
Ligand effects enhancing low-temperature oxygen reduction kinetics in neutral conditions†
Yongjian
Zhao
ab,
Murong
Huang
a,
Yongshuai
Kang
a,
Yong
Fang
a,
Tianyou
Zhao
a,
Hu
Wang
a,
Jiayi
Ou
a,
Jiajun
Liu
a,
Meixi
Zhong
a,
Tao
Wang
d,
Xianhu
Sun
 *bc,
Chenyang
Zhao
*a and
Dan
Wang
*a
*bc,
Chenyang
Zhao
*a and
Dan
Wang
*a
aCollege of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen 518071, Guangdong, China. E-mail: cyzhao@szu.edu.cn; danwang@szu.edu.cn; danwang@ipe.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 101408, China. E-mail: sunxianhu@ucas.ac.cn
cBinzhou Institute of Technology, Weiqiao-UCAS Science and Technology Park, Binzhou 256606, Shandong, China. E-mail: sunxianhu@ucas.ac.cn
dCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: twangxm@xmu.edu.cn
First published on 17th April 2025
Abstract
The sluggish oxygen reduction kinetics, resulting from ineffective O2 activation and hydrogenation, has hindered the performance improvement of self-breathing zinc–air batteries (ZABs), especially in harsh environments with low temperatures and low proton concentrations. Herein, we report a series of N-, P-doped carbon catalysts with distinct coordination topologies and structural characteristics. The combination of in situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS), in situ Raman spectroscopy and density functional theory (DFT) calculations collaboratively reveals that the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ligands effectively regulate the charge density and spin states around carbon sites and activate O–O bonds through bridge chemisorption (Yeager model), shifting the reaction kinetics to a favorable reaction pathway. As a result, the P, N co-doped carbon materials (CNP-900) display remarkable half-wave potentials, fast kinetic and minimal degradation over a wide pH and temperature range. Moreover, flexible zinc–air batteries (FZABs) based on CNP-900 exhibit maximum power densities of 104.2 and 47.1 mW cm−2 under alkaline and neutral conditions, respectively, at a temperature of −20 °C. These results provide new perspectives on the kinetic enhancement of metal-free oxygen reduction catalysts and emphasize the significance of O2 adsorption/activation in harsh environments.
O ligands effectively regulate the charge density and spin states around carbon sites and activate O–O bonds through bridge chemisorption (Yeager model), shifting the reaction kinetics to a favorable reaction pathway. As a result, the P, N co-doped carbon materials (CNP-900) display remarkable half-wave potentials, fast kinetic and minimal degradation over a wide pH and temperature range. Moreover, flexible zinc–air batteries (FZABs) based on CNP-900 exhibit maximum power densities of 104.2 and 47.1 mW cm−2 under alkaline and neutral conditions, respectively, at a temperature of −20 °C. These results provide new perspectives on the kinetic enhancement of metal-free oxygen reduction catalysts and emphasize the significance of O2 adsorption/activation in harsh environments.
Broader contextFlexible zinc–air batteries (FZABs), as promising energy storage devices, have attracted increasing attention. However, their wide application is hindered by the sluggish kinetics resulting from ineffective O2 activation on the air cathode, especially in neutral and low-temperature conditions. The “end-on” fashion (Pauling model), as a common O2 adsorption structure, is the most studied in the oxygen reduction reaction (ORR). In contrast, the “bridge” adsorption (Yeager model) has a smaller O–O cleavage energy barrier and faster electron transfer kinetics, and thus the Yeager model seems more efficient to catalyze/activate O2. In this work, we provide new insights into the regulation of oxygen adsorption patterns by switching from the Pauling model to the Yeager one to enhance the kinetics of heteroatom-doped carbon catalysts. This is achieved by the development of P–N co-doped metal-free carbon catalysts, based on which the unique PO ligand regulates the charge and spin states of adjacent carbon sites, enabling a transition from “end-on” to the “bridge” chemisorption of O2 on C/P pairs, exhibiting outstanding ORR performance in neutral media.
|
Introduction
Self-breathing wearable flexible zinc–air batteries (FZABs) have attracted increasing attention due to their high energy density, safety, affordability, and eco-friendliness.1–3 Acting as an intermediary between the electrolyte and the atmosphere, the air electrode is considered as the most significant component of FZABs, with multiple functionalities in promoting mass transfer, accelerating charge transport, and minimizing reaction energy barriers to improve the electrochemical performance of the devices. The oxygen reduction reaction (ORR) is a crucial electrochemical process that occurs at the air electrode and involves multiple electron/charge transfers.4 The complexity of the ORR results in sluggish reaction kinetics and an insurmountable high energy barrier, especially at low temperatures, which is one of the biggest obstacles to limiting the practical application of FZABs.5 To tackle these challenges, noble metal catalysts represented by platinum (Pt) are widely used. However, a profitable balance between cost and performance is still challenging to reach. The low-temperature performance of the Pt-based ORR catalysts is far away from scalable applications, as evidenced by the notable increase in overpotential and kinetic hindrance. Therefore, the exploration of efficient ORR catalysts with high intrinsic activity and low-temperature adaptability is essential for promoting the practicality of FZABs.Recently, metal-free heteroatom-doped carbon nanomaterials have emerged as one of the most promising catalyst materials to replace scarce and precious metals, since they have merits of low-cost, high durability, and structure adjustability.6–8 Their intrinsic activity originates from the regulation of charge, spin and coordination states by heteroatom doping with improved proton-coupled electron transfer (PCET) capability in late steps of the catalytic cycles.9–12 However, contrary to the widespread notion that a PCET step with the highest energy barrier limits the reaction rate, recent research progress has increasingly pointed out the decisive role of oxygen adsorption/activation on the catalyst surface.13,14 The O2 adsorption with “end-on” fashion (Pauling model) is the most widely studied model in the ORR.15–18 Nevertheless, the bridge adsorption of O2 (Yeager model) has a smaller O–O cleavage energy barrier and faster PCET kinetics process than the Pauling model, accelerating reaction, especially under harsh pH and temperature conditions.19,20 Although the Yeager model possesses intrinsic kinetics advantages that should be adopted for efficiently catalyzing oxygen, the modulation of oxygen adsorption mode by heteroatom doping is rarely reported.
Among the various dual-heteroatom combinations, phosphorus (P) mixing with N could achieve large structural aberration (C: 77 pm; N: 70 pm; P: 110 pm), low electronegativity and outstanding electron transfer ability with O2 molecules,21,22 which enable spin modulation and charge redistribution of active sites to enhance the ORR performance.23 This is manifested by P–N co-doped graphene that is reported to exhibit a half-wave potential (E1/2) of 0.845 V in an alkaline medium.24
Despite this, there is an ongoing debate on the precise arrangement of N–P dopants and their specific roles in the ORR. Firstly, the large atomic radius of P will give rise to severe lattice repulsion once P atoms are inserted into the carbon lattice. This is energetically unfavorable, together with an inappropriate estimation of the P contribution. Thus, P prefers to locate at the surface edges or defects of the carbon matrix rather than bulk area,25 guided by either sp3 or sp3d hybridization. Secondly, because of the strong oxophilicity from P, the oxidation of P is unavoidable during the catalyst preparation, and the resultant various P–O configurations are often ignored due to the complex bonding environments and technical difficulties in characterization.26–29 Such PO configurations are more withstanding than P–O/OH structure units under high temperature treatments, a typical synthesis step of heteroatom-doped carbons. Thus, the attribution of enhanced ORR activity in P, N co-doped carbons to P–O/OH may be questionable, and a deserved research topic should be clarified regarding the role of the chemically stable and more prevalent PO.
Upon P doping, a positive charge center is created at the P sites due to the electronegativity difference.30,31 It is generally believed that the adsorption and dissociation of O2 mainly occur at the positively charged sites, i.e., P sites. However, some recent reports show that the ORR activity of carbon sites is not only controlled by their charge, but also by their spin density, and coordination state.32–34 Therefore, through precise adjustment of the local microenvironment, the carbon atoms connected to P may also be activated, creating additional reactive sites for the ORR. It is noted that the cooperative effect of C–P atoms could offer an opportunity to finely tune the O2 adsorption/activation process, which is regarded as the real rate-limiting step of the ORR if taking the solvent effect into account,35 thereby enabling favorable ORR kinetics even under hostile pH and temperature conditions. However, whether the carbon atoms adjacent to the P dopants participate in the ORR process remains unclear.
Herein, we report P–N co-doped metal-free carbon catalysts with an optimized local coordination environment for robust ORR across wide pH and temperature windows. The elaborately designed PO ligands are found to modify the charge and spin states of adjacent carbons, which successfully activates the catalysts for promoting ORR kinetics and induces the end-on chemisorption of O2 transition to the bridge ones with favorable 4e− pathway energetics. As a result, the P–N co-doped metal-free carbon catalysts demonstrate an impressive ORR performance with an E1/2 of 0.77 V and minimal attenuation after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential cycles in 0.1 M PBS solution. Moreover, the assembled neutral FZABs based on the catalysts show exceptional rate capability, cycling life and temperature adaptability, achieving maximum power densities of 47.1, 35.4 and 25.6 mW cm−2 at 20 °C, 0 °C and −20 °C, respectively.
000 potential cycles in 0.1 M PBS solution. Moreover, the assembled neutral FZABs based on the catalysts show exceptional rate capability, cycling life and temperature adaptability, achieving maximum power densities of 47.1, 35.4 and 25.6 mW cm−2 at 20 °C, 0 °C and −20 °C, respectively.
Results and discussion
As illustrated in Fig. 1a, P, N co-doped carbon catalysts are synthesized via a molten-salt-mediated approach.36–38 The synthesis begins with preparing ZIF-8(Cl) by mixing organic–inorganic precursors, which self-assemble into a circular shape disorderly stacked under Cl− coordination (Fig. 1a, left). The mixture is then mixed with KCl/ZnCl2 salts and heated at 900 °C under Ar flow for 2 hours, forming hierarchical N-doped C materials (Fig. 1a, middle) due to Zn evaporation and carbon matrix etching. Finally, triphenylphosphine as a P source is added to the N-doped C materials. At this moment, the abundant defective sites act as “magnets” to capture the P atoms, forming P, N co-doped carbon materials (CNPs) (right panel in Fig. 1a). Similar synthesis procedures are used to prepare a series of samples for comparison (including CNP-800, CNP-900, and CNP-1000, NC and PC, detailed in the Experimental section in the ESI†). These comparison samples are designed to build underlying relationships in between microstructure and ORR performances.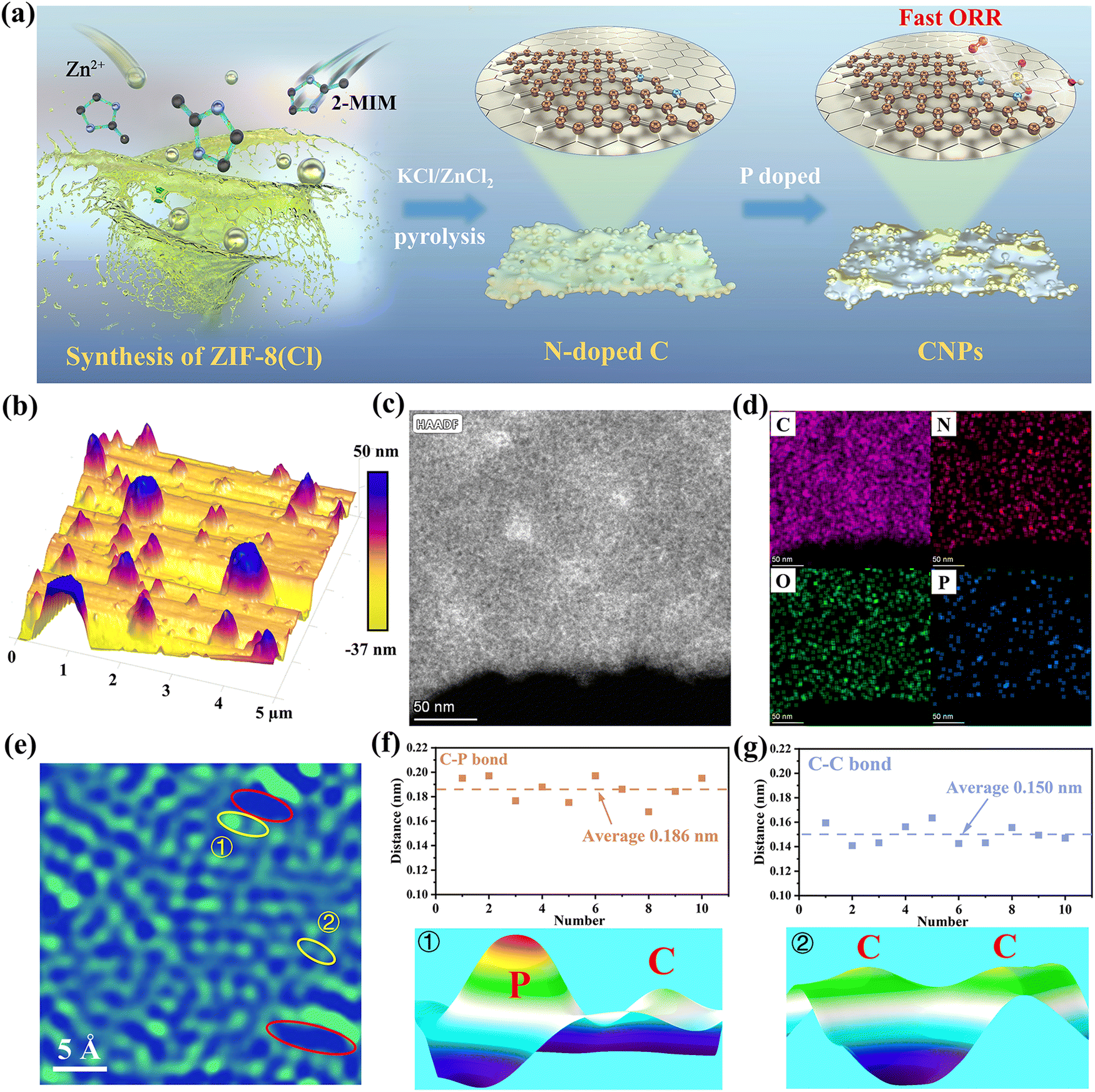 | ||
| Fig. 1 Synthesis and characterization of P, N co-doped carbon catalysts. (a) Schematics of the synthesis procedures of P, N co-doped carbon catalysts. (b) AFM image showing the surface morphology of the as-prepared catalyst. (c) HAADF-STEM image of the as-prepared well-continuous catalyst. (d) EDS elemental mappings of C, N, O, and P. (e) Zoom-in HAADF-STEM image of CNP-900, in which red circles represent void defects, and zones 1 and 2 suggest P–C, and C–C coordination bonds, respectively. (f) and (g) Statistic measurements of the distance of C–P bonds and C–C bonds (upper panel) and corresponding 3D surface plots (bottom panels) extracted from zones 1 and 2, respectively. | ||
Scanning electron microscopy (SEM) images reveal that both NC and CNP materials have lamellar structures decorated by micro-scale spheres (inset in Fig. S1, ESI†). Such morphology is probably caused by strong polarity and high Zn vapor pressure in the molten salt system during pyrolysis (Fig. S1, ESI†). The lamellar-spherical structure, exemplified by CNP-900, is further confirmed by atomic force microscopy (AFM) images (Fig. 1b and Fig. S2, ESI†), which show numerous spheres on a rough substrate. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images show bright imaging contrast domains represent the spheres due to thickness variation (Fig. 1c). The interplanar spacing of CNP-900 is measured at 0.43 nm, larger than that of graphitic carbon (0.34 nm), indicating a disordered turbostratic graphitic structure (Fig. S3 and S4, ESI†). Energy dispersive X-ray spectroscopy (EDS) mappings reveal that C, N, O, and P are distributed throughout CNP-900, but with P being more concentrated at substrate edges and/or decorated sphere edges (Fig. 1c and d). In addition, TEM analysis (Fig. 1c and Fig. S3a and b, ESI†) shows numerous defects with dark imaging contrast, and a zoomed-in HAADF-STEM image (Fig. 1e) highlights topological defects (red circles), disrupting π-bonds and facilitating electron transfer.39 The average distance between two neighboring atoms is around 0.186 nm, which is larger than the in-plane C–C distance (0.150 nm) but closer to the theoretical in-plane P–C distance (0.181 nm). For example, the intensity profile (Fig. 1f) drawn along zone 1 near the nano-crack in Fig. 1e shows the distance of the bright and dim atomic contrast is 0.186 nm, while the distance of two adjacent atoms far away from the nano-crack is 0.150 nm (Fig. 1g). These results verify that the P atoms are successfully trapping at abundant edge sites of the nano-cracks (Fig. 1g).26,40 Intensity profiles (Fig. 1f and g) confirm that P atoms at nano-crack edges validate their successful incorporation into the material structure.
The X-ray diffraction (XRD) patterns of the as-synthesized CNPs and NC materials are depicted in Fig. 2a. These peaks of CNPs at 21.2° and 43.6° are attributed to the (002) and (101) planes of graphite-like microcrystals, respectively (Fig. 2a). The left shifting of the peaks confirms the expansion of the carbon lattice induced by heteroatom doping and nanopores generation. The N2 adsorption/desorption isotherms are used to confirm the existence of great amounts of nanopores. Results show both NC and CNPs exhibit high adsorption capacity at P/P0 < 0.1 and a steep slope between 0.1–0.3P/P0, suggesting the co-existence of micropores and smaller mesopores (Fig. 2b). Further analysis reveals that NC and CNPs have a narrow pore-size distribution less than 5 nm (Fig. 2c). The specific surface areas of NC, CNP-800, CNP-900, and CNP-1000 are 1793.8, 1686.4, 1679.7, and 2288.1 m2 g−1, respectively (Fig. S5, ESI†), ensuring efficient mass transport during the ORR.37,41 Numerous micropores and defects may break electronic symmetry, consequently regulating reaction thermodynamics.
 | ||
| Fig. 2 Identification of coordination environments of as-synthesis catalysts. (a) XRD patterns of as-prepared CNPs and NC materials. (b) Nitrogen adsorption/desorption isotherms of CNP-900 and NC materials. (c) Pore size distributions of CNP-900 and NC. (d) XPS P 2p spectra of CNPs. (e) The percentages of P species in CNPs. (f) XPS O 1s spectra of CNPs and NC. Normalized XANES spectra at the (g) C K-edge, (h) N K-edge and (i) P K-edge of CNP-900. | ||
The elemental compositions and valence states of the samples are analyzed by X-ray photoelectron spectroscopy (XPS) (Fig. S6–S9, ESI†) to explore the species of C, N, and P on the surface. In the C 1s region, the three characteristic peaks at 284.7, 285.7, and 287.7 eV correspond to CC, C–N, and C–O/CO, respectively (Fig. S6b–S9b, ESI†). The high-resolution N 1s spectra show four peaks at 398.4, 399.5, 400.9, and 402.7 eV, corresponding to pyridinic-N, pyrrolic-N, graphitic-N, and oxidized-N, respectively (Fig. S6c–S9c, ESI†). The dominant graphitic-N and pyridinic-N species are conductive to enhance the 4e− pathway selectivity and stability of CNPs.42,43 The P 2p spectrum of CNP-800 can be deconvoluted into three peaks at 132.8, 134.1 and 135.3 eV, corresponding to P–C, P–O and PO, respectively (Fig. 2d).44 When temperature increases, the P–O species disappear; while, the proportion of PO species increases (Fig. S7d–S9d, ESI†). The percentage of PO species in CNP-900 reaches up to 66%, which is twice that of P–C (34%), suggesting the presence of the C–(PO)2 local structure (Fig. 2e). The transition from P–O to PO is also confirmed by the O 1s spectrum due to an increase in O 1s binding energies with increased temperature (Fig. 2f). The fine structure of CNP-900 is further characterized by X-ray absorption near edge structure (XANES) spectrum. As shown in Fig. 2g, the C K-edge absorption spectrum exhibits three characteristic resonances at 286, 289 and 293.5 eV, attributed to the π* antibonding state of sp2 CC, π* antibonding state of sp2 C–N–C and σ* antibonding state of C–C configurations, respectively.17 Compared to highly oriented pyrolytic graphite, the right shifting in resonance positions indicates a transition from 1s to π*/σ* as a result of the heteroatom doping, which modifies the local electronic structure of adjacent carbons.45 In the N K-edge spectrum, the two dominant peaks at 400.6 and 402.6 eV are associated with the π* antibonding states of pyridinic-N with e2u and b2g symmetries and/or sp2 hybridized graphitic-N, respectively (Fig. 2h).46 The shoulder peak adjacent to pyridinic-N indicates a relatively low percentage of pyrrolic-N in CNP-900, consistent with the XPS results in Fig. S8c (ESI†). The broad feature centered at 410 eV is associated with the transition to unfilled σ* orbitals of C–N. The P K-edge spectrum shown in Fig. 2i displays a sharp peak at 2153.2 eV and a broad peak at 2169.6 eV. The former is associated with the σ* antibonding states of P–C/P–O, which overlap in binding energy due to structural symmetry. The latter is associated with the π* antibonding states of PO, further confirming the dominance of the PO configuration.47
The combination of effective dual-heteroatom doping, plentiful edge sites and unique coordination environment significantly improves the ORR performance of CNP-900 under various temperature and pH conditions. The real potential of the reference electrode is calibrated in different solutions before the electrochemical performance testing (Fig. S10–S12, ESI†). In 0.1 M PBS (pH = 7), the cyclic voltammograms (CV) of CNPs show a positively shifted reduction peak compared to NC, indicating enhanced oxygen sensitivity due to N, P co-doping (Fig. S13a, ESI†). Among the samples, CNP-900 exhibits the most positive wave, consistent with the linear sweep voltammetry (LSV) results shown in Fig. S13b (ESI†). Considering the similar specific surface area and pore characteristics, this difference can be attributed to the variation in coordination environment and content of dopants of P (Fig. S14, ESI†). The onset potential (Eonset, defined as the potential at which the current density reaches 0.1 mA cm−2) and half-wave potential (E1/2) of CNP-900 are 0.94 V and 0.77 V, respectively, surpassing those of Pt/C (Eonset = 1.00 V and E1/2 = 0.68 V), positioning it one of the state-of-the-art non-noble ORR catalysts reported (Fig. 3a and Tables S1 and S2, ESI†). The Tafel slope of CNP-900 is 86 mV dec−1, lower than that of NC (93 mV dec−1), indicating enhanced reaction kinetics after P doping (Fig. 3b). The slope deviation from the theoretical values may arise from the presence of adsorbates and the outer-sphere nature of the first electron transfer step.48,49 The Tafel slopes of CNP-900 remain between 99–70 mV dec−1 from 0 to 60 °C, indicating that the rate-determining step is almost temperature-independent (Fig. 3c). This rapid ORR kinetics gives CNP-900 the capability to drive ZABs over a wide temperature range. The rotating ring-disk electrode (RRDE) measurement shows that CNP-900 undergoes an efficient four-electron pathway, consistent with the results calculated from the Koutecký–Levich (K–L) equation (Fig. 3d and Fig. S13c, ESI†). Impressively, the H2O2 yield over CNP-900 (1.5–4.1%) is even lower than that of Pt/C, implying that the N/P dopants with specific configurations facilitate the cleavage of O2 at an accelerated rate. Previous studies have demonstrated that alterations in orbital interactions and adsorption sites can modify the adsorption model and reaction pathway of the ORR, thus influencing the reaction kinetics and selectivity.50–54 Therefore, the extremely low H2O2 yield may be related to a change in the chemisorption model that enables the breaking of O–O bond. As an important evaluation criterion for ORR catalysts under actual operating conditions, the stability of the samples is systematically studied through accelerated aging tests (AAT) and chronoamperometric tests. As shown in Fig. 3e, CNP-900 retains 76.5% of its initial current after 70000 s at 0.6 V, outperforming Pt/C, which retains 67.8% after 40000 s. After 10000 repeated CV cycles, E1/2 of Pt/C reduces by 32 mV, while only a negligible degradation (13 mV) is observed for CNP-900 after 10000 CV cycles, confirming the excellent stability of CNP-900 under different working conditions (Fig. 3f and Fig. S15, ESI†). This outstanding stability is attributed to the robust carbon skeleton and reduced oxygen radical release. Compared to noble metals, carbon-based ORR catalysts demonstrate inherent inertness to shuttle species in fuel cells. Taking methanol as an example, the response current of CNP-900 shows little change with the addition of methanol, while that of Pt/C decreases by about 70% (Fig. S16, ESI†). Poisoning experiments reveal that P plays a significant role in enhancing ORR activity, as indicated by a larger E1/2 shift in CNP-900 when SCN− and Cl− are added.55–57 Typically, SCN− anions function as strongly adsorbing ligands that induce steric hindrance at axial positions, thereby blocking oxygen access to all potential active sites (positively charged C and P centers) in CNP-900 that may catalyze ORR under neutral conditions. In contrast, Cl− anions exhibit competitive adsorption with both O2 molecules and *OOH intermediates specifically at the more positively charged P sites. This distinct adsorption behavior enables precise identification of the true active centers through comparative poisoning experiments.
 | ||
| Fig. 3 Performance testing of catalysts. (a) LSV polarization curves and (b) corresponding Tafel slopes of CNP-900, NC and Pt/C in 0.1 M PBS solutions at room temperature. (c) Tafel slopes of CNP-900 and Pt/C at 0 °C, 20 °C, 40 °C and 60 °C. (d) H2O2 yield and (e) chronoamperometric plots of CNP-900 and Pt/C in 0.1 M PBS solutions at room temperature. (f) LSV polarization curves of CNP-900 before and after the accelerated aging tests. (g) Comparison of E1/2 of CNP-900 with the previously reported non-noble-metal catalysts in neutral, alkaline and acidic media, respectively. (h) Comparison of the E1/2 in neutral, alkaline and acidic media of CNP-900 and the previously reported pH-universal catalysts. | ||
Furthermore, CNP-900 also exhibits exceptional ORR activity in both alkaline (pH = 13) and acidic (pH = 1) media. As shown in Fig. S18 and S21 (ESI†), the CV curves of CNP-900 display the most positive reduction peak. E1/2 of CNP-900 in 0.1 M KOH reaches 0.91 V, superior to that of NC and Pt/C (E1/2 = 0.86 V), showing CNP-900 is one of the best noble-metal-free ORR catalysts under alkaline conditions (Fig. S18b and Tables S3 and S4, ESI†). Besides, the lower Tafel slope of 64 mV dec−1 enables CNP-900 to rapidly split O2 (Fig. S19a, ESI†). E1/2 of CNP-900 is 0.78 V in 0.1 M HClO4, which is comparable to that of Pt/C (E1/2 = 0.84 V) and is better than the majority of documented noble-metal-free catalysts (Fig. S21b and Tables S5 and S6, ESI†). Additionally, the Tafel slope of CNP-900 as low as 84 mV dec−1 is better than that of Pt/C (97 mV dec−1) (Fig. S22a, ESI†). These results enable CNP-900 standing out as one of the top-rated pH-universal, noble-metal-free ORR catalysts (Fig. 3g and h).41,58–71 The Koutecky–Levich (K–L) and RRDE analysis confirm that CNP-900 undergoes an efficient four-electron pathway during ORR (Fig. S18c and S19b, ESI†). The current loss of CNP-900 in the chronoamperometric tests is significantly lower than that of Pt/C, either in 0.1 M KOH (8.6% vs. 29.1%, Fig. S19c, ESI†) or in 0.1 M HClO4 (27.5% vs. 46.4%, Fig. S22b, ESI†). After 35000 CV cycles, the E1/2 of CNP-900 shows a negligible shift of only 8 mV, further demonstrating robust ORR performance compared to NC and Pt/C (Fig. S19d–f, ESI†). Additionally, CNP-900 exhibits strong resistance to methanol poisoning in both neutral and acidic media, with no significant current decay upon methanol addition (Fig. S20 and S22c, ESI†).
As demonstrated above, P atoms prefer to be located at the edges by adopting PO and P–C configurations. Hence, an edge C–(PO)2 model is constructed to mimic the structure characteristics of CNP-900 (Fig. S23, ESI†). For comparison, two edge models, C–PO/OH and C–PO, are also built to clarify the structure–performance relationship of CNPs. Generally, the reaction kinetics of heteroatom-doped carbon catalysts is hindered by the sluggish generation of OOH*, which involves two successive steps, the adsorption of O2 and following hydrogenation.72 While a large number of works has pointed out that the thermodynamics of the latter can be optimized by enhancing the OOH* adsorption through scaling relation, the impact of the former on ORR performance has not received enough attention, which is recently recognized as the real rate-determining step of ORR.73–76 We thus first examine the adsorption energy of O2 on different models.
Generally, O2 adsorption occurs in three modes: end-on (Pauling model), side-on (Griffiths model), and bridge (Yeager model),77–79 with the end-on mode being most common in heteroatom-doped carbon catalysts.80–83 The calculated adsorption energies of O2via the end-on fashion are inconsistent with the fast reaction kinetics shown earlier (Fig. S24, ESI†). We then infer a transition of the adsorption model due to P doping and extended the calculation to the other fashions to examine the possible collaboration between P and C (Fig. 4a). As expected, the adsorption energy of O2 on C–(PO)2, C–PO/OH and C–PO via the bridge fashion decreases to −1.13, −0.21 and −1.76 eV, respectively, suggesting that the activation and involvement of adjacent C effectively enhance the chemisorption of O2 (Fig. S24 and S25, ESI†). In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) is employed to investigate the O2 chemisorption behavior and the evolution of oxygen species under realistic catalytic conditions. As shown in Fig. 4b, NC exhibits a clearly identifiable *OOH peak at 1234 cm−1 over a wide potential range (1.1–0.5 V), indicating that *OOH dissociation is the rate-limiting step for NC. The peak at 1466 cm−1 corresponds to the stretching vibration of the O–O bond in the end-on orientation. This suggests that O2 adsorbs on the C site according to the Pauling model, and is subsequently protonated to form *OOH (*O2 + H+ + e− → *OOH). Notably, the introduction of P induces significant changes in the O2 adsorption mode and reaction kinetics. A prominent PO absorption peak at 1120 cm−1 is observed for CNP-900, which is consistent with the XPS results (Fig. 4c). Additionally, the O–O bond stretching vibration of CNP-900 shifts to 1432 cm−1, indicating a transition from the “end-on” to the bridge mode.84 This suggests that the O2 adsorption is modulated by the PO ligands, making the Yeager pathway thermodynamically more favorable. The intensity of the *OOH peak is reduced, demonstrating enhanced dissociation kinetics for *OOH. Furthermore, in situ Raman spectroscopy is used to detect oxygen-containing intermediates (Fig. S26, ESI†). The *OOH peak for CNP-900 at 1156 cm−1 is slightly red-shifted compared to that of NC at 1181 cm−1, indicating a lowered dissociation barrier for *OOH upon P introduction, which is consistent with the in situ ATR-SEIRAS results.
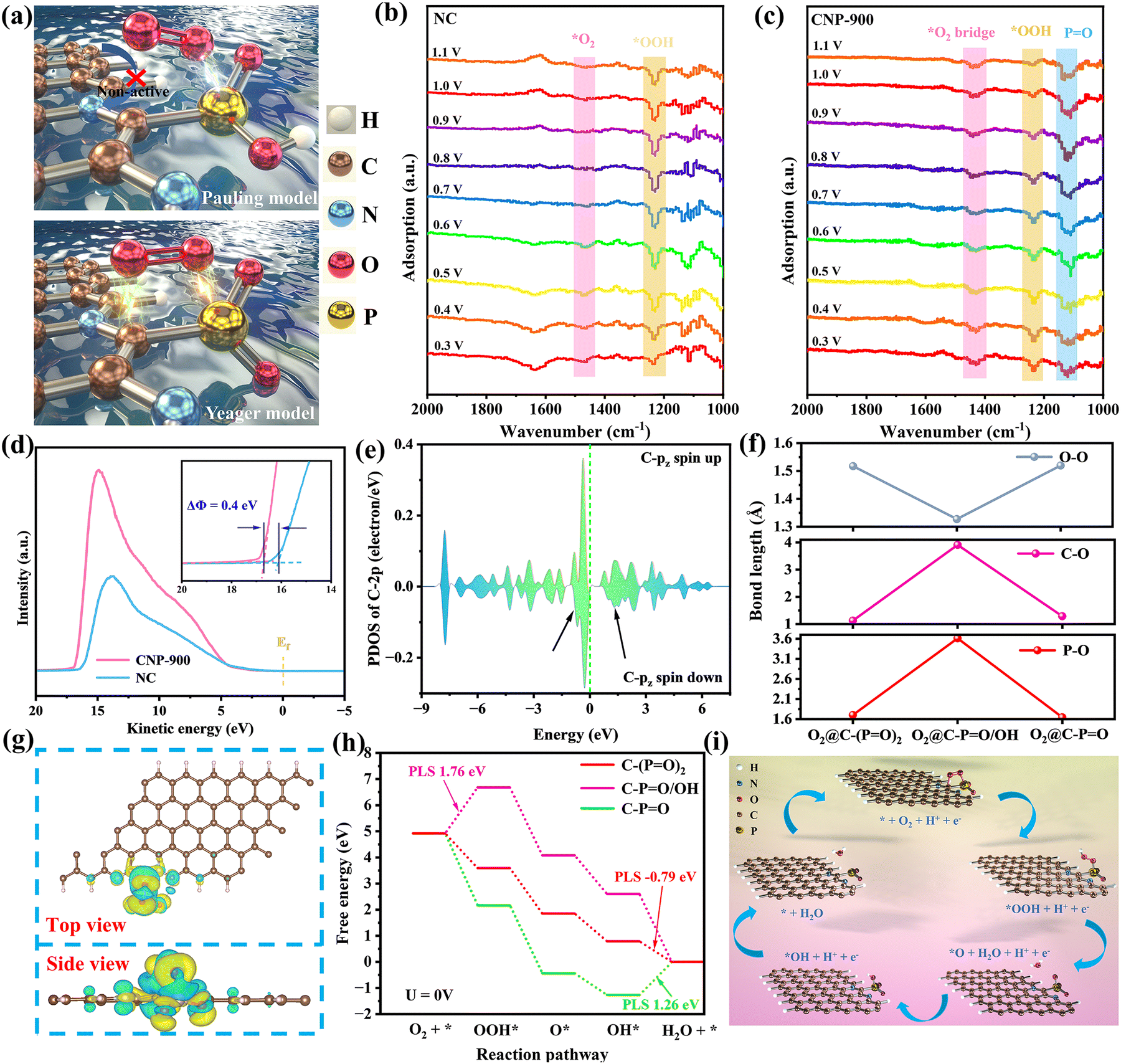 | ||
| Fig. 4 Proposed theoretical models for the structure characteristics of CNP-900. (a) Schematic representation of the PO ligand induced activation toward O2 adsorption: Pauling model and Yeager model. In situ ATR-SEIRAS testing for (b) NC and (c) CNP-900 at various potentials (vs. RHE) in O2-saturated 0.1 M PBS. (d) UPS of CNP-900 and NC. (e) PDOS of C 2pz, showing spin polarization of the carbon atom connected to P. (f) The O–O, C–O and P–O bond lengths after O2 chemisorption. (g) Differential charge density diagram of O2@C–(PO)2. Yellow and cyan represent charge accumulation and depletion, respectively. (h) Gibbs free energy profiles of the four elemental steps. (i) Diagram of ORR catalytic mechanism on C–(PO)2. | ||
Notably, the C 2pz orbital shows electronic asymmetry below the Fermi level, which could be a significant contributing factor for the enhanced bridge adsorption of O2 (Fig. 4e). As previously reported, spin regulation is an effective strategy to enhance the ORR kinetics because of the paramagnetic nature of triplet O2.41,51 The degree of activation of O2 can be estimated by the O–O (LO–O) bond length.85 As shown in Fig. 4f and Table S7 (ESI†), LO–O elongates by 0.307, 0.048 and 0.309 Å for O2@C–(PO)2, O2@C–PO/OH and O2@C–PO, respectively, indicative of a reduced bond strength due to the electron transfer from C/P to the  orbitals of O2, especially for O2@C–(PO)2. As shown in Fig. 4d, Φ of CNP-900 is 0.4 eV lower than that of NC, indicating a stronger driving force for electron transfer to adsorbed O2, which facilitates O2 splitting to speed up the ORR kinetics.86,87 Basically, the bond lengths of C–O (LC–O) and P–O (LP–O) are highly correlated to the participation and cooperation degree of C/P pairs. The strong p-orbital coupling between C/P and O2 in O2@C–(PO)2 significantly shortens LP–O and LC–O to 1.70 Å and 1.42 Å, respectively, resulting in the most favorable bridge chemisorption of O2 (Fig. S27, ESI†). In comparison, the reduced orbital coupling between C and O2 (LC–O = 1.58 Å) in O2@C–PO increases the electron-donating barrier from C to O2, most likely due to the excessive chemisorption of O2 at the P site and the formation of delocalized π electrons. For O2@C–PO/OH, the longest LP–O (3.6 Å) and LC–O (4.2 Å) implies insignificant O2 activation and weak interaction with C/P pairs, consistent with the almost unchanged PDOS of O2 after chemisorption. The “bond lengths-activity” correlation plots clearly demonstrate that ORR activity is positively related with the participation and cooperation degree of C/P pairs (Fig. S28, ESI†). The Bader charge shows the electrons are transferred from the C/P pair to *O2 (Fig. 4g).
orbitals of O2, especially for O2@C–(PO)2. As shown in Fig. 4d, Φ of CNP-900 is 0.4 eV lower than that of NC, indicating a stronger driving force for electron transfer to adsorbed O2, which facilitates O2 splitting to speed up the ORR kinetics.86,87 Basically, the bond lengths of C–O (LC–O) and P–O (LP–O) are highly correlated to the participation and cooperation degree of C/P pairs. The strong p-orbital coupling between C/P and O2 in O2@C–(PO)2 significantly shortens LP–O and LC–O to 1.70 Å and 1.42 Å, respectively, resulting in the most favorable bridge chemisorption of O2 (Fig. S27, ESI†). In comparison, the reduced orbital coupling between C and O2 (LC–O = 1.58 Å) in O2@C–PO increases the electron-donating barrier from C to O2, most likely due to the excessive chemisorption of O2 at the P site and the formation of delocalized π electrons. For O2@C–PO/OH, the longest LP–O (3.6 Å) and LC–O (4.2 Å) implies insignificant O2 activation and weak interaction with C/P pairs, consistent with the almost unchanged PDOS of O2 after chemisorption. The “bond lengths-activity” correlation plots clearly demonstrate that ORR activity is positively related with the participation and cooperation degree of C/P pairs (Fig. S28, ESI†). The Bader charge shows the electrons are transferred from the C/P pair to *O2 (Fig. 4g).
The chemisorbed O2 then decomposes following an association pathway (Fig. S29, ESI†). Fig. 4h displays the energy profiles of the four elemental steps on different models. At U = 0 V, the C–(PO)2 model exhibits four successive exothermal proton/electron-coupled processes, and the energy barrier of the potential-limiting step (PLS) is only 0.44 eV. In contrast, the *OOH formation and *OH removal are energetically unfavorable on the C–PO/OH and C–PO models, respectively, resulting in huge energy barriers that inhibit the occurrence of ORR. Since the adsorption energies of *OOH, *O and *OH are scaling related, ΔG*OH can be utilized as a descriptor to assess the activity of a catalyst, and an ideal one should have a ΔG*OH of 0.7–1.0 eV.9,88 ΔG*OH of the C–PO/OH and C–PO models are 2.7 eV and −1.15 eV respectively, suggesting either excessively weak or strong *OH adsorption. On the other hand, the C–(PO)2 model exhibits a best fit ΔG*OH of 0.79 eV, emphasizing the significance of ligand modulation in altering the interaction of P with reaction intermediates to achieve optimal energetics. Fig. 4i shows the ORR catalytic mechanism on the C–(PO)2 model.
In view of the rapid reaction kinetics of CNP-900 under wide pH and temperature conditions, we assemble sandwich-type neutral and alkaline FZABs to assess its practicability in real devices using catalyst-leaded carbon cloth as the air cathode and PVA/SA hydrogel as the electrolyte (Fig. 5a). As shown in Fig. 5b, the CNP-900-based neutral FZAB exhibits a stable open-circuit voltage (OCV) up to 1.37 V for 13 hours at 20 °C. The OCVs only slightly decrease at 0 °C (1.36 V) and −20 °C (1.34 V), demonstrating excellent subzero adaptability. Compared with its Pt/C-based counterpart, the CNP-900-based neutral FZAB exhibits superior rate capability with substantially reduced polarization (Fig. 5c). The battery voltage maintains above 1.0 V even at a high current density of 20 mA cm−2. As shown in Fig. 5d, the power density of CNP-900-based FZAB reaches 47.1, 36.4 and 25.6 mW cm−2 at 20, 0 and −20 °C respectively, which surpasses that of Pt/C-based FZAB (30 mW cm−2 at 20 °C, Fig. S30, ESI†). Table S8 (ESI†) highlights the advanced nature of the CNP-900-based neutral FZAB by comparing its performance to those reported FZABs using a noble-metal-free air cathode. Two CNP-900-based FZABs in series can turn on a yellow LED or a mini-fan in real-life scenarios (Fig. 5e and Fig. S31, ESI†). Under a variety of bending circumstances (spanning from 0 to 180° in angle), the FZAB maintains consistent OCV and output, demonstrating excellent tolerance to mechanical deformation (Fig. 5f).
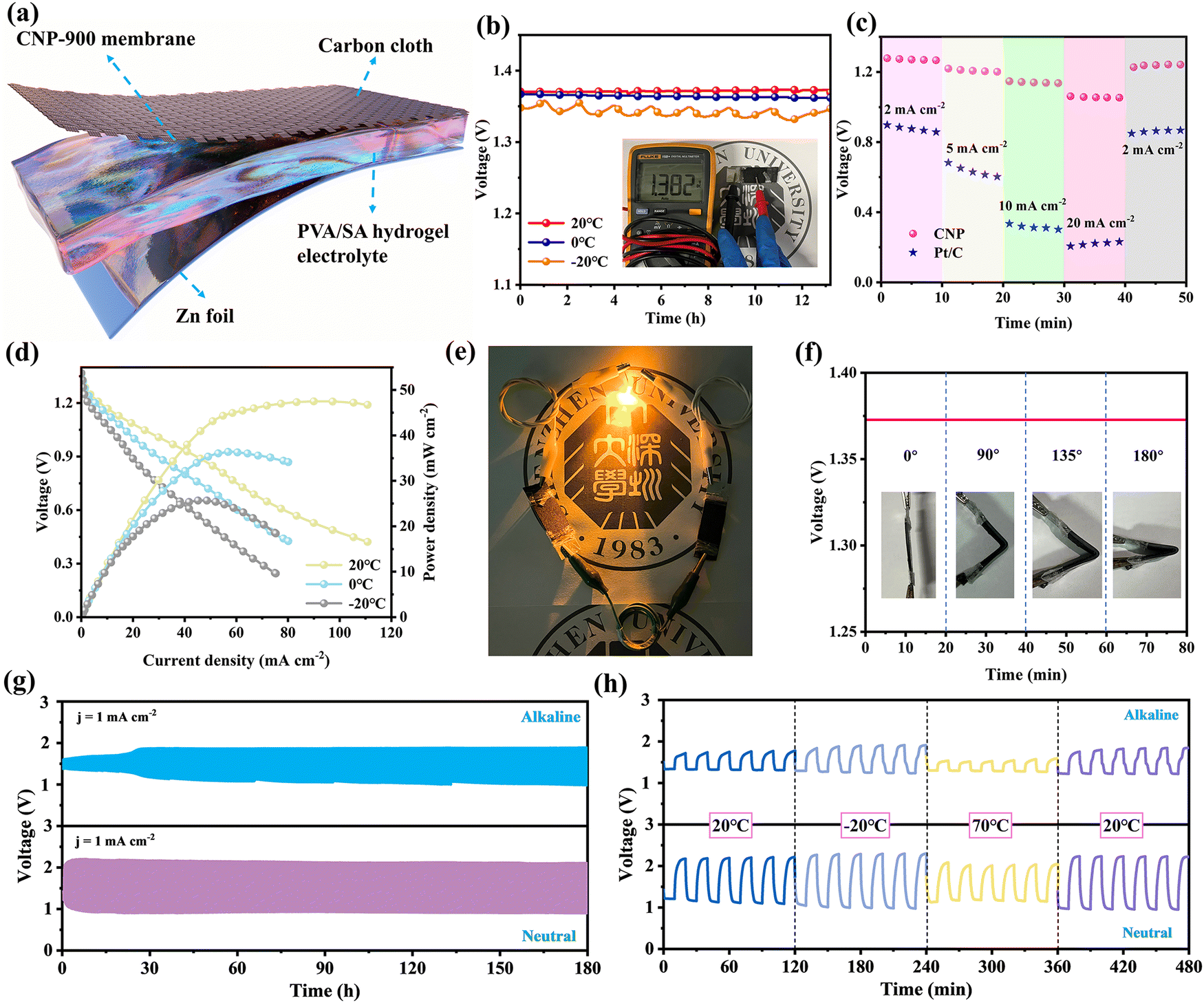 | ||
| Fig. 5 Performance testing of CNP-900-based FZABs. (a) Schematic diagram of FZAB. (b) OCV, (c) rate capabilities, (d) polarization and power density curves of CNP-900-based neutral FZABs at 20, 0 and −20 °C. (e) Photograph of a yellow LED powered by two CNP-900-based neutral FZABs in series. (f) OCV of CNP-900-based neutral FZABs at various bend angles. (g) Cycling stability of the CNP-900-based FZABs at alkaline and neutral media. (h) Wide temperature range adaptability of the CNP-900-based FZABs at alkaline and neutral media. | ||
By introducing RuO2 into the air cathode, the long-term cycling stability of the FZABs is achieved. As shown in Fig. 5g, the CNP-900-based FZAB exhibits a narrow charge/discharge gap with a round-trip efficiency of 58.6%. Steady charge/discharge profiles are maintained for at least 180 hours, especially in neutral media. Notably, the CNP-900-based FZABs exhibit exceptional cycling performance over a wide temperature range (−20 to 70 °C). The discharge plateau only slightly decreases to 1.02 V at −20 °C and rebounds to 1.17 V at 70 °C, demonstrating good wide-temperature endurance (Fig. 5h). The CNP-900-based alkaline FZAB exhibits similar temperature-dependence to its neutral counterpart. Its OCV and maximum power density are 1.47 V/104.2 mW cm−2, 1.45 V/70.4 mW cm−2 and 1.43 V/53.0 mW cm−2 at 20 °C, 0 °C and −20 °C, respectively, which outperform those of Pt/C-based FZAB (1.27 V/52 mW cm−2 at 20 °C), and are among the best for alkaline FZABs reported (Fig. S32 and Table S8, ESI†). The aforementioned findings suggest that CNP-900 enhances the reaction adaptability of FZABs even in hostile environments with low temperatures and proton concentrations.
Conclusion
In this work, we successfully address the difficulty of O–O bond activation on metal-free carbon catalysts, accelerating the reaction kinetics even at limited proton supply and low-temperature conditions. The C 2pz orbital occupation state of CNP-900 is well-regulated by adjacent PO ligands, leading to a transition from end-on to bridge chemisorption of O2, which is followed by an energetically favorable proton/electron coupling process. As a result, the E1/2 of CNP-900 reaches 0.77 V with negligible degradation of E1/2 (13 mV) after 10000 CV cycles in a neutral medium. The assembled neutral FZABs exhibit a stable OCV of 1.34 V and a maximum power density of 25.6 mW cm−2 even under harsh conditions (−20 °C). In situ ATR-SEIRAS, in situ Raman spectroscopy and DFT calculations are adopted to reveal the efficient O2 adsorption/activation on C/P pairs. This work provides new insights into regulating oxygen adsorption patterns to enhance the kinetics of heteroatom-doped carbon catalysts.
Author contributions
Y. J. Z. designed and finished the most experiments. Y. J. Z. wrote the manuscript. M. R. H. performed the in situ Raman experiments. Y. S. K. and Y. F. carried out the XRD characterization and analysis. Y. J. Z., T. Y. Z., H. W., J. J. L. and M. X. Z. carried out the zinc–air batteries experiments. Y. J. Z. and J. Y. O. carried out the TEM analysis. T. W. carried out the in situ ATR-SEIRAS experiments. D. W., X. H. S. and C. Y. Z. were in charge of this scientific research project, and the leaders of the actual coordination of the contributions.Data availability
The data that support the findings of this study are included in the published article and its ESI.† These data are also available from the corresponding authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Key Research and Development Program of China (2024YFA1509400), the Natural Science Foundation of Guangdong province (2024A1515010320) and the Natural Science Foundation of Shenzhen (20231120215210002). Dan Wang acknowledges the funding support from the National Key R&D Program of China (2024YFA1509401), and Shenzhen University 2035 Program for Excellent Research, Grant No. 2024B005. Xianhu Sun acknowledges the funding support from the National Key R&D Program of China (2024YFA1509402), and the Institute of Weiqiao UCAS Science and Technology (GYY-GDHX-2024-ZY-007). The authors acknowledge the TEM assistance received from the Electron Microscope Center and Instrument Analysis Center of Shenzhen University.Notes and references
- M. Jiao, L. Dai, H. R. Ren, M. Zhang, X. Xiao, B. Wang, J. Yang, B. Liu, G. Zhou and H. M. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202301114 CrossRef CAS PubMed.
- Z. Pei, Z. Yuan, C. Wang, S. Zhao, J. Fei, L. Wei, J. Chen, C. Wang, R. Qi, Z. Liu and Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 4793–4799 CrossRef CAS PubMed.
- F. Meng, H. Zhong, D. Bao, J. Yan and X. Zhang, J. Am. Chem. Soc., 2016, 138, 10226–10231 CrossRef CAS PubMed.
- E. C. M. Tse, C. J. Barile, N. A. Kirchschlager, Y. Li, J. P. Gewargis, S. C. Zimmerman, A. Hosseini and A. A. Gewirth, Nat. Mater., 2016, 15, 754–759 CrossRef CAS PubMed.
- Y. Zhang, Y.-P. Deng, J. Wang, Y. Jiang, G. Cui, L. Shui, A. Yu, X. Wang and Z. Chen, Energy Storage Mater., 2021, 35, 538–549 CrossRef.
- X. Feng, Y. Bai, M. Liu, Y. Li, H. Yang, X. Wang and C. Wu, Energy Environ. Sci., 2021, 14, 2036–2089 RSC.
- J. Zhang and L. Dai, ACS Catal., 2015, 5, 7244–7253 CrossRef CAS.
- X. Wang, M. Yu and X. Feng, eScience, 2023, 3, 100141 CrossRef.
- N. Yang, L. Li, J. Li, W. Ding and Z. Wei, Chem. Sci., 2018, 9, 5795–5804 RSC.
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Nat. Catal., 2021, 4, 615–622 CrossRef CAS.
- R. E. Warburton, A. V. Soudackov and S. Hammes-Schiffer, Chem. Rev., 2022, 122, 10599–10650 CrossRef CAS PubMed.
- Y. Wu, Y. Zhang, L. Lan, T. Hu, S. Tang, D. Lützenkirchen-Hecht, K. Yuan and Y. Chen, Angew. Chem., Int. Ed., 2025, e202502019 Search PubMed.
- Z. Wang, M. Cheng, Y. Liu, Z. Wu, H. Gu, Y. Huang, L. Zhang and X. Liu, Angew. Chem., Int. Ed., 2023, 62, e202301483 CrossRef CAS PubMed.
- Y. Zhang, J. Ma, Z. Zhuang, A. Huang, S. Zhang, W. Zhai, Y. Yu, Q. Peng, H. Xiao, C. Nan and C. Chen, ACS Catal., 2024, 14, 18851–18860 CrossRef CAS.
- J. Zhao, C. Fu, K. Ye, Z. Liang, F. Jiang, S. Shen, X. Zhao, L. Ma, Z. Shadike, X. Wang, J. Zhang and K. Jiang, Nat. Commun., 2022, 13, 685 CrossRef CAS PubMed.
- J. Du, G. Han, W. Zhang, L. Li, Y. Yan, Y. Shi, X. Zhang, L. Geng, Z. Wang, Y. Xiong, G. Yin and C. Du, Nat. Commun., 2023, 14, 4766 CrossRef CAS PubMed.
- H. Zou, S. Shu, W. Yang, Y.-C. Chu, M. Cheng, H. Dong, H. Liu, F. Li, J. Hu, Z. Wang, W. Liu, H. M. Chen and L. Duan, Nat. Commun., 2024, 15, 10818 CrossRef CAS PubMed.
- W. Niu, S. Pakhira, G. Cheng, F. Zhao, N. Yao, J. L. Mendoza-Cortes and B. E. Koel, Nat. Mater., 2024, 23, 1704–1711 CrossRef CAS PubMed.
- P. Guo, B. Liu, F. Tu, Y. Dai, Z. Zhang, Y. Xia, M. Ma, Y. Zhang, L. Zhao and Z. Wang, Energy Environ. Sci., 2024, 17, 3077–3087 RSC.
- D. Deng, J. Qian, X. Liu, H. Li, D. Su, H. Li, H. Li and L. Xu, Adv. Funct. Mater., 2022, 32, 2203471 CrossRef CAS.
- F. Yang, X. Fan, C. Wang, W. Yang, L. Hou, X. Xu, A. Feng, S. Dong, K. Chen, Y. Wang and Y. Li, Carbon, 2017, 121, 443–451 CrossRef CAS.
- M. A. Patel, F. Luo, M. R. Khoshi, E. Rabie, Q. Zhang, C. R. Flach, R. Mendelsohn, E. Garfunkel, M. Szostak and H. He, ACS Nano, 2016, 10, 2305–2315 CrossRef CAS PubMed.
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- G.-L. Chai, K. Qiu, M. Qiao, M.-M. Titirici, C. Shang and Z. Guo, Energy Environ. Sci., 2017, 10, 1186–1195 RSC.
- Z. W. Liu, F. Peng, H. J. Wang, H. Yu, W. X. Zheng and J. Yang, Angew. Chem., Int. Ed., 2011, 50, 3257–3261 CrossRef PubMed.
- W. Xue, Q. Zhou, X. Cui, S. Jia, J. Zhang and Z. Lin, Nano Energy, 2021, 86, 106073 CrossRef CAS.
- H. Sun, S. Liu, M. Wang, T. Qian, J. Xiong and C. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 33054–33061 CrossRef CAS PubMed.
- Y. Li, J. Han, Z. Xu, R. Zhao, Y. Wang and R. Guo, Langmuir, 2021, 37, 2001–2010 CrossRef CAS PubMed.
- J. Wang, R. Gao, L. Zheng, Z. Chen, Z. Wu, L. Sun, Z. Hu and X. Liu, ACS Catal., 2018, 8, 8953–8960 CrossRef CAS.
- J. Gu, S. Magagula, J. Zhao and Z. Chen, Small Methods, 2019, 3, 1800550 CrossRef.
- T. Najam, S. S. A. Shah, W. Ding, J. Jiang, L. Jia, W. Yao, L. Li and Z. Wei, Angew. Chem., Int. Ed., 2018, 57, 15101–15106 CrossRef CAS PubMed.
- J. Li, S. Yin, F. Dong, W. Cen and Y. Chu, ACS Appl. Mater. Interfaces, 2017, 9, 19861–19869 CrossRef CAS PubMed.
- Q. Guo, J. Feng, D. Chen, N. Song, H. Dong, L. Yu and L. Dong, J. Phys. Chem. C, 2021, 125, 9747–9755 CrossRef CAS.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS PubMed.
- S. Yu, Z. Levell, Z. Jiang, X. Zhao and Y. Liu, J. Am. Chem. Soc., 2023, 145, 25352–25356 CrossRef CAS PubMed.
- D. Chen and S. Mu, Adv. Mater., 2024, 36, 2408285 CrossRef CAS PubMed.
- H. Wang, Y. Zhao, J. Li, X. Wang, M. Huang, L. Zhang and C. Zhao, Small Struct., 2023, 4, 2300007 CrossRef CAS.
- L. Lan, Y. Wu, Y. Pei, Y. Wei, T. Hu, D. Lützenkirchen-Hecht, K. Yuan and Y. Chen, Adv. Mater., 2025, 37, 2417711 CrossRef CAS PubMed.
- A. Shen, Y. Zou, Q. Wang, R. A. W. Dryfe, X. Huang, S. Dou, L. Dai and S. Wang, Angew. Chem., Int. Ed., 2014, 53, 10804–10808 CrossRef CAS PubMed.
- X. Zhang, Z. Lu, Z. Fu, Y. Tang, D. Ma and Z. Yang, J. Power Sources, 2015, 276, 222–229 CrossRef CAS.
- Y. Zhao, H. Wang, J. Li, Y. Fang, Y. Kang, T. Zhao and C. Zhao, Adv. Funct. Mater., 2023, 33, 2305268 CrossRef CAS.
- K. Takeyasu, M. Furukawa, Y. Shimoyama, S. K. Singh and J. Nakamura, Angew. Chem., Int. Ed., 2021, 60, 5121–5124 CrossRef CAS PubMed.
- L. Zhang, T. Gu, K. Lu, L. Zhou, D. S. Li and R. Wang, Adv. Funct. Mater., 2021, 31, 2103187 CrossRef CAS.
- J. Xiao, Y. Wang, T. C. Zhang, L. Ouyang and S. Yuan, J. Power Sources, 2022, 517, 230727 CrossRef CAS.
- S. C. Ray, C. W. Pao, H. M. Tsai, J. W. Chiou, W. F. Pong, C. W. Chen, M. H. Tsai, P. Papakonstantinou, L. C. Chen, K. H. Chen and W. G. Graham, Appl. Phys. Lett., 2007, 90, 192107 CrossRef.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- I. Persson, W. Klysubun and D. Lundberg, J. Mol. Struct., 2019, 1179, 608–611 CrossRef CAS.
- J. A. Behan, E. Mates-Torres, S. N. Stamatin, C. Domínguez, A. Iannaci, K. Fleischer, M. K. Hoque, T. S. Perova, M. García-Melchor and P. E. Colavita, Small, 2019, 15, 1902081 CrossRef CAS PubMed.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- S. Zhou, S. Huang, X. Wang, S. Chen, L. Wang, P. Zhang and C. Zhao, Chem. Eng. J., 2023, 472, 145129 CrossRef CAS.
- X. Wei, S. Song, W. Cai, X. Luo, L. Jiao, Q. Fang, X. Wang, N. Wu, Z. Luo, H. Wang, Z. Zhu, J. Li, L. Zheng, W. Gu, W. Song, S. Guo and C. Zhu, Chem, 2023, 9, 181–197 CAS.
- C. Tang, L. Chen, H. Li, L. Li, Y. Jiao, Y. Zheng, H. Xu, K. Davey and S.-Z. Qiao, J. Am. Chem. Soc., 2021, 143, 7819–7827 CrossRef CAS PubMed.
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS.
- Z. Chen, X. Liao, C. Sun, K. Zhao, D. Ye, J. Li, G. Wu, J. Fang, H. Zhao and J. Zhang, Appl. Catal., B, 2021, 288, 120021 CrossRef CAS.
- X. Wang, Z. Chen, Z. Han, H. Gai, J. Zhou, Y. Wang, P. Cui, J. Ge, W. Xing, X. Zheng, M. Huang and H. Jiang, Adv. Funct. Mater., 2022, 32, 2111835 CrossRef CAS.
- D. Jain, Q. Zhang, V. Gustin, J. Hightower, S. Gunduz, A. C. Co, J. T. Miller, A. Asthagiri and U. S. Ozkan, J. Phys. Chem. C, 2020, 124, 10324–10335 CrossRef CAS.
- K. Holst-Olesen, M. Reda, H. A. Hansen, T. Vegge and M. Arenz, ACS Catal., 2018, 8, 7104–7112 CrossRef CAS.
- H. Yang, Z. Li, S. Kou, G. Lu and Z. Liu, Appl. Catal., B, 2020, 278, 119270 CrossRef CAS.
- M. Zhao, H. Liu, H. Zhang, W. Chen, H. Sun, Z. Wang, B. Zhang, L. Song, Y. Yang, C. Ma, Y. Han and W. Huang, Energy Environ. Sci., 2021, 14, 6455–6463 RSC.
- L. Xie, X. Li, B. Wang, J. Meng, H. Lei, W. Zhang and R. Cao, Angew. Chem., Int. Ed., 2019, 58, 18883–18887 CrossRef CAS PubMed.
- L. An, Z. Zhang, J. Feng, F. Lv, Y. Li, R. Wang, M. Lu, R. B. Gupta, P. Xi and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17624–17631 CrossRef CAS PubMed.
- Y. Zan, Z. Zhang, M. Dou and F. Wang, Catal. Sci. Technol., 2019, 9, 5906–5914 RSC.
- X. Zheng, J. Wu, X. Cao, J. Abbott, C. Jin, H. Wang, P. Strasser, R. Yang, X. Chen and G. Wu, Appl. Catal., B, 2019, 241, 442–451 CrossRef CAS.
- M. Tong, F. Sun, G. Xing, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2023, 62, e202314933 CrossRef CAS PubMed.
- E. Luo, H. Zhang, X. Wang, L. Gao, L. Gong, T. Zhao, Z. Jin, J. Ge, Z. Jiang, C. Liu and W. Xing, Angew. Chem., Int. Ed., 2019, 131, 12599–12605 CrossRef.
- J.-C. Li, X. Qin, F. Xiao, C. Liang, M. Xu, Y. Meng, E. Sarnello, L. Fang, T. Li, S. Ding, Z. Lyu, S. Zhu, X. Pan, P.-X. Hou, C. Liu, Y. Lin and M. Shao, Nano Lett., 2021, 21, 4508–4515 CrossRef CAS PubMed.
- F. Luo, A. Roy, L. Silvioli, D. A. Cullen, A. Zitolo, M. T. Sougrati, I. C. Oguz, T. Mineva, D. Teschner, S. Wagner, J. Wen, F. Dionigi, U. I. Kramm, J. Rossmeisl, F. Jaouen and P. Strasser, Nat. Mater., 2020, 19, 1215–1223 CrossRef CAS PubMed.
- Y. Li, J. Huang, X. Hu, L. Bi, P. Cai, J. Jia, G. Chai, S. Wei, L. Dai and Z. Wen, Adv. Funct. Mater., 2018, 28, 1803330 CrossRef.
- M. Liu, J. Lee, T. C. Yang, F. Zheng, J. Zhao, C. M. Yang and L. Y. S. Lee, Small Methods, 2021, 5, 2001165 CrossRef CAS PubMed.
- Z. Yao, Y. Li, D. Chen, Y. Zhang, X. Bao, J. Wang and Q. Zhong, Chem. Eng. J., 2021, 415, 129033 CrossRef CAS.
- L. Yan, L. Xie, X. L. Wu, M. Qian, J. Chen, Y. Zhong and Y. Hu, Carbon Energy, 2021, 3, 856–865 CrossRef CAS.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- M. Zhang, H. Li, J. Chen, F. X. Ma, L. Zhen, Z. Wen and C. Y. Xu, Adv. Funct. Mater., 2022, 33, 2209726 CrossRef.
- F. Luo, J. Zhu, S. Ma, M. Li, R. Xu, Q. Zhang, Z. Yang, K. Qu, W. Cai and Z. Chen, Energy Storage Mater., 2021, 35, 723–730 CrossRef.
- H. Jiang, J. Xia, L. Jiao, X. Meng, P. Wang, C.-S. Lee and W. Zhang, Appl. Catal., B, 2022, 310, 121352 CrossRef CAS.
- R. Gao, J. Wang, Z.-F. Huang, R. Zhang, W. Wang, L. Pan, J. Zhang, W. Zhu, X. Zhang, C. Shi, J. Lim and J.-J. Zou, Nat. Energy, 2021, 6, 614–623 CrossRef CAS.
- C. He, C. Xia, F. M. Li, J. Zhang, W. Guo and B. Y. Xia, Adv. Energy Mater., 2023, 14, 2303233 CrossRef.
- D. Zhang, E. Mitchell, X. Lu, D. Chu, L. Shang, T. Zhang, R. Amal and Z. Han, Mater. Today, 2023, 63, 339–359 CrossRef CAS.
- Y. Tian, D. Deng, L. Xu, M. Li, H. Chen, Z. Wu and S. Zhang, Nano-Micro Lett., 2023, 15, 122 CrossRef CAS PubMed.
- S. Liu, Y. Zhang, B. Ge, F. Zheng, N. Zhang, M. Zuo, Y. Yang and Q. Chen, Adv. Mater., 2021, 33, 2103133 CrossRef CAS PubMed.
- Y. Li, G. Zhong, H. Yu, H. Wang and F. Peng, Phys. Chem. Chem. Phys., 2015, 17, 21950–21959 RSC.
- G. Ye, S. Liu, K. Huang, S. Wang, K. Zhao, W. Zhu, Y. Su, J. Wang and Z. He, Adv. Funct. Mater., 2022, 32, 2111396 CrossRef CAS.
- X. Feng, G. Chen, Z. Cui, R. Qin, W. Jiao, Z. Huang, Z. Shang, C. Ma, X. Zheng, Y. Han and W. Huang, Angew. Chem., Int. Ed., 2023, 63, e202316314 CrossRef PubMed.
- D. Xue, Y. Yuan, Y. Yu, S. Xu, Y. Wei, J. Zhang, H. Guo, M. Shao and J.-N. Zhang, Nat. Commun., 2024, 15, 5990 CrossRef CAS PubMed.
- K. Liu, J. Fu, Y. Lin, T. Luo, G. Ni, H. Li, Z. Lin and M. Liu, Nat. Commun., 2022, 13, 2075 CrossRef CAS PubMed.
- L. Lin, R. Jacobs, T. Ma, D. Chen, J. Booske and D. Morgan, Phys. Rev. Appl., 2023, 19, 037001 CrossRef CAS.
- L. Lin, R. Jacobs, D. Chen, V. Vlahos, O. Lu-Steffes, J. A. Alonso, D. Morgan and J. Booske, Adv. Funct. Mater., 2022, 32, 2203703 CrossRef CAS.
- W. Liang, J. Chen, Y. Liu and S. Chen, ACS Catal., 2014, 4, 4170–4177 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee01407g |
| This journal is © The Royal Society of Chemistry 2025 |