
Defective 1T-VS2 with fibonacci pattern unlocking high mass-loading and self-charging cathodes for aqueous zinc-ion batteries†
Tao
Li
a,
Xinji
Dong
a,
Hange
Yang
a,
Jianwei
Zhang
b,
Rong
Huang
b,
Zhuoran
Lv
a,
Yueyue
Li
a,
Shicong
Zhang
a,
Fuqiang
Huang
 *a and
Tianquan
Lin
*a
*a and
Tianquan
Lin
*a
aSchool of Materials Science and Engineering, Zhangjiang Institute for Advanced Study (ZIAS), Shanghai Jiao Tong University, Shanghai, P. R. China
bKey Laboratory of Polar Materials and Devices (MOE), Department of Electronics, East China Normal University, Shanghai, China
First published on 5th March 2025
Abstract
Cathode materials that exhibit high capacity, rapid charging, and long lifespan at high mass loading are crucial for the commercialization of aqueous zinc-ion batteries (AZIBs). However, challenges such as sluggish electrochemical kinetics and structural degradation during cycling often lead to low specific capacity and poor cycling stability, especially under high mass loading conditions, hindering their practical application. In this study, we introduce a novel defective 1T-VS2 micro-rose material with a Fibonacci golden pattern structure, engineered to optimize the electrochemical performance of AZIBs. The unique rose-like morphology of the material promotes both a uniform and enriched electric field and concentration distribution, facilitating efficient ion and electron transport. This architecture, combined with abundant sulfur vacancies and vanadium intercalation, enhances structural stability, reduces cation migration barriers, and accelerates electrochemical kinetics. At high mass loading (up to 30 mg cm−2), the defective 1T-VS2 cathode demonstrates excellent capacity retention (220 mA h g−1, 83% retention), remarkable cycling stability (80% retention over 400 cycles at 20 mA cm−2), and superior rate capability. Notably, the material also exhibits outstanding self-charging performance, with a high self-charging efficiency and an impressive self-charging rate, even at a high mass loading of 10 mg cm−2. This work not only underscores the exceptional electrochemical properties of the defective 1T-VS2 cathode but also presents a design strategy that integrates macro-to-micro-scale structural optimization, offering a promising direction for the development of high-performance cathodes in energy storage applications.
Broader contextSafe and reliable batteries are indispensable in modern life, powering everything from smartphones to wearable devices. However, traditional batteries face critical challenges in outdoor environments, particularly when depleted, as they rely on fixed infrastructure for recharging. This limitation can jeopardize safety in emergencies, restricting vital communication when it is needed most. Inspired by the natural Fibonacci patterns found in flowers, we envisioned that precise micro- and nanoscale structural customization could yield a material capable of both efficient electrochemical cycling and self-charging. Here, we present a defective 1T-VS2 micro-rose material, designed as a high mass-loading cathode for aqueous zinc-ion batteries through a multi-scale approach spanning from the macro to atomic level. At the macroscopic scale, the material adopts a rose-like morphology that follows the Fibonacci sequence, generating a uniformly enriched electric field and ion distribution—critical for efficient charge transport. At the microscopic level, sulfur vacancies and vanadium intercalation enhance structural stability, accelerate electron and ion transport, and lower cation migration energy barriers. These structural advantages not only enable outstanding electrochemical performance in traditional charge–discharge processes but also unlock exceptional air-driven self-charging capabilities under extreme conditions. |
Introduction
Aqueous zinc-ion batteries (AZIBs) are promising candidates for large-scale energy storage due to the high theoretical capacity of the zinc anode (820 mA h g−1), combined with inherent safety, cost-effectiveness, and environmental sustainability.1–5 To achieve their commercial viability, it is essential to develop cathode materials that deliver both high capacity and long cycle life. Among existing cathode options, manganese-based compounds are cost-effective and environmentally friendly but suffer from Mn2+ dissolution and structural degradation.6,7 In contrast, vanadium-based compounds offer high capacity and structural versatility but struggle with cycling stability.8–10 Vanadium disulfide (VS2) has emerged as a promising cathode material for AZIBs due to its unique layered structure and favourable electrochemical properties. VS2 exhibits a high theoretical capacity attributed to the multiple oxidation states of vanadium (V4+/V3+). Its two-dimensional layered structure provides open channels for Zn2+ diffusion, promoting fast ion transport and enhanced rate performance. Additionally, the metallic conductivity of VS2 ensures superior electron transport compared to many vanadium oxides (e.g., V2O5).11 Furthermore, VS2 exhibits a suitable oxidation–reduction potential12 that enables spontaneous recharging upon exposure to atmospheric oxygen, a feature beneficial for self-charging AZIBs in energy storage and emergency power supply applications.13 Nonetheless, practical implementation of VS2 remains limited due to sluggish electrochemical kinetics and structural instability. These factors prevent VS2 from achieving optimal capacity and lead to rapid capacity decay throughout the cycling process. which exacerbates as mass loading continue to increase in accordance with commercial requirement (>10 mg cm−2).To address these challenges, extensive efforts have been devoted to macro-scale strategies,14 such as compositing VS2 with conductive materials like carbon-based substances15–17 and MXene,18–20 as well as morphological optimizations.21,22 These approaches have indeed improved electron conductivity and mitigated volume changes during cycling. However, they fail to sufficiently address the lattice distortions caused by repeated Zn2+ insertion/extraction and fall short in enhancing the intrinsic electrochemical kinetics, thus limiting specific capacity and long-term cycling stability, especially under high mass-loading conditions.
Nature often inspires efficient design principles, as seen in the Fibonacci patterns exhibited by sunflower and rose petals.23 These patterns optimize space utilization, enhance light capture, and minimize resource overlap, leading to superior structural stability and efficiency. Additionally, the visually appealing patterns may attract pollinators more effectively, and the optimized growth dynamics ensure minimal overlap, supporting better access to resources like air and nutrients.24
Inspired by these natural structures, we propose a multi-scale design strategy involving atomic, crystal, and microstructural engineering to synthesize defective rose-like 1T-VS2 (Rose D-VS2) with a Fibonacci golden pattern structure as a high mass-loading cathode with self-charging capabilities for AZIBs. As shown in Fig. 1a, the rose-like microstructure follows the Fibonacci spiral, ensuring both uniformly enriched electric field and ion distribution, which facilitates rapid and homogeneous cation insertion/extraction. This structure enables dense electrode stacking while maintaining effective ion transport channels and external energy exchange. Unlike conventional VS2, our defect-engineered VS2 incorporates intercalated vanadium atoms forming strong bonds with sulfur, which stabilize the interlayer structure and preserve lattice integrity during cycling. Simultaneously, the presence of abundant sulfur vacancies and V intercalation synergistically enhances ion/electron transport while reducing cation migration energy barriers. As a result of these structural advantages, the rose-like defective VS2 cathode delivers a high specific capacity of 262 mA h g−1 at 0.2 A g−1 with a mass loading of 10 mg cm−2 and retains 220 mA h g−1 with 83% capacity retention even at a mass loading of 30 mg cm−2. Regardless of the mass loading, the cathode consistently exhibits excellent cycling stability at low current densities of 0.2 A g−1. Impressively, even at an ultra-high current density of 20 mA cm−2, this rose-like VS2 cathode with a mass loading of 20 mg cm−2 achieves an 80% capacity retention over 400 cycles. The energy storage mechanism involves a hybrid Zn2+/H+ co-insertion process. Notably, such cathode also demonstrates efficient self-charging performance under high mass-loading conditions, making it highly suitable for both energy storage and emergency power applications.
 | ||
| Fig. 1 Structure and morphology characterization of the Rose D-VS2. (a) Design concept of defect-rich VS2 micro-rose materials. Inset: The optical photograph of a rose. (b) SEM images illustrating the morphological evolution process of the Rose D-VS2. (c) Cross-section SEM image showing the morphology of the Rose D-VS2. (d) The HADDF-STEM image revealing the atomic structure of the Rose D-VS2. (e) Rietveld refinement of the XRD pattern of the Rose D-VS2. (f) High-resolution V 2p XPS spectrum of the Rose D-VS2. (g) High-resolution S 2p XPS spectrum of the Rose D-VS2. | ||
Results and discussion
Structure and morphology characterization of the Rose D-VS2
The Rose D-VS2 material was synthesized via a hydrothermal reaction followed by thermal treatment in an Ar/H2 atmosphere, with detailed procedures described in the ESI.† To compare the performance of our prepared VS2, we also synthesized VS2 nanocrystals using the same method but with a shorter reaction time. The micro-rose morphology formation of Rose D-VS2 was examined through scanning electron microscopy (SEM). As shown in Fig. 1b and Fig. S1, S2 (ESI†), VS2 nanoparticles progressively evolve from seed-like structures into aesthetically appealing micro-rose formations with a Fibonacci golden spiral as hydrothermal treatment duration increases. Despite its small specific surface area (Fig. S3, ESI†), cross-sectional SEM image (Fig. 1c) reveals non-overlapping VS2 leaves, each fully exposed to the electrolyte, enhancing wettability (Fig. S4, ESI†). This architecture facilitates effective ion transport channels, minimizes cation diffusion distances, and optimizes space for electrochemical reactions. Transmission electron microscopy (TEM) further confirmed the well-crystallized, micron-sized sheets forming the Rose D-VS2 structure (Fig. S5, ESI†). Spherical aberration-corrected TEM (AC-TEM) imaging (Fig. 1d and Fig. S6, ESI†) provided additional insights into the atomic structure, clearly displaying V self-intercalation and S vacancies within the Rose D-VS2.X-ray diffraction (XRD) patterns (Fig. S7, ESI†) confirm that both Rose D-VS2 and nano VS2 exhibit characteristic peaks consistent with 1T-VS2 (hexagonal crystal structure, P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1). In contrast, inductively coupled plasma (ICP) results (Fig. S8, ESI†) reveal a vanadium-to-sulfur ratio of 5
m1). In contrast, inductively coupled plasma (ICP) results (Fig. S8, ESI†) reveal a vanadium-to-sulfur ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 in the Rose D-VS2, denoted as V4+1S8+1, indicating the presence of intrinsic structural defects. Based on these results, Rietveld profile refinement of the XRD data for the Rose D-VS2 material was conducted. The refinements (Fig. 1e and Fig. S9, ESI†) confirm a single hexagonal crystal phase (Pm1) with lattice parameters a = b = 3.22735 Å and c = 5.74741 Å. To further investigate the chemical structure in greater detail, X-ray photoelectron spectroscopy (XPS) was performed. The V 2p spectrum (Fig. 1f) reveals the presence of V4+ (516.8 eV and 524.5 eV),25 V3+ (514 eV and 522.4 eV), and V2+ (513 eV and 520.8 eV), while the S 2p spectrum shows the presence of S0 (Fig. 1g). The formation of V3+ is attributed to the sulfur vacancies generated during thermal treatment in an Ar/H2 atmosphere and V2+ arises from vanadium self-intercalation. These findings are corroborated by electron paramagnetic resonance (EPR) spectroscopy (Fig. S10, ESI†).25–27
:9 in the Rose D-VS2, denoted as V4+1S8+1, indicating the presence of intrinsic structural defects. Based on these results, Rietveld profile refinement of the XRD data for the Rose D-VS2 material was conducted. The refinements (Fig. 1e and Fig. S9, ESI†) confirm a single hexagonal crystal phase (Pm1) with lattice parameters a = b = 3.22735 Å and c = 5.74741 Å. To further investigate the chemical structure in greater detail, X-ray photoelectron spectroscopy (XPS) was performed. The V 2p spectrum (Fig. 1f) reveals the presence of V4+ (516.8 eV and 524.5 eV),25 V3+ (514 eV and 522.4 eV), and V2+ (513 eV and 520.8 eV), while the S 2p spectrum shows the presence of S0 (Fig. 1g). The formation of V3+ is attributed to the sulfur vacancies generated during thermal treatment in an Ar/H2 atmosphere and V2+ arises from vanadium self-intercalation. These findings are corroborated by electron paramagnetic resonance (EPR) spectroscopy (Fig. S10, ESI†).25–27
Based on the above analyses, it can be concluded that during thermal treatment, sulfur atoms migrate from lattice positions, forming S0 and leaving vacancies in their original sites. This disruption weakens vanadium–sulfur bonds, allowing some vanadium atoms to migrate into the interlayer space, where they form new stable V–S bonds. These strong V–S bonds28 play a crucial role in stabilizing the interlayer structure, maintaining crystal integrity during entire cation insertion and extraction processes.
Electrochemical reaction kinetics of the Rose D-VS2
The electrochemical reaction kinetics of the Rose D-VS2 and nano VS2 were systematically investigated, considering their variations in morphology and crystal structure. To explore these differences, we employed COMSOL multiphysics simulations, modelling macrostructures based on the morphologies of both materials and examining their influence on electrochemical performance. As illustrated in Fig. 2a, the Rose D-VS2, with its Fibonacci golden spiral architecture, exhibits a more uniform and denser distribution of both the electric field and concentration field compared to the nano VS2. This enhanced structural uniformity promotes faster and more homogeneous ion and electron transport, contributing to superior energy storage capacity and structural stability during charge–discharge cycles.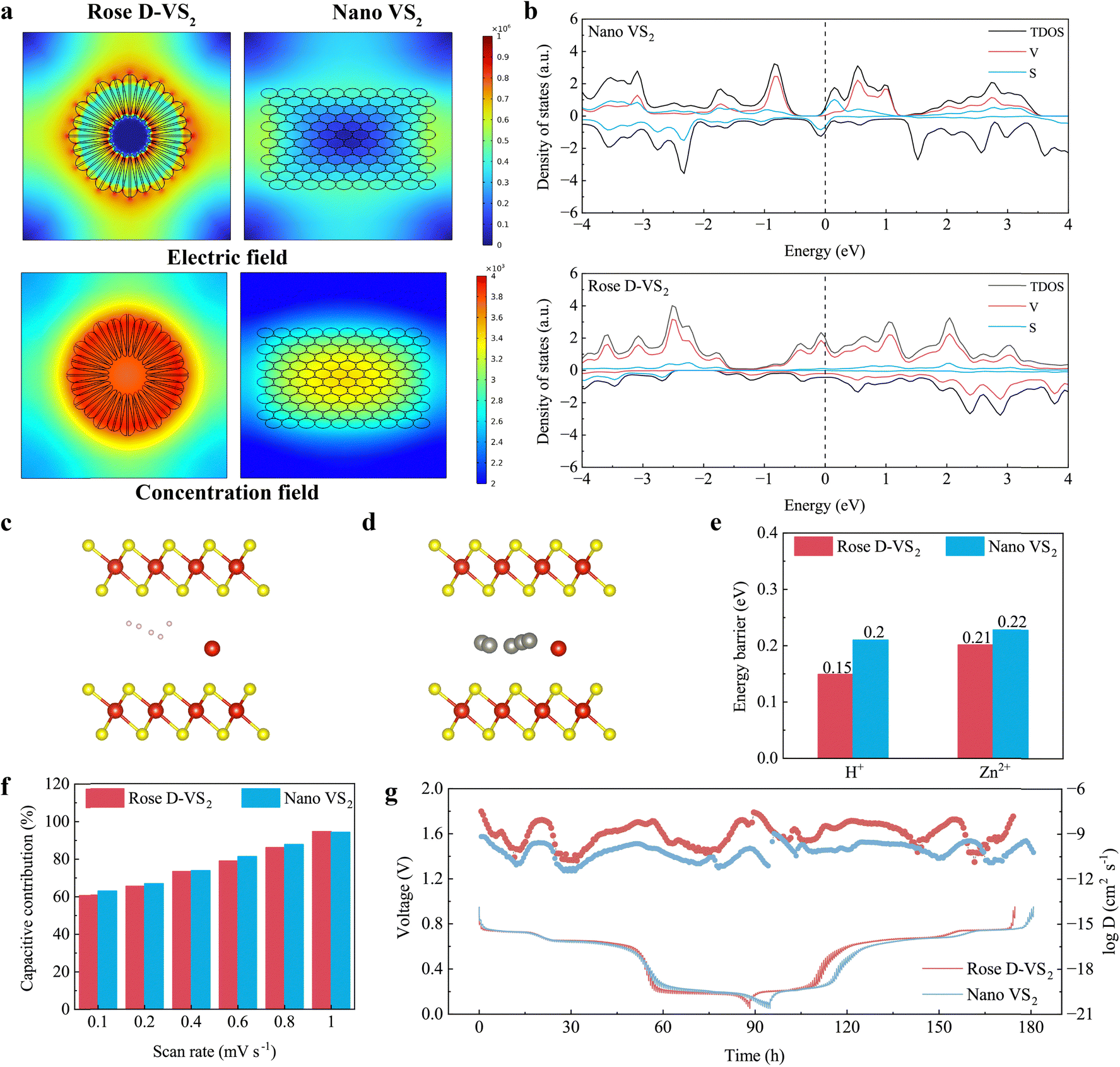 | ||
| Fig. 2 Electrochemical reaction kinetics of the Rose D-VS2. (a) COMSOL Multiphysics simulation illustrating the variations in the electric field and the concentration field across different VS2 materials. (b) Density of states (DOS) of different VS2 materials. (c) The migration pathways of protons in the interlayer of the Rose D-VS2. (d) The migration pathways of Zn2+ ions in the interlayer of the Rose D-VS2. (e) Maximum migration energy barriers for various cations in different VS2 materials. (f) Contribution ratios of capacitive-controlled behaviors at varying scanning rates. The contributions of pseudo-capacitance (k1v) and diffusion (k2v1/2) to the overall capacity are quantified using the equation of i(V) = k1v + k2v1/2, where i(V) is the current at a fixed voltage (V). (g) Charge–discharge GITT profiles at 0.02 A g−1 between 0.05 V and 0.95 V in the third cycle and corresponding cation diffusion coefficients for different VS2 materials. | ||
To gain further insight into the impact of S vacancies and V intercalation on electron and ion transport, density functional theory (DFT) simulations were performed. The density of states (DOS) and band structure results (Fig. 2b and Fig. S11, ESI†) indicate that the Rose D-VS2, with abundant defects, displays increased electronic states at the Fermi level, primarily associated with V 3d orbitals. This increase enhances the electronic conductivity (9300 S m−1) compared to the nano VS2 (38 S m−1), as shown in Fig. S12 (ESI†). The calculated migration pathways of H+ and Zn2+ ions within the interlayer region of different VS2 materials are shown in Fig. 2c and d and Fig. S13 (ESI†). Notably, the calculation results (Fig. 2e and Fig. S14, ESI†) reveal a lower migration energy barrier for the Rose D-VS2 with a significant decrease in the proton migration energy (0.15 eV vs. 0.2 eV), indicating facilitated cation diffusion.
To validate these theoretical findings, cyclic voltammetry (CV) and galvanostatic intermittent titration technique (GITT) measurements were performed. CV curves at various scan rates (Fig. S15, ESI†) demonstrate that the electrochemical reaction is governed by either diffusion-controlled or capacitance-controlled processes, described by the equation ip = avb (logip = b × logv + loga), where ip is the peak current, v is the scan rate, and a and b are adjustable constants.29 The b values for the Rose D-VS2 range between 0.55 and 0.66, signifying a synergistic control of faradaic intercalation and pseudocapacitance mechanisms. Lower b values compared to the nano VS2 suggest a higher contribution from faradaic processes, likely due to the smaller specific surface area and enhanced ion/electron transport efficiency of the Rose D-VS2. This interpretation is supported by the reduced pseudocapacitance contribution observed in the Rose D-VS2 (Fig. 2f).
Further insights were obtained from GITT analysis (Fig. 2g),30 revealing the cation diffusion coefficients (D) for the Rose D-VS2 range from 1.3 × 10−11 cm2 s−1 to 3.05 × 10−8 cm2 s−1, significantly higher than those measured for the nano VS2 (6.41 × 10−10–1.11 × 10−9 cm2 s−1). This enhanced diffusion performance can be attributed to the more uniform concentration field and lower energy barriers for cation migration in the Rose D-VS2. These experimental results align well with theoretical simulations, confirming that the Rose D-VS2 achieves superior electron and ion transport kinetics, thereby enhancing electrochemical performance across both macroscopic and microscopic scales.
Hybrid ion storage mechanism of the Rose D-VS2
To elucidate the energy storage mechanism of the Rose D-VS2 electrode, systematic ex situ characterizations were performed at selected charge/discharge states during the initial cycles (Fig. 3a). Differential capacity (dQ/dV) curves reveal three pairs of reversible redox peaks: 0.68 V/0.76 V (1st pair), 0.6 V/0.68 V (2nd pair), and 0.14 V/0.21 V (3rd pair). Ex situ XRD patterns (Fig. 3b) show no new peaks when discharging the Rose D-VS2 to 0.67 V, indicating that the 1st pair of peaks corresponds to Zn2+ insertion/extraction. However, further discharging to 0.4 V results in the emergence of new peaks at approximately 12.9° and 23°, associated with Zn(OTf)x(OH)y due to H+ intercalation.31 These peaks persist until recharging to 0.7 V, suggesting that the 2nd and 3rd pairs of peaks are influenced by H+ insertion/extraction. Additionally, the peak at 35.7° (for the (112) plane) shifts left during discharge and returns to its original position during charge, reflecting interlayer expansion and contraction caused by cation insertion/extraction. The reversible appearance and disappearance of peaks, coupled with peak shifts, highlight the excellent reversibility of electrochemical reactions.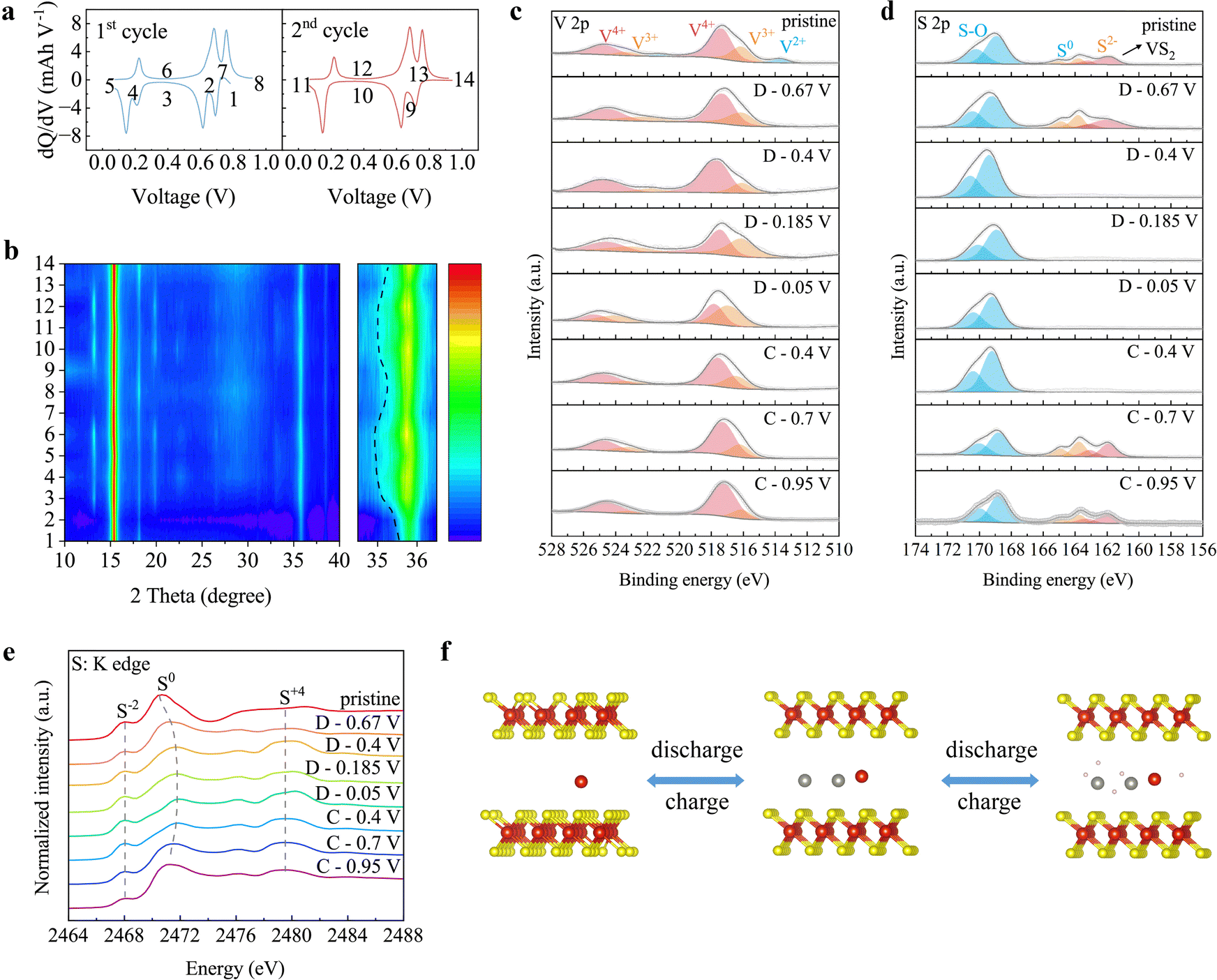 | ||
| Fig. 3 Hybrid ion storage mechanism of the Rose D-VS2. (a) Differential capacity curves for the 1st and 2nd cycle of the Rose D-VS2 cathode. (b) Corresponding ex situ XRD pattern demonstrating the energy storage mechanism of the Rose D-VS2. (c) and (d) Ex situ (c) V 2p spectra and (d) S 2p spectra of the Rose D-VS2 cathodes under varying voltage conditions. (e) Ex situ XAFS spectra of the Rose D-VS2 cathode under different voltages at the S K-edge. (f) A schematic diagram depicts the Zn2+/H+ hybrid ion energy storage mechanism of the Rose D-VS2. The mass loading of the Rose D-VS2 cathodes is 10 mg cm−2, and it is cycled in a 2 M Zn(OTf)2 aqueous electrolytes. | ||
Ex situ V 2p XPS spectra (Fig. 3c) primarily reveal a reversible conversion between V4+ and V3+, indicating a single-electron electrochemical reaction process. Similarly, ex situ S 2p spectra (Fig. 3d) demonstrate that after discharging through the 1st reduction peak, the VS2 signal disappears due to the formation of Zn(OTf)x(OH)y on the electrode surface from H+ insertion. Upon H+ release, Zn(OTf)x(OH)y vanishes, and the VS2 signal reappears, corroborating XRD results. Synchrotron-based near-edge X-ray absorption fine structure (XAFS) spectra (Fig. 3e) further confirm the formation of high-valence S (2479.7 eV) associated with Zn(OTf)x(OH)y during H+ insertion. Within the voltage range of 0.95–0.67 V, the spectra also reveal reversible redox reactions of S0, indicating its contribution to the reversible capacity.32,33
As summarized in Fig. 3f, the energy storage mechanism of Rose D-VS2 involves a Zn2+/H+ hybrid ion process, enabling efficient and reversible charge storage. Within the voltage range of 0.67–0.95 V, Zn2+ ions are inserted and extracted, accompanied by minor S0 redox reactions. In the lower voltage range of 0.05–0.67 V, H+ ions are inserted and extracted, resulting in the reversible formation and dissolution of Zn(OTf)x(OH)y. This dual-ion mechanism, supported by ex situ XRD, XPS, and XAFS analyses, highlights the exceptional electrochemical dynamics of Rose D-VS2, including high reversibility and capacity contributions from both zinc ions and protons.
Electrochemical performance of the Rose D-VS2
The Zn2+/H+ hybrid ion storage mechanism of the Rose D-VS2 enables it to achieve outstanding electrochemical performance under high mass loading conditions (>10 mg cm−2), a critical factor for commercialization. This mechanism ensures efficient charge storage and reversibility, contributing to the material's superior capacity, cycling stability, and self-charging capabilities, making it a promising candidate for advanced energy storage applications. As shown in Fig. 4a and Fig. S16 (ESI†), the Rose D-VS2 cathode with a mass loading of 10 mg cm−2 exhibits exceptional rate performance, achieving specific discharge capacities of 285 mA h g−1 at 0.1 A g−1, 186 mA h g−1 at 1 A g−1 and 121 mA h g−1 at 2 A g−1, outperforming the nano VS2 cathode. Furthermore, the Rose D-VS2 cathode exhibits a superior cycling stability, retaining a specific discharge capacity of 165 mA h g−1 (82% capacity retention) over 200 cycles at 0.5 A g−1, alongside an impressive initial Coulombic efficiency (CE) of 96.5% and a high average CE of 99.8% (Fig. 4b and Fig. S17, ESI†). In contrast, nano VS2 cathode only achieves only 134.4 mA h g−1 (79.8% capacity retention) after 30 cycles, experiencing significant fluctuations after 80 cycles and ultimately failing rapidly. It also exhibits a low initial CE of 76.4% and highly variable CE values throughout its operational lifespan. The Rose D-VS2 outperforms VS2 materials reported in the literature (Table S1, ESI†). The corresponding post-cycling SEM images (Fig. S18, ESI†) reveal that the Rose D-VS2 cathode retains its flower-like morphology, while the nano VS2 structure disintegrates. During cycling, in contrast to the continuously increasing impedance observed in nano VS2, the Rose D-VS2 exhibits a remarkably low and stable impedance throughout (Fig. S19, ESI†), underscoring its interface stability and exceptional electrochemical reaction kinetics during the cycling process. These findings underscore the superiority of the Rose D-VS2 as an advanced material for electrochemical applications.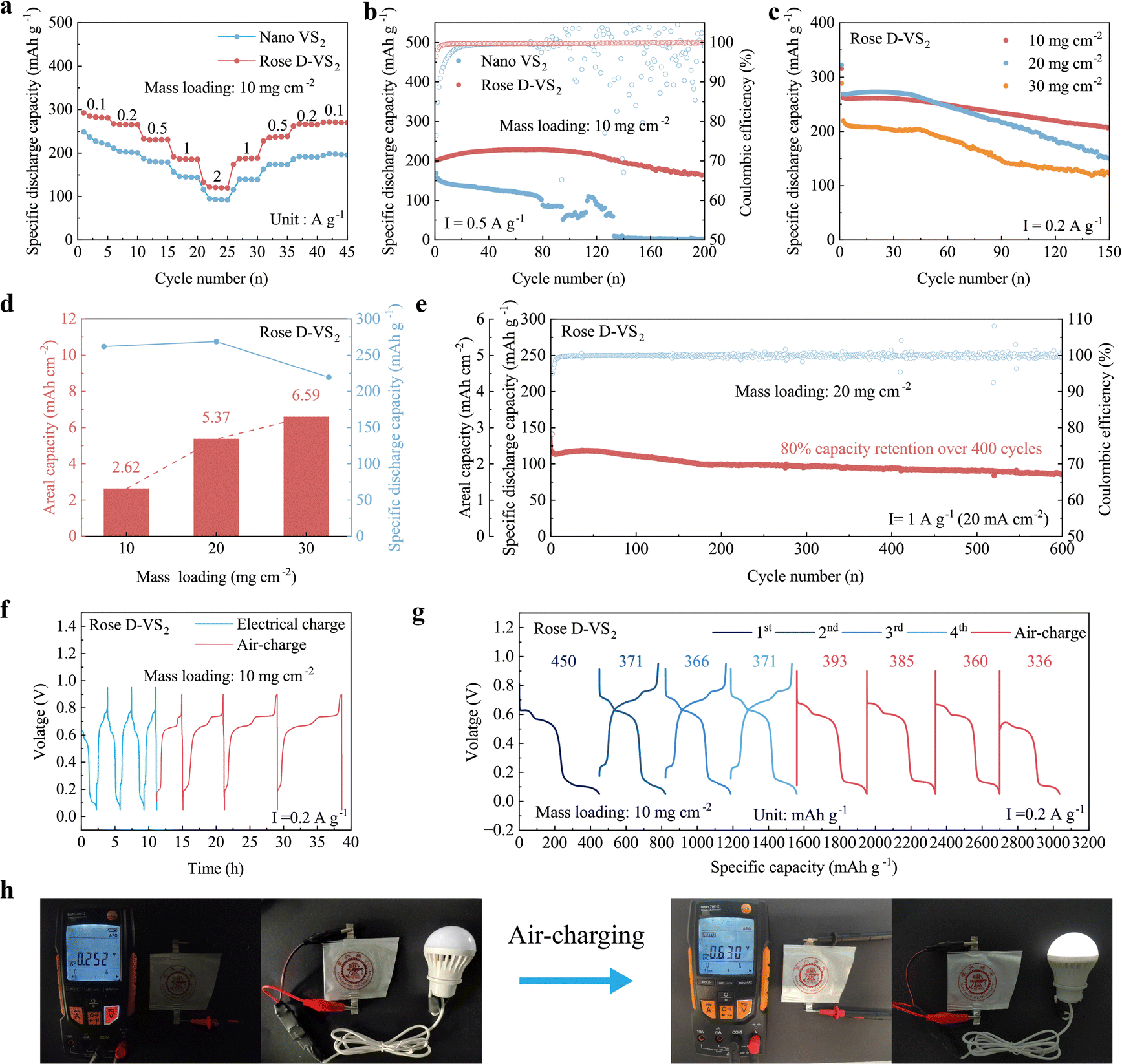 | ||
| Fig. 4 Electrochemical performance of the Rose D-VS2. (a) Rate performance of various VS2 cathodes with a mass loading of 10 mg cm−2. (b) Cycling performance of different VS2 cathodes with a mass loading of 10 mg cm−2. (c) Cycling performance of the Rose D-VS2 cathode under varying mass loadings. (d) The correlation between specific capacity and areal capacity for the Rose D-VS2 cathode across different mass loadings. (e) Long-term cycling stability of the Rose D-VS2 cathode with the mass loading of 20 mg cm−2 at a high current density of 20 mA cm−2. (f) Time–voltage curve showing the air self-charging rate of the Rose D-VS2. (g) Capacity-voltage curve showing the air-charge performance of the Rose D-VS2. (h) Schematic representation of the air-charging process in devices. | ||
Further investigations examined the electrochemical performance of the Rose D-VS2 under higher mass loading conditions. At a mass loading of 10 mg cm−2, the cathode demonstrates remarkable cycling stability, achieving a specific capacity of 206.4 mA h g−1 (2.06 mA h cm−2) over 150 cycles at a low current density of 0.2 A g−1 (Fig. 4c and Fig. S20a, ESI†). Below 20 mg cm−2, specific discharge capacity remains unaffected by mass loading variations, while areal capacity increases linearly with mass loading (Fig. 4d and Fig. S21, ESI†). Notably, at 20 mg cm−2, the cathode demonstrates outstanding cycling stability, retaining 80% capacity over 400 cycles at an ultra-high current density of 20 mA cm−2 (Fig. 4e and Fig. S20b, ESI†). When mass loading increased up to 30 mg cm−2, the cathode achieved an impressive areal capacity of 6.59 mA h cm−2 and retained excellent stability with 3.68 mA h cm−2 over 150 cycles.
In the 2 M Zn(OTf)2 electrolyte (pH = 4.6), the Rose D-VS2 exhibited a charging platform voltage below 0.136 V (νs. SHE), significantly lower than the oxygen reduction potential (O2 + 4H+ → 2H2O, 0.9586 V νs. SHE). This characteristic indicates that in an open system, the dissolution of atmospheric oxygen into an acidic aqueous electrolyte initiates a spontaneous redox reaction at the three-phase interface involving discharged VS2, the electrolyte, and O2—specifically occurring on the cathode surface, thus generating a self-charging effect.34,35
To explore this self-charging capability, a pouch cell with a 10 mg cm−2 Rose D-VS2 cathode was assembled and subjected to air-charging experiments. Following conventional charge–discharge cycles, the cell was exposed to air for self-charging. Remarkably, the Rose D-VS2 with such a high mass loading achieved full charge within 4 hours without external assistance, delivering a specific discharge capacity of 393 mA h g−1. It exhibited a self-charging efficiency  36 exceeding 100% and an average self-charging rate
36 exceeding 100% and an average self-charging rate  of 53.7 mA h g−1 h−1 (Fig. 4f–g). The pouch cell successfully recharged through air exposure, lighting a small bulb again (Fig. 4h). This carefully engineered Rose D-VS2 demonstrates exceptional self-charging capabilities, surpassing previously reported materials (Table S2, ESI†), and holds significant potential for emergency energy storage applications.
of 53.7 mA h g−1 h−1 (Fig. 4f–g). The pouch cell successfully recharged through air exposure, lighting a small bulb again (Fig. 4h). This carefully engineered Rose D-VS2 demonstrates exceptional self-charging capabilities, surpassing previously reported materials (Table S2, ESI†), and holds significant potential for emergency energy storage applications.
Conclusions
In conclusion, we have successfully synthesized a defect-rich 1T-VS2 micro-rose material by employing synergistic structural regulation spanning from macro to micro scales. The rose-like morphology of the VS2 material forms a Fibonacci golden pattern structure, which generates a uniform and concentrated electric and ionic field distribution. This unique configuration facilitates highly uniform and rapid electrochemical processes, thereby enhancing the structural stability of the Rose D-VS2 material during prolonged electrochemical cycling. At the microstructural level, the Rose D-VS2 material features abundant sulfur vacancies and vanadium intercalation. These intrinsic defects significantly improve ion and electron transport, reduce cation migration energy barriers, and bolster overall structural integrity. As a result, the Rose D-VS2 cathode exhibits remarkable electrochemical performance, achieving an impressive capacity retention of 83% (220 mA h g−1) as the mass loading increases from 10 mg cm−2 to 30 mg cm−2. Under high mass loading conditions of 20 mg cm−2, the cathode retains 80% of its capacity over 400 cycles, even at an ultra-high current density of 20 mA cm−2. Furthermore, the favourable redox potential and unique structural attributes of the Rose D-VS2 enable efficient self-charging capabilities with superior self-charging efficiency and fast self-charging rate, particularly under a high mass loading of 10 mg cm−2. Beyond its exceptional electrochemical performance, this work exemplifies a scalable and innovative design approach that integrates structural optimization from macro to micro levels. This strategy not only advances the development of high-performance cathodes for aqueous zinc-ion batteries but also provides a conceptual framework for the future design of next-generation energy storage materials.Author contributions
T. Q. L., F. Q. H., and T. L. conceived the project and designed the experiments. T. L. completed the electrochemical experiment and data analysis. X. J. D. and H. G. Y. prepared and synthesized the electrode materials. J. W. Z. and R. H. characterized the microstructure of the material. Z. R. L and Y. Y. L. characterized the energy storage mechanism of the material. S. C. Z. provided recommendations for separator analysis. T. Q. L., F. Q. H., and T. L. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (Grant No. 2019YFA0210600), the Shanghai Pilot Program for Basic Research-Shanghai Jiao Tong University, and the National Natural Science Foundation of China (Grant No. BC0500463).Notes and references
- J. Song, K. Xu, N. Liu, D. Reed and X. Li, Mater. Today, 2021, 45, 191–212 CrossRef CAS.
- Y. Shang and D. Kundu, Joule, 2023, 7, 244–250 CrossRef.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- S. W. D. Gourley, R. Brown, B. D. Adams and D. Higgins, Joule, 2023, 7, 1415–1436 CrossRef CAS.
- Y. Xu, T. Li, S. Zhang, Y. Shen, F. Huang and T. Lin, J. Mater. Chem. A, 2024, 12, 8254–8261 RSC.
- Y. Zhang, S. Zhang, Z. Chen, T. Li, Y. Zhao, F. Huang and T. Lin, J. Mater. Chem. A, 2022, 10, 9402–9407 Search PubMed.
- S. Zeng, W. Xu, D. Zheng, H. Zhang, F. Wang, X. Liu and X. Lu, J. Mater. Chem. A, 2021, 9, 5053–5059 RSC.
- X. Dou, X. Xie, S. Liang and G. Fang, Sci. Bull., 2024, 69, 833–845 CrossRef CAS.
- T. Zhou, L. Xie, Q. Han, X. Qiu, Y. Xiao, X. Yang, X. Liu, S. Yang, L. Zhu and X. Cao, Coord. Chem. Rev., 2024, 498, 215461 CrossRef CAS.
- X. Chen, H. Zhang, J.-H. Liu, Y. Gao, X. Cao, C. Zhan, Y. Wang, S. Wang, S.-L. Chou, S.-X. Dou and D. Cao, Energy Storage Mater., 2022, 50, 21–46 CrossRef CAS.
- P. He, M. Yan, G. Zhang, R. Sun, L. Chen, Q. An and L. Mai, Adv. Energy Mater., 2017, 7, 1601920 CrossRef.
- Y. Zhang, F. Wan, S. Huang, S. Wang, Z. Niu and J. Chen, Nat. Commun., 2020, 11, 2199 CrossRef CAS PubMed.
- C. Liu, W. Xu, C. Mei, M. Li, W. Chen, S. Hong, W. Y. Kim, S. Y. Lee and Q. Wu, Adv. Energy Mater., 2021, 11, 2003902 CrossRef CAS.
- T. Jiao, Q. Yang, S. Wu, Z. Wang, D. Chen, D. Shen, B. Liu, J. Cheng, H. Li, L. Ma, C. Zhi and W. Zhang, J. Mater. Chem. A, 2019, 7, 16330–16338 RSC.
- J. Liu, W. Peng, Y. Li, F. Zhang and X. Fan, J. Mater. Chem. C, 2021, 9, 6308–6315 RSC.
- Y. Mao, B. Zhao, J. Bai, H. Ma, P. Wang, W. Li, K. Xiao, S. Wang, X. Zhu and Y. Sun, Small, 2023, 19, 2207998 CrossRef CAS PubMed.
- T. Chen, X. Zhu, X. Chen, Q. Zhang, Y. Li, W. Peng, F. Zhang and X. Fan, J. Power Sources, 2020, 477, 228652 CrossRef CAS.
- P. Meng, W. Wang, J. Shang, P. Liu, H. Xu, Q. Wang, S. Wang, F. Wang and X. Wang, Small Methods, 2023, 7, 2201471 CrossRef CAS PubMed.
- L. Zhang, Y. Li, X. Liu, R. Yang, J. Qiu, J. Xu, B. Lu, J. Rosen, L. Qin and J. Jiang, Adv. Sci., 2024, 11, 2401252 CrossRef CAS PubMed.
- Y. Mao, J. Bai, S. Lin, P. Wang, W. Li, K. Xiao, S. Wang, X. Zhu, B. Zhao and Y. Sun, Small, 2023, 20, 2306615 CrossRef PubMed.
- H. Sun, L. Yang, E. Hu, M. Feng, C. Fan, W. Wang, H. Li, X. Wang and Z. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 40247–40256 CrossRef CAS PubMed.
- T. Wang, W. Gao, Y. Zhao, S. Wang and W. Huang, J. Mater. Sci. Technol., 2024, 173, 107–113 CrossRef CAS.
- S. Ghoddousi, M. Mohammadnejad, M. Safarabadi and M. Haghighi-Yazdi, Int. J. Mech. Sci., 2025, 285, 109853 CrossRef.
- L. B. Grassi, D. P. A. Marins, J. F. Paim, L. B. Palaoro, M. E. V. Segatto and M. H. M. Paiva, Renewable Energy, 2024, 229, 120646 CrossRef.
- Y. Zhao, D. Yang, T. He, J. Li, L. Wei, D. Wang, Y. Wang, X. Wang, G. Chen and Y. Wei, Chem. Eng. J., 2021, 421, 129715 CrossRef CAS.
- Q. Li, Y. Guo, Y. Tian, W. Liu and K. Chu, J. Mater. Chem. A, 2020, 8, 16195–16202 RSC.
- L. He, X. Zhang, D. Yang, J. Li, M. Wang, S. Liu, J. Qiu, T. Ma, J. Ba, Y. Wang and Y. Wei, Nano Lett., 2023, 23, 7411–7418 CrossRef CAS.
- T. P. Owzarski and H. F. Franzen, J. Chem. Phys., 1974, 60, 1113–1117 CrossRef CAS.
- L. Yang, Y.-J. Zhu, F. Zeng, H.-P. Yu, L.-Y. Dong, J. Tao, G. He and H. Li, Energy Storage Mater., 2024, 65, 103162 CrossRef.
- K. Zhu, S. Wei, H. Shou, F. Shen, S. Chen, P. Zhang, C. Wang, Y. Cao, X. Guo, M. Luo, H. Zhang, B. Ye, X. Wu, L. He and L. Song, Nat. Commun., 2021, 12, 6878 CrossRef CAS PubMed.
- Y. Dai, J. Li, L. Chen, K. Le, Z. Cai, Q. An, L. Zhang and L. Mai, ACS Energy Lett., 2021, 6, 684–686 CrossRef CAS.
- H. Li, R. Meng, Y. Guo, B. Chen, Y. Jiao, C. Ye, Y. Long, A. Tadich, Q.-H. Yang, M. Jaroniec and S.-Z. Qiao, Nat. Commun., 2021, 12, 5714 CrossRef CAS PubMed.
- Y. Feng, X. Yu, B. Wang, T. Zhang, W. Zhou, Y. Wang, X. Li, J. Zhao, J. Zhao, W. Li, C. Ma, D. Zhao and D. Chao, Angew. Chem., Int. Ed., 2024, 137(8), e202420514 CrossRef.
- Y. Zhang, F. Wan, S. Huang, S. Wang, Z. Niu and J. Chen, Nat. Commun., 2020, 11, 2199 CrossRef CAS PubMed.
- L. Zhong, J. Li, C. Liu, L. Fang, Z. Yuan, D. Yu and X. Chen, Adv. Funct. Mater., 2023, 33, 2215133 CrossRef CAS.
- L. Zhong, C. Wang, J. He, Z. lin, X. Yang, R. Li, S. Zhan, L. Zhao, D. Wu, H. Chen, Z. Tang, C. Zhi and H. Lv, Adv. Mater., 2024, 36, 2470209 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00612k |
| This journal is © The Royal Society of Chemistry 2025 |