. E. Schaumann, Interactions of dissolved organic matter with natural and engineered inorganic colloids: a review, Environ. Sci. Technol., 2014, 48, 8946–8962 CrossRef CAS PubMed.
- S. Dultz, H. Steinke, R. Mikutta, S. K. Woche and G. Guggenberger, Impact of organic matter types on surface charge and aggregation of goethite, Colloids Surf., A, 2018, 554, 156–168 CrossRef CAS.
- C. Mikutta and R. Kretzschmar, Spectroscopic evidence for ternary complex formation between arsenate and ferric iron complexes of humic substances, Environ. Sci. Technol., 2011, 45, 9550–9557 CrossRef CAS PubMed.
- G. Albarran, W. Boggess, V. Rassolov and R. H. Schuler, Absorption spectrum, mass spectrometric properties, and electronic structure of 1,2-benzoquinone, J. Phys. Chem. A, 2010, 114, 7470–7478 CrossRef CAS PubMed.
- J. P. Gustafsson, I. Persson, D. B. Kleja and J. W. Van Schaik, Binding of iron(III) to organic soils: EXAFS spectroscopy and chemical equilibrium modeling, Environ. Sci. Technol., 2007, 41, 1232–1237 CrossRef CAS PubMed.
- E. Orlowska, A. Roller, M. Pignitter, F. Jirsa, R. Krachler, W. Kandioller and B. K. Keppler, Synthetic iron complexes as models for natural iron-humic compounds: synthesis, characterization and algal growth experiments, Sci. Total Environ., 2017, 577, 94–104 CrossRef CAS PubMed.
- A. Vilgé-Ritter, J. Rose, A. Masion, J.-Y. Bottero and J.-M. Lainé, Chemistry and structure of aggregates formed with Fe-salts and natural organic matter, Colloids Surf., A, 1999, 147, 297–308 CrossRef.
- H. Guénet, M. Davranche, D. Vantelon, M. Pédrot, M. Al-Sid-Cheikh, A. Dia and J. Jestin, Evidence of organic matter control on As oxidation by iron oxides in riparian wetlands, Chem. Geol., 2016, 439, 161–172 CrossRef.
- L. K. ThomasArrigo, C. Mikutta, J. Byrne, K. Barmettler, A. Kappler and R. Kretzschmar, Iron and arsenic speciation and distribution in organic flocs from streambeds of an arsenic-enriched peatland, Environ. Sci. Technol., 2014, 48, 13218–13228 CrossRef CAS PubMed.
- W. Yang, W. Xiang, Z. Bao, C. Huang, M. Ma, X. Lu, L. Yao and Y. Wang, Phosphorus sorption capacity of various iron-organic matter associations in peat soils, Environ. Sci. Pollut. Res., 2022, 29, 77580–77592 CrossRef CAS PubMed.
- C. Moens and E. Smolders, Suwannee River Natural Organic Matter concentrations affect the size and phosphate uptake of colloids formed by iron oxidation, Geochim. Cosmochim. Acta, 2021, 312, 375–391 CrossRef CAS.
- Y. Audette, D. S. Smith, C. T. Parsons, W. Chen, F. Rezanezhad and P. Van Cappellen, Phosphorus binding to soil organic matter via ternary complexes with calcium, Chemosphere, 2020, 260, 127624 CrossRef CAS PubMed.
- A. Kleeberg, C. Herzog and M. Hupfer, Redox sensitivity of iron in phosphorus binding does not impede lake restoration, Water Res., 2013, 47, 1491–1502 CrossRef CAS PubMed.
- J. Cosmidis, K. Benzerara, G. Morin, V. Busigny, O. Lebeau, D. Jézéquel, V. Noël, G. Dublet and G. Othmane, Biomineralization of iron-phosphates in the water column of Lake Pavin (Massif Central, France), Geochim. Cosmochim. Acta, 2014, 126, 78–96 CrossRef CAS.
- J. Frommer, A. Voegelin, J. Dittmar, M. A. Marcus and R. Kretzschmar, Biogeochemical processes and arsenic enrichment around rice roots in paddy soil: results from micro-focused X-ray spectroscopy, Eur. J. Soil Sci., 2011, 62, 305–317 CrossRef.
- B. W. Strobel, Influence of vegetation on low-molecular-weight carboxylic acids in soil solution - a review, Geoderma, 2001, 99, 169–198 CrossRef CAS.
- J. Dessureault-Rompré, B. Nowack, R. Schulin and J. Luster, Spatial and temporal variation in organic acid anion exudation and nutrient anion uptake in the rhizosphere of Lupinus albus L, Plant Soil, 2007, 301, 123–134 CrossRef.
- L. Curti, O. W. Moore, P. Babakhani, K.-Q. Xiao, C. Woulds, A. W. Bray, B. J. Fisher, M. Kazemian, B. Kaulich and C. L. Peacock, Carboxyl-richness controls organic carbon preservation during coprecipitation with iron(oxyhydr)oxides in the natural environment, Commun. Earth Environ., 2021, 2, 229 CrossRef.
- C. Moens, D. Montalvo and E. Smolders, The concentration and size distribution of iron-rich colloids in pore waters are related to soil organic matter content and pore water calcium concentration, Eur. J. Soil Sci., 2021, 72, 2199–2214 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00313f |
| This journal is © The Royal Society of Chemistry 2025 |