
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of strong and weak intermolecular interactions to engineer buried interfaces in inverted wide-bandgap perovskite solar cells†
Hui
Li
*a,
Davide
Regaldo
a,
Chun-Sheng Jack
Wu
 a,
Mirko
Prato
b,
Antonella
Treglia
a,
Heyong
Wang
a,
Wolfram
Hempel
c,
Michele
Sessolo
d,
Yang
Zhou
e,
Andrea
Olivati
a and
Annamaria
Petrozza
*a
a,
Mirko
Prato
b,
Antonella
Treglia
a,
Heyong
Wang
a,
Wolfram
Hempel
c,
Michele
Sessolo
d,
Yang
Zhou
e,
Andrea
Olivati
a and
Annamaria
Petrozza
*a
aCenter for Nano Science and Technology, Istituto Italiano di Tecnologia, via Rubattino 81, 20134 Milano, Italy. E-mail: hui.li@iit.it; annamaria.petrozza@iit.it
bMaterials Characterization Facility, Istituto Italiano di Tecnologia, via Morego 30, 16163 Genova, Italy
cZentrum für Sonnenenergie- und Wasserstoff-Forschung Baden-Württemberg (ZSW), Meitnerstr.1, 70563 Stuttgart, Germany
dInstituto de Ciencia Molecular, Universidad de Valencia, C/Catedrático J. Beltrán 2, 46980 Paterna, Spain
eWuhan National Laboratory for Optoelectronics, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China
First published on 6th May 2025
Abstract
The interfaces between the charge extraction layers and the perovskite layer are critical in defining the performance and stability of wide-bandgap (WBG) perovskite solar cells (PSCs). They govern multiple critical factors affecting the operation of photovoltaic devices such as the energetics of the contact, and the crystallization process of the thin film, thus its structural and electronic quality. Self-assembled monolayers (SAMs) have emerged as promising candidates as hole-selective materials for inverted PSCs, thanks to the flexibility provided by the large library of their functional groups. Herein, we outline a molecular hybridization strategy through the incorporation of the histamine molecule into the [4-(3,6-dimethyl-9H-carbazol-9yl)butyl]phosphonic acid (Me-4PACz), which is one of the most common hole extracting layers. Playing with intermolecular strong and weak interactions, we can contextually act on multiple processes. The proton transfer from the phosphonic acid group of the Me-4PACz to the ethylamine functional group of histamine enables the design of the interface dipole to facilitate hole extraction and minimize recombination losses. Then, the protonated amines balance the nucleation of halide components and stabilize the halide ions in the perovskite, avoiding their migration. Thus, three-dimensional nanovoids and tensile stress at the bottom surface were reduced, stabilizing the buried interface. Finally, the π–π interactions between the imidazole moiety and Me-4PACz improve the molecular assembling of the SAM, reducing disorder at the interfacial contact. The general impact of these results has been tested on PSCs based on lead mix-halide perovskites with two different bandgaps. The inverted WBG PSCs with 1.77 eV bandgap present a power conversion efficiency (PCE) of 20.34%, and maintain 95.5% of the initial PCE after 1000 hours of continuous illumination. The highly challenging WBG PSCs with 1.83 eV bandgap deliver a PCE of 18.99% with a Voc as high as 1.364 V-ranking among the highest reported PCEs and Voc values for such large bandgap.
Broader contextAs a critical constituent of tandem photovoltaics, wide-bandgap (WBG) perovskite solar cells (PSCs) suffer from large open-circuit voltage (Voc) deficit and inferior operational stability. The interfaces between the charge extraction layers and the perovskite govern multiple critical factors affecting the operation of devices including contact energetics, perovskite crystallization, thus its structural and electronic quality, especially for WBG perovskites with more complex composition. Self-assembled monolayers (SAMs) have emerged as promising candidates as hole-selective materials, thanks to their molecular flexibility and designability. Herein, we grow a widely used SAM molecule, Me-4PACz, together with an organic molecule Histamine, which is able to drive weak and strong chemical interaction with the functional groups of SAM molecule, in order to act on the quality of the SAM formation on substrate, design of the interface dipole and WBG perovskite crystallization. When applied to WBG PSCs, our approach yields a PCE of 20.34% (1.77 eV) with >1000 hours operational stability, and a PCE of 18.99% with a Voc of 1.364 V for the 1.83 eV case-among the highest reported for this bandgap. This work highlights an effective and generalizable route toward efficient interface of inverted PSCs, contributing to the advancement of tandem photovoltaics. |
Introduction
Over the past decade, metal halide perovskite (MHP) based photovoltaic (PVs) has rapidly advanced in power conversion efficiency (PCE), gaining a leading position among the next-generation solar technologies. A significant advantage of MHPs over traditional semiconductors is their easily tunable bandgap (Eg), which can be adjusted by designing the chemical composition of the crystal unit. Lead halide perovskites can span their bandgap from approximately 1.5 eV to 2.3 eV by varying the I− to Br− ratio. This tunability makes them ideal for creating absorbers with optimized Eg which is suitable for sub-cells in tandem solar cells and semitransparent building-integrated PVs.1–5 These commonly referred to as wide-bandgap (WBG) perovskite solar cells (PSCs) can efficiently harvest high-energy photons while delivering high open-circuit voltage (Voc). Hence, improving the efficiency and stability of WBG PSCs is pivotal for maximizing the photovoltaic performance of tandem solar cells. Currently, the PCE of mid-bandgap (MBG, ∼1.5 eV) MHPs-based devices have achieved an impressive PCE of 26.9% along with less than 1% performance decay after operation at maximum power point (MPP) for more than 4500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) hours, yielding a theoretical operational T80 lifetime of over nine years.6,7 In striking contrast, WBG PSCs still suffer from much higher Voc deficit exceeding hundreds of millivolts and inferior operational stability than MBG counterparts (T80 lifetime usually lower than 1000 hours).8,9 Snaith et al. identified the nonradiative recombination at the interfaces between the perovskite layer and charge transport layers (CTL) as the major factor limiting the Voc, especially for high-bromide content WBG PSCs.10 On the one hand, perovskite surfaces are typically more defective than the bulk, fostering the formation of deep-level point defects that intensify interfacial non-radiative recombination losses.11 On the other hand, the energy-level misalignment at the perovskite/CTL interface exacerbates the mismatch between quasi-Fermi level splitting (QFLS) and Voc, further constraining the Voc of WBG PSCs.12
hours, yielding a theoretical operational T80 lifetime of over nine years.6,7 In striking contrast, WBG PSCs still suffer from much higher Voc deficit exceeding hundreds of millivolts and inferior operational stability than MBG counterparts (T80 lifetime usually lower than 1000 hours).8,9 Snaith et al. identified the nonradiative recombination at the interfaces between the perovskite layer and charge transport layers (CTL) as the major factor limiting the Voc, especially for high-bromide content WBG PSCs.10 On the one hand, perovskite surfaces are typically more defective than the bulk, fostering the formation of deep-level point defects that intensify interfacial non-radiative recombination losses.11 On the other hand, the energy-level misalignment at the perovskite/CTL interface exacerbates the mismatch between quasi-Fermi level splitting (QFLS) and Voc, further constraining the Voc of WBG PSCs.12
Carbazole-phosphonic acid-based self-assembled monolayers (SAMs) have swiftly emerged as highly effective hole extracting layers (HTLs) in inverted single-junction PSCs owing to their superior hole extraction, cost-effectiveness, facile synthesis and excellent compatibility with tandem solar cells architectures.13–15 Recently, these SAMs have played a pivotal role in mitigating the Voc losses in WBG PSCs by optimizing the energetic alignment at the perovskite/HTL interface.10 However, despite their remarkable charge extraction properties, SAMs still face notable limitations. Specifically, the widespread application of [4-(3,6-dimethyl-9H-carbazol-9-yl)butyl]phosphonic acid (Me-4PACz) has been hindered by its inhomogeneous distribution on the substrate and its poor wettability with perovskite precursors. These characteristics limit the direct deposition of high-quality perovskite films onto Me-4PACz and cause undesirable interfacial losses at the buried interfaces, impeding further performance improvement of devices. The uncontrollable crystallization process in WBG perovskites, given its higher level of chemical inhomogeneity, remains a significant challenge, which not only results in suboptimal film quality but also creates initial sites for halide segregation. These defects further deteriorate the interface contact, ultimately compromising the overall performance and reliability of the devices.16 Tailoring the properties of buried SAMs/perovskite interface is essential for overcoming these challenges and maximizing the potential of WBG PSCs in high-performance tandem solar cell.
It is clear that the interface must be optimized taking into account its multi-functional role. Here we engineer the Me-4PACz SAM by the introduction of an organic molecule, Histamine (2-(1H-imidazol-4-yl)ethanamine), which is able to drive weak and strong chemical interaction with the functional groups of SAM molecule in order to act on the quality of the SAM formation on the substrate, on the design of the interface dipole and on the crystallization of the WBG perovskite thin film at the same time. Consequently, the 1.77 eV perovskite based inverted PSCs deliver a champion PCE of 20.34%, along with a T95 operational lifetime exceed 1000 hours under ISOS-L-1I protocol (25 °C, AM 1.5G) and a T80 operational lifetime over 600 hours under ISOS-L-2I protocol (65 °C, AM 1.5G), representing one of the highest operational stabilities among WBG PSCs with a 1.75–1.80 eV perovskite absorber. Moreover, the general implication of this strategy is demonstrated by testing it in the more complex device embodying the WBG with 1.83 eV bandgap. The PSCs reach a PCE of 18.99% and a Voc of 1.364 V, one of the highest PCE and Voc among the reported >1.8 eV based WBG PSCs.
Result and discussion
Herein, we fabricate WBG PSCs with a p–i–n inverted architecture of ITO/NiOx/Me-4PACz/perovskite/C60/BCP/Au as shown in Fig. 1(a). The selected WBG perovskite composition for this study is formamidinium-cesium lead iodide-bromide [FA0.83Cs0.17Pb(I0.6Br0.4)3], chosen for its optimal bandgap (∼1.77 eV), making it ideal for all-perovskite tandem solar cells, as well as its superior material stability. The organic molecule, Histamine (2-(1H-imidazol-4-yl)ethanamine) is dissolved as an additive in the Me-4PACz solution (referred to as Hi–Me). The photocurrent density–voltage (J–V) characteristics of the 1.77 eV-perovskite based devices are shown in Fig. 1(b). The control devices based on Me-4PACz exhibit a power conversion efficiency (PCE) of 18.77% with a Voc of 1.304 V. The target devices based on Hi–Me achieved a champion PCE of 20.34%, along with a Voc of 1.324 V, a short-circuit current density (Jsc) of 18.42 mA cm−2, and a fill factor (FF) of 83.38%. Moreover, the devices based on Hi–Me exhibit a stabilized power output of 20.10% (Fig. S1, ESI†). Fig. S2 (ESI†) compares the photovoltaic performance of the control and Hi–Me based devices (15 devices for each), in which the consistent PCE, Voc, Jsc, and FF values across devices further validate the high reproducibility of the molecular hybridization approach. Fig. S3 (ESI†) shows the external quantum efficiency (EQE) spectra of the best Hi–Me device, with the integrated Jsc value of 18.33mA cm−2, in good agreement with the J–V characteristics. Building on these findings, we further investigate the universality of Hi–Me SAM by optimizing the performance of 1.83 eV based WBG devices, specifically those utilizing the perovskite composition FA0.83Cs0.17Pb(I0.5Br0.5)3, a widely adopted material for fabricating perovskite/organic tandem solar cells. Fig. 1(c) displays that the optimized devices based on Hi–Me delivered a champion PCE of 18.99%, with a high Voc of 1.364 V, which is one of the highest PCE and Voc among the reported >1.8 eV based WBG PSCs (Table S1, ESI†). A stabilized power output of 18.75% is delivered (Fig. S4, ESI†), and the superior photovoltaic performance reproducibility of Hi–Me devices is also validated (Fig. S5, ESI†). The EQE spectra of 1.83 eV based WBG PSCs is also shown in Fig. S6 (ESI†). Notably, the Hi–Me devices based on both bandgaps show an enhancement both of the Voc and FF with respect to the control samples, indicating that the modification of the buried interface mitigates the non-radiative recombination and facilitates the charge transport within the devices (Fig. S6, ESI†).
 | ||
| Fig. 1 Photovoltaic performance of devices. (a) Schematic diagram of device configuration of inverted WBG PSCs. (b) Champion J–V curves of 1.77 eV WBG PSCs based on Me-4PACz and Hi–Me (The solid line is the reverse scan, and the dotted line is the forward scan). (c) Champion J–V curves of 1.83 eV devices based on Me-4PACz and Hi–Me (The solid line is the reverse scan, and the dotted line is the forward scan). Long-term MPP tracking of the encapsulated 1.77 eV-based devices under simulated AM 1.5G illumination (100 mW cm−2 without UV filter) in an N2-filled chamber at (d) 25 °C, and (e) 65 °C. | ||
Then the 1.77 eV-perovskite based devices were subjected to maximum power point tracking (MPPT) under simulated AM 1.5G illumination (AM1.5 G-100 mW cm−2 without UV filter) in an N2-filled chamber. In comparison to mid-bandgap PSCs, WBG PSCs display significantly inferior operational stability, primarily due to their lower defect tolerance, light-induced phase segregation, and inefficient charge extraction. As demonstrated in Fig. 1(d), the devices based on Me-4PACz exhibit a rapid degradation under aging at 25 °C (ISOS-L-1I protocol of the IEC61215:2016 standard),17 with a T95 lifetime of less than 100 hours. In striking contrast, the efficiency of devices based on Hi–Me retain 95.5% after aging for 1000 hours. This represents one of the highest operational stabilities reported for devices based on WBG perovskites with the bandgap of 1.75–1.80 eV, an optimal range for all-perovskite tandem solar cells (Table S2, ESI†). Furthermore, the ISOS-L-2I protocol (MPPT under 65 °C, AM 1.5G) was performed for the devices, in which the target devices exhibited a T80 lifetime exceeding 600 hours while the T80 of control devices remain less than 100 hours (Fig. 1(e)). The significant enhancement in operational stability, achieved through the incorporation of Histamine into Me-4PACz, is equally evident in devices based on a 1.83 eV bandgap (Fig. S7, ESI†). The markedly improved operational stability of the devices is attributed to the synergistic effects of the higher quality perovskite film, fewer voids at the buried interface, and more efficient charge transport, enabled by the tailored buried interface, which will be further elaborated below. Collectively, the devices based on Hi–Me delivered both high PCE and superior operational stability.
Histamine molecule consists of an aromatic imidazole ring with a weakly basic nitrogen (pKa of 5.8) and (a delocalized π-electron system), and an ethylamine chain with a strongly basic primary amine (pKa of 9.4) that is readily protonated under physiological pH, making it positively charged and highly reactive in biological environments. As reported, the amine head of amine molecules would react with the phosphonic acid (–PO(OH)2) group of Me-4PACz.18 As shown in Fig. 2(a), when mixed with the Me-4PACz SAM solution, the proton transfer is favorable at the ethylamine side of Histamine molecule but not at the imidazole moiety, due to the distinct basicities and proton affinities of the two functional groups. To verify the acid–base interaction, we prepared the Histamine: Me-4PACz mixture (1:1.5 molar ratio, the best molar ratio for the devices) which was dried under vacuum and subsequently annealed at 100 °C in a nitrogen glovebox to ensure complete solvent removal. Fourier infrared transform spectroscopy (FTIR) of Histamine, Me-4PACz, and Hi–Me in solid-state powder are shown in Fig. 2(b). The shift of the P–OH peaks from approximately ∼969.3 cm−1 and ∼1015.1 cm−1 to higher wavenumber in the Hi–Me mixture compound indicates strengthened bonding, resulting from the deprotonation of the first P–OH group of the phosphonic acid in Me-4PACz.18,19 In contrast, the N–H bending peaks of the ethylamine group in Histamine, originally observed at ∼1643.7 cm−1 and ∼1611.5 cm−1, shift to lower wavenumbers in the Hi–Me mixture compound, likely due to the protonation of –NH2 to –NH3+. Moreover, the stretching peaks of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group of Histamine located at 1591.6 cm−1 shift to lower wavenumber, which might be attributed to the π–π interaction between aromatic imidazole ring and Me-4PACz. X-Ray photoelectron spectroscopy (XPS) measurements of spectra collected on Me-4PACz and Hi–Me films on NiOx are shown in Fig. S9 (ESI†). The N 1s spectrum of Hi–Me-4PACz exhibits, with respect to the Me-4PACz case, the additional three peaks due to the presence of Histamine in the film. In particular, the peak located at 401.2 eV, corresponds to the protonated –NH2 (NH3+).20 And interestingly, in the presence of Histamine, the XPS of P 2p XPS peaks of Me-4PACz shift to lower binding energy, which is attributed to the increased electron density on phosphorus atoms due to the proton removal from P–OH. Thus, we can conclude the proton transfer between –PO(OH)2 group of Me-4PACz and Histamine and a π–π interaction between the two molecules.
N group of Histamine located at 1591.6 cm−1 shift to lower wavenumber, which might be attributed to the π–π interaction between aromatic imidazole ring and Me-4PACz. X-Ray photoelectron spectroscopy (XPS) measurements of spectra collected on Me-4PACz and Hi–Me films on NiOx are shown in Fig. S9 (ESI†). The N 1s spectrum of Hi–Me-4PACz exhibits, with respect to the Me-4PACz case, the additional three peaks due to the presence of Histamine in the film. In particular, the peak located at 401.2 eV, corresponds to the protonated –NH2 (NH3+).20 And interestingly, in the presence of Histamine, the XPS of P 2p XPS peaks of Me-4PACz shift to lower binding energy, which is attributed to the increased electron density on phosphorus atoms due to the proton removal from P–OH. Thus, we can conclude the proton transfer between –PO(OH)2 group of Me-4PACz and Histamine and a π–π interaction between the two molecules.
 | ||
| Fig. 2 The intermolecular interaction mechanism of buried interface. (a) Schematic diagram of selective acid–base interaction between Histamine and Me-4PACz. (b) FTIR spectra of Histamine, Me-4PACz, and mixture (Hi–Me, 1:1.5 molar ratio) in solid-state powder. (c) KPFM images and CPD distribution of Me-4PACz and Hi–Me film. (d) Schematic diagram of molecular hybridization engineering buried interface and the formation mechanism of interfacial dipole. | ||
Then we investigate how the bonding between the SAMs and the oxide is affected by the presence of the Histamine. The attenuated total reflectance FTIR (ATR-FTIR) of SAM films in Fig. S10 (ESI†) presents that the PO32− stretching (at 1024.6 cm−1 for the control sample) exhibits a blueshift of around 6cm−1 with the addition of Histamine, suggesting the improved surface binding of Me-4PACz on NiOx.21 Atomic force microscopy (AFM) and Kelvin probe force microscopy (KPFM) were performed to elucidate the distribution and surface potential of SAMs on NiOx. As demonstrated in Fig. S11 (ESI†), uneven dots appearing on the Me-4PACz film suggest the presence of aggregated molecules. In contrast, the Hi–Me film presents much more homogeneous contact potential distribution (CPD) (Fig. 2(c)), indicating the facilitated uniform coverage of Me-4PACz on NiOx. Moreover, the average CPD of Hi–Me film was enhanced from around −300 mV to 0 mV compared with Me-4PACz film, which corresponds to a reduced work function.22,23 The change in work function is also corroborated by Kelvin probe measurements (Fig. S12, ESI†), suggesting the formation of surface negative dipole. The scheme in Fig. 2(d) shows how the acid–base interaction between Histamine and Me-4PACz induces the asymmetric charge distribution of charge across the Histamine molecule and alters its dipole moment (from 2.05 D to 12.39 D, Fig. S13, ESI†), thus leading to the formation of a negative dipole layer that enhances the hole extraction efficiency while blocking the electron transport across the perovskite/SAMs interface. This is well in agreement with acid–basic reaction verified above between the SAM molecule and the Histamine.
The amine side of protonated Histamine is capable of occupying the formamidinium vacancies, thus serving as the crystallization template, realizing defect passivation as well as stabilizing the [PbX6]4− octehedras.24–26 We use the density functional theory (DFT) to simulate the filling of formamidinium vacancies (VFA) by the protonated Histamine on the perovskite surface (Fig. S15, ESI†). The absorption energy of –NH3+ inserted in VFA is −7.36 eV, suggesting the stable absorption. We thus moved to investigate the nature of interface with the WBG perovskite. The better quality of the perovskite/SAM interface in presence of Histamine is observed through the scanning electron microscopy (SEM) images of the bottom surface film which is obtained using a scatheless peel-off. Numerous nanovoids were observed on the bottom surface of perovskite film grown on the Me-4PACz while reduced in the perovskite film deposited on Hi–Me (Fig. S16, ESI†). The removal of three-dimensional nanovoids at the buried interface is further substantiated by cross-sectional SEM analysis presented in Fig. 3(a). These nanovoids can serve as reservoirs for decomposition byproducts such as iodine vapor generated during light-soaking, thus initializing the structural evolution and degradation of perovskite films and devices.27 Their elimination enhances the interfacial contact and stabilizes the buried interface. Moreover, grazing-incidence X-ray diffraction (GIXRD) patterns of bottom surface with 0.01° incident angle in Fig. S17 (ESI†) shows that the crystallinity of perovskite film prepared on Hi–Me increases by 8-times compared with control film, indicating the greatly improved quality of the buried interface. The absence of peak at the low angles suggests that no 2D phase is formed in the perovskite film prepared on Hi–Me. A shift of the (100) diffraction peak towards lower angles is observed in the GIXRD patterns with the incident angle of 0.3°, which is attributed to the VFA occupation by the protonated Histamine. The interfacial strain distribution is also studied using the depth-dependent GIXRD. As shown in Fig. 3(c), the characteristic peaks shift to lower angles as the incident angle increases from 0.3° to 1.1°, indicating a gradual increase in crystal plane distance along the direction perpendicular to the substrate, consistent with Bragg's Law.28 The residual strain is lower in the top region of the perovskite film compared to the bottom interface, due to the difference in α values between the HTL and perovskite, leading to constrained lattice contraction during cooling. It is shown that the perovskite film prepared on Hi–Me reduces the diffraction peak shift and exhibits a smaller slope (−0.0048) than the control film (−0.019), suggesting the relived tensile strain and reduced lattice distortion in the bottom region. The depth-resolved data of the perovskite film deposited on NiOx/Hi–Me was further obtained via time-of-flight secondary ion mass spectrometry (ToF-SIMS). As shown in Fig. S18 (ESI†), the CsCH3N2H+, Pb+, CsI+, and CsBr+ singles refer to the FA+, Pb2+, I− and Br− of perovskite film, respectively. The Ni+ and Cs2PO2+ singles correspond to the NiOx and Me-4PACz, respectively. The C5H7N2+ signal, attributed to Histamine, is predominantly localized at the buried Me-4PACz/perovskite interface, with a notable presence of the upper surface of Me-4PACz. And this additive can also penetrate into the perovskite film, thus passivating the grain boundaries. This observation underscores that Histamine is not entirely removed during the spin-coating process, highlighting its strong interfacial interaction with both perovskite and Me-4PACz, which contribute to the stabilization and functionalization of the buried interface.
 | ||
| Fig. 3 The characteristics of buried interface and perovskite. (a) Cross-sectional SEM images of perovskite film deposited on Me-4PACz (upper) and Hi–Me (lower). (the scale bar is 400 nm). (b) Depth-dependent GIXRD patterns of the perovskite films, and d-spacing values obtained from perovskite (110) plane as a function of incidence angle. (c) In situ PL spectra and (d) PL peak position evolution during spin-coating process of perovskite film deposited on Me-4PACz and Hi–Me. (e) SEM images of perovskite film deposited on Me-4PACz (left) and Hi–Me (right) (the scale bar is 2 μm). (f) XPS spectra of Br 3d and Pb 4f XPS spectra of bottom surface of perovskite film deposited on Me-4PACz and Hi–Me. | ||
The role of Histamine in perovskite crystallization was also explored by monitoring the crystallization process in real time using in situ photoluminescence (PL) spectroscopy. Fig. 3(d) and (e) illustrate the evolution of PL intensity and peak position during the spin-coating process. Upon the addition of antisolvent, the PL peak quickly shifting to 670 nm indicates the start of nucleation. Owing to the lower formation energy, Br-rich nuclei is formed first, causing a progressive blue-shift of PL peak position in control film during spin-coating process.16 In contrast, the perovskite film prepared on Hi–Me presents a comparatively stable PL peak and stronger PL intensity, which emphasizes the role of Histamine in balancing the nucleation rates between I-rich and Br-rich components. The balanced nucleation rate between I/Br components benefits the homogeneous halide distribution in perovskite film. The halide distribution within the perovskite films is then demonstrated by the hyperspectral microscope which combines the advantages of microscopy imaging and spectral analysis. The PL peak emission wavelength map in Fig. S19 (ESI†) displays that some regions with a wide distribution ranging from around 680 nm to 750 nm are observed in control film, which is attributed to the halide phase segregation.29 The perovskite film prepared on Hi–Me presents the relatively symmetric PL peak distribution around 700 nm, illustrating the homogeneous halide distribution within the perovskite film. The higher degree of chemical homogeneity over the crystallization process is also reflected in the reduced PbI2 residues at the surface and grain boundaries (Fig. S20, ESI†), which is of course beneficial for the stabilization of perovskite film and devices.30,31 GIXRD patterns of top surface also illustrate the reduction of PbI2 in the perovskite film deposited on Hi–Me (Fig. S21, ESI†).
In order to illuminate the mechanism of crystallization modulation, XPS measurements were conducted on bottom surface of perovskite film, again obtained using a scatheless peel-off technique (Fig. S3e and S22, ESI†). Compared with the control film, the Br 3d XPS spectra of the perovskite film prepared on Hi–Me exhibits a shift to lower binding energy, while the I 3d XPS peaks remain unchanged, which is ascribed to the stronger interaction between the –NH3+ of protonated Histamine and Br− ions than I− ions. Therefore, –NH3+ of protonated Histamine modulates the coordination environment by preferentially interacting with Br−, thereby reducing its chemical availability and slowing down the nucleation rate of Br-rich phase. This results in a more balanced nucleation kinetics between Br- and I-rich phase, leading to the more homogeneous perovskite crystallization process, which is in well agreement with the in situ PL measurement. Moreover, the XPS spectra of Pb 4f show that the peaks observed at 138.0 eV and 142.9 eV correspond to Pb 4f, respectively. The control film exhibited two additional peaks at 136.4 eV and 141.3 eV, attributed to the presence of metallic Pb0 which introduces deep-level energy states in the semiconductor bandgap.32 In contrast, the Pb0/(Pb0 + Pb2+) ratio decreased markedly from 4.5% to a negligible level for the perovskite film prepared on the Hi–Me.
We conclude our investigations by looking at how the charge carrier dynamics are influenced by the nature of the interface, which we see having a strong influence on the quality of the entire semiconductor thin film. As shown in Fig. 4(a), the excitation density dependent photoluminescence quantum yield (PLQY) measurement provides insights into the radiative recombination dynamics. At low excitation densities, the PLQY is primarily governed by nonradiative recombination processes, such as defect-assisted recombination, resulting in a relatively low PLQY. As the excitation power density increases, the PLQY gradually rises, indicating a transition towards radiative recombination dominance. This behavior reflects the saturation of nonradiative pathways as the carrier population increases, effectively enhancing the radiative recombination efficiency. The perovskite film prepared on Hi–Me exhibits much higher PLQY values in comparison with the control film across a wide range of excitation density, illustrating the suppressed defect-assisted nonradiative recombination and enhanced radiative recombination efficiency.33,34 Photoluminescence (PL) intensity mapping of the exposed bottom surface is presented in Fig. 4(b) and (c), with the corresponding PL spectra of the marked region demonstrated in Fig. S23 (ESI†). Notably, the PL intensity of the perovskite film prepared on Hi–Me is twice as high as that of the film prepared on Me-4PACz. Laser confocal fluorescence lifetime mapping obtained by exciting the full devices from the bottom surface exhibits the increased average lifetime from 4.5 ns of control devices to 6.7 ns of Hi–Me devices (Fig. S24, ESI†). Then we also check the light stability of the perovskite film deposited on Me-4PACz and Hi–Me. Fig. 4(d) demonstrates the evolution of the PL spectra under continuous laser exposure. After 15 minutes aging, the control film exhibited the developed new PL peaks at around 760 nm due to the light-induced halide phase segregation, while the perovskite film prepared on Hi–Me displayed significant resistance to such segregation. The more stabilized perovskite film is due to the higher film-quality with less trap density combined with the more efficient buried interface, benefiting the stability enhancement of the devices.
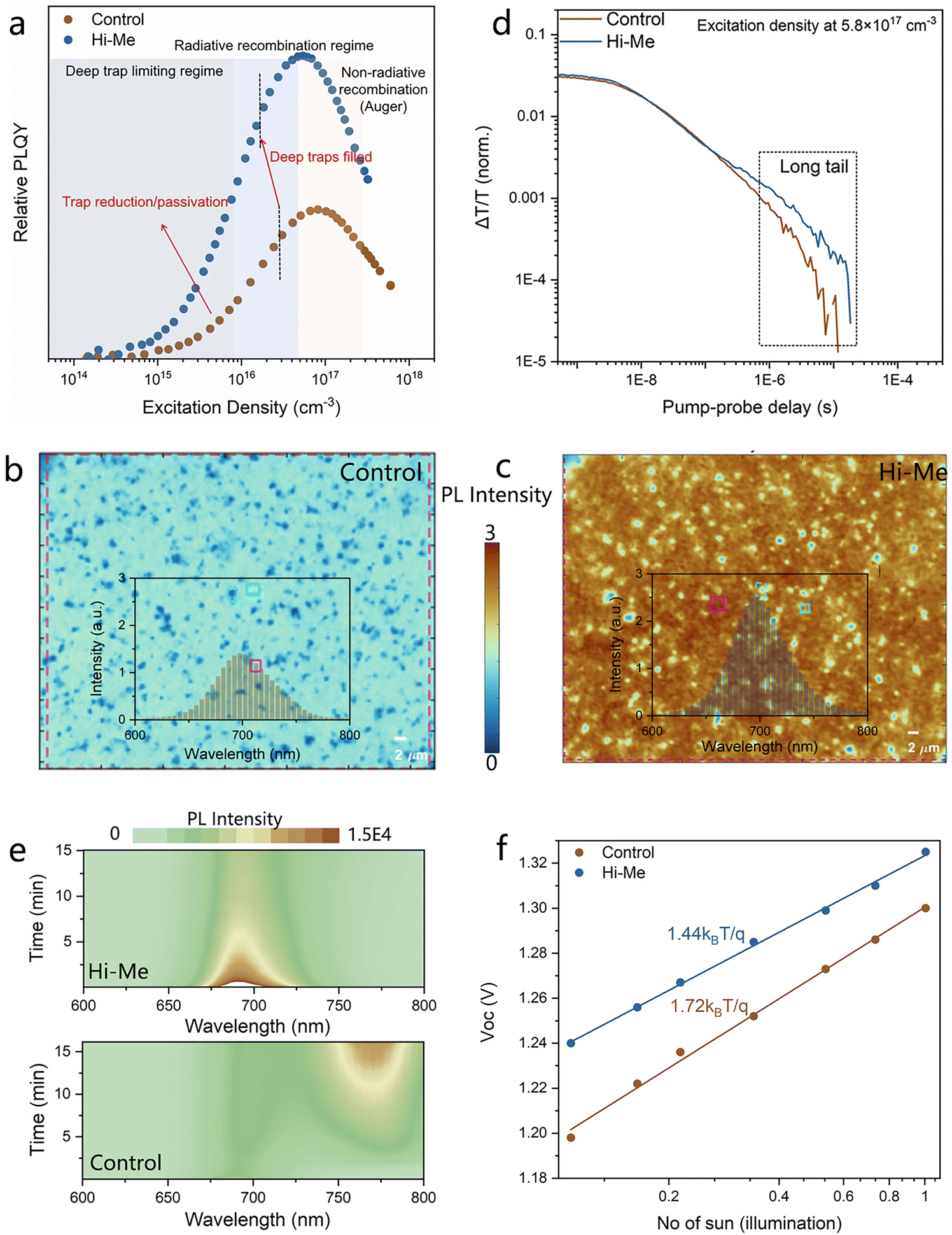 | ||
| Fig. 4 Charge carrier dynamics. (a) Relative PLQY of perovskite films deposited on Me-4PACz and Hi–Me. PL intensity mapping of the bottom surface of perovskite film deposited on (b) Me-4PACz and (c) Hi–Me. (d) TA dynamics plots of photo-blenching peaks, excitation density ≈5.8 × 1017 cm−3. (e) Contour plots showing PL evolutions under continuous laser excitation of perovskite film deposited on Me-4PACz (lower) and Hi–Me (upper). (f) Linear relationship of Voc of the devices with the natural logarithmic of light intensity. | ||
Transient absorption (TA) measurement of the half devices with the structure of ITO/NiOx/SAM/perovskite was performed at excitation density of 5.8 × 1017 cm−3, where the trap mediated dynamics can be ignored. This allows us to mainly focus on the charge transfer dynamics at the perovskite/SAMs interface (spectra in Fig. S25, ESI†). As depicted in Fig. 4(e), the longer-decay TA dynamics (long tail) of the photo-bleaching (PB) peak at the ns–μs regime is observed in the perovskite film prepared on Hi–Me. The long tail is assigned to a long-living trapped carriers, which means the reduced recombination rate of their free counterparts.35 Given the sample architecture, we can infer that more efficient is the hole extraction, longer will be the tail, thus confirming, once again, the improved electronic quality of the interface. Fig. S26 (ESI†) presents the photoemission spectra (raw signal and its cube root) obtained from ambient photoemission spectroscopy, which are employed to determine the ionization energy of the samples: the highest occupied molecular orbital (HOMO) of SAMs and the valence band maximum (VBM) of the perovskite layers.36 These measurements reveal the presence of a 0.12 eV energy barrier for hole extraction at the Me-4PACz/perovskite interface. The incorporation of Histamine eliminates this barrier and creates a 0.15 eV energy cliff, which facilitates more efficient hole extraction across the buried Hi- Me/perovskite interface.
The relationship between photovoltaic parameters and illumination intensity provides an effective method for evaluating the internal recombination mechanisms within devices. As shown in Fig. 4(f), the linear relationship of Voc of the devices with the natural logarithmic of light intensity shows that the devices based on Hi–Me exhibit a lower ideality factor (n) value of 1.44 compared to 1.72 for the devices based on Me-4PACz. An n value approaching 1 suggests that Shockley–Read–Hall recombination is significantly suppressed within devices.37 Meanwhile, the devices based on Hi–Me show a higher FF under low illumination intensity, as a result of the suppressed bimolecular recombination as the carrier density decreases (Fig. S27, ESI†).38 These results confirm the reduced nonradiative recombination and improved charge transport efficiency within the devices based on Hi–Me, highlighting its pivotal role in optimizing optoelectronic performance. The space-charge limited current (SCLC) measurements using hole-only devices with the structure of ITO/NiOx/SAMs/perovskite/PTAA/Au also reveal the less defects distributed within the devices based on Hi–Me (Fig. S28, ESI†).
Conclusion
In this work, we utilized a molecular hybridization strategy to optimize the formation of the SAM based on Me-4PACz and modify the buried interface for inverted WBG PSCs. The mixed SAM, featuring enhanced interfacial contact, facilitated the controlled growth of perovskite crystals, mitigated interface recombination losses, alleviated residual tensile stress, and efficiently promoted hole extraction across the perovskite/Me-4PACz interface. The developed interface engineering enabled the 1.77 eV inverted WBG PSCs with a champion PCE of 20.34%. These devices demonstrated exceptional operational stability, with a T95 lifetime exceeding 1000 hours under ISOS-L-1I protocol and a T80 lifetime surpassing 600 hours under ISOS-L-2I protocol. Furthermore, the versatility of the approach was validated with 1.83 eV WBG PSCs, which delivered a remarkable PCE of 18.99% and an impressive Voc of 1.364 V, achieving the top-tier PCE and Voc among reported >1.8 eV WBG PSCs. This study underscores the critical role of buried interface engineering in advancing high-performance WBG inverted PSCs and can be extended to tandem solar cells.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the support from the projects supported by the HORIZON Grants VALHALLA (no. 101082176) and the Italian Ministry of Environment and Energy Security in the framework of the Project GoPV (CSEAA_00011) for Research on the Electric System. H.W. is a Marie Skłodowska-Curie Fellow (no. 101105123). M. S. acknowledges financial support from the Comunitat Valenciana (CISEJI/2022/43).References
- Y. Wang, R. Lin, C. Liu, X. Wang, C. Chosy, Y. Haruta, A. D. Bui, M. Li, H. Sun, X. Zheng, H. Luo, P. Wu, H. Gao, W. Sun, Y. Nie, H. Zhu, K. Zhou, H. T. Nguyen, X. Luo, L. Li, C. Xiao, M. I. Saidaminov, S. D. Stranks, L. Zhang and H. Tan, Nature, 2024, 635, 867–873 CrossRef PubMed.
- X. Jiang, S. Qin, L. Meng, G. He, J. Zhang, Y. Wang, Y. Zhu, T. Zou, Y. Gong, Z. Chen, G. Sun, M. Liu, X. Li, F. Lang and Y. Li, Nature, 2024, 635, 860–866 CrossRef PubMed.
- Z. Zhang, W. Chen, X. Jiang, J. Cao, H. Yang, H. Chen, F. Yang, Y. Shen, H. Yang, Q. Cheng, X. Chen, X. Tang, S. Kang, X.-M. Ou, C. J. Brabec, Y. Li and Y. Li, Nat. Energy, 2024, 9, 592–601 CrossRef CAS.
- S. Liu, Y. Lu, C. Yu, J. Li, R. Luo, R. Guo, H. Liang, X. Jia, X. Guo, Y.-D. Wang, Q. Zhou, X. Wang, S. Yang, M. Sui, P. Müller-Buschbaum and Y. Hou, Nature, 2024, 628, 306–312 Search PubMed.
- J. Liu, Y. He, L. Ding, H. Zhang, Q. Li, L. Jia, J. Yu, T. W. Lau, M. Li, Y. Qin, X. Gu, F. Zhang, Q. Li, Y. Yang, S. Zhao, X. Wu, J. Liu, T. Liu, Y. Gao, Y. Wang, X. Dong, H. Chen, P. Li, T. Zhou, M. Yang, X. Ru, F. Peng, S. Yin, M. Qu, D. Zhao, Z. Zhao, M. Li, P. Guo, H. Yan, C. Xiao, P. Xiao, J. Yin, X. Zhang, Z. Li, B. He and X. Xu, Nature, 2024, 635, 596–603 Search PubMed.
- H. Chen, C. Liu, J. Xu, A. Maxwell, W. Zhou, Y. Yang, Q. Zhou, A. S. R. Bati, H. Wan, Z. Wang, L. Zeng, J. Wang, P. Serles, Y. Liu, S. Teale, Y. Liu, M. I. Saidaminov, M. Li, N. Rolston, S. Hoogland, T. Filleter, M. G. Kanatzidis, B. Chen, Z. Ning and E. H. Sargent, Science, 2024, 384, 189–193 CrossRef CAS PubMed.
- J. Suo, B. Yang, E. Mosconi, D. Bogachuk, T. A. S. Doherty, K. Frohna, D. J. Kubicki, F. Fu, Y. Kim, O. Er-Raji, T. Zhang, L. Baldinelli, L. Wagner, A. N. Tiwari, F. Gao, A. Hinsch, S. D. Stranks, F. De Angelis and A. Hagfeldt, Nat. Energy, 2024, 9, 172–183 Search PubMed.
- Z. Cheng, M. Zhang, Y. Zhang, W. Qi, Z. Wang, B. Liu and D. Di, Nano Energy, 2024, 127, 109708 CrossRef CAS.
- A. J. Ramadan, R. D. J. Oliver, M. B. Johnston and H. J. Snaith, Nat. Rev. Mater., 2023, 8, 822–838 Search PubMed.
- P. Caprioglio, J. A. Smith, R. D. J. Oliver, A. Dasgupta, S. Choudhary, M. D. Farrar, A. J. Ramadan, Y.-H. Lin, M. G. Christoforo, J. M. Ball, J. Diekmann, J. Thiesbrummel, K.-A. Zaininger, X. Shen, M. B. Johnston, D. Neher, M. Stolterfoht and H. J. Snaith, Nat. Commun., 2023, 14, 932 CrossRef CAS PubMed.
- J. Xue, R. Wang and Y. Yang, Nat. Rev. Mater., 2020, 5, 809–827 CrossRef CAS.
- M. Stolterfoht, P. Caprioglio, C. M. Wolff, J. A. Márquez, J. Nordmann, S. Zhang, D. Rothhardt, U. Hörmann, Y. Amir, A. Redinger, L. Kegelmann, F. Zu, S. Albrecht, N. Koch, T. Kirchartz, M. Saliba, T. Unold and D. Neher, Energy Environ. Sci., 2019, 12, 2778–2788 RSC.
- M. Li, M. Liu, F. Qi, F. R. Lin and A. K. Y. Jen, Chem. Rev., 2024, 124, 2138–2204 CrossRef CAS PubMed.
- X. Yu, X. Sun, Z. Zhu and Z. A. Li, Angew. Chem., Int. Ed., 2025, 64, e202419608 CrossRef CAS PubMed.
- S. Liu, J. Li, W. Xiao, R. Chen, Z. Sun, Y. Zhang, X. Lei, S. Hu, M. Kober-Czerny, J. Wang, F. Ren, Q. Zhou, H. Raza, Y. Gao, Y. Ji, S. Li, H. Li, L. Qiu, W. Huang, Y. Zhao, B. Xu, Z. Liu, H. J. Snaith, N.-G. Park and W. Chen, Nature, 2024, 632, 536–542 CrossRef PubMed.
- M. Chen, Y. Li, Z. Zeng, M. Liu, T. Du, X. Huang, L. Bi, J. Wang, W. Jiang, Y. An, S.-W. Tsang, J. Yin, S. Wu and A. K. Y. Jen, Energy Environ. Sci., 2024, 17, 9580–9589 RSC.
- M. V. Khenkin, E. A. Katz, A. Abate, G. Bardizza, J. J. Berry, C. Brabec, F. Brunetti, V. Bulović, Q. Burlingame, A. Di Carlo, R. Cheacharoen, Y.-B. Cheng, A. Colsmann, S. Cros, K. Domanski, M. Dusza, C. J. Fell, S. R. Forrest, Y. Galagan, D. Di Girolamo, M. Grätzel, A. Hagfeldt, E. von Hauff, H. Hoppe, J. Kettle, H. Köbler, M. S. Leite, S. Liu, Y.-L. Loo, J. M. Luther, C.-Q. Ma, M. Madsen, M. Manceau, M. Matheron, M. McGehee, R. Meitzner, M. K. Nazeeruddin, A. F. Nogueira, Ç. Odabaşı, A. Osherov, N.-G. Park, M. O. Reese, F. De Rossi, M. Saliba, U. S. Schubert, H. J. Snaith, S. D. Stranks, W. Tress, P. A. Troshin, V. Turkovic, S. Veenstra, I. Visoly-Fisher, A. Walsh, T. Watson, H. Xie, R. Yıldırım, S. M. Zakeeruddin, K. Zhu and M. Lira-Cantu, Nat. Energy, 2020, 5, 35–49 CrossRef.
- R. Azmi, D. S. Utomo, B. Vishal, S. Zhumagali, P. Dally, A. M. Risqi, A. Prasetio, E. Ugur, F. Cao, I. F. Imran, A. A. Said, A. R. Pininti, A. S. Subbiah, E. Aydin, C. Xiao, S. I. Seok and S. De Wolf, Nature, 2024, 628, 93–98 CrossRef CAS PubMed.
- Z. Li, X. Sun, X. Zheng, B. Li, D. Gao, S. Zhang, X. Wu, S. Li, J. Gong, J. M. Luther, Z. A. Li and Z. Zhu, Science, 2023, 382, 284–289 Search PubMed.
- L. Caprile, A. Cossaro, E. Falletta, C. Della Pina, O. Cavalleri, R. Rolandi, S. Terreni, R. Ferrando, M. Rossi, L. Floreano and M. Canepa, Nanoscale, 2012, 4, 7727–7734 Search PubMed.
- S. M. Park, M. Wei, N. Lempesis, W. Yu, T. Hossain, L. Agosta, V. Carnevali, H. R. Atapattu, P. Serles, F. T. Eickemeyer, H. Shin, M. Vafaie, D. Choi, K. Darabi, E. D. Jung, Y. Yang, D. B. Kim, S. M. Zakeeruddin, B. Chen, A. Amassian, T. Filleter, M. G. Kanatzidis, K. R. Graham, L. Xiao, U. Rothlisberger, M. Grätzel and E. H. Sargent, Nature, 2023, 624, 289–294 CrossRef CAS PubMed.
- J. Qi, J. Liu, Y. Ma, Y. Cheng, K. Chen, W. Hu, J. Xiang, X. Wang, J. Zhao, Y. Zhou, A. Mei and H. Han, Adv. Energy Mater., 2024, 14, 2402344 CrossRef CAS.
- F. Du, H. Gu, W. Jiang, W. Yang, Y. Lin, W. Zhu, X. Qin, X. Xie, L. Bu, X. Liu, S. Yang and C. Liang, Adv. Funct. Mater., 2025, 35, 2413281 CrossRef CAS.
- S. Teale, M. Degani, B. Chen, E. H. Sargent and G. Grancini, Nat. Energy, 2024, 9, 779–792 CrossRef CAS.
- Z. Liang, H. Xu, Y. Zhang, G. Liu, S. Chu, Y. Tao, X. Xu, S. Xu, L. Zhang, X. Chen, B. Xu, Z. Xiao, X. Pan and J. Ye, Adv. Mater., 2022, 34, 2110241 CrossRef CAS PubMed.
- M. Lee, L. Wang, D. Zhang, J. Li, J. Kim, J. S. Yun and J. Seidel, Adv. Mater., 2024, 36, 2407291 CrossRef CAS PubMed.
- S. Chen, X. Dai, S. Xu, H. Jiao, L. Zhao and J. Huang, Science, 2021, 373, 902–907 CrossRef CAS PubMed.
- Y. Zhou, E. L. Wong, W. Mróz, G. Folpini, S. Martani, J. Jiménez-López, A. Treglia and A. Petrozza, ACS Energy Lett., 2024, 9, 1666–1673 CrossRef CAS.
- X. Guo, Z. Jia, S. Liu, R. Guo, F. Jiang, Y. Shi, Z. Dong, R. Luo, Y.-D. Wang, Z. Shi, J. Li, J. Chen, L. K. Lee, P. Müller-Buschbaum, D. S. Ginger, D. J. Paterson and Y. Hou, Joule, 2024, 8, 2554–2569 CrossRef CAS.
- Q. Zhou, J. Duan, X. Yang, Y. Duan and Q. Tang, Angew. Chem., Int. Ed., 2020, 59, 21997–22001 CrossRef CAS PubMed.
- G. Li, Y. Hu, M. Li, Y. Tang, Z. Zhang, A. Musiienko, Q. Cao, F. Akhundova, J. Li, K. Prashanthan, F. Yang, P. Janasik, A. N. S. Appiah, S. Trofimov, N. Livakas, S. Zuo, L. Wu, L. Wang, Y. Yang, B. Agyei-Tuffour, R. W. MacQueen, B. Naydenov, T. Unold, E. Unger, E. Aktas, S. Eigler and A. Abate, Angew. Chem., Int. Ed., 2023, 62, e202307395 CrossRef CAS PubMed.
- J. Liang, X. Hu, C. Wang, C. Liang, C. Chen, M. Xiao, J. Li, C. Tao, G. Xing, R. Yu, W. Ke and G. Fang, Joule, 2022, 6, 816–833 Search PubMed.
- J. Qin, X.-K. Liu, C. Yin and F. Gao, Trends Chem., 2021, 3, 34–46 Search PubMed.
- W. Xu, Q. Hu, S. Bai, C. Bao, Y. Miao, Z. Yuan, T. Borzda, A. J. Barker, E. Tyukalova, Z. Hu, M. Kawecki, H. Wang, Z. Yan, X. Liu, X. Shi, K. Uvdal, M. Fahlman, W. Zhang, M. Duchamp, J.-M. Liu, A. Petrozza, J. Wang, L.-M. Liu, W. Huang and F. Gao, Nat. Photonics, 2019, 13, 418–424 CrossRef CAS.
- F. J. Berger, I. Poli, E. Aktas, S. Martani, D. Meggiolaro, L. Gregori, M. D. Albaqami, A. Abate, F. De Angelis and A. Petrozza, ACS Energy Lett., 2023, 8, 3876–3882 Search PubMed.
- H. D. Pham, S. M. Jain, M. Li, S. Manzhos, K. Feron, S. Pitchaimuthu, Z. Liu, N. Motta, H. Wang, J. R. Durrant and P. Sonar, J. Mater. Chem. A, 2019, 7, 5315–5323 Search PubMed.
- L. Huang, H. Cui, W. Zhang, D. Pu, G. Zeng, Y. Liu, S. Zhou, C. Wang, J. Zhou, C. Wang, H. Guan, W. Shen, G. Li, T. Wang, W. Zheng, G. Fang and W. Ke, Adv. Mater., 2023, 35, 2301125 CrossRef CAS PubMed.
- X. Wu, D. Zhang, B. Liu, Y. Wang, X. Wang, Q. Liu, D. Gao, N. Wang, B. Li, L. Wang, Z. Yu, X. Li, S. Xiao, N. Li, M. Stolterfoht, Y.-H. Lin, S. Yang, X. C. Zeng and Z. Zhu, Adv. Mater., 2024, 36, 2410692 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 28 Figures and 2 Tables. See DOI: https://doi.org/10.1039/d5ee01110h |
| This journal is © The Royal Society of Chemistry 2025 |