
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic ultrasound-driven synthesis of syngas from CO2 saturated water†
Lina
Chen
a,
Yi
Qin
b,
Claire T.
Coulthard
a,
Zoë R.
Turner
 a,
Chunping
Chen
*a,
James
Kwan
*b and
Dermot
O’Hare
*a
a,
Chunping
Chen
*a,
James
Kwan
*b and
Dermot
O’Hare
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Oxford, OX1 3TA, UK
bDepartment of Engineering Science, University of Oxford, Oxford, OX1 3PJ, UK. E-mail: james.kwan@eng.ox.ac.uk
First published on 6th May 2025
Abstract
Conventional catalytic CO2 reduction into value-added products often encounters challenges such as high energy barriers and complex operational setups. Here, we introduce a sonocatalysis approach to CO2 reduction in water under ambient conditions. In an acoustic cavitation-induced high-energy local environment, the Cu nanoparticles incorporated on the ZnAl-layered double oxide create a favorable energy barrier for CO2 reduction in water, a CO production rate of 23.8 μmolCO g−1 h−1 with over 85% selectivity was achieved by ultrasonic irradiation of a CO2-saturated aqueous solution at room temperature. Furthermore, more acoustic cavitation was produced with 5% CO2 in argon dissolved in water, resulting in a higher CO productivity of 252.7 μmolCO g−1 h−1, 11 times larger than pure CO2. Hydrogen production also increased with acoustic cavitation, creating a syngas mixture with a CO to H2 ratio of 1.2 to 2.2. This approach produces a high sonochemical efficiency of 211.1 μmol kJ−1 g−1 L−1 for the ultrasound-driven fuel production from CO2 and water. These results highlight the use of cavitation to provide an alternative approach to CO2 conversion.
Broader contextIn this manuscript we describe an application of the emerging technology of sonocatalysis. We have found that in an acoustic cavitation-induced high-energy local environment, a solid catalyst comprised of metallic Cu nanoparticles incorporated on the ZnAl-layered double oxide creates a favourable energy barrier for CO2 reduction in water. Currently, our best catalyst produces CO at a rate of 23.8 μmolCO g−1 h−1 with over 85% selectivity upon ultrasonic irradiation of a CO2-saturated aqueous solution at room temperature. We found that more acoustic cavitation can be produced with 5% CO2 in argon dissolved in water, which resulted in a higher CO productivity of 252.7 μmolCO g−1 h−1, 11 times larger than in pure CO2. Hydrogen production also increased with acoustic cavitation, creating a syngas mixture with a CO to H2 ratio of 1.2 to 2.2. This approach produces an outstanding sonochemical efficiency of 211.1 μmol kJ−1 g−1 L−1 for the ultrasound-driven fuel production from CO2 and water. Overall, these results dramatically highlight the use of cavitation to provide an alternative approach for CO2 conversion. |
Introduction
Upgrading CO2 to value-added fuels and chemicals will be a key component of our future circular economy particularly if it can be achieved under mild conditions.1,2 Among the well-established technologies, electrocatalysis and photocatalysis can enable CO2 reduction in water.3,4 However, electrocatalysis requires complex setups and high overpotentials to drive the reaction, while photocatalysis depends on an intermittent solar flux and suffers from low efficiency. Both methods typically utilise media such as KHCO3 or organic additives, which despite enhancing CO2 solubility and activation, increases costs and pose environmental risks.5,6 Therefore, developing alternative approaches to achieve CO2 conversion with green, safe, and low-cost methods in pure water under ambient conditions would be highly desirable.Sonochemistry employs ultrasound to generate cavitation bubbles in water, whose collapse creates a local high-energy microenvironment to drive chemical reactions.7–9 The local microenvironment may reach temperatures and pressures up to 5000 K and 1000 atm respectively at the moment of collapse.10,11 This high-energy microenvironment enables the sonochemical reduction of CO2, this non-catalytic process has been termed CO2 sonolysis and can be traced back to 1985 and 1998.12,13 CO2 sonolysis is typically an inefficient process or requires an additional agent (e.g. H2).12,14 The introduction of a catalyst to a sonochemical system is termed sonocatalysis, in the case of CO2 sonocatalysis, the catalyst not only provides nucleation sites for cavitation bubbles but also provides chemically active sites for CO2 reduction.15–17 Moreover, the cavitation bubbles potentially act as microreactors, which experience rapid oscillating temperatures enabling environmental conditions far from equilibrium, thereby potentially inhibiting reverse reactions and promoting product formation.11 In this regard, CO2 sonocatalysis has the potential to emerge as a promising carbon fixation technology. Recently, our group reported a novel sonochemical cylindrical reactor design that enables an intense localised acoustic field with spontaneous cavitation, advancing sonocatalysis research.18 However, CO2 sonocatalysis, remains in its nascent stages and necessitates further research to fully comprehend its potential applications. Like other catalysis approaches, the efficiency of CO2 conversion in sonocatalysis depends heavily on the presence of catalytically active sites. Therefore, a critical focus in its development is the exploration of catalysts that can effectively harness ultrasound energy to convert CO2 efficiently and selectively, which is a subject of significant interest and priority, despite its challenging nature.
Inspired by extensive studies on CO2 catalysis systems, copper (Cu0) has been widely identified as a metal that provides active sites for CO2 conversion.19,20 However, nanoscale metallic Cu-containing materials are prone to oxidation, leading to deactivation. To address this, various methods have been explored to stabilise metallic Cu, including the introduction of capping agents, alloying with other metals, and anchoring on supports.21–24 Among these, metal oxides and metal hydroxides have shown promise as supports due to their ability to enhance its resistance to oxidation and also facilitate CO2 adsorption.23,24 Layered double hydroxides (LDHs) are a large class of lamellar materials that are commonly represented by the formula, [M1–xM′x(OH)2]a+[An−a/n]·mH2O (M and M′ are commonly divalent and trivalent cations respectively, An− is the interlayer anion, and 0 < x < 1).25,26 In particular, Cu-containing LDHs have been shown to offer great promise as precursors to Cu-based catalysts for thermal CO2 reduction using H2.27,28
In this study, CuxZnAl-LDHs were prepared by co-precipitation method. Upon calcination in air, it produces uniform mixed metal oxides, also referred to as layered double oxide (LDO). The Cu0 nanoparticles that form following a subsequent reduction step are securely supported on a ZnAl-LDO, designated as r-Cux/ZnAl-LDO (Fig. 1a). ZnAl-LDO can serve to both stabilise, geometrically control and electronically promote Cu. The Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn:Al ratio has been varied to produce an optimum catalytic performance. These catalysts facilitate the sonocatalytic CO2-to-CO conversion under 1 MHz ultrasound irradiation, the best of which was identified as r-Cu0.8/ZnAl-LDO with a CO productivity of 23.8 μmolCO g−1 h−1 and selectivity of over 85% in pure CO2 saturated water under 1 atm CO2. Based on mechanistic insights we suggest that the active metallic Cu sites, within the ultrasound-induced micro-high-energy environment, synergistically promote the sonocatalytic CO2 reduction process. It is noteworthy that dissolved gases influence acoustic cavitation, with previous research indicating that the highest cavitation was found under Ar.13,14 Accordingly, positive correlation of the cavitation activity and CO2 conversion was revealed and showed that the heightened cavitation in 5% CO2/Ar leads to both enhancements of CO (252.7 μmolCO g−1 h−1) and H2 (150.1 μmolH2 g−1 h−1) for syngas production, achieving a superior sonochemical efficiency (SE) of 211.1 μmol kJ−1 g−1 L−1.
:Zn:Al ratio has been varied to produce an optimum catalytic performance. These catalysts facilitate the sonocatalytic CO2-to-CO conversion under 1 MHz ultrasound irradiation, the best of which was identified as r-Cu0.8/ZnAl-LDO with a CO productivity of 23.8 μmolCO g−1 h−1 and selectivity of over 85% in pure CO2 saturated water under 1 atm CO2. Based on mechanistic insights we suggest that the active metallic Cu sites, within the ultrasound-induced micro-high-energy environment, synergistically promote the sonocatalytic CO2 reduction process. It is noteworthy that dissolved gases influence acoustic cavitation, with previous research indicating that the highest cavitation was found under Ar.13,14 Accordingly, positive correlation of the cavitation activity and CO2 conversion was revealed and showed that the heightened cavitation in 5% CO2/Ar leads to both enhancements of CO (252.7 μmolCO g−1 h−1) and H2 (150.1 μmolH2 g−1 h−1) for syngas production, achieving a superior sonochemical efficiency (SE) of 211.1 μmol kJ−1 g−1 L−1.
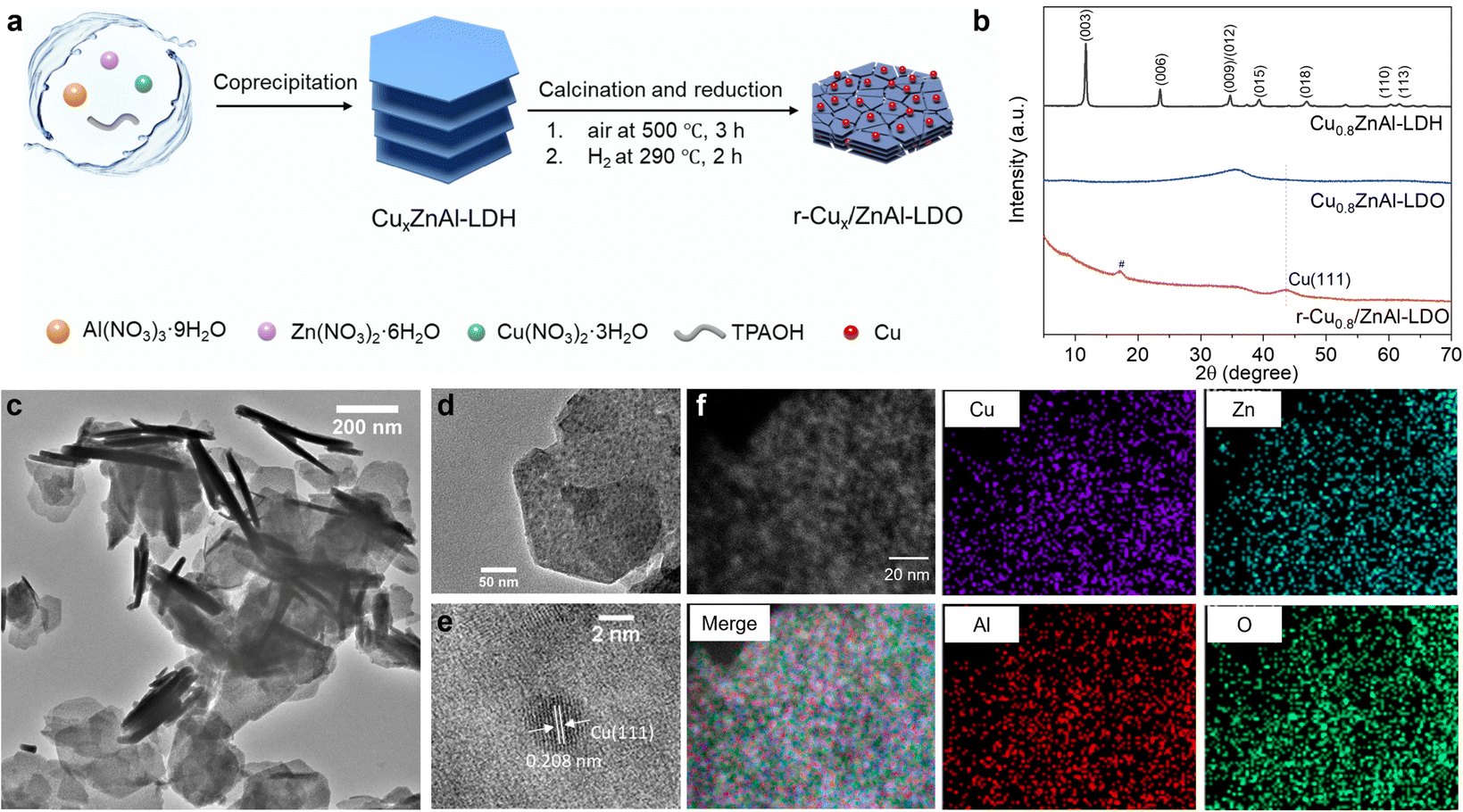 | ||
| Fig. 1 Synthesis and characterisation of r-Cu0.8/ZnAl-LDO. (a) Schematic illustration of the synthesis of Cu0.8/ZnAl-LDO catalyst. (b) XRD patterns of Cu0.8ZnAl-LDH, Cu0.8ZnAl-LDO, and r-Cu0.8/ZnAl-LDO. # peak from PMMA holder. (c) and (d) TEM image of r-Cu0.8/ZnAl-LDO. (e) HRTEM image of r-Cu0.8/ZnAl-LDO. (f) STEM image of r-Cu0.8/ZnAl-LDO with its elemental mapping results of Cu (purple), Zn (light blue), Al (red), and O (green). | ||
Results and discussion
As depicted in Fig. 1a, CuxZnAl-LDHs were synthesised as precursors that form r-Cux/ZnAl-LDO catalysts following calcination and reduction treatments. To analyze these materials, powder X-ray diffraction (XRD) studies were conducted (Fig. 1b and Fig. S1, ESI†). The XRD pattern of Cu0.8ZnAl-LDH exhibits the characteristic Bragg reflections of an LDH, including a both a harmonic (00l) series, and (110), (113) Bragg reflections. The interlayer anion was identified as CO32− using IR spectroscopy (Fig. S2, ESI†). After calcination in air at 500 °C, the characteristic LDH Bragg reflections disappear, and a broad feature appears that can be assigned to an amorphous mixed metal oxide (Cu0.8ZnAl-LDO). Further reduction in H2 at 290 °C resulted in the appearance of a new Bragg reflection at 2θ = 44°, corresponding to Cu0 (111),29 indicating the formation of metallic Cu in the r-Cu0.8/ZnAl-LDO catalyst. A series of r-Cux/ZnAl-LDOs with different Cu loadings was synthesised, where x represents the stoichiometry of Cu in the catalyst. The amount of Cu in the samples was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES) (x = 0, 0.4, 0.8, 1.4, 2.8) (Table S1, ESI†). The introduction of different Cu loadings did not affect the formation of LDH precursors, and a similar Cu0 (111) Bragg reflection was observed after reduction among r-Cux/ZnAl-LDOs but additional CuO and Cu2O were found in r-Cu2.8ZnAl-LDO with higher Cu loading (Fig. S1, ESI†).Transmission electron microscopy (TEM) revealed that the nanoplatelet morphology of r-Cux/ZnAl-LDO remains (Fig. 1c and Fig. S3, ESI†). Enlarged images of the nanoplatelets show well-dispersed dark dots embedded on the nanoplatelet (Fig. 1d and Fig. S3, ESI†). As Cu is more readily reduced than Zn and Al, it is predicted that these well-dispersed dark dots are metallic Cu nanoparticles formed after the reduction step in H2 at 290 °C. HRTEM image confirms that these dark dots are indeed metallic Cu as evidenced by characteristic (111) lattice fringes of metallic Cu with d spacing of 0.208 nm30 (Fig. 1e). It can be seen that the resultant size of metallic Cu nanoparticles in the r-Cux/ZnAl-LDOs (x = 0, 0.4, 0.8, 1.4, 2.8) increases with increasing Cu loading (Fig. S2, ESI†). Additionally, energy-dispersive X-ray spectroscopy (EDS) images demonstrate the uniform distribution of Cu, Zn, Al, and O elements in the r-Cux/ZnAl-LDOs (Fig. 1f and Fig. S4, ESI†).
The elemental composition and chemical oxidation states were characterised by X-ray photoelectron spectroscopy (XPS). The XPS results demonstrate the coexistence of Cu, Zn, Al, and O in the r-Cux/ZnAl-LDOs (Fig. S5, ESI†). The XPS spectrum of Cu 2p photoemission for r-Cu0.8/ZnAl-LDO shows characteristic peaks centred at 932.20 eV for Cu 2p5/2 and 952.00 eV for Cu 2p3/2, with a spin–orbit splitting distance of 19.8 eV.31 These values are close to those of metallic Cu (932.05 eV for Cu 2p5/2 and 952.05 eV for Cu 2p3/2), significantly differing from the Cu 2p spectra of CuO and Cu0.8ZnAl-LDO (Fig. 2a). This indicates that the copper in r-Cu0.8/ZnAl-LDO is primarily in a metallic state rather than in an oxidised form. Similar results for Cu 2p photoemission were also found in the r-Cu0.4/ZnAl-LDO, but with a slightly positive shift for r-Cu1.4/ZnAl-LDO (Fig. S6, ESI†). The Zn 2p photoemission spectrum for r-Cu0.8/ZnAl-LDO showed a slight shift to lower binding energy compared to Cu0.8ZnAl-LDO, indicating a slight reduction in Zn due to the H2 reduction (Fig. 2b and Fig. S6b, ESI†). Conversely, the Al XPS spectra showed little change in binding energy in the r-Cux/ZnAl-LDOs, suggesting that the oxidation state of Al remains largely unchanged after H2 reduction (Fig. S6c and d, ESI†). X-ray absorption fine structure (XAFS) spectroscopy was further performed to confirm the chemical state and coordination environment of Cu atoms. As shown in Fig. 2c, the normalised Cu K-edge X-ray absorption near-edge structure (XANES) data of r-Cu0.8/ZnAl-LDO most closely resembles metallic Cu. The oxidation state of Cu was determined using the first derivative of the XANES spectra (Fig. S7, ESI†), confirming the dominance of metallic Cu in r-Cu0.8/ZnAl-LDO. Extended X-ray absorption fine structure (EXAFS) spectra were analyzed using k2χ(k) weighted Fourier transform (FT) spectra (Fig. 2d and Fig. S8, ESI†). A notable peak centred at approximately 2.14 Å corresponds to the Cu–Cu scattering path, which is comparable to that of Cu foil (2.22 Å). Additionally, a very weak peak at 1.45 Å is attributed to Cu–O bonding, with CuO (1.49 Å) serving as a reference (Fig. 2d). This result supports the presence of primarily metallic copper with minor contributions from copper oxides in the r-Cu0.8/ZnAl-LDO catalyst. The wavelet transforms (WTs) were also conducted to further elucidate the Cu atomic coordination structures (Fig. 2e–g). The WT maximum intensities at 7.1 and 5.3 Å−1 correspond to the Cu–Cu bonds for Cu foil and Cu–O for CuO, respectively. In r-Cu0.8/ZnAl-LDO, the main WT maximum intensities centered at 7.1 Å−1 could be assigned to Cu–Cu bonding. Together, the results suggest that Cu nanoparticles incorporated on the ZnAl-LDO were stabilised primarily in the metallic oxidation state, which is important for CO2 conversion.
 | ||
| Fig. 2 Structural characterisation of r-Cu0.8/ZnAl-LDO. (a) and (b) High-resolution XPS spectra of Cu 2p and Zn 2p spectra of r-Cu0.8/ZnAl-LDO catalyst. (c) Cu K-edge X-ray absorption near-edge structure (XANES) spectra and (d) the Fourier transforms of EXAFS spectra of r-Cu0.8/ZnAl-LDO, Cu foil and CuO. Wavelet transforms (WTs) of Cu in Cu foil (e), r-Cu0.8/ZnAl-LDO (f), and CuO (g). | ||
Sonocatalytic testing was carried out in a custom-made chamber placed in a sonochemical cylindrical reactor that directs acoustic waves to the reactor vessel (Fig. S9 and Table S2, ESI†). 10 mg catalyst and 2.5 mL DI water were placed in the sealed chamber, followed by CO2 purging under room temperature and pressure. Detailed procedures for the sonocatalytic CO2 reduction reaction are described in the ESI.† The gaseous and liquid products were determined by gas chromatography and 1H NMR spectroscopy, respectively. No liquid products were detected whereas gaseous products, including H2, CO and CH4, were observed when using the r-Cux/ZnAl-LDOs catalysts (Fig. S10 and S11, ESI†). To probe the origin of the gaseous products, a series of control experiments was conducted (Fig. 3a). No impurities are detected in the CO2 gas supply, and no gaseous products were detectable without ultrasound irradiation or without addition of a catalyst. Therefore, we conclude that the presence of the Cu-based catalyst, in combination with ultrasound, is necessary to obtain gaseous products. Upon substitution of the CO2 supply with pure inert gases, such as N2 and Ar, no CO was observed but H2 and CH4 were detected (Fig. S12, ESI†). The H2 production is due to the well documented enhanced cavitation-induced H2O sonolysis.32 Further 13C isotope labelling experiments were performed to identify the origins of carbon-containing gaseous products. Our results demonstrate that CO2 is responsible for the formation of CO, as m/z = 29, corresponding to 13CO, was consistently detected in repeated 13CO2 isotope labelling experiments (Fig. 3b and Fig. S13, ESI†). In contrast, CH4 was found to originate from the catalyst itself rather than from CO2 (Fig. S14, ESI†). Combined with the observations in Fig. S11–S13 (ESI†), CO production originates from CO2 conversion rather than decomposition of carbon-based residues within the catalysts. In addition, both CuxZnAl-LDH precursors and CuxZnAl-LDOs produce negligible gaseous products after the CO2 sonocatalysis, indicating that H2 reduction to produce metallic Cu0 nanoparticles immobilised on a ZnAl-LDO support (r-Cux/ZnAl-LDO catalysts) is essential for successful sonocatalytic CO2-to-CO conversion.
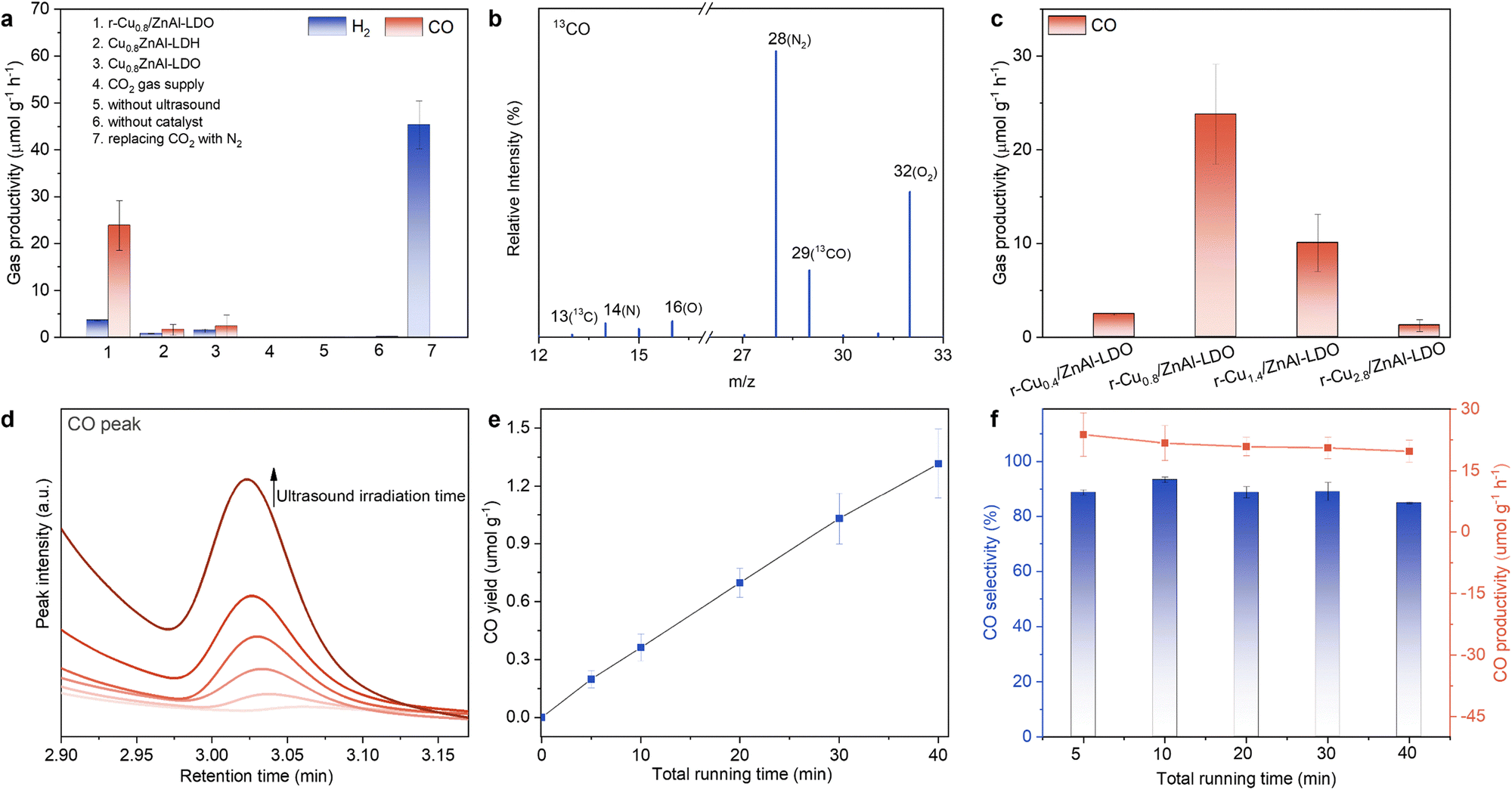 | ||
| Fig. 3 Sonocatalytic CO2 reduction performance of r-Cux/ZnAl-LDO catalysts. (a) Evolution of products in a series of control experiments under various reaction conditions. (b) Mass spectrum of produced 13CO (m/z = 29) via isotopic 13CO2 reduction under ultrasound treatment over r-Cu0.8/ZnAl-LDO catalyst. (c) CO production among r-Cux/ZnAl-LDOs with different Cu loadings. (d) Peak intensity of CO produced over r-Cu0.8/ZnAl-LDO at different total running time. (e) Time-dependent CO yield of r-Cu0.8/ZnAl-LDO at 1 MHz and 0.212 kW. (f) Time-dependent CO productivity and selectivity for r-Cu0.8/ZnAl-LDO catalyst. The quantitative analysis was derived from the ultrasound irradiation time, calculated using duty cycles relative to the total running time (see ESI† for details). | ||
To gain an overall view of the active sites for this reaction, the evolution of sonocatalytic CO2-to-CO conversion was tested as a function of Cu participation. No CO production was detected using a Cu-free, r-ZnAl-LDO catalyst, and only a trace amount of CO was detected using a H2 reduced copper oxide catalyst (r-CuO) (Fig. S15, ESI†). The r-CuO catalyst without ZnAl-LDO may not be stable, resulting in low catalytic CO2-to-CO activity (Fig. S16, ESI†). In contrast, the catalytic activity significantly changed when the trimetallic r-Cux/ZnAl-LDO catalysts were tested as shown in Fig. 3c, whereby CO was the dominant carbonaceous product. Further optimization of Cu loadings indicates that CO productivity initially increases (x = 0.4 to 0.8) with increased Cu loading and then decreases (x > 0.8). Peak CO productivity of 23.8 μmolCO g−1 h−1 (or 95.6 mmolCO gCu−1 h−1 when normalised against Cu loadings) was found for the r-Cu0.8/ZnAl-LDO catalyst (Fig. 3c). We presume the reduction in catalytic activity at higher Cu loadings is likely related to the increased average diameter of the Cu nanoparticles.25 Using r-Cu0.8/ZnAl-LDO catalyst, CO production continuously increased with increasing ultrasound irradiation exposure (Fig. 3d). Quantitative analysis for CO indicates that the yield has a linear relationship to ultrasound time (Fig. 3e and Fig. S17, ESI†). Over the ultrasound time range examined, the production rate of CO was calculated to be 19.7 to 23.8 μmolCO g−1 h−1 with a selectivity consistently above 85% (Fig. 3f). It is noted that quantitative analysis for the productivity calculation is based on the time with ultrasound irradiation exposure rather than the total running time. This is because the sonochemical reactor emitted pulsed ultrasound (as opposed to continuous wave ultrasound) to prevent the ultrasound transducer from overheating. Detailed calculations are given in the ESI.† In addition, physical mixtures with calcination treatment, including a r-(Cu + ZnO + Al2O3) mixture and a r-(Cu + ZnAl-LDO) mixture, show poorer CO and H2 productivity in comparison to r-Cu0.8/ZnAl-LDO (Fig. S18, ESI†). These results confirm that the sonocatalytic performance is related to the critical size of active metallic Cu nanoparticles stabilised on the ZnAl-LDO.
Above a critical ultrasound intensity threshold, acoustic cavitation occurs and emits noise distinct from the acoustic field.33,34 Cavitation and its subsequent chemical effects are influenced by the nature of the local liquid environment such as dissolved gas. Thus, the gas mixture may critically impact sonocatalytic CO2 performance since cavitation may create extreme conditions that could facilitate CO2 catalysis. Here, we investigated two gas atmospheres, pure CO2 and a 5% CO2 in Ar mixture. The inert gas Ar was chosen because it is known to enhance sonochemical activity owing to the low heat capacity. We measured the cavitation threshold as a metric for cavitation activity, and thus sonocatalytic CO2-to-CO conversion. Noise emitted by cavitation bubbles was detected with a passive cavitation detector and analyzed for its frequency content. This power spectral density curve was summed to calculate a metric for power. Values with a signal greater than 6 dB above noise from water under identical acoustic conditions (an arbitrary threshold) was defined as emissions from a cavitation event.35 Detailed cavitation detection and analysis were given in the ESI,† Fig. S19 and Table S3. As shown in Fig. 4a, the probability of cavitation events in both pure CO2 and 5% CO2/Ar are not enhanced until the peak-to-peak drive voltage exceeds 103 V. Interestingly, the likelihood for cavitation in 5% CO2/Ar was consistently higher than that in pure CO2 above 103 V. Similarly, higher performance in CO2 conversion was found with the gas supply of 5% CO2/Ar compared to pure CO2 (Fig. 4b and inset of Fig. 4b). Using a 5% CO2/Ar gas mixture, the productivity of H2 and CO was measured at 150.1 and 252.7 μmol g−1 h−1 respectively, along with the sonochemical efficiency (SE) of 211.1 μmol kJ−1 g−1 L−1 for the total product formation from CO2 and water. Both SE and CO productivity of r-Cu0.8/ZnAl-LDO in 5% CO2/Ar rank top among the ultrasound-triggered catalysis and recently reported photocatalytic CO2-to-CO research (Tables S5 and S6, ESI†). In addition, the H2 and CO productivity represents a ∼36-fold increase and a ∼11-fold enhancement with respect to pure CO2, respectively (Fig. 4b). High productivity of CO and H2 was achieved under 5% CO2/Ar; for example, H2 and CO yields reach ∼0.10 and ∼0.17 μmol, respectively, with 10 mg of catalyst for a running time of 40 min. It is noteworthy that the production of CO:H2 ratio using 5% CO2/Ar lies in the range 1.2 to 2.2 (Table S4, ESI†), demonstrating its potential in syngas production. These results confirm a significant impact of introducing Ar on boosting catalytic CO2-to-CO conversion, providing a clear trend linking the cavitation to sonocatalytic CO2 conversion.
 | ||
| Fig. 4 Correlation of cavitation and sonocatalytic CO2 reduction reaction performance in different gas atmospheres. (a) The comparison of probability for cavitation between pure CO2 and 5% CO2/Ar with catalyst in 2.5 mL DI water. (b) The comparison of the gaseous productivities from sonocatalysis between pure CO2 and 5% CO2/Ar. | ||
In addition, we further investigate the influence of cavitation on CO2 sonolysis, the experiment was conducted in the 5% CO2/Ar atmosphere with the absence of a catalyst. Fig. S20 (ESI†) shows very small amounts of H2 and CO were detected, greatly lower than that with the presence of r-Cu0.8/ZnAl-LDO catalyst. Previously, Koblov et al. reported the introduction of RANEY® Ni-Ru/C as catalyst for CO2 to syngas sonocatalysis results in the production of H2 (0.79 μmol g−1 h−1) and CO (0.894 μmol g−1 h−1) (Table S5, ESI†).36 Although this work presented the enhancement with the presence of catalyst, its performance showed around 239 times lower by comparing tor-Cu0.8/ZnAl-LDO catalyst. These results suggest that the enhanced cavitation for CO2 conversion still requires the presence of a suitable catalyst, underscoring the critical role of catalysts.
We note that previous studies have demonstrated ultrasound-triggered catalysis for CO2 conversion. These examples are based on piezocatalysis or triboelectric nanogenerators.37–39 Piezocatalysis relies on an ultrasound-triggered spontaneous polarisation in piezoelectric materials (eg. Nb-doped lead zirconate titanate) due to the acoustic pressure from the application of ultrasound in the vicinity of Curie temperature (Tc) creates free charges for CO2 reduction. Whereas triboelectric nanogenerators (TENG) convert mechanical energy into electrical energy by utilising the charge difference generated by friction. Both required the charge difference on the catalyst to access CO2 reduction reaction.40 To examine whether electron transfer is involved during sonocatalytic CO2 to CO conversion, we added 1 mM AgNO3 solution as electron sacrificial agent.41 As shown in Fig. 5a, the production of CO has little change by introducing AgNO3 sacrificial agent. This result suggests that the CO2-to-CO reduction might not involve electron transfer during sonocatalysis, potentially excluding the contribution from piezocatalysis and contact-electro-catalysis.
 | ||
| Fig. 5 Investigation on the mechanism of sonocatalytic CO2-to-CO conversion using r-Cu0.8/ZnAl-LDO. (a) The comparison of CO production in the presence of radical or electron trap. (b) Measured electron paramagnetic resonance (EPR) spectrum of the solution with addition of DMPO after ultrasound irradiation. (c) Proposed mechanism for CO production. | ||
Further investigation on radical involvement during CO2 sonocatalysis was conducted. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) was added with a view to capture radicals produced during the ultrasound irradiation.42 Our results show CO production increased by the addition of DMPO (Fig. 5a). Analysis of the electron paramagnetic resonance (EPR) spectrum taken after ultrasound irradiation reveals a quadruplet characteristic of DMPO-˙OH. This result shows that the capture of ˙OH radical leads to an increase of CO production. Considering the cavitation-induced bubble collapse results in the formation of ˙H and ˙OH radicals. The capture of ˙OH radical potentially leads to the enrichment of ˙H for CO2 reduction reaction. Thus, a possible mechanism was proposed as shown in Fig. 5c. The CO2 adsorbed (*CO2) on the metallic Cu active sites could be protonated by ˙H with the formation of *CO(OH); accompany with the local high-energy environment from the moment of bubble collapse could thermodynamically facilitate the breakage of C–OH bonding to form adsorbed CO (*CO) and ˙OH radicals. In the final step, the desorption of *CO leads to CO production and regenerates the active sites.
The evolution of the catalyst before and after CO2 sonocatalysis was investigated. TEM images show that the nanoplatelet morphology of r-Cu0.8/ZnAl-LDO is largely preserved, although slight cracking into smaller pieces was observed under intense ultrasonication. Element mapping analysis showed little change after the catalytic process (Fig. S21, ESI†). The crystallinity and phase composition were analysed using XRD. As shown in Fig. 6a, the XRD data shows that r-Cu0.8/ZnAl-LDO was unchanged after CO2 purging whereas new diffraction features with characteristic Bragg peaks of an LDH were found in the catalyst after CO2 sonocatalysis. This is due to the well-documented reconstruction of the LDO back to an LDH structure in the presence of water and air. Interestingly, the Bragg peak at 2θ = 44° corresponding to Cu (111) remained before and after sonocatalysis, indicating a good stability of Cu nanoparticles during the reaction. This result suggests the reconstruction of LDO might occur within Zn and Al elements as the presence of Jahn–Teller distortion of Cu in LDH makes it less prone to reconstruct. Cu 2p XPS spectra further confirms that the binding energy of Cu has negligible change before and after the sonocatalysis (Fig. 6b). Although a slight oxidation of Cu was found in the Cu XANES spectra after sonocatalysis, the majority of Cu was still in the metallic state with the main WT maximum intensities centered at 7.1 Å−1, corresponding to Cu–Cu bonding (Fig. 6c and inset of Fig. 6c). These results indicate the Cu nanoparticles on ZnAl-LDO are relatively stable, which is crucial for maintaining catalytic efficiency.
 | ||
| Fig. 6 Evolution of the catalyst before and after CO2 sonocatalysis. (a) The XRD patterns and (b) XPS spectra of r-Cu0.8/ZnAl-LDO before and after the CO2 sonocatalysis with the specific air-sensitive chambers. (c) Cu K-edge X-ray absorption near-edge structure (XANES) spectra before and after the CO2 sonocatalysis, inset is Wavelet transforms (WTs) of catalyst after sonocatalysis. (i): r-Cu0.8/ZnAl-LDO; (ii): r-Cu0.8/ZnAl-LDO-after CO2 purging; (iii): r-Cu0.8/ZnAl-LDO-after CO2 sonocatalysis. | ||
Conclusions
We have demonstrated that sonocatalysis has the potential to become an appealing technology for CO2 reduction under mild and green conditions. This approach creates a local high-energy environment surrounding a suitably active catalyst favorable for CO2 reduction. r-Cu0.8/ZnAl-LDO has been shown to be active for CO2 reduction in water at room temperature and ambient pressure with a CO productivity of 23.8 μmolCO g−1 h−1 and > 85% selectivity using pure CO2. Detailed experiments on the catalysts reveal that the active sites for CO2 reduction are metallic Cu0 nanoparticles stabilised by ZnAl-LDO. Our proposed mechanism suggests that metallic Cu sites synergistically couple with ultrasound-induced micro-high-energy environment to enable the sonocatalytic CO2 reduction process. This novel approach creates a local high-energy environment driven by acoustic cavitation that can be tuned using different gas atmospheres. Further study on the correlation between CO2-to-CO conversion and cavitation reveals that higher cavitation likelihood leads to higher CO productivity. A ∼11-fold enhancement in the CO production with the yield of 252.7 μmolCO g−1 h−1 was achieved in 5% CO2 in Ar gas. This result indicates that high CO productivity can be achieved at low CO2 concentrations, potentially offering an alternative approach to utilising CO2 feedstock. In addition, the CO:H2 ratio produced in 5% CO2/Ar lies in the range of 1.2 to 2.2, demonstrating its potential for syngas production. This approach achieved a high sonochemical efficiency of 211.1 μmol kJ−1 g−1 L−1 for ultrasound-driven product formation from CO2 and water. This work not only advanced CO2 reduction technologies but also provides an innovative approach that may have significant impact across multiple fields.
Author contributions
This project was conceived by C. C. J. K. and D. O. H.; L. C. performed the synthesis and characterisation of catalysts; L. C. and Y. Q. conducted the sonochemical measurements; C. T. C. assisted with the electron microscopy studies; L. C., J. K. and D. O. H. wrote the paper; all authors discussed the results and edited the manuscript.Data availability
The data presented in this study are supplemented in the ESI.† Further data may be obtained by contacting the corresponding authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. C., C. C., Z. R. T., C. T. C. and D. O. H. would like to thank SCG Chemicals Public Co., Ltd (Thailand) for funding. The authors thank UK Catalysis Hub block allocation for beamtime (SP34632-2). The authors also acknowledge Diamond light source for allocation of beam times MG37405-1 on ePSIc. This work was supported by the Engineering and Physical Sciences Research Council (Grant Reference EP/W012316/1) and EPSRC UKRI Impact Acceleration Account Award (Grant Reference EP/X525777/1).Notes and references
- M. Meinshausen, N. Meinshausen, W. Hare, S. C. Raper, K. Frieler, R. Knutti, D. J. Frame and M. R. Allen, Nature, 2009, 458, 1158–1162 CrossRef CAS PubMed
.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. Kenis, C. A. Kerfeld, R. H. Morris, C. H. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- M. H. Islam, O. S. Burheim and B. G. Pollet, Ultrason. Sonochem., 2019, 51, 533–555 CrossRef CAS PubMed
-
T. J. Mason, Sonochemistry, Royal Society of Chemistry, 1990 Search PubMed
- R. Mettin, C. Cairós and A. Troia, Ultrason. Sonochem., 2015, 25, 24–30 CrossRef CAS PubMed
-
T. Leighton, The acoustic bubble, Academic press, 2012 Search PubMed
- A. Dehane, S. Merouani, A. Chibani, O. Hamdaoui and M. Ashokkumar, Chem. Eng. Process., 2022, 179, 109075 CrossRef CAS
- A. Henglein, Z. Naturforsch. B, 1985, 40, 100–107 CrossRef
- H. Harada, Ultrason. Sonochem., 1998, 5, 73–77 CrossRef CAS PubMed
- M. H. Islam, O. S. Burheim, J. Y. Hihn and B. G. Pollet, Ultrason. Sonochem., 2021, 73, 105474 CrossRef CAS PubMed
- P. Gholami, A. Khataee, R. D. C. Soltani and A. Bhatnagar, Ultrason. Sonochem., 2019, 58, 104681 CrossRef CAS PubMed
- U. S. Jonnalagadda, X. Su and J. J. Kwan, Ultrason. Sonochem., 2021, 73, 105530 CrossRef CAS PubMed
- X. Su, U. S. Jonnalagadda, L. D. Bharatula and J. J. Kwan, Ultrason. Sonochem., 2021, 79, 105753 CrossRef CAS PubMed
- C. C. Y. Wong, J. L. Raymond, L. N. Usadi, Z. Zong, S. C. Walton, A. C. Sedgwick and J. Kwan, Ultrason. Sonochem., 2023, 99, 106559 CrossRef CAS PubMed
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC
- M. Jin, G. He, H. Zhang, J. Zeng, Z. Xie and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 10560–10564 CrossRef CAS PubMed
- R. Long, Y. Li, Y. Liu, S. Chen, X. Zheng, C. Gao, C. He, N. Chen, Z. Qi, L. Song, J. Jiang, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2017, 139, 4486–4492 CrossRef CAS PubMed
- M. Luo, Z. Wang, Y. C. Li, J. Li, F. Li, Y. Lum, D. H. Nam, B. Chen, J. Wicks, A. Xu, T. Zhuang, W. R. Leow, X. Wang, C. T. Dinh, Y. Wang, Y. Wang, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5814 CrossRef CAS PubMed
- L. Dai, Q. Qin, P. Wang, X. Zhao, C. Hu, P. Liu, R. Qin, M. Chen, D. Ou, C. Xu, S. Mo, B. Wu, G. Fu, P. Zhang and N. Zheng, Sci. Adv., 2017, 3, e1701069 CrossRef PubMed
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed
- X. Xu, J. Wang, A. Zhou, S. Dong, K. Shi, B. Li, J. Han and D. O'Hare, Nat. Commun., 2021, 12, 3069 CrossRef CAS PubMed
- M. M. J. Li, C. Chen, T. Ayvalı, H. Suo, J. Zheng, I. F. Teixeira, L. Ye, H. Zou, D. O’Hare and S. C. E. Tsang, ACS Catal., 2018, 8, 4390–4401 CrossRef CAS
- M. Lyu, J. Zheng, C. Coulthard, J. Ren, Y. Zhao, S. C. E. Tsang, C. Chen and D. O'Hare, Chem. Sci., 2023, 14, 9814–9819 RSC
- H. Cho, Y. Park, S. Kim, T. Ahn, T.-H. Kim and H. C. Choi, npj 2D Mater. Appl., 2020, 4, 35 CrossRef CAS
- K. Chung, J. Bang, A. Thacharon, H. Y. Song, S. H. Kang, W. S. Jang, N. Dhull, D. Thapa, C. M. Ajmal, B. Song, S. G. Lee, Z. Wang, A. Jetybayeva, S. Hong, K. H. Lee, E. J. Cho, S. Baik, S. H. Oh, Y. M. Kim, Y. H. Lee, S. G. Kim and S. W. Kim, Nat. Nanotechnol., 2022, 17, 285–291 CrossRef CAS PubMed
-
J. Moulder, W. Stickle, P. Sobol, K. Bomben and J. Chastain, Handbook of X-ray photoelectron spectroscopy, 1992 Search PubMed
- N. Merabet and K. Kerboua, Int. J. Hydrogen Energy, 2022, 47, 17879–17893 CrossRef CAS
- V. Bull, J. Civale, I. Rivens and G. Ter Haar, Ultrasound Med. Biol., 2013, 39, 2406–2421 CrossRef PubMed
- J. Haller, V. Wilkens and A. Shaw, Ultrasound Med. Biol., 2018, 44, 377–396 CrossRef PubMed
- J. J. Kwan, S. Graham, R. Myers, R. Carlisle, E. Stride and C. C. Coussios, Phys.
Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 92, 023019 CrossRef CAS PubMed
-
A. Koblov, Sonochemical Reduction of Carbon Dioxide, PhD thesis, Curtin University, 2011 Search PubMed
- Y. Zhang, P. T. Thuy Phuong, N. P. Hoang Duy, E. Roake, H. Khanbareh, M. Hopkins, X. Zhou, D. Zhang, K. Zhou and C. Bowen, Nanoscale Adv., 2021, 3, 1362–1374 RSC
- P. T. T. Phuong, D.-V. N. Vo, N. P. H. Duy, H. Pearce, Z. M. Tsikriteas, E. Roake, C. Bowen and H. Khanbareh, Nano Energy, 2022, 95, 107032 CrossRef CAS
- N. Wang, W. Jiang, J. Yang, H. Feng, Y. Zheng, S. Wang, B. Li, J. Z. X. Heng, W. C. Ong, H. R. Tan, Y.-W. Zhang, D. Wang, E. Ye and Z. Li, Nat. Commun., 2024, 15, 5913 CrossRef CAS PubMed
- F.-R. Fan, S. Xie, G.-W. Wang and Z.-Q. Tian, Sci. China: Chem., 2021, 64, 1609–1613 CrossRef CAS
- J. Zhao, X. Zhang, J. Xu, W. Tang, Z. L. Wang and F. R. Fan, Angew. Chem., Int. Ed., 2023, e202300604 CAS
- Y. Wang, Y. Xu, S. Dong, P. Wang, W. Chen, Z. Lu, D. Ye, B. Pan, D. Wu, C. D. Vecitis and G. Gao, Nat. Commun., 2021, 12, 3508 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee01202c |
| This journal is © The Royal Society of Chemistry 2025 |