
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into catalyst degradation during alkaline water electrolysis under variable operation†
Raul A. Marquez a,
Jay T. Benderb,
Ashton M. Alemancd,
Emma Kalokowskia,
Thuy Vy Lea,
Chloe L. Williamsonb,
Morten Linding Frederiksene,
Kenta Kawashimaa,
Chikaodili E. Chukwunekea,
Andrei Dolocanf,
Delia J. Millironabgh,
Joaquin Resascob,
Thomas F. Jaramillocd and
C. Buddie Mullins*abfgh
a,
Jay T. Benderb,
Ashton M. Alemancd,
Emma Kalokowskia,
Thuy Vy Lea,
Chloe L. Williamsonb,
Morten Linding Frederiksene,
Kenta Kawashimaa,
Chikaodili E. Chukwunekea,
Andrei Dolocanf,
Delia J. Millironabgh,
Joaquin Resascob,
Thomas F. Jaramillocd and
C. Buddie Mullins*abfgh
aDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: mullins@che.utexas.edu
bMcKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712, USA
cDepartment of Chemical Engineering, Stanford University, Stanford, CA 94305, USA
dSUNCAT Center for Interface Science and Catalysis, SLAC National Accelerator Laboratory, Menlo Park, CA 94025, USA
eDepartment of Biological & Chemical Engineering, Aarhus University, Aarhus N 8200, Denmark
fTexas Materials Institute, The University of Texas at Austin, Austin, Texas 78712, USA
gAllen J. Bard Center for Electrochemistry, The University of Texas at Austin, Austin, Texas 78712, USA
hH2@UT, The University of Texas at Austin, Austin, Texas 78712, USA
First published on 12th June 2025
Abstract
Energy conversion technologies that are key to decarbonization efforts face significant durability challenges due to variable operation. Understanding the impact of variable operation on catalytic stability and identifying the key variables that dictate degradation is crucial for developing robust technologies. Here, we present a comprehensive investigation of the effects of variable operation on liquid alkaline water electrolysis. Our findings reveal that variable operation induces severe degradation of Ni, Fe, and Co catalytic films that is not observed during steady-state operation. By systematically interrogating the electrode discharge during simulated shutdown tests using Raman spectroscopy and mass spectrometry techniques, we uncover significant alterations caused by reverse currents in real time. These include changes in crystal structure, composition, film thickness, electronic conductivity, and dissolution rates. Lab-scale electrolyzer experiments further highlight the impact of variable operation on catalyst materials under relevant conditions. Finally, we provide guidelines for leveraging these insights to advance electrocatalysis research. This work underscores the importance of integrating realistic stressors into stability testing and offers practical guidelines for catalyst design, performance evaluation, and industrial implementation. Collectively, insights from this study will drive the development of more resilient energy conversion technologies.
Broader contextMeeting society's growing energy demands while minimizing environmental impact requires transformative breakthroughs. Leveraging cost-effective renewable electricity from variable sources, such as wind and solar, offers a promising path to producing essential chemicals and energy carriers. Hydrogen, already key in many industries, plays a pivotal role in this transition. However, hydrogen is currently produced primarily from fossil fuels, necessitating a shift to more sustainable alternatives. Global investment in hydrogen technologies has surged, but success depends on developing efficient carbon-neutral solutions. Green hydrogen production via water electrolysis is a mature technology; however, integrating it with variable renewable energy presents new challenges. Electrolyzers must operate efficiently despite fluctuating power and withstand frequent startups and shutdowns, which are detrimental. Electrocatalysts, the core components of electrolyzers, are vulnerable to these conditions, experiencing corrosion and deactivation. Left unaddressed, variable operation can degrade performance, increase operating costs, and limit the deployment of water electrolysis. Therefore, understanding how variable operation affects electrocatalyst stability is crucial for developing resilient materials, designing robust devices, and standardizing accelerated stress tests. To underscore the impact of dynamic operating conditions and clarify the factors driving catalyst degradation, we investigate how transition metal-based electrocatalysts degrade during variable operation in liquid alkaline water electrolysis. |
Introduction
Transitioning to a low-carbon economy requires robust energy conversion technologies to support electrification and decarbonization efforts.1 Hydrogen is a key player in this transition due to its versatility and potential to reduce emissions in hard-to-electrify sectors, such as heavy industry and transportation.2–4 However, despite increasing investments,2 approximately 96% of hydrogen produced globally still comes from fossil fuels,5 underscoring the need to develop carbon-neutral alternatives. A promising approach is green hydrogen production via water electrolysis powered by renewable electricity. This technology is expected to expand rapidly, with global electrolysis capacity projected to grow a thousandfold by 2030.6Water electrolysis involves two reactions, each requiring catalysts: the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode.7,8 To circumvent the use of precious metals that are required when operating in acidic media,9 liquid alkaline water electrolysis (LAWE) offers a cost-effective solution by employing earth-abundant catalysts, such as nickel and iron, which are stable in alkaline conditions.10,11 LAWE has been under constant development since the early 20th century and is currently deployed at scale.9,12 However, integrating LAWE with variable renewable energy introduces challenges related to long-term stability and performance under dynamic operating conditions.13
Electrolyzers must operate efficiently despite fluctuating power inputs, which are inherent to solar and wind energy sources.14–16 Although modern LAWE systems can adapt to fluctuating loads, rapid changes and intermittent standby periods significantly stress electrolyzer components.5,6 Variable operation also increases safety risks, particularly when gas crossover exceeds critical thresholds at low current densities.6,17 Additional complexities arise under industrially relevant conditions, including impure feedstocks,18,19 temperature variations,6,20 and frequent startup/shutdown cycles.21 Thus, electrolyzers must be resilient to dynamic operating environments to achieve viability at scale.1
Among the stressors induced by variable operation, shutdown processes are particularly detrimental to electrode durability.5,21,22 Modern LAWE systems operate in a bipolar configuration, where the cathode and anode of adjacent cells are electrically connected through bipolar plates and ionically linked via electrolyte manifolds.6 Shutdowns trigger reverse currents, which arise from the electromotive force generated as electrodes return to their original chemical states.15 These reverse currents, also known as shunt currents, invert the roles of the anode and cathode, causing the bipolar plate to act as a galvanic cell.17,21,22 Reverse currents are prone to occur as the load decreases, primarily due to a decreased ionic resistance when gases are absent in the circulating electrolyte. This phenomenon leads to current efficiency losses, non-uniform stack performance, hydraulic failures, and gas purity fluctuations.17 Electrodes are particularly vulnerable to reverse currents that induce deep discharge states,6,22 electrocatalyst dissolution, and structural transformations, ultimately shortening electrode lifespan.22 Given the growing deployment of fluctuating energy sources, mitigating the impact of shutdown-induced reverse currents is critical for long-term electrolyzer stability.17
Despite its technological maturity, LAWE research has historically prioritized catalytic activity over long-term stability.23 This is in contrast to studies for acidic electrolyzer systems, which have placed greater emphasis on degradation mechanisms given the scarcity of noble metals.5,15,24,25 Electrode stability is equally critical in LAWE, particularly if intended to operate in the harsh conditions imposed by variable operation.26 LAWE systems employ strategies such as protective polarization during standby conditions. However, these increase capital costs and do not fully resolve the challenges posed by reverse currents.6,15,27 A more effective approach involves developing inherently resilient electrodes. Industry experts have underscored this need,6 emphasizing the importance of ensuring long-term stability under real-world operating conditions.
Understanding electrocatalyst degradation under industrially relevant conditions is essential for advancing LAWE.28–31 However, studying degradation is challenging due to the highly dynamic nature of the electrocatalytic interface,32,33 and the multiple pathways leading to catalyst degradation.4,30,31 Reverse currents exacerbate several of these mechanisms, particularly in transition metal-based electrodes.22,34 Addressing this challenge requires careful electrode material selection, as reverse currents can induce reaction conditions for which the original materials were not designed.34 Additionally, in situ and operando analytical tools are crucial for capturing real-time degradation mechanisms.35,36 These efforts will ultimately support the development of accelerated stress tests (ASTs) incorporating variability, allowing catalysts to be evaluated under more realistic, demanding conditions.6,29
Here, we comprehensively study the effects of variable operation on the stability of transition metal oxyhydroxide electrocatalysts for LAWE. Using electrochemical, Raman spectroscopy, and mass spectrometry techniques, we systematically examine how reverse currents during simulated shutdown conditions induce phase transformations, change the catalytic film composition and thickness, modify electronic conductivity, accelerate metal dissolution, and impact electrode stability and electrolyzer performance. Our analytical approach also offers a framework for future studies on catalyst degradation and stability. Based on these insights, we provide guidelines for advancing LAWE research. This study highlights the need to integrate stability considerations and real-world operating conditions into electrocatalysis research to accelerate the large-scale deployment of water electrolysis technologies.
Results and discussion
Operating conditions alter electrode discharge during shutdown
To understand electrode behavior during shutdown, we simulated electrode discharge under open-circuit conditions. We used transition metal hydroxides deposited on Ti foil (TF) substrates as catalysts and tested them in a three-electrode cell (Fig. S1, ESI†). In a typical experiment, the open-circuit potential (OCP) was monitored, followed by a charging step at a constant current density to simulate OER (anode) or HER (cathode) conditions during water electrolysis.34 Galvanostatic polarization via chronopotentiometry (CP) was used to ensure a constant charging rate and align with industrial operating conditions.21,30The electrode charging process is shown in Fig. 1a, where applying a constant current density shifts the electrode potential from its initial OCP (∼0.6 V vs. RHE). For a Ni(OH)2 film acting as an anode, the feature observed between 1.3 and 1.5 V vs. RHE corresponds to the transition of Ni2+ to Ni3+ (i.e., hydroxide-to-oxyhydroxide phase).32,37–39 Beyond this point, the electrode potential plateaus. This indicates complete oxidation of the material, after which all charge is directed toward the OER. When acting as a cathode, the electrode rapidly polarizes to −0.2 V vs. RHE. These electrode potentials serve as reference points for the metal hydroxide/oxyhydroxide phase transformations, which can be used to analyze electrode discharge profiles (see below).
 | ||
| Fig. 1 Examining electrode discharge during simulated shutdown tests. (a) Electrode potential profiles for Ni(OH)2 films acting as the anode/O2 electrode (red) or cathode/H2 electrode (blue) during open circuit condition (OCP), followed by galvanostatic polarization using at j = ±5 mA cm−2. Potentials on the right depict events of interest associated with the electrode potential profiles. The anodic charge (Q) is decoupled into contributions from the Ni2+ to Ni3+ transition and water oxidation. (b) OCP decay profiles following abruptly interrupting galvanostatic polarization at 25 mA cm−2 in 0.1 M KOH electrolyte. Electrodes consist of Ni(OH)2 films deposited on Ti foil substrates. | ||
Electrode discharge profiles do not exhibit significant differences between purified and unpurified KOH (e.g., compare Fig. 1a and Fig. S2, ESI†), which is attributed to the low Fe concentration (<10 ppb) in 0.1 M KOH (Table S1, ESI†). However, repeated charge/discharge cycling is expected to accelerate Fe incorporation and cause potential shifts if impurity concentrations are significant.10,37,39 The incorporation rate depends on the catalyst composition, oxyhydroxide layer thickness, and Fe concentration in the electrolyte.38,40,41 Given that CP minimizes Fe incorporation,10 and working with unpurified KOH better aligns with industrial conditions,6,19 we opted to conduct electrode discharge experiments in unpurified KOH electrolyte, carefully controlling Fe content to ensure reproducibility (Table S1, ESI†).
To simulate electrolyzer shutdown, the current was abruptly stopped after the charging step. This results in an electromotive force that drives a thermodynamically spontaneous self-discharge process.26 Fig. 1b illustrates this phenomenon for the anode. Note that electrodes momentarily switch identities during discharge. Therefore, we avoid referring to them as cathode or anode and instead refer to them as the H2 and O2 electrodes, respectively. Upon current interruption, the electrode potential drops rapidly due to capacitive discharge, which occurs on a milliseconds timescale (see Fig. S2a, ESI†).42 The rapid discharge is followed by a slower OCP decay, revealing a redox feature between ∼1.4 and ∼0.9 V vs. RHE, which persists for ∼13 s. This event corresponds to the pseudo-capacitive self-discharge of nickel oxyhydroxide into hydroxide:22,26
| NiOOH + H2O + e− ⇄ Ni(OH)2 + OH− | (1) |
A sequence of physical processes underlies the observed electrode self-discharge. First, the discharge of the electrical double layer generates a rapid potential drop.42 Concomitantly, this fast discharge is influenced by the electrodes catalyzing fuel cell reactions. Here, residual gases present in the electrolyte or trapped within the film result in oxygen reduction being catalyzed at the O2 electrode and hydrogen oxidation at the H2 electrode.22 The impact of these reactions is most pronounced immediately after the current is switched off because the driving force for these reactions decreases as the electrode potential continues to drop. The rates of these fuel cell reactions become limited by reactant diffusion once the gases are depleted at the electrode surface. Next, the slow redox discharge occurs as the electrodes gradually revert to their original oxidation state, dominating the OCP decay profile.21
Multiple factors govern the rate of the self-discharge reaction (eqn (1)). Primarily, the potential difference between the O2 and H2 electrodes serves as a major influence; lower plateau potentials decrease the driving force (relative to the H2 electrode potential), thereby prolonging the discharge.34 Furthermore, note that eqn (1) involves a proton transfer, making it sensitive to the interfacial pH.42 These processes underscore the complexity of electrode behavior during shutdown and the formidable challenge it presents.
We systematically examined the influence of operating conditions on the discharge rate. As shown in Fig. S2 (ESI†), increasing the applied current from 5 to 100 mA cm−2 accelerates the discharge rate during the OCP step. The initial rapid discharge phase remains nearly unchanged, indicating that capacitive discharge and fuel cell reactions occur quickly, regardless of the applied current. Since the electrolyte was saturated with O2 gas when the current was switched off, the rates of the fuel cell reactions are expected to be rapidly limited by diffusion in all cases. In contrast, the rate of the redox self-discharge varied significantly, and was particularly slow at 5 and 10 mA cm−2.
In addition to the potential difference between the O2 and H2 electrodes, the pH that the catalyst experiences may also play a significant role in the rate of this phase transition. The OER consumes hydroxide ions, lowering the interfacial pH.42,43 Given that eqn (1) is pH-dependent, an increasingly acidic environment present at higher currents would accelerate the self-discharge reaction, aligning with our observations. Increasing the KOH concentration from 0.1 to 7 M prolongs the OCP decay during the redox transition (Fig. S3, ESI†), likely due to the pH dependence of eqn (1) and restricted mobility of OH− ions,44 indicating that concentrated alkaline media slows the self-discharge rate. Raising the temperature from 20 to 50 °C accelerates the redox discharge (Fig. S4a, ESI†) and the OCP stabilization to ∼0.6 V vs. RHE (Fig. S4b, ESI†). This behavior is expected due to enhanced discharge kinetics at elevated temperatures.22,34,44 The accelerated OCP stabilization at high temperatures may be linked to changes in the corrosion potential (see below).
The kinetics of electrode discharge also varied for Co- and Fe-containing bimetallic Ni hydroxides (Fig. S5, ESI†). Adding Fe and Co increased the discharge capacity during the redox transition, consistent with previous reports.32,34 In addition to differences in OER activity during the galvanostatic step (OER overpotential at 50 mA cm−2: Ni > NiCo > NiFe), variations in the shape and onset of the redox transitions are attributed to shifts caused by Fe and Co incorporation.32,38,39 Note also that the final OCP differs slightly among these compositions. Comparing Ni(OH)2 with a Ni foil electrode also reveals only slight variations in redox discharge (Fig. S6, ESI†). Differences in surface area and morphology can lead to differences in the observed discharge, as it occurs primarily on the surface.34 The Ni foil electrode is expected to develop a more compact oxyhydroxide layer during CP.10 In contrast, the ∼550 nm-thick Ni(OH)2 film is expected to have a smaller grain size and undergo near-complete conversion to NiOOH, which could explain the subtle differences in discharge capacity between both electrodes.
Next, we examined the discharge behavior of H2 electrodes. Fig. S7 (ESI†) shows that increasing the applied current from −5 to −100 mA cm−2 results in a deeper discharge, shifting the OCP towards more positive potentials (∼0.4 V vs. RHE). Similar to the O2 electrode, the rapid initial discharge behavior remains identical regardless of the applied current. A few seconds after initiating the discharge, the OCP profiles following −50 and −100 mA cm−2 steps exhibit subtle distortions. Conversely, the OCP profiles for lower current densities plateau quickly. This distortion becomes more pronounced as current density increases.
While Ni(OH)2 might be partially reduced during the HER, there is no visible redox transition that is comparable in magnitude to the Ni2+/3+ redox peak that could explain this distortion. Instead, we attribute this behavior to interfacial pH swings generated at high currents, a recently studied phenomenon.42 A control experiment using Pt as the H2 electrode (Fig. S8, ESI†), which exhibits no redox behavior, shows this distortion and aligns with the response reported previously.42 This suggests that, much like weakly-buffered electrolytes, electrodes in 0.1 M KOH experience significant interfacial pH fluctuations during discharge. The effect is strongly influenced by KOH concentration (Fig. S9, ESI†). While the OCP rapidly stabilizes in 7 M KOH, indicating that the interfacial pH swing is minimal, profiles in 1 M and 0.1 M KOH are distorted. These results suggest that the interfacial pH influences electrode discharge kinetics. Further studies are necessary to examine this phenomenon in detail.
The equilibrium potential reached during open circuit conditions also reflects, in part, the corrosion potential.45 No external current is applied under this condition. However, oxidation and reduction reactions still occur at equal rates, resulting in no net current flow. To assess corrosion behavior after electrode discharge, we conducted potentiodynamic polarization experiments. As shown in Fig. S10 (ESI†), Pt and Ni foil electrodes exhibit corrosion potentials of 825 and 840 mV, respectively, which are more positive than the Ni(OH)2 O2 electrode (767.3 mV). More positive corrosion potentials indicate a more oxidizing environment at the electrode, meaning it is more likely to accept electrons than lose them—characteristic of noble metals. In contrast, more negative corrosion potentials indicate a reducing environment, making the electrode more prone to corrosion.45 The lower corrosion potential of Ni(OH)2 indicates that it is more susceptible to corrosion than Pt and Ni metals. Increasing the KOH electrolyte concentration from 0.1 to 7 M shifts the corrosion potential from 767.3 to 66.5 mV (Fig. S11, ESI†) while raising the temperature from 20 to 50 °C lowers it to 635 mV (Fig. S12, ESI†). These results indicate that corrosion is accelerated in concentrated KOH electrolytes and at high temperatures. Corrosion potentials follow the trend Ni(OH)2 < NiFe(OH)2 < NiCo(OH)2 (Fig. S13, ESI†), implying that Co- and Fe-containing films are more resistant to corrosion.
Altogether, these findings demonstrate that electrode discharge is strongly influenced by intrinsic factors (e.g., catalyst composition) and extrinsic conditions (e.g., electrolyte concentration, applied current, and temperature). These conditions also impact electrode corrosion. However, while working under open circuit conditions provides insights into spontaneous transformations, it is essential to recognize that the electrode potential changes experienced by H2 and O2 electrodes on the same bipolar plate depend on the balance of their discharge capacities.34 Thus, an open circuit condition will not always occur in practice, and deeper discharge scenarios must be considered (see below).
Variable operation intensifies loss of performance
A key challenge in evaluating electrocatalyst stability is determining the severity of the conditions to which materials should be exposed. The community increasingly acknowledges that testing under steady-state conditions yields different outcomes than dynamic operation.5,25 Some ASTs have incorporated OCP steps to simulate electrolyzer shutdown.27,29 However, industrial electrolyzers can experience deeper electrode discharge during shutdown, potentially beyond 0 V vs. RHE,22,34 and reverse currents on the order of 0.1–1% of the total current are often experienced by the middle cells in bipolar stacks.21,22,46We compared the stability of metal hydroxide films deposited on TF under steady-state and dynamic conditions. Galvanostatic polarization via CP was used to simulate constant and reverse current conditions, with on/off intervals adapted from AST protocols.27 Electrochemical impedance spectroscopy (EIS) was used to measure the high-frequency resistance (HFR) and charge transfer resistance (Rct) of the electrodes during operation. Since the same electrolyte was used throughout, changes in HFR were attributed to variations in the electronic resistance of the electrocatalytic film and contact resistances.29,47 Meanwhile, Rct, determined from the diameter of the semicircle in Nyquist plots, served as a proxy for catalytic activity. The electrode potential was decoupled from the total cell potential using a reference electrode.
First, we evaluated the stability of Ni, Co, NiCo, and NiFe hydroxides under steady-state operating conditions. These tests involved a series of CP steps (50 mA cm−2, 15 min), each followed by a potentiostatic EIS measurement (Fig. S14a, ESI†). Steady-state operation led to a gradual increase in cell and electrode potentials, with minimal or negligible variation in the HFR. Only Ni(OH)2/TF exhibits an initial stabilization phase, where the electrode potential increases at 46.9 mV h−1 before slowing to 4.7 mV h−1 (Fig. S14, ESI†). The HFR remained constant at 13.1 Ω cm2, and the potential during the CP step increased by 246 mV after 7.5 h. Co (Fig. S15, ESI†) and NiCo hydroxides (Fig. S16, ESI†) show an increase in electrode potential at 3.1 mV h−1 for Co(OH)2/TF and 3.3 mV h−1 for NiCo(OH)2/TF, with a gradual increase in HFR. In contrast, NiFe(OH)2/TF (Fig. S17, ESI†) exhibits a more pronounced potential increase (47.8 mV h−1) and a stable HFR of 11.5 Ω cm2, leading to a total potential increase of 331 mV after 7.5 h.
Next, we evaluated variable operation by adding an OCP period between constant current steps (Fig. S18a, ESI†). Similar to steady-state operation, the electrodes exhibited a small increase in the electrode potential during the CP step and minimal changes in HFR. The electrode potential and HFR increase at about the same rate for all the electrodes (Fig. S18, S20, S22, and S24, ESI†), and OCP decay profiles throughout cycling are nearly identical, with the distinct redox M2+/3+ transition following shutdown (Fig. S19, S21, S23, and S25, ESI†). The primary difference was the final OCP value, which varied most notably for NiFe(OH)2/TF (Fig. S25d, ESI†).
To study the role of reverse currents, we then replaced the OCP step with a reverse current of −5 mA cm−2 (Fig. S26a, ESI†), representing 10% of the current load during the galvanostatic step. This condition was deliberately exaggerated to accelerate degradation and highlight the severe impact of reverse currents on device performance. In contrast to steady-state and variable OCP operation tests, the electrodes deteriorated rapidly under reverse currents, with degradation rates varying with film composition. The Ni(OH)2/TF (Fig. S26 and S27, ESI†), Co(OH)2/TF (Fig. S28 and S29, ESI†), NiCo(OH)2/TF (Fig. S30 and S31, ESI†), and NiFe(OH)2/TF electrodes (Fig. S32 and S33, ESI†) exhibit a similar degradation pattern, characterized by an initial rise in overpotential, followed by a sharp increase of several hundred mV per hour.
The HFR remains stable within the 11–14 Ω cm2 range across all electrodes. Nyquist plots initially display two semicircles attributed to the contact resistance that develops between the catalytic film and the Ti foil support.48 Eventually, a single semicircle develops with a drastic rise in the Rct, consistent with the loss of electrocatalytic activity. Furthermore, the overlay of potential decay profiles during the reverse current step shows a progressive disappearance of the M2+/3+ redox feature, suggesting a decrease in the number of available redox active sites (Fig. S27d, S29d, S31d, and S33d, ESI†).
To contrast steady-state and variable operation, Fig. 2 displays illustrative stability data for NiCo(OH)2/TF. Under steady-state and open circuit conditions, the electrode potentials increase by a few mV per hour (Fig. 2a and b), whereas reverse current tests exhibit more pronounced increase rates, especially after ∼3 hours (Fig. 2c). Note that electrodes do not experience deep discharge during variable OCP operation, and electrode potentials remain above 1.0 V vs. RHE (Fig. 2d), while reverse currents induce more negative potentials close to −0.5 V vs. RHE (Fig. 2e). To identify the cause of the severe deterioration observed during reverse current cycling, we tested bare TF as an electrode. Ti is a poor OER catalyst, requiring ∼6 V vs. RHE to reach a current density of just 0.1 mA cm−2 and exhibiting a severe overpotential increase rate of 831 mV h−1 (Fig. S34, ESI†). Despite this instability, its HFR remains constant at 12.5 Ω cm2. Ti foil lacks noticeable redox features during the reverse current step and is an inefficient HER catalyst, with its cathodic potential shifting from −0.60 to −0.65 V vs. RHE (Fig. S35, ESI†). Given its poor catalytic activity and similar HFR range (11–14 Ω cm2) as other films, these findings suggest that the Ti foil substrate increasingly influences the response during reverse current cycling.
 | ||
| Fig. 2 Steady-state, variable OCP, and variable reverse current operation modes deteriorate NiCo catalytic films at different rates. Electrode potential as a function of time at a (a) constant current density of 50 mA cm−2, (b) variable operation using an OCP step, and (c) variable operation using a reverse current step (j = −5 mA cm−2). The arrow depicts the average potential increase rate. Overlays of (d) OCP decay and (e) reverse current steps. The arrows depict the progression of the experiment. Experiments were conducted in a three-electrode cell setup. | ||
A possible explanation for the severe deterioration is the gradual loss of catalyst material during cycling, leading to exposure of the underlying support. To test this hypothesis, we tested Ni foil as an electrode. In contrast to Ti-supported catalytic films and bare Ti foil, the Ni foil electrode exhibits a stable response during reverse current tests (Fig. S36, ESI†). The electrode potential initially decreases, likely due to the formation of the OER-active NiOOH layer, followed by a steady overpotential increase at 3.2 mV h−1. The HFR initially decreases, then stabilizes at ∼14 Ω cm2, and finally increases at a slow rate. Nyquist plots reveal a single, well-defined semicircle, confirming the absence of contact resistance. In contrast to the Ti-supported catalysts, the performance of the Ni foil electrode improves over cycling, and the Ni2+/3+ redox feature during the reverse current step grows over time (Fig. S37d, ESI†). These results suggest that the NiOOH layer grows during cycling, and because the electrode consists of bulk Ni metal, active sites are continuously replenished after dissolution or detachment of surface sites. These observations support that reverse currents expose electrodes to severe redox cycles, leading to loss of the catalytic film and eventual substrate exposure (see below).
Variable operation induces phase transformations of the O2 electrode
To gain deeper insights into the transformations induced by reverse currents, we systematically examined catalytic films using a combination of analytical techniques. We first used in situ surface-enhanced Raman spectroscopy (SERS) to track phase transformations in metal oxide/hydroxide OER catalysts. Films were deposited on Au screen-printed electrodes (SPEs) and tested in a custom-made spectroelectrochemical cell (Fig. S38, ESI†). Using this approach, we are able to observe the hydroxide-to-oxyhydroxide phase transition as the potential increases. Using a Ni(OH)2 film as a control (Fig. S39, ESI†), we observe the characteristic Raman bands of the hydroxide phase at 458, 495, and 694 cm−1 shift to those of oxyhydroxide at 477 and 558 cm−1 at potentials positive of the Ni2+/3+ redox peak.32,38,49 We optimized the reverse step conditions to minimize bubble interference with the Raman measurement. Discharge at open circuit and a reverse current density of −0.05 mA cm−2 led to a gradual potential decay that did not reach reducing potentials (Fig. S40, ESI†). The oxyhydroxide phase remains detectable under both conditions (Fig. S41, ESI†). Only a reverse current density of −0.25 mA cm−2 discharges the electrode to −0.2 V vs. RHE within 10 min (Fig. S40, ESI†).Fig. 3 shows an in situ SERS measurement during the discharge of Ni(OH)2 at −0.25 mA cm−2. The electrode potential drops rapidly following the galvanostatic step, exhibiting the Ni2+/3+ redox transition before stabilizing after 380 s at ∼0.8 V vs. RHE (Fig. 3a). Raman spectra collected at regular intervals consistently show oxyhydroxide bands throughout this period (Fig. 3b). Then, the potential continues to decline steadily until ∼560 s, coinciding with the emergence of hydroxide bands. The potential sharply decreases to reducing potentials near −0.2 V vs. RHE, where it stabilizes. Corresponding Raman spectra reveal a marked decrease in the intensity of hydroxide bands, suggesting the electrochemical reduction of Ni(OH)2 into a more metallic state.
 | ||
| Fig. 3 In situ SERS reveals phase transitions during deep discharge. (a) Electrode potential decay during the discharge of Ni(OH)2 under a reverse current condition of −0.25 mA cm−2. (b) Corresponding Raman spectra collected at different points during the electrode discharge displayed in (a). The discharge step followed a constant anodic potential step of 1.5 V vs. RHE. Dashed grey lines in (b) serve as visual guides for peak positions of notable Raman bands. The corresponding species of the nickel electrode are shown in (b). Experiments were conducted in unpurified 0.1 M KOH electrolyte at 20 °C. The Ni(OH)2 film was deposited on a roughened Au screen-printed electrode (SPE). | ||
We also examined phase transitions during the discharge of bimetallic films. As shown in Fig. S42 (ESI†), in situ SERS captures the hydroxide-to-oxyhydroxide transition in NiCo(OH)2, aligning with the M2+/3+ redox peak. From Fig. S43 (ESI†), the discharge profile closely resembles that of Ni(OH)2, with subtle differences arising from the redox peak shift due to Co incorporation. Despite these nuances, the trends remain consistent: oxyhydroxide bands disappear after the redox transition (∼0.8 V vs. RHE, 500 s), hydroxide bands emerge during the steady potential decline (0.6–0.2 V vs. RHE, 660 s), and finally, deep discharge eventually drives the electrode to −0.2 V vs. RHE, coinciding with the emergence of a more metallic phase. The redox peak shifts anodically in NiFe(OH)2 (Fig. S44, ESI†), yet in situ SERS still captures the oxyhydroxide-to-hydroxide transition. While the discharge profile exhibits slight differences due to the redox peak shift (Fig. S45, ESI†), the same phase transition occurs. The hydroxide phase attenuates at 0.2 V vs. RHE, higher than the −0.2 V vs. RHE observed for the other films.
We further examined the reversibility of these phase transformations using in situ SERS during variable operation tests. Raman spectra were collected during potentiostatic steps under OER conditions and variable operation under open circuit or deep discharge conditions at a negative potential. For Ni(OH)2 (Fig. S46, ESI†), NiCo(OH)2 (Fig. S47, ESI†), and NiFe(OH)2 (Fig. S48, ESI†) films, the oxyhydroxide phase consistently appears under OER potentials, while hydroxide bands emerge during the reverse step under OCP (∼0.8 V vs. RHE). In contrast, variable operation with reverse current steps induces a deep electrode discharge, resulting in the formation of a more metallic phase. For example, in NiCo(OH)2, the hydroxide bands are significantly attenuated, and even oxyhydroxide bands during OER steps fail to fully develop in the last two cycles (Fig. S49, ESI†). Similarly, NiFe(OH)2 (Fig. S50, ESI†) exhibits weak hydroxide bands during the reverse step, and the oxyhydroxide bands during OER steps show inconsistent intensity throughout cycling. Compared to OCP, the weaker oxyhydroxide band intensity observed under deep discharge suggests that reverse current conditions strongly reduce the O2 electrode surface, and the transition back to the oxyhydroxide phase is incomplete.
Variable operation modifies the chemical composition of the O2 electrode
We further investigated composition changes in catalytic films induced by reverse currents using time-of-flight secondary ion mass spectrometry (ToF-SIMS). This technique enables analysis of the film's composition as a function of depth. Given the severe degradation observed in variable operation tests involving reverse currents, we analyzed the NiCo(OH)2/TF (Fig. S30, ESI†) and NiFe(OH)2/TF (Fig. S32, ESI†) electrodes described in Section 2 ex situ and compared them with pristine samples. ToF-SIMS measurements yield profiles showing the relative abundance of secondary ion fragments as a function of depth (Fig. S51–S53, ESI†). These profiles were used to construct depth distribution ranges, representing regions with 68.3% of the total secondary-ion fragment concentration nominally within the catalytic film. Fig. 4a and b depict these depth distribution ranges for NiCo(OH)2 and NiFe(OH)2 films before and after reverse current tests, with the bars indicating their respective distribution depths. | ||
Fig. 4 Variable operation tests induce chemical composition changes in O2 electrodes. ToF-SIMS depth distribution ranges of various secondary ion fragments before (BC) and after (AC) variable operation tests for (a) NiCo(OH)2/TF and (b) NiFe(OH)2/TF electrodes. A depiction of the apparent catalyst film thickness before and after testing is shown on the right of each plot. Effects of the Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co ratio on NiCo(OH)2 films: (c) Linear sweep voltammetry scans and (d) in-plane effective conductivities at increasing Co percentages in the film. The onset potential for conductivity in (d) aligns with the position of the hydroxide/oxyhydroxide redox peak in (c). The gray-shaded area in (d) indicates a region with conductivity below the detection limit of our measurement. Electrode potentials are not corrected for series resistance, and current densities are not normalized for differences in film thickness. Conductivity experiments were conducted on interdigitated array (IDA) electrodes in unpurified 1 M KOH electrolyte at 20 °C. :Co ratio on NiCo(OH)2 films: (c) Linear sweep voltammetry scans and (d) in-plane effective conductivities at increasing Co percentages in the film. The onset potential for conductivity in (d) aligns with the position of the hydroxide/oxyhydroxide redox peak in (c). The gray-shaded area in (d) indicates a region with conductivity below the detection limit of our measurement. Electrode potentials are not corrected for series resistance, and current densities are not normalized for differences in film thickness. Conductivity experiments were conducted on interdigitated array (IDA) electrodes in unpurified 1 M KOH electrolyte at 20 °C. | ||
Secondary ion fragments containing Ni, Co, and O are primarily distributed within the top ∼110 nm of the pristine NiCo(OH)2/TF electrode (Fig. 4a). We use this range to represent the apparent catalytic film thickness (i.e., the blue bar on the right side in Fig. 4a). This assumption is consistent with the location of the Ti substrate, as Ti− fragments do not appear until a depth of ∼120 nm (i.e., gray bar in Fig. 4a). After variable operation, the depth distribution ranges decrease significantly, while the Ti− fragment now emerges at just ∼20 nm from the electrode surface, indicating substantial thinning of the catalytic film (i.e., from ∼110 to ∼40 nm). Note that FeOH− fragments detected after cycling are attributed to Fe incorporation near the electrode surface, aligning with our previous findings.32 Similarly, the NiFe(OH)2/TF electrode undergoes thinning of the catalytic film, decreasing from ∼160 to ∼130 nm (Fig. 4b).
Although ex situ ToF-SIMS does not fully capture the mechanism behind catalyst loss, the results confirm that variable operation exacerbates degradation. Despite the severe performance decline (Fig. S30 and S32, ESI†), secondary ion fragments from the catalyst remain detectable after testing, indicating that the film is not entirely stripped from the support. This finding contrasts with previous studies reporting complete catalytic film detachment.50 We speculate that both degradation mechanisms may occur depending on the specific testing conditions.51 Further insights into the catalyst degradation mechanism are provided (see below).
Another observation from ex situ ToF-SIMS is that Ni, Co, and Fe have similar depth distributions in the pristine samples, but their profiles diverge after testing. This change becomes clearer when comparing the integrated secondary-ion yields for Ni, Co, and Fe fragments within the confidence interval before and after variable operation tests (Fig. S54 and S55, ESI†). As shown in Fig. S56 (ESI†), the NiCo(OH)2/TF electrode lost 62.6% of Ni and 11.5% of Co, while the NiFe(OH)2/TF electrode lost 98.2% of Ni and 74.7% of Fe. These findings suggest that variable operation alters the Ni:M ratio in the catalytic film, which is expected to influence the electronic conductivity. This property critically influences device performance,32,52,53 and demands greater attention.26 Inspired by previous studies on in-plane conductivity and Fe content,39,52 we performed in situ measurements on NiCo(OH)2 to examine the effect of Co content.
NiCo(OH)2 films with varying Co content were deposited on gold interdigitated array (IDA) electrodes, and their in-plane conductivity was measured in situ (Fig. S57, ESI†). As shown in Fig. 4c and Fig. S58a (ESI†), increasing the Co content shifts the M2+/3+ redox peak cathodically, with a corresponding rise in the conductivity (Fig. 4d). The magnitude of this increase depends on the Co content; for instance, films with 13 mol% Co exhibit an effective conductivity nearly four orders of magnitude higher than pure Ni films above 0.4 V vs. Hg/HgO. Note that the conductivity plateaus above 0.54 V vs. Hg/HgO for all NiCo(OH)2 films exceeding this Co content (Fig. S58b, ESI†). This sharp increase in electronic conductivity following the redox peak is characteristic of partially filled, low-spin Co3+ and Ni3+,32,54 with Co doping further enhancing conductivity.34,50
Collectively, these findings reveal that variable operation changes the metal composition in the catalyst film, which in turn affects its electronic conductivity. Variations in the Ni/Co ratio also alter the discharge capacity,32,34 suggesting that endurance to reverse currents may degrade nonlinearly over time. Since deep discharge during variable operation alters the film's composition, shifts in the HFR and overpotential increase during cycling may result from conductivity and discharge capacity changes, in addition to contact resistances.34,51 Lastly, the differences in metal composition suggest that transition metals are affected at different rates, possibly by dissolution (see below).
Variable operation induces catalyst and substrate dissolution
In situ and operando characterization techniques are powerful tools for studying degradation in real time,4,30 a critical capability for understanding the effects of dynamic environments. To further investigate catalyst stability under variable conditions, we used on-line inductively coupled plasma mass spectrometry (ICP-MS), an analytical platform with isotopic sensitivity that enables trace-level elemental detection.55 This technique has been instrumental in linking transient electrochemical events with metal dissolution,23,33,56 making it particularly suited to revealing the specific impacts of variable operation on catalyst film degradation.27 Experiments were conducted using a custom electrochemical flow cell positioned upstream of the ICP-MS instrument (Fig. S59, ESI†).57 NiCo(OH)2/TF and NiFe(OH)2/TF electrodes were tested as O2 electrodes. Variable operation tests were performed using CP at 1.35 mA cm−2, followed by a reverse step at −1.35 mA cm−2. Concomitantly, Ni, Co, Fe, and Ti concentrations in the KOH electrolyte exiting the flow cell were measured. As shown in Fig. 5, these conditions resulted in electrode potentials that varied between 1.5 and −0.5 V vs. RHE, inducing deep discharge of the material. Metal dissolution is anticipated within this potential range due to the thermodynamic instability of Ni, Co, and Fe, as indicated by Pourbaix diagrams (Fig. S60, ESI†).22,30,32,58,59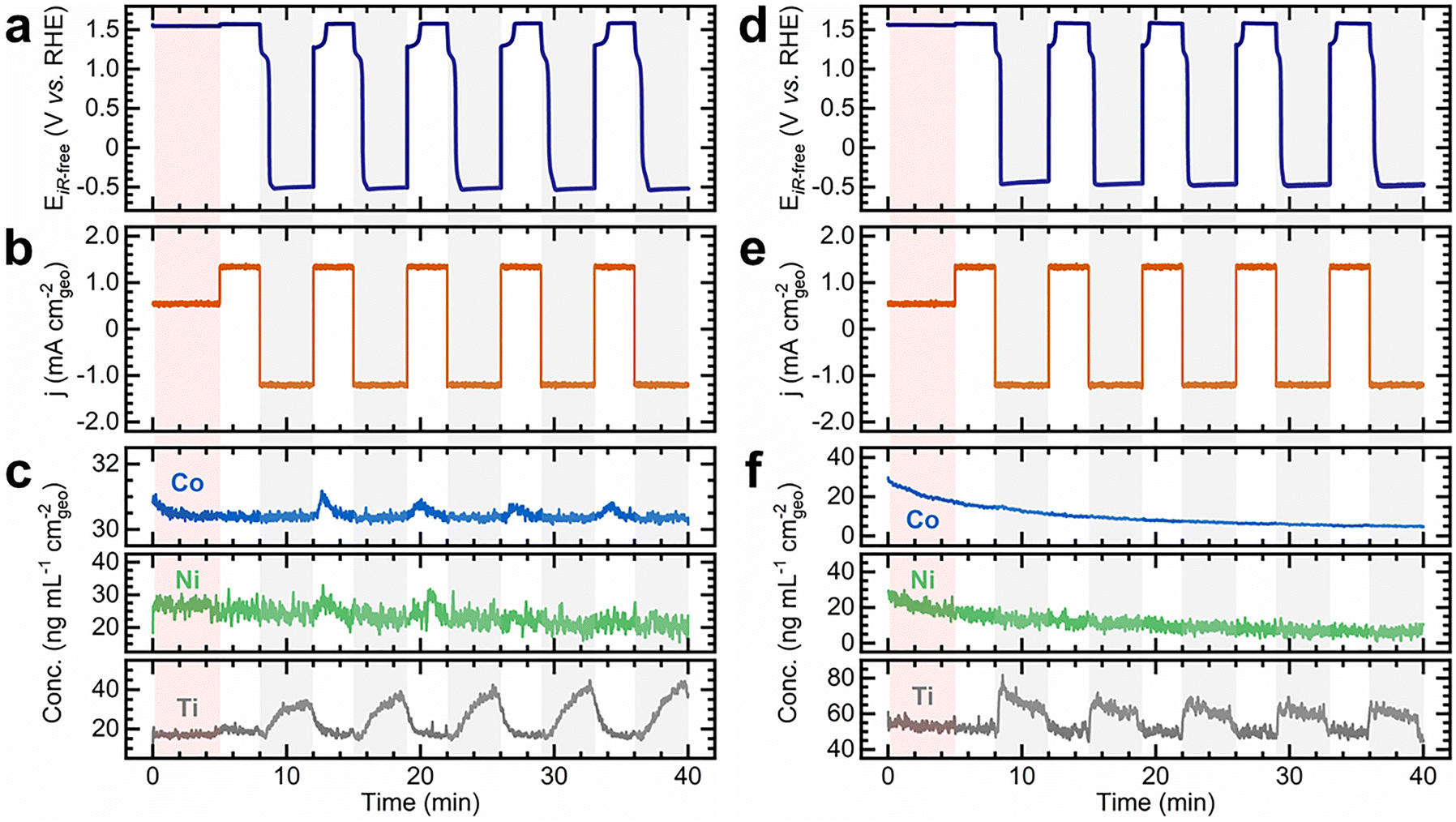 | ||
| Fig. 5 On-line ICP-MS reveals catalyst and Ti support dissolution during variable operation tests. (a) iR-corrected electrode potential profiles, (b) corresponding current density steps (±1.35 mA cm−2), and (c) resulting concentration profiles for Co, Ni, and Ti during variable operation tests of the NiCo(OH)2/TF electrode in unpurified 0.1 M KOH electrolyte. (d) iR-corrected electrode potential profiles, (e) corresponding current density steps (±1.35 mA cm−2), and (f) resulting concentration profiles for Co, Ni, and Ti during variable operation tests of the NiCo(OH)2/TF electrode in unpurified 0.1 M KOH electrolyte spiked with 5 mM KPi. The red-shaded area represents the initial preconditioning step at 0.5 mA cm−2 for 5 min to oxidize the hydroxide film into oxyhydroxide. The grey-shaded areas highlight the reverse current steps and serve as visual guides. Currents and concentrations are normalized to the geometric surface area of the electrode exposed to the electrolyte in the flow cell (0.197 cm2). Experiments were carried out in unpurified 0.1 M KOH electrolyte at 20 °C with a flow rate of 2 mL min−1. | ||
The electrode potential, current density, and metal concentrations in the electrolyte are synchronized in Fig. 5 to correlate electrochemical and dissolution events. Fig. 5a–c show the results for the NiCo(OH)2/TF electrode. After initial conditioning, the electrode potential alternates between oxidizing and reducing conditions (Fig. 5a), displaying the charging and discharging redox features observed in Fig. 1. The oxyhydroxide self-discharge appears during the reverse step, followed by stabilization at −0.5 V vs. RHE. Returning the electrode to OER conditions reoxidizes the film, transforming the mixed hydroxide/metallic phase formed during deep discharge to the oxyhydroxide phase. While the current density remains stable (Fig. 5b), concentration profiles are dynamic and differ for each metal (Fig. 5c). The Co concentration declines rapidly during conditioning before stabilizing at ∼30.5 ng mL−1 cm−2 (equivalent to a dissolution rate, rds, of 1.01 ng s−1 cm−2). Sharp Co concentration spikes consistently appear upon reoxidation of the catalytic film, coinciding with the M2+/3+ redox transition. The Ni concentration steadily decreases from ∼30 to ∼20 ng mL−1 cm−2 throughout the test, with subtle spikes emerging during reoxidation. Ti dissolution occurs at a nearly constant rate (rds: ∼0.57 ng s−1 cm−2) during conditioning and OER steps. However, the concentration of Ti rises under reverse currents to nearly twice its baseline value—a trend that repeats throughout cycling.
The NiFe(OH)2/TF electrode exhibits a similar trend (Fig. S61, ESI†) but with three distinct differences: (1) the Fe baseline concentration is significantly higher (∼120 ng mL−1 cm−2, rds: 4.3 ng s−1 cm−2), (2) Fe dissolution during reoxidation is more pronounced, nearly doubling its baseline and persisting throughout the entire OER step, and (3) Ni and Ti dissolution are notably attenuated. The reverse current step also suppresses Fe dissolution, as the concentration rapidly declines once the electrode is negatively polarized. These findings indicate that variable operation intensifies catalyst dissolution, particularly during reoxidation, with Ni, Co, and Fe exhibiting distinct dissolution rates (Fe > Co ∼ Ni).
Prior studies show consistent behavior with our observed dissolution results. First, the sharp dissolution upon reoxidation aligns with evidence that redox cycling induces mechanical stress in Ni, Co, and Fe oxyhydroxides.60 Although Co and Fe are expected to exhibit high structural stability in NiO6,32 our findings indicate that dynamic operation disrupts stability through repeated phase transitions. Second, Ni, Co, and Fe dissolve at different rates, consistent with our ToF-SIMS results showing changes in film composition (Fig. 4a and b) and mass loss, with Fe showing the highest loss (Fig. S56, ESI†). Metals with different thermodynamic stabilities (i.e., corrosion potentials) are expected to corrode at varying rates under identical discharge conditions, leading to metal-specific dissolution spikes in potentiodynamic conditions.4 Third, we find that dissolution is primarily exacerbated during reoxidation and not during gas evolution steps, indicating that chemical dissolution, rather than mechanical detachment, is most important to catalyst stability. However, bubble-induced mechanical degradation is still possible at higher current densities.50 On-line ICP-MS cannot distinguish between dissolution mechanisms without using specialized methodologies.55 Future studies should consider differentiating between these various mechanisms. Lastly, the dissolution trend follows the reported stability order: NiOxHy > CoOxHy ≫ FeOxHy, the inverse of the OER activity trend, reinforcing the notion that active OER catalysts are often inherently less stable.33
Our findings reveal that the catalytic support is also vulnerable to degradation under reverse currents. The electrolyte can penetrate through the porous oxyhydroxide film,10,44,61 reaching the support if the film is thin enough or if sufficient time is allowed for complete oxidation. We attribute Ti dissolution to the destabilization of the Ti oxide passivation layer:27 reverse currents chemically reduce the passivation layer formed under oxidizing conditions, exposing unstable Ti metal and exacerbating Ti dissolution. Returning the electrode to OER conditions forms the protective layer again and suppresses dissolution. These results suggest that Ti may not be an optimal catalytic support for LAWE, and its instability prevents the accurate measurement of stability metrics.4,31 We note, however, that using Ti in this study enabled us to distinguish catalyst dissolution from support degradation.
Interestingly, Fe dissolution is apparently suppressed during reverse steps, possibly due to metal redeposition. This phenomenon has been implicated to explain the behavior of self-healing catalysts, particularly in the presence of phosphate.62–64 To investigate this self-healing behavior, we used on-line ICP-MS to compare metal dissolution in the presence and absence of potassium phosphate (KPi). In unpurified KOH electrolytes spiked with KPi, the NiCo(OH)2/TF electrode exhibits the same electrode potential and current response as in phosphate-free KOH (Fig. 5d and e); however, metal concentration profiles differ significantly (Fig. 5f). Despite starting at similar concentrations (∼30 ng mL−1 cm−2), Co and Ni concentrations steadily decline, reaching a baseline nearly six times lower by the end of the test. The dissolution spikes during reoxidation disappear, resulting in a more stable response. Ti dissolution increases nearly threefold compared to phosphate-free KOH (rds: ∼1.67 ng s−1 cm−2), with dissolution spikes during the reverse current step amplifying the dissolution rate ∼1.5 times. The dissolution pattern shifts in KPi, with Ti spiking abruptly before gradually declining, in contrast to the steady increase in phosphate-free KOH.
Ni and Fe dissolution remains stable without spikes for the NiFe(OH)2/TF electrode in KPi (Fig. S62, ESI†), while Ti shows an elevated baseline and a decreasing dissolution rate during the reverse step. Ni and Fe dissolution baselines remain comparable to those in phosphate-free KOH. These trends align with proposed self-healing mechanisms,63,64 where corrosion is suppressed by proton-accepting electrolytes that facilitate metal redeposition. However, the increased Ti dissolution in KPi demands further investigation, as corrosion control strategies beneficial for the catalyst may inadvertently affect other components.
These results reaffirm that metal oxyhydroxide catalysts are not entirely stable in alkaline media,30 with increased rates of dissolution resulting from exposure of the material to deep discharge conditions. While Pourbaix diagrams are often used to predict catalyst dissolution from a thermodynamic standpoint,4,42 our findings underscore the need to consider dissolution kinetics for a comprehensive understanding of catalyst degradation under dynamic conditions.57 Lastly, variable operation tests with KPi show that self-healing agents influence dissolution kinetics under these conditions.
Decoupling O2 and H2 electrode degradation in a zero-gap electrolyzer configuration
To develop practical insights into device performance, considering commercial electrolyzer architectures is crucial. Membrane-electrode assembly (MEA) configurations alter the reaction environment that catalysts are exposed to relative to conventional cells, and industrially relevant conditions are expected to impact degradation.4,29,65,66 Building on our fundamental insights into variable operation and reverse currents, we conducted long-term tests using an MEA electrolyzer to track electrode potential evolution under industrially relevant conditions. These tests used a zero-gap electrolyzer setup and protocols described in our previous work (Fig. S63, ESI†).29 The design was modified to accommodate a reference electrode in the working electrode endplate, allowing decoupling of electrode potentials (Fig. S63a, ESI†).21 Given the rapid degradation of thin catalytic films and the unsuitability of Ti foil as a support for practical stability tests, we employed Ni mesh electrodes instead.Electrodes were operated at steady state at 120 mA cm−2, followed by discharge at a constant reverse current of −0.25 mA cm−2, based on the estimated ionic resistance through a simulated manifold (Fig. S64, ESI†). For a galvanostatic step at 120 mA cm−2, the reverse current density used represents ∼0.2% of the total current, aligning with the range of severe shutdown conditions experienced by bipolar electrolyzers.21,22,46 Following previous guidelines,29 the electrolyzer was conditioned under operational conditions (unpurified 7 M KOH, 50 °C, electrolyte flow rate: 200 mL min−1) until a stable response was achieved. The electrolyzer's performance was systematically monitored via full-cell and iR-corrected polarization curves for the electrolyzer (Fig. S65, ESI†), the O2 electrode (Fig. S66, ESI†), and the H2 electrode (Fig. S67, ESI†), along with galvanostatic EIS to monitor the HFR.5,24,29
Fig. 6 summarizes the electrolyzer's performance during variable operation, with supplementary data in Fig. S68–S72 (ESI†). The total cell potential increases at a rate of ∼0.2 mV h−1, that is, ∼0.035 and ∼0.049 mV h−1 for the O2 and H2 electrodes, respectively (Fig. 6a–c). Nyquist plots for the O2 (Fig. S69, ESI†) and H2 electrodes (Fig. S71, ESI†) remain largely unchanged. However, the H2 electrode shows a more pronounced increase in the Rct. The O2 electrode's HFR initially decreases before stabilizing, while the H2 electrode remains stable throughout the test. The O2 electrode potential decreases by 19 mV between initial and final cycles (Fig. S70, ESI†), whereas the H2 electrode potential increases by 65 mV (Fig. S72, ESI†).
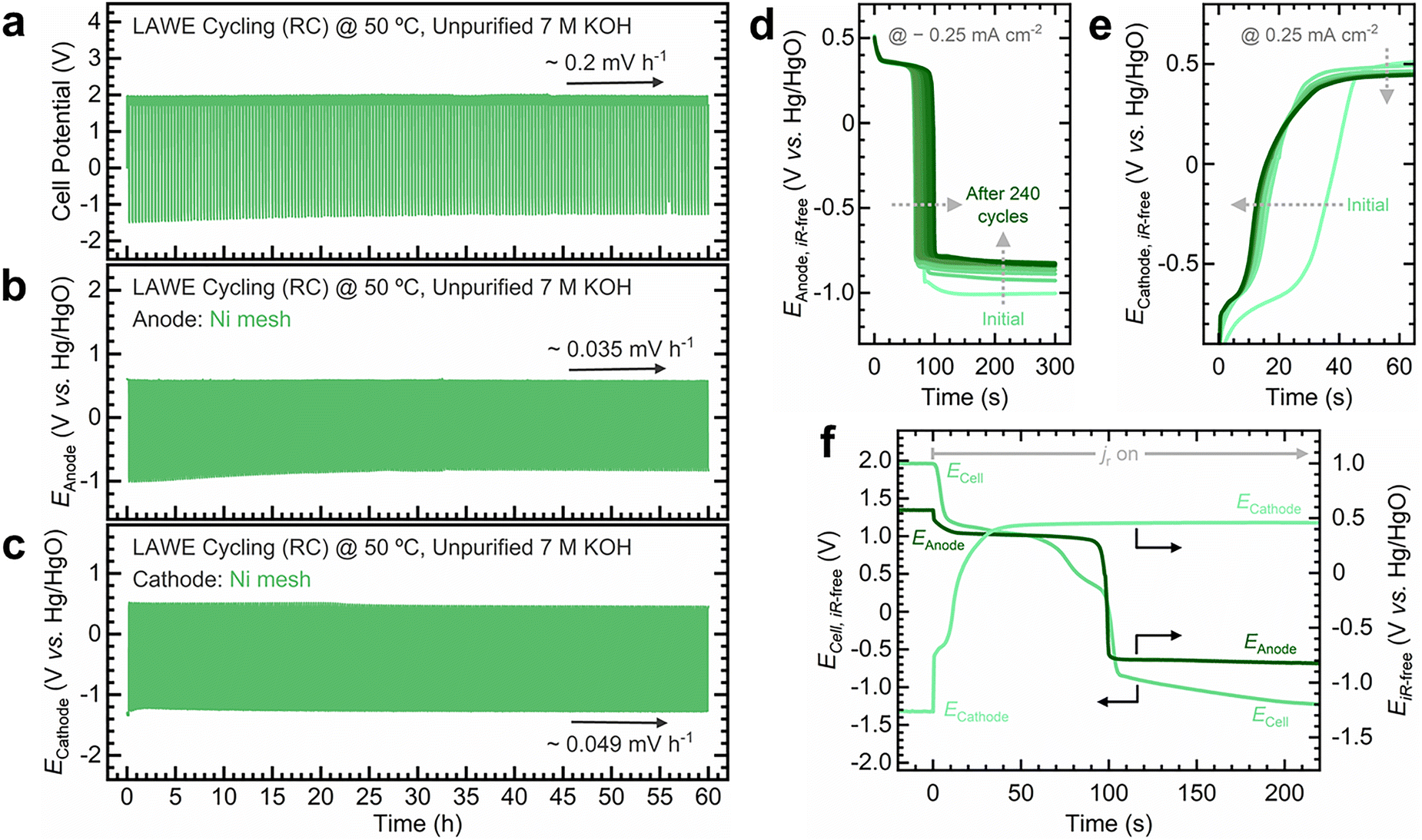 | ||
| Fig. 6 Ni mesh electrodes show a stable performance during variable operation. (a) Total cell, (b) O2 electrode (anode), and (c) H2 electrode (cathode) potential profiles during variable operation tests in a zero-gap alkaline water electrolyzer. The arrows in (a), (b), and (c) indicate the potential increase rate during the highlighted intervals. Comparison of the iR-corrected (d) anode (−0.25 mA cm−2) and (e) cathode (0.25 mA cm−2) potentials during the reverse current step across cycles. The arrows in (d) and (e) indicate the progression of potential profiles during cycling. (f) Comparison of the total cell, anode, and cathode potentials during the reverse current step after 240 cycles. Experimental conditions: unpurified 7 M KOH electrolyte at 50 °C, electrolyte flow rate: 200 mL min−1, projected electrode area: 4 cmgeo2. | ||
During the reverse current step, the O2 electrode discharges to negative potentials (Fig. 6d), exhibiting two key trends: the redox transition stretches over cycling (i.e., self-discharge takes longer), and the potential becomes less negative over cycling. In contrast, the H2 electrode initially displays a pronounced redox feature during oxidation, which diminishes with cycling (Fig. 6e). These changes become more apparent when decoupling the electrode potential from the total cell potential: the O2 electrode discharge extends from 80 to 100 s after 240 cycles (Fig. S73, ESI†), whereas the H2 electrode discharge shortens from 50 to 30 s (Fig. S74, ESI†). Comparing the discharge profiles of both electrodes highlights the distinct impact of variable operation on each (Fig. 6f and Fig. S75, ESI†).
Similar to the variable operation test using a Ni foil O2 electrode in a three-electrode cell (Fig. S36, ESI†), Ni mesh electrodes in a zero-gap electrolyzer exhibit a stable response under variable operation. The growth of the redox transition on the O2 electrode suggests the formation of an increasingly thick NiOOH layer due to continuous redox cycling induced by reverse currents. This behavior resembles the oxyhydroxide layer growth induced by cyclic voltammetry (CV),10 although not directly comparable to CV since reverse currents operate under galvanostatic control (i.e., constant discharge). Ex situ ICP-MS measurements also reveal the continuous dissolution of Ni throughout cycling (Fig. S76, ESI†). While variable operation induces phase transformations and dissolution, as our in situ/operando measurements demonstrate, the Ni mesh electrode supplies fresh active sites from the bulk as the porous oxyhydroxide layer expands, yielding a stable electrochemical response.
Despite maintaining overall performance, the H2 electrode exhibits signs of degradation, with a slightly higher overpotential increase rate and HFR than the O2 electrode (Fig. S71, ESI†). The shift in the redox transition upon oxidation during the reverse step suggests that the oxyhydroxide layer is electrochemically reduced to a blend of hydroxide/metallic phases (Fig. S72d, ESI†).26 Postmortem X-ray photoelectron spectroscopy (XPS) analysis reveals a higher Ni(OH)2 content at the H2 electrode compared to the O2 electrode (Fig. S77–S79, ESI†). Ni(OH)2 acts as an insulator,32,39 which explains the higher HFR and overpotential increase of the H2 electrode after testing.27,32 These findings suggest that the H2 electrode transforms into a less active but more stable phase,30 consistent with previous reports arguing that the H2 electrode is more vulnerable to reverse currents than the O2 electrode.21
Ex situ ICP-MS and XPS measurements reveal that Fe incorporates into the O2 electrode during variable operation, as expected.10,19,32,39 Adding Fe to Ni-based OER catalysts has been leveraged to enhance their OER activity,65 although studies also suggest that Fe undergoes dynamic exchange and may be lost during cycling.32,33,65 To understand this behavior under variable operating conditions, we tested Fe-doped Ni mesh as the O2 electrode in the zero-gap electrolyzer. Instead of depositing a NiFe film, which risks detachment from the substrate, we incorporated Fe into the porous Ni oxyhydroxide layer via CV cycling in a Fe-containing electrolyte (Fig. S80, ESI†).
The full-cell performance remains nearly unchanged (Fig. S81, ESI†) and resembles that of the electrolyzer with Ni mesh electrodes (Fig. S65, ESI†). However, the Fe-doped Ni mesh (Fig. S82, ESI†) shows slightly higher electrode potentials, a greater HFR, and a steeper Tafel slope than the bare Ni mesh electrode (Fig. S66, ESI†). The total cell and O2 electrode potentials increase at 0.7 mV s−1 (Fig. S83, ESI†) and 0.1 mV s−1 (Fig. S84, ESI†), respectively, compared to 0.2 and 0.035 mV s−1 for the bare Ni mesh (Fig. S68 and S69, ESI†). The HFR is ∼1.3 times higher than for Ni mesh (Fig. S84 and S69, ESI†). While the Fe-doped Ni mesh shows a smaller potential decrease (∼9 mV) after cycling (Fig. S85c, ESI†), its redox feature extends up to ∼250 s (Fig. S85d and S86, ESI†). The redox self-discharge time increases ten-fold for Fe-doped Ni mesh but only 1.25 times for Ni mesh, indicating greater oxyhydroxide growth in Fe-doped Ni electrodes.
Ex situ ICP-MS shows an increase in Fe concentration in the electrolyte during cycling (Fig. S87, ESI†), while ex situ XPS reveals a significant decrease in Fe content in the Fe-doped Ni mesh electrode after cycling (Fig. S88–S90, ESI†), reaching a composition similar to that of Ni mesh after cycling (Fig. S79, ESI†). These results confirm Fe loss from the Fe-doped Ni mesh O2 electrode during variable operation, indicating that rapid Fe dissolution stabilizes through dynamic exchange with the catalyst.32,33 Fe deposition on the cathode has been linked to degradation and poor HER performance.18,65 Although this effect was not confirmed by ex situ XPS (Fig. S90c, ESI†), ex situ ICP-MS analysis of the catholyte (Fig. S87b, ESI†) confirmed Fe crossover. It remains unclear whether reverse currents prevent Fe deposition by forming a thinner oxyhydroxide layer on the H2 electrode or reverse it through Fe redissolution during polarity reversal. Further investigation is needed, as metal cations in the electrolyte can induce other undesirable effects.4,27
We also conducted variable operation tests at 1 A cm−2 to better reflect industrially relevant conditions (Fig. S91–S94, ESI†). Compared to the test at 120 mA cm−2, cell and electrode potentials increased at a faster rate, likely due to the greater stress imposed by the higher currents. The larger reverse current also accelerated the discharge of the NiOOH phase, quickly driving the electrode to more reducing conditions (Fig. S93 and S94, ESI†). The more pronounced reduction of the NiOOH layer at the O2 electrode and concurrent formation of NiOOH at the H2 electrode likely account for the stronger degradation observed at higher current densities.
Collectively, the lab-scale electrolyzer tests offer valuable insights into Ni electrode behavior. Bulk Ni maintains stability under variable operation by replenishing active sites. However, electrodes undergo phase transformations and metal dissolution during deep discharge, with the H2 electrode appearing slightly more prone to degradation. These processes can eventually lead to electrode deactivation or undesirable effects from cation leaching. Furthermore, while Fe–Ni electrodes remain stable, they offer no significant advantage over bulk Ni electrodes in unpurified KOH.
A framework for LAWE testing
Based on our analysis of variable operation impacts on LAWE, we outline guidelines for approaching catalyst stability testing and highlight opportunities for future research.Furthermore, standardized cell designs are recommended to improve reproducibility and capture processes under relevant settings.29,51,73 Electrode potentials can be decoupled using reference electrodes but must be used carefully.21,74 We note that developing universal ASTs is currently unrealistic due to the complexity of catalyst degradation and the need for greater mechanistic understanding. Nevertheless, tailoring ASTs to specific situations or degradation mechanisms can be a more effective near-term strategy.31 The chlor-alkali industry, which employs standardized monitoring methods, offers valuable insights.22,28 Researchers are also encouraged to adopt established ASTs from the literature as a starting point.5,12,25
Conclusions
In this study, we systematically investigated the effects of variable operation in LAWE, uncovering critical insights into the stability of transition metal oxyhydroxide electrocatalysts. In contrast to the stable behavior observed during steady-state or open circuit conditions, we showed that reverse currents during simulated shutdowns expose catalytic films to deep discharge conditions, which severely degrade catalytic films.Using a combination of analytical techniques, we identified specific transformations induced by variable operation: in situ SERS revealed phase transitions during deep electrode discharge; ToF-SIMS confirmed catalyst degradation via film thinning, changes in composition, and uneven metal loss; in situ conductivity measurements highlighted changes in electronic conductivity with metal composition; and on-line ICP-MS revealed increased catalyst dissolution during reoxidation and of the catalytic support during reverse currents. Zero-gap electrolyzer experiments further demonstrated that O2 and H2 electrodes undergo distinct potential fluctuations, evolving differently in conductivity, redox behavior, and dissolution rates. Altogether, these results confirm that catalyst dissolution and degradation correlate with the hydroxide/oxyhydroxide phase transitions caused by reverse currents.
We aim to raise awareness of the impact of variable operation on material degradation and incentivize more research focused on understanding catalytic stability under real-world operating conditions. Using the insights developed from our in situ/operando techniques and variable operation tests, we offer guidelines and opportunities for designing more resilient electrode materials, developing effective strategies to counteract the effects of reverse currents, standardizing ASTs, and refining catalyst degradation studies. We encourage work to expand on these findings and further elucidate unresolved aspects, such as electrolyte composition effects.
Emerging fields with similar chemistries, such as anion-exchange membrane water electrolysis, can also investigate the impact of variable operation on the degradation pathways of critical components, including anion-exchange polymers used in membranes and gas diffusion electrodes.75–77 Our analytical approach for tracking catalyst transformations and electrode dynamics could also be extrapolated to other contexts in catalysis research. Our work highlights the need to place stability as a central part of electrocatalysis research. By bridging fundamental science and industrial-scale operations, we can accelerate the development of durable and cost-effective hydrogen production technologies.
Experimental
Electrode and electrolyte preparation
Ti foil (Thermo Scientific) was used as the catalytic support unless otherwise indicated. Electrocatalytic films were cathodically deposited on Ti foil using a two-electrode configuration following previous guidelines.39,52,78 Films were deposited galvanostatically at −1 mA cm−2 (thickness: ∼110 nm). KOH electrolytes were prepared from reagent-grade potassium hydroxide (Sigma Aldrich, 89.5%) and CO2-free deionized water (18.2 MΩ cm). Tests were performed using unpurified electrolytes unless otherwise indicated. KOH electrolytes were prepared and purified following established protocols.37,39 Note: most experiments were conducted in 0.1 M KOH instead of 1 M to maintain consistency, as this condition was necessary for in situ and operando measurements. Further details on substrate preparation, film electrodeposition, and electrode nomenclature can be found in the ESI.†Electrochemical methods
A 100 mL PTFE cell (Fig. S1, ESI†) was used in three-electrode cell experiments. Graphite rods (Gamry) were used as counter electrodes in three-electrode cell tests to prevent incidental Pt incorporation.79 Electrode potentials were measured against Hg/HgO reference electrodes (CH Instruments). Potentials were converted to the reversible hydrogen electrode (RHE), corrected for the liquid junction potential, and periodically monitored to ensure stability.74 Unless otherwise indicated, measurements were conducted using a Gamry Reference 620 potentiostat/galvanostat. All current densities were calculated based on the projected geometric area. Electrode potentials were corrected for the uncompensated resistance (Ru).48,80 Prior to experiments, electrodes were conditioned via CP to form a thin oxyhydroxide layer.10 Further details on the cell setup, reference electrodes, iR compensation, and electrochemical conditioning can be found in the ESI.†Simulated shutdown tests to study electrode discharge
The OCP decay was measured as follows: a CP step was applied for 480 s at the specified current density. Next, a fast two-step measurement, combining a CP step (10 s, same current density) followed by an OCP step (30 s), was performed to capture the rapid discharge. Then, an OCP measurement (1 h) was recorded. Together, these steps constitute an electrode discharge test. Following the electrode discharge experiment, a potentiodynamic polarization test was carried out according to corrosion testing protocols.45,81–83 Further details on the experimental setup, OCP decay measurements, and potentiodynamic polarization tests can be found in the ESI.†Accelerated catalyst degradation tests in a three-electrode cell
Experiments were conducted as an automated sequence with a stability cycling loop, each consisting of a galvanostatic step at 50 mA cm−2 (10 min) followed by a reverse current at −5 mA cm−2 (5 min). For steady-state current tests, the cycling loop included only the 50 mA cm−2 step extended to 15 min; for reverse steps under open circuit conditions, the second step was conducted at 0 mA. The loop was repeated 30 times for steady-state current tests and 60 times for reverse current tests. Further experimental details can be found in the ESI.†In situ SERS measurements
Catalytic films were deposited on screen-printed gold electrodes (Metrohm) at −1 mA cm−2 (thickness: ∼90 nm). Experiments were conducted in a custom electrochemical cell (Fig. S38, ESI†). Raman measurements were carried out using a Horiba LabRAM ARAMIS confocal Raman microscope. Spectra were collected using a 633 nm laser (2.5 mW) at a 1 cm−1 resolution and optimized following previous guidelines.84 Electrode discharge experiments consisted of an initial constant potential step followed by a reverse current step via CP at −0.25 mA cm−2. Spectra were acquired in a single accumulation of 10 s. Further details on electrode preparation, spectra collection, and electrochemical measurements can be found in the ESI.†ToF-SIMS measurements
Depth profiles were acquired using an IONTOF GmbH TOF-SIMS M6 instrument in negative polarity (Cs+ beam, 40 nA, 0.5 kV). An area of 300 × 300 μm2 was sputtered, followed by raster-scanning a 100 × 100 μm2 area within the sputtered crater using a Bi+ analysis beam (4 pA, 30 keV). High-resolution imaging was performed in fast-imaging mode. A statistical depth profile analysis was performed to estimate the depth distribution ranges of secondary ion fragments. Further experimental details can be found in the ESI.†In-plane effective conductivity measurements
Films were deposited on IDA gold electrodes (Metrohm) at −1 mA cm−2. Deposition times were optimized to ensure consistent film thicknesses and prevent detachment. Film thickness was measured via non-contact profilometry (Keyence VK-X1100). Electrochemical measurements were done in a three-electrode electrochemical cell (Fig. S52a, ESI†) using a CH Instruments 832 bipotentiostat. Constant potential steps from −0.02 to 0.64 V vs. Hg/HgO were applied with 20 mV increments. Potential stepping was performed with a 10 mV offset between working electrodes. The in-plane effective conductivity was calculated as described elsewhere.52 Further details on film preparation and conductivity measurements can be found in the ESI.†On-line ICP-MS measurements
Catalytic films deposited on Ti foil (thickness: ∼550 nm) were tested in an in-house electrochemical flow cell (Fig. S54, ESI†) described elsewhere.57 Electrochemical measurements were done using a BioLogic VSP-300 potentiostat, a Pt wire counter electrode placed downstream of the flow cell, and an Ag/AgCl reference electrode (BASi). Current densities were optimized to minimize interference from gas bubbles. Solution-mode ICP-MS was used to measure metal concentrations in unpurified 0.1 M KOH electrolytes using an iCAP RQ ICP-MS (Thermo Fisher Scientific). The spectrometer was configured to detect 60Ni, 48Ti, 57Fe, and 59Co isotopes. Calibration curves were prepared from reference standards (Sigma-Aldrich) in semiconductor-grade KOH at concentrations ranging from 2.5 to 500 ppb. For variable operation experiments, a single stability cycling loop consisted of a CP step (1.35 mA cm−2, 3 min) followed by a reverse current step (−1.35 mA cm−2, 3 min). Further details on the experimental setup, electrochemical measurements, and data analysis can be found in the ESI.†Zero-gap electrolyzer experiments
Ni gauze (Thermo Scientific) was used as the H2 and O2 electrodes. Fe-doped Ni mesh anodes were prepared by conditioning a Ni gauze strip in KOH electrolyte spiked with Fe (1 mM). We used the zero-gap alkaline water electrolyzer reported in our previous study,29 incorporating an endplate with a dedicated port for the Hg/HgO reference electrode (Fig. S58, ESI†). ZIRFON PERL UTP 500 (Agfa) was used as the separator. Electrochemical tests were conducted with two potentiostats operating simultaneously (Fig. S58d, ESI†). Experiments were conducted in 7 M KOH electrolyte at 50 °C and a flow rate of 200 mL min−1. Unpurified KOH was used to maintain industrial relevance. Fe impurity concentrations (∼550 ppb) were consistent across experiments, as recommended.6,18,19 Variable operation tests were conducted using an automated sequence adapted from our previous work,29 designed to perform conditioning, polarization curves, and stability cycling loops. Galvanostatic EIS was used to measure the HFR. Further details on the electrode preparation, electrolyzer operation, estimation of the reverse current, and ex situ XPS and ICP-MS analysis can be found in the ESI.†Author contributions
R. A. M.: conceptualization, methodology, formal analysis, investigation, writing – original draft, visualization. J. T. B.: conceptualization, investigation, formal analysis, writing – review & editing. A. M. A.: conceptualization, investigation, formal analysis, writing – review & editing. E. K.: investigation, writing – review & editing. T. V. L.: investigation, writing – review & editing. C. L. W.: investigation, writing – review & editing. M. L. F.: conceptualization, investigation, writing – review & editing. K. K.: conceptualization, writing – review & editing. C. E. C.: conceptualization, writing – review & editing. A. D.: investigation, formal analysis. D. J. M.: resources, writing – review & editing, supervision, funding acquisition. J. R.: conceptualization, writing – review & editing, supervision, funding acquisition. T. F. J.: conceptualization, resources, writing – review & editing, supervision, funding acquisition. C. B. M.: conceptualization, resources, writing – review & editing, project administration, funding acquisition. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article, including 3D models for custom electrochemical cells and tabulated data from experiments, are available at GitHub at: https://doi.org/10.5281/zenodo.15252231.Acknowledgements
The authors gratefully acknowledge funding from the National Science Foundation (NSF) via Grant 2102307, MRI NSF Grant 2117623 for the acquisition of the VersaProbe 4 scanning XPS instrument, and the Welch Foundation through Grants F-2076 and F-1848 for their generous support. D. J. M. and J. R. acknowledge support from the UT Austin Energy Institute. A. M. A. and T. F. J. were supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, Catalysis Science Program to the SUNCAT Center for Interface Science and Catalysis for contributions to the experimental design and analysis, in particular for ICP-MS studies. R. A. M. acknowledges CONAHCYT for his Doctoral Scholarship Award (CVU 919871) and the generous support from the Electrochemical Society through the 2024 Edward G. Weston Summer Fellowship. We thank David Gray and Shallaco McDonald for manufacturing the in situ SERS measurement cell and Dr. Egil Rasten, Dr. Michaela Burke Stevens, and Dr. Grace Lindquist for their valuable guidance and feedback.References
- W. J. Shaw, M. K. Kidder, S. R. Bare, M. Delferro, J. R. Morris, F. M. Toma, S. D. Senanayake, T. Autrey, E. J. Biddinger, S. Boettcher, M. E. Bowden, P. F. Britt, R. C. Brown, R. M. Bullock, J. G. Chen, C. Daniel, P. K. Dorhout, R. A. Efroymson, K. J. Gaffney, L. Gagliardi, A. S. Harper, D. J. Heldebrant, O. R. Luca, M. Lyubovsky, J. L. Male, D. J. Miller, T. Prozorov, R. Rallo, R. Rana, R. M. Rioux, A. D. Sadow, J. A. Schaidle, L. A. Schulte, W. A. Tarpeh, D. G. Vlachos, B. D. Vogt, R. S. Weber, J. Y. Yang, E. Arenholz, B. A. Helms, W. Huang, J. L. Jordahl, C. Karakaya, K. (Cyrus) Kian, J. Kothandaraman, J. Lercher, P. Liu, D. Malhotra, K. T. Mueller, C. P. O’Brien, R. M. Palomino, L. Qi, J. A. Rodriguez, R. Rousseau, J. C. Russell, M. L. Sarazen, D. S. Sholl, E. A. Smith, M. B. Stevens, Y. Surendranath, C. J. Tassone, B. Tran, W. Tumas and K. S. Walton, Nat. Rev. Chem., 2024, 8, 376–400 CrossRef PubMed.
- D. Castelvecchi, Nature, 2022, 611, 440–443 CrossRef CAS PubMed.
- Z. J. Schiffer and K. Manthiram, Joule, 2017, 1, 10–14 CrossRef.
- J. Edgington and L. C. Seitz, ACS Catal., 2023, 13, 3379–3394 CrossRef CAS.
- P. Aßmann, A. S. Gago, P. Gazdzicki, K. A. Friedrich and M. Wark, Curr. Opin. Electrochem., 2020, 21, 225–233 CrossRef.
- J. C. Ehlers, A. A. Feidenhans’l, K. T. Therkildsen and G. O. Larrazábal, ACS Energy Lett., 2023, 8, 1502–1509 CrossRef CAS.
- K. Kawashima, R. A. Marquez, L. A. Smith, R. R. Vaidyula, O. A. Carrasco Jaim, Z. Wang, Y. J. Son, C. L. Cao and C. B. Mullins, Chem. Rev., 2023, 123, 12795–13208 CrossRef CAS PubMed.
- Y. Zhou and H. J. Fan, ACS Mater. Lett., 2021, 3, 136–147 CrossRef CAS.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- Y. J. Son, S. Kim, V. Leung, K. Kawashima, J. Noh, K. Kim, R. A. Marquez, O. A. Carrasco-Jaim, L. A. Smith, H. Celio, D. J. Milliron, B. A. Korgel and C. B. Mullins, ACS Catal., 2022, 12, 10384–10399 CrossRef CAS.
- C.-Z. Yuan, K. S. Hui, H. Yin, S. Zhu, J. Zhang, X.-L. Wu, X. Hong, W. Zhou, X. Fan, F. Bin, F. Chen and K. N. Hui, ACS Mater. Lett., 2021, 3, 752–780 CrossRef CAS.
- S. Appelhaus, L. Ritz, S.-V. Pape, F. Lohmann-Richters, M. R. Kraglund, J. O. Jensen, F. Massari, M. Boroomandnia, M. Romanò, J. Albers, C. Kubeil, C. Bernäcker, M. S. Lemcke, N. Menzel, G. Bender, B. Chen, S. Holdcroft, R. Delmelle, J. Proost, J. Hnát, P. Kauranen, V. Ruuskanen, T. Viinanen, M. Müller, T. Turek and M. Shviro, Int. J. Hydrogen Energy, 2024, 95, 1004–1010 CrossRef CAS.
- A. A. Samu, A. Kormányos, E. Kecsenovity, N. Szilágyi, B. Endrődi and C. Janáky, ACS Energy Lett., 2022, 7, 1859–1861 CrossRef CAS PubMed.
- K. M. K. Yap, W. J. Wei, M. R. Pabón, A. J. King, J. C. Bui, L. Wei, S.-W. Lee, A. Z. Weber, A. T. Bell, A. C. Nielander and T. F. Jaramillo, Energy Environ. Sci., 2024, 17, 2453–2467 RSC.
- H. Kojima, K. Nagasawa, N. Todoroki, Y. Ito, T. Matsui and R. Nakajima, Int. J. Hydrogen Energy, 2023, 48, 4572–4593 CrossRef CAS.
- D. G. Nocera, Energy Environ. Sci., 2010, 3, 993–995 RSC.
- G. Sakas, A. Ibáñez-Rioja, S. Pöyhönen, A. Kosonen, V. Ruuskanen, P. Kauranen and J. Ahola, Renewable Energy, 2024, 225, 120266 CrossRef CAS.
- M. Demnitz, Y. M. Lamas, R. L. Garcia Barros, A. de Leeuw den Bouter, J. van der Schaaf and M. Theodorus de Groot, iScience, 2023, 27, 108695 CrossRef PubMed.
- M. T. de Groot, Curr. Opin. Chem. Eng., 2023, 42, 100981 CrossRef.
- M. Schalenbach, A. Zeradjanin, O. Kasian, S. Cherevko and K. Mayrhofer, Int. J. Electrochem. Sci., 2018, 13, 1173–1226 CrossRef CAS.
- N. Guruprasad, J. van der Schaaf and M. T. de Groot, J. Power Sources, 2024, 613, 234877 CrossRef CAS.
- E. Rasten, ECS Trans., 2024, 113, 25–41 CrossRef CAS.
- D. Hochfilzer, I. Chorkendorff and J. Kibsgaard, ACS Energy Lett., 2023, 8, 1607–1612 CrossRef CAS PubMed.
- M. Demnitz, J. van der Schaaf and T. de Groot, J. Electrochem. Soc., 2025, 172, 014504 CrossRef CAS.
- A. Abdel Haleem, K. Nagasawa, Y. Kuroda, Y. Nishiki, A. Zaenal and S. Mitsushima, Electrochemistry, 2021, 89, 186–191 CrossRef CAS.
- Y. Kim, S.-M. Jung, K.-S. Kim, H.-Y. Kim, J. Kwon, J. Lee, H.-S. Cho and Y.-T. Kim, JACS Au, 2022, 2, 2491–2500 CrossRef CAS PubMed.
- A. Weiß, A. Siebel, M. Bernt, T.-H. Shen, V. Tileli and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, F487 CrossRef.
- A. R. Zeradjanin, Nat. Catal., 2023, 6, 458–459 CrossRef CAS.
- R. A. Marquez, M. Espinosa, E. Kalokowski, Y. J. Son, K. Kawashima, T. V. Le, C. E. Chukwuneke and C. B. Mullins, ACS Energy Lett., 2024, 9, 547–555 CrossRef CAS.
- L. Du and W. Zheng, APL Energy, 2024, 2, 021501 CrossRef CAS.
- M. Risch, Curr. Opin. Electrochem., 2023, 38, 101247 CrossRef CAS.
- R. A. Marquez, E. Kalokowski, M. Espinosa, J. T. Bender, Y. J. Son, K. Kawashima, C. E. Chukwuneke, L. A. Smith, H. Celio, A. Dolocan, X. Zhan, N. Miller, D. J. Milliron, J. Resasco and C. B. Mullins, Energy Environ. Sci., 2024, 17, 2028–2045 RSC.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- K. Oda, Y. Kuroda and S. Mitsushima, Electrocatalysis, 2023, 14, 499–510 CrossRef CAS.
- L. Bui, M. Joswiak, I. Castillo, A. Phillips, J. Yang and D. Hickman, ACS Eng. Au, 2022, 2, 17–26 CrossRef CAS.
- A. J. Martín, S. Mitchell, C. Mondelli, S. Jaydev and J. Pérez-Ramírez, Nat. Catal., 2022, 5, 854–866 CrossRef.
- R. A. Márquez, K. Kawashima, Y. J. Son, G. Castelino, N. Miller, L. A. Smith, C. E. Chukwuneke and C. B. Mullins, ACS Energy Lett., 2023, 8, 1141–1146 CrossRef.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- M. Mattinen, J. Schröder, G. D’Acunto, M. Ritala, T. F. Jaramillo, M. B. Stevens and S. F. Bent, Cell Rep. Phys. Sci., 2024, 5, 102284 CrossRef CAS.
- C. F. Crago, S. Li, A. M. Aleman, T. Siboonruang, M. Rojas Mendoza, T. F. Jaramillo and M. B. Stevens, J. Am. Chem. Soc., 2025, 147, 3925–3930 CrossRef CAS PubMed.
- E. R. Sauvé, B. Y. Tang, N. K. Razdan, W. L. Toh, S. Weng and Y. Surendranath, Joule, 2024, 8, 728–745 CrossRef.
- R. A. Marquez, J. T. Bender, S. C. da Cunha, A. M. Aleman, A. Sahu, V. Ganesan, D. J. Milliron, J. Resasco, T. F. Jaramillo and C. B. Mullins, ACS Energy Lett., 2025, 10, 2075–2083 CrossRef CAS.
- E. S. Davydova, Y. Halpern, A. Breytus and A. Rothschild, Solid State Ion., 2025, 422, 116815 CrossRef CAS.
- Practice for Conventions Applicable to Electrochemical Measurements in Corrosion Testing. ASTM G3-14. ASTM International, 2014.
- A. Abdel Haleem, J. Huyan, K. Nagasawa, Y. Kuroda, Y. Nishiki, A. Kato, T. Nakai, T. Araki and S. Mitsushima, J. Power Sources, 2022, 535, 231454 CrossRef CAS.
- M. Bernt and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, F3179 CrossRef CAS.
- Y. J. Son, R. A. Marquez, K. Kawashima, L. A. Smith, C. E. Chukwuneke, J. Babauta and C. B. Mullins, ACS Energy Lett., 2023, 8, 4323–4329 CrossRef CAS.
- J. F. Jackovitz, Proceedings of the Symposium on the Nickel Electrode, The Electrochemical Society, New Jersey, 1982.
- N. Todoroki, K. Nagasawa, H. Enjoji and S. Mitsushima, ACS Appl. Mater. Interfaces, 2023, 15, 24399–24407 CrossRef CAS PubMed.
- S. M. Alia, K. S. Reeves, D. A. Cullen, H. Yu, A. J. Kropf, N. Kariuki, J. H. Park and D. J. Myers, J. Electrochem. Soc., 2024, 171, 044503 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- K. Yan, S.-W. Lee, K. M. K. Yap, A. S. Mule, R. T. Hannagan, J. E. Matthews, G. A. Kamat, D. U. Lee, M. B. Stevens, A. C. Nielander and T. F. Jaramillo, J. Am. Chem. Soc., 2025, 147, 4079–4088 CrossRef CAS PubMed.
- C. Luan, D. Escalera-López, U. Hagemann, A. Kostka, G. Laplanche, D. Wu, S. Cherevko and T. Li, ACS Catal., 2024, 14, 12704–12716 CrossRef CAS.
- A. M. Aleman, C. F. Crago, G. A. Kamat, A. S. Mule, J. E. Avilés Acosta, J. E. Matthews, N. Keyes, R. T. Hannagan, A. C. Nielander, M. B. Stevens and T. F. Jaramillo, J. Am. Chem. Soc., 2025, 147, 10309–10319 CrossRef CAS PubMed.
- L.-F. Huang, M. J. Hutchison, R. J. Santucci, J. R. Scully and J. M. Rondinelli, J. Phys. Chem. C, 2017, 121, 9782–9789 CrossRef CAS.
- R. A. Marquez, E. E. Oefelein, T. V. Le, K. Kawashima, L. A. Smith and C. B. Mullins, ACS Mater. Lett., 2024, 6, 2905–2918 CrossRef CAS.
- C. Dette, M. R. Hurst, J. Deng, M. R. Nellist and S. W. Boettcher, ACS Appl. Mater. Interfaces, 2019, 11, 5590–5594 CrossRef CAS PubMed.
- R. A. Marquez-Montes, K. Kawashima, Y. J. Son, J. A. Weeks, H. H. Sun, H. Celio, V. H. Ramos-Sánchez and C. B. Mullins, J. Mater. Chem. A, 2021, 9, 7736–7749 RSC.
- W. Liu, X. Ding, J. Cheng, J. Jing, T. Li, X. Huang, P. Xie, X. Lin, H. Ding, Y. Kuang, D. Zhou and X. Sun, Angew. Chem., 2024, 136, e202406082 CrossRef.
- D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 3838–3839 CrossRef CAS PubMed.
- C. Costentin and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13380–13384 CrossRef CAS PubMed.
- F. D. Speck, K. E. Dettelbach, R. S. Sherbo, D. A. Salvatore, A. Huang and C. P. Berlinguette, Chem, 2017, 2, 590–597 CAS.
- P. C. Jiménez, G. K. H. Wiberg, G. W. Sievers, V. Brüser and M. Arenz, J. Mater. Chem. A, 2023, 11, 20129–20138 RSC.
- B. R. Wygant, K. Kawashima and C. B. Mullins, ACS Energy Lett., 2018, 3, 2956–2966 CrossRef CAS.
- K. Kawashima, R. A. Márquez-Montes, H. Li, K. Shin, C. L. Cao, K. M. Vo, Y. J. Son, B. R. Wygant, A. Chunangad, D. H. Youn, G. Henkelman, V. H. Ramos-Sánchez and C. B. Mullins, Mater. Adv., 2021, 2, 2299–2309 RSC.
- M. Schreier, P. Kenis, F. Che and A. S. Hall, ACS Energy Lett., 2023, 8, 3935–3940 CrossRef CAS.
- J. T. Bender, A. S. Petersen, F. C. Østergaard, M. A. Wood, S. M. J. Heffernan, D. J. Milliron, J. Rossmeisl and J. Resasco, ACS Energy Lett., 2023, 8, 657–665 CrossRef CAS.
- G. A. Lindquist, J. C. Gaitor, W. L. Thompson, V. Brogden, K. J. T. Noonan and S. W. Boettcher, Energy Environ. Sci., 2023, 16, 4373–4387 RSC.
- B. Delmon, Appl. Catal., 1985, 15, 1–16 CrossRef CAS.
- M. Geuß, M. Milosevic, M. Bierling, L. Löttert, D. Abbas, D. Escalera-López, V. Lloret, K. Ehelebe, K. J. J. Mayrhofer, S. Thiele and S. Cherevko, J. Electrochem. Soc., 2023, 170, 114510 CrossRef.
- K. Kawashima, R. A. Márquez, Y. J. Son, C. Guo, R. R. Vaidyula, L. A. Smith, C. E. Chukwuneke and C. B. Mullins, ACS Catal., 2023, 13, 1893–1898 CrossRef CAS.
- G. A. Lindquist, Q. Xu, S. Z. Oener and S. W. Boettcher, Joule, 2020, 4, 2549–2561 CrossRef CAS.
- G. A. Lindquist, J. C. Gaitor, W. L. Thompson, V. Brogden, K. J. T. Noonan and S. W. Boettcher, Energy Environ. Sci., 2023, 16, 4373–4387 RSC.
- R. A. Krivina, G. A. Lindquist, S. R. Beaudoin, T. N. Stovall, W. L. Thompson, L. P. Twight, D. Marsh, J. Grzyb, K. Fabrizio, J. E. Hutchison and S. W. Boettcher, Adv. Mater., 2022, 34, 2203033 CrossRef CAS PubMed.
- M. B. Stevens, L. J. Enman, A. S. Batchellor, M. R. Cosby, A. E. Vise, C. D. M. Trang and S. W. Boettcher, Chem. Mater., 2017, 29, 120–140 CrossRef CAS.
- R. Chen, C. Yang, W. Cai, H.-Y. Wang, J. Miao, L. Zhang, S. Chen and B. Liu, ACS Energy Lett., 2017, 2, 1070–1075 CrossRef CAS.
- W. Zheng, ACS Energy Lett., 2023, 8, 1952–1958 CrossRef CAS.
- Reference Test Method for Making Potentiodynamic Anodic Polarization Measurements. ASTM G5-14. ASTM International, 2014.
- Test Method for Conducting Potentiodynamic Polarization Resistance Measurements. ASTM G59-97 (Reapproved 2014). ASTM International, 2014.
- Practice for Calculation of Corrosion Rates and Related Information from Electrochemical Measurements. ASTM G102-89 (Reapproved 2015). ASTM International, 2015.
- W. Zheng, Chem.:Methods, 2022, 3, e202200042 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and procedures, supporting figures and tables, additional characterization data. See DOI: https://doi.org/10.1039/d5ee02194d |
| This journal is © The Royal Society of Chemistry 2025 |