
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-site passivation by heterocycle functionalized amidinium cations toward high-performance inverted perovskite solar cells and modules†
Menghan
Lu‡
a,
Jike
Ding‡
a,
Quanxing
Ma‡
a,
Zuolin
Zhang
a,
Mengjia
Li
a,
Wenhuan
Gao
a,
Wenlong
Mo
 bc,
Boxue
Zhang
d,
Thierry
Pauporté
d,
Jiajia
Zhang
*e,
Yang
Wang
*f,
Jian-Xin
Tang
*g,
Jiangzhao
Chen
*h and
Cong
Chen
*ag
bc,
Boxue
Zhang
d,
Thierry
Pauporté
d,
Jiajia
Zhang
*e,
Yang
Wang
*f,
Jian-Xin
Tang
*g,
Jiangzhao
Chen
*h and
Cong
Chen
*ag
aState Key Laboratory of Reliability and Intelligence of Electrical Equipment, School of Materials Science and Engineering, Hebei University of Technology, Tianjin 300401, China. E-mail: chencong@hebut.edu.cn
bCollege of Chemical and Textile Engineering, Xinjiang University of Science & Technology, Korla, Xinjiang 841000, China
cState Key Laboratory of Chemistry and Utilization of Carbon Based Energy Resources, College of Chemical Engineering and Technology, Xinjiang University, Urumqi 830017, China
dChimie ParisTech, PSL Research University, CNRS, Institut de Recherche de Chimie Paris (IRCP), UMR8247, 11 rue P. et M. Curie, Paris F-75005, France
eAnhui Provincial Key Laboratory of Green Carbon Chemistry, College of Chemistry and Materials Engineering, Fuyang Normal University, Fuyang 236037, China. E-mail: jjzh@mail.ustc.edu.cn
fCAS Key Laboratory of Green Printing, Beijing National Laboratory for Molecular Science (BNLMS), Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: wangyang@iccas.ac.cn
gMacao Institute of Materials Science and Engineering (MIMSE), Faculty of Innovation Engineering, Macau University of Science and Technology, Taipa, Macao 999078, China. E-mail: jxtang@must.edu.mo
hFaculty of Materials Science and Engineering, Kunming University of Science and Technology, Kunming 650093, China. E-mail: jzchen@kust.edu.cn
First published on 14th May 2025
Abstract
Presently, most high-efficiency inverted perovskite solar cells (PSCs) are fabricated using toxic antisolvent and in a nitrogen-filled glovebox, which results in increased cost, reduced reproducibility, and incompatibility with large-area modules. In addition, interfacial trap-assisted nonradiative recombination impedes the further advancement of power conversion efficiency (PCE) and long-term operational stability of p–i–n inverted PSCs. Herein, we report a dual-site passivation of anionic and cationic defects through heterocycle functionalized amidinium cations, which stabilizes the interface between perovskite films and electron transport layers and minimizes interfacial nonradiative recombination loss. The dual-site passivation of amidinium cations is accomplished through precisely modulating the distance between two anchoring sites and spatial conformation. The dual-site passivation endows pyridine-functionalized amidinium salt 4-amidinopyridinium chloride (APCl) with an exceptional defect passivation ability. The APCl modulation enables high-efficiency inverted PSCs with a champion PCE of 26.83% (certified steady-state PCE of 26.32%), which is the best PCE ever reported for PSCs fabricated based on vacuum flash and in ambient air. The APCl-modulated devices could retain 95.8% of their initial performance after 2000 h of continuous maximum power point tracking. Moreover, the high-efficiency large-area module with a PCE of 19.83% (aperture area of 40.1 cm2) is obtained by this dual-site passivation technique of amidinium cations.
Broader contextThe development of high-efficiency and stable perovskite solar cells (PSCs) represents a critical step toward achieving widespread adoption of renewable energy technologies. This study addresses key challenges in the fabrication of inverted PSCs, particularly the reduction of interfacial defects that hinder power conversion efficiency (PCE) and stability. By introducing dual-site passivation using heterocycle-functionalized amidinium cations, the research highlights a novel strategy for simultaneously passivating anionic and cationic defects at the perovskite interface. The innovation enables record-setting PCE values of 26.83% for devices fabricated in ambient air, along with enhanced operational stability and scalability for large-area modules. These advancements provide a scalable and environmentally friendly pathway for PSC manufacturing, aligning with global efforts to transition to sustainable energy solutions. The demonstrated techniques show promise for accelerating the commercial deployment of perovskite-based photovoltaics, offering a compelling solution to current energy and environmental challenges. |
Introduction
Organic–inorganic metal halide perovskite solar cells (PSCs) are one of the foremost contenders for future photovoltaics due to their low cost and high power conversion efficiency (PCE).1–8 However, most high-efficiency PSCs are fabricated using toxic antisolvent and in a nitrogen-filled glovebox, which results in increased costs, reduced reproducibility, and incompatibility with large-area modules.1,3,5,9–11 To overcome the above problems, PSCs prepared using vacuum flash and/or in ambient air are preferred.12–15 In recent years, although p–i–n inverted PSCs have demonstrated significant advances in operational stability and PCE,1,3,5,9,13,16,17 maximizing their potential is still highly imperative from the perspective of science and industrial production. On the one hand, the PCEs of large-area and small-area devices need to be further increased to narrow the gap with a theoretical limit value. On the other hand, the commercial deployment of perovskite photovoltaics is still hindered by its poor long-term operational stability.18,19 Unlike perovskite oxides, relatively weak chemical interactions exist in organic–inorganic hybrid metal halide perovskites, which results in the formation of a high concentration of defects in the final perovskite films during rapid crystallization and high-temperature annealing.20–22 Defects in perovskite films are mainly concentrated at surfaces and interfaces, with densities approximately 100 times higher than that in the bulk.23,24 The defects at the surface and interface of perovskite films can not only act as carrier recombination centers but also deteriorate device longevity. Moreover, the defects provide pathways for ion migration, and the external stimulus (e.g., water, oxygen, and heat) would accelerate ion migration.14,25–27 The intrinsic instability induced by trap states poses enormous challenges for realizing long-term operationally stable PSCs because it cannot be addressed through encapsulation technology.It is well-known that passivating defects at the surface and interface through interface engineering is a popular and effective approach to enhance device PCE and durability.21 To heal interfacial defects, various passivators have been developed, primarily including Lewis acids,28 Lewis bases,5,29,30 wide bandgap tunnelling layers,31,32 low-dimensional perovskites,16,33,34 and organic/inorganic salts.27,35–37 Among them, organic or inorganic salts exhibit immense potential on account of their ability to simultaneously passivate positively and negatively charged defects within perovskite films.11,12,27,38 The rational design of organic cations and anions is of crucial importance for more efficient organic salt passivators. Since chloridion (Cl−) has been certified to be very effective in passivating the defects at grain boundaries and interfaces,39,40 the design and optimization of organic cations are critical in developing excellent passivating agents. The presently reported organic cations mainly include ammonium38 and amidine11 cations. Recently, Sargent et al. revealed that amidinium cations have much better thermal stability than ammonium cations because of much-suppressed deprotonation, which resulted in exceptional operational stability.11 Consequently, the combination of amidinium cations with Cl− anions is highly desirable to passivate interfacial defects. However, most cations presently reported can only passivate negatively charged defects (e.g., FA+ vacancies) because of a very limited number of active sites.41,42 Developing organic cations, which can synchronously passivate different charged defects, is still an enormous puzzle. One feasible and effective method should be to employ heteroatoms (e.g., N, O and S) or functional groups with strong coordination capacity to functionalize organic cations because electron-donating heteroatom or functional groups can passivate positively charged defects (like halide vacancies and undercoordinated Pb2+) and the cations can passivate negatively charged defects (like FA+ vacancies). However, the orientation, arrangement and spatial conformation of organic cations determine whether low-dimensional perovskites can form at the surface of 3D perovskite films.12 This indicates that appropriate orientation and spatial conformation of functionalized organic cations are required to ensure successful dual-site anchoring of organic cations at the surface of perovskites instead of forming low-dimensional perovskites.
Perovskite film deposition under ambient conditions often leads to a higher density of interfacial anionic (I− vacancies) and cationic (FA+ vacancies) defects,43,44 resulting in increased non-radiative recombination. Unlike conventional glovebox-based fabrication using antisolvent methods, our approach addresses this issue by integrating a dual-site passivation strategy that compensates for these interfacial defects. The dual-site anchoring mechanism of heterocycle-functionalized amidinium cations allows for simultaneous passivation of anionic and cationic defects, effectively mitigating the detrimental effects caused by ambient fabrication conditions.
In this work, we propose a dual-site anchoring strategy through heterocycle functionalized amidinium cations, which synchronously passivated FA+ and I− vacancies. It was uncovered that appropriate orientation and spatial conformation of organic amidinium cations are necessary for dual-site passivation. Among investigated passivators, the optimal device performance was accomplished after 4-amidinopyridinium chloride (APCl) treatment, which is due to horizontal binding and dual-site anchoring induced by the appropriate distance between two binding functional groups. The APCl-modulated inverted PSCs and modules achieved a PCE of 26.83% and 19.83% (aperture area of 40.1 cm2), respectively. In particular, a certified PCE value of 26.32% is the highest steady-state PCE ever reported for PSCs fabricated based on vacuum flash evaporation and in ambient air. The APCl passivation also resulted in excellent device light stability, maintaining 95.8% of their initial performance after 2000 h of continuous maximum power point tracking (MPPT). This work provides insights into strengthening the defect passivation effect of organic cations by dual-site anchoring via rational engineering of the functional group and spatial conformation.
Results
Theoretical investigation on the interaction between organic cations and perovskite
In this study, a perovskite precursor solution was spin-coated onto ITO/NiOx/Me-4PACz films without using antisolvent but employing vacuum flash technology for perovskite crystallization. Three kinds of functionalized amidine salts, including benzamidine hydrochloride (phFACl), APCl, and 2-thiazolecarboxamidine hydrochloride (TFACl), were utilized to treat the surface of perovskite films in inverted PSCs. These salts have the same Cl− anions but different organic amidinium cations. Since Cl− anions have been certified to be effective defect passivators,39,40 the present work will focus on modulating and optimizing organic cations. To unlock the full potential of the defect passivation of organic cations and achieve dual-site anchoring, the functionalization by functional groups is essential. The phFA+, AP+ and TFA+ were obtained by using a benzene ring, pyridine ring and thiazole ring to functionalize FA+, respectively. Fig. 1a–c show the chemical structures and electrostatic potential (ESP) of three sorts of cations. Obviously, the positive charge mainly concentrates on the amidinium cation section while the negative charge is distributed on N atoms at pyridine and thiazole rings, suggesting that pyridine and thiazole rings show Lewis base properties. Compared with the benzene ring, it was speculated that N atoms at pyridine and thiazole rings could heal undercoordinated Pb2+ defects via coordination bonding. It was reported that pyridine and thiazole heterocycles can effectively passivate undercoordinated Pb2+ defects.45–47 We conjectured that dual-site binding can be realized for AP+ and TFA+, which will be systematically studied by theoretical and experimental evidence. | ||
| Fig. 1 Theoretical investigation on the interaction between organic cations and perovskite. (a)–(c) The chemical structures and electrostatic potential of (a) phFA+, (b) AP+ and (c) TFA+. (d)–(f) The binding energy of (d) phFA+, (e) AP+ and (f) TFA+ with FAPbI3 containing VI and VFA defects in a parallel configuration. (g) The summarized binding energy values of phFA+, AP+ and TFA+ with FAPbI3 containing VFA and VI simultaneously. (h) Distance between the amidinium cation and N atom on the pyridine ring in AP+ (∼4.655 Å). (i) Distance between the amidinium cation and N atom on the thiazole ring in TFA+ (∼2.717 Å). (j) Schematic diagram of the dual-site passivation of AP+ for perovskite surfaces containing VFA and VI. | ||
Density functional theory (DFT) simulations were performed to explore the interaction of three organic amidinium cations with perovskite FAPbI3 containing both VFA and VI defects. The three organic cations were positioned on the perovskite surface in a vertical or parallel orientation configuration. We then calculated the binding energies (Eb) between organic cations and perovskite. As exhibited in Fig. 1d–g and Fig. S1 (ESI†), for the parallel orientation configuration, phFA+, AP+, and TFA+ showed an Eb of −1.91 eV, −2.22 eV and −1.99 eV, respectively. For the vertical orientation configuration, the Eb was −1.65 eV, −1.48 eV, and −1.35 eV, respectively. This indicates that regardless of the organic cation type, the parallel orientation exhibited larger binding energy and stronger interaction with perovskite as compared to the vertical orientation. In other words, the former is more thermodynamically advantageous than the latter. This verified that benzene, pyridine, and thiazole rings on the other end of the organic cations participated in the chemical interaction with perovskites.
Compared with phFA+, AP+ and TFA+ cations possessed higher binding energy, which could be attributed to the coordination interaction of pyridine or thiazole heterocycles with positively charged undercoordinated Pb2+ and/or iodide vacancy defects. Interestingly, we observed that AP+ exhibited a much larger binding energy with the perovskite surface than TFA+. This can be attributed to the appropriate distance of ∼4.7 Å between the –FA+ group and N atom on the pyridine ring in AP+ (Fig. 1h), which matches well with the distance of ∼4.5 Å between VFA and VI on the perovskite surface. In contrast, the distance between the –FA+ group and the N atom on the thiazole ring in TFA+ (∼2.7 Å) is much shorter than the distance of ∼4.5 Å between VFA and VI (Fig. 1i). This implies that dual-site anchoring is feasible for AP+ but it is not allowed for TFA+ from the perspective of spatial conformation and steric hindrance. As illustrated in Fig. 1j, the amidinium cation at one end of AP+ was bonded at the FA+ vacancy via hydrogen bonding and ionic bonding while the N atom on the pyridine ring at the other end of AP+ was chemically anchored at undercoordinated Pb2+ and/or I− vacancy via coordination bonding. It is concluded that AP+ can effectively passivate the FA+ vacancy and undercoordinated Pb2+ and/or I− vacancy through dual-site chemical anchoring.
Experimental investigation on the interaction between organic cations and perovskite
To experimentally confirm the chemical interaction between perovskite and organic cations, Fourier-transform infrared spectra (FTIR), nuclear magnetic resonance (NMR) and X-ray photoelectron spectroscopy (XPS) were performed. As shown from the FTIR in Fig. 2a, after mixing phFACl with PbI2, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration was shifted from 1685.2 cm−1 to 1673.3 cm−1. For APCl, the CN bond of the FA group shifts from 1691.3 cm−1 to 1671.6 cm−1 with an offset of 19.7 cm−1. Meanwhile, the CN bond in pyridine shifts from 1654.6 cm−1 to 1638.9 cm−1, with an offset of 15.7 cm−1 (Fig. 2b). When mixing TFACl with PbI2, we can observe that the CN stretching vibration of the FA group was shifted from 1683.6 cm−1 to 1670.0 cm−1 with an offset of 13.6 cm−1 (Fig. 2c). These results revealed that CN in three ammonium salts can bond with Pb2+. Moreover, after mixing the TFACl with PbI2, the C–S stretching vibration peak located near 689.4 cm−1 was shifted to 759.8 cm−1, indicating the interaction between S at the thiazole ring and Pb2+. Additionally, we investigated the interactions of phFACl, APCl, and TFACl with the perovskite film (Fig. 2d). The phFACl, APCl, and TFACl modulated perovskite film exhibited the CN bond shifted from 1711.5 cm−1 to 1707.5 cm−1, 1701.5 cm−1, and 1710.5 cm−1. The largest displacement in the APCl modulated perovskite film indicates the strongest binding ability for trap defects. Using 1H NMR spectroscopy, we demonstrated the strong NH⋯I hydrogen bonding interactions between –C(NH2)2+ on three organic cations and PbI2 (Fig. 2e–g). For all salts, the 1H NMR peak of –C(NH2)2+ was moved toward a low chemical shift, confirming NH⋯I hydrogen bonding interactions of –C(NH2)2+ with PbI2.48
N stretching vibration was shifted from 1685.2 cm−1 to 1673.3 cm−1. For APCl, the CN bond of the FA group shifts from 1691.3 cm−1 to 1671.6 cm−1 with an offset of 19.7 cm−1. Meanwhile, the CN bond in pyridine shifts from 1654.6 cm−1 to 1638.9 cm−1, with an offset of 15.7 cm−1 (Fig. 2b). When mixing TFACl with PbI2, we can observe that the CN stretching vibration of the FA group was shifted from 1683.6 cm−1 to 1670.0 cm−1 with an offset of 13.6 cm−1 (Fig. 2c). These results revealed that CN in three ammonium salts can bond with Pb2+. Moreover, after mixing the TFACl with PbI2, the C–S stretching vibration peak located near 689.4 cm−1 was shifted to 759.8 cm−1, indicating the interaction between S at the thiazole ring and Pb2+. Additionally, we investigated the interactions of phFACl, APCl, and TFACl with the perovskite film (Fig. 2d). The phFACl, APCl, and TFACl modulated perovskite film exhibited the CN bond shifted from 1711.5 cm−1 to 1707.5 cm−1, 1701.5 cm−1, and 1710.5 cm−1. The largest displacement in the APCl modulated perovskite film indicates the strongest binding ability for trap defects. Using 1H NMR spectroscopy, we demonstrated the strong NH⋯I hydrogen bonding interactions between –C(NH2)2+ on three organic cations and PbI2 (Fig. 2e–g). For all salts, the 1H NMR peak of –C(NH2)2+ was moved toward a low chemical shift, confirming NH⋯I hydrogen bonding interactions of –C(NH2)2+ with PbI2.48
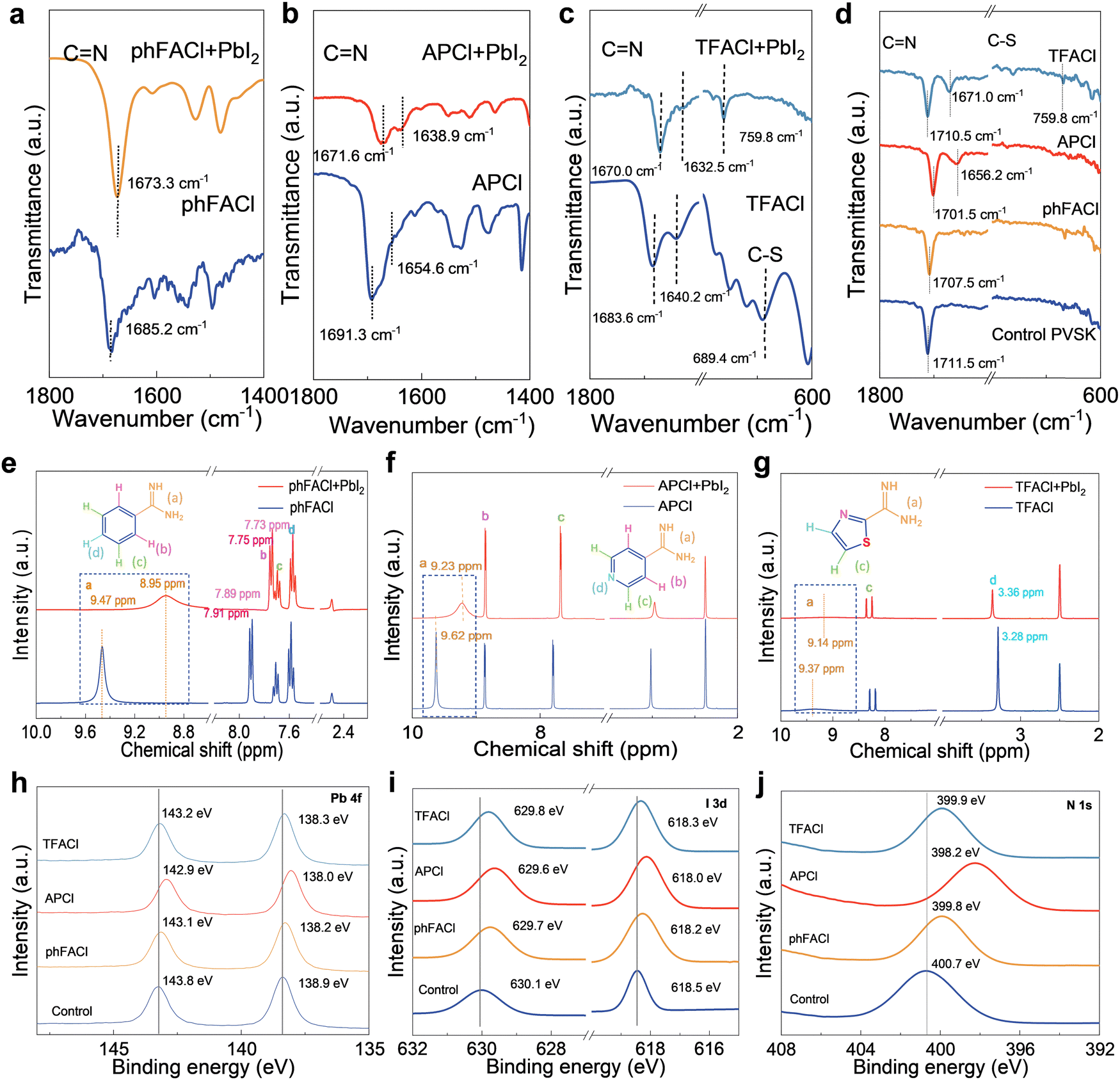 | ||
| Fig. 2 Characterization of the interaction between organic cations and perovskite. (a) FTIR of bare phFACl powder and phFACl (5 mg ml−1 in IPA) modulated PbI2 films. (b) FTIR of bare APCl powder and APCl (5 mg ml−1 in IPA) modulated PbI2 films. (c) FTIR of bare TFACl powder and TFACl (5 mg ml−1 in IPA) modulated PbI2 films. (d) FTIR of the control, phFACl, APCl, and TFACl-modulated perovskite films. (e) 1H NMR spectra of bare phFACl and phFACl + PbI2. (f) 1H NMR spectra of bare APCl and APCl + PbI2. (g) 1H NMR spectra of bare TFACl and TFACl + PbI2. XPS spectra of Pb 4f (h), I 3d (i), and N 1s (j) for the control, phFACl, APCl, and TFACl modulated perovskite films. | ||
Fig. 2h–j show the XPS spectra of the control and modulated perovskite films. As shown in Fig. 2h, the control film exhibited Pb 4f5/2 and Pb 4f7/2 peaks at 143.8 and 138.9 eV, respectively. After modulation with phFACl, APCl, and TFACl, these peaks shifted to lower binding energies, which confirmed the interaction between organic salts and perovskite. On the one hand, the Cl− in these organic salts would interact with undercoordinated Pb2+ and/or halide vacancy defects. On the other hand, the cations in three salts would interact with FA+ vacancy by hydrogen bonding and ionic bonding. The above simultaneous interaction of anion and cation resulted in the shift of Pb 4f peaks. It can be easily found that APCl-modulated perovskite film showed the largest shift of Pb 4f peaks among all investigated samples, which should be put down to dual-site binding resulting from the interaction between the N atom on pyridine and undercoordinated Pb2+ and/or I− vacancy. Similar to Pb 4f peaks, the greatest shift in I 3d peaks was also observed for the APCl-modulated film among all samples (Fig. 2i), demonstrating its strongest interaction with perovskite because of dual-site chemical anchoring of pyridine-functionalized organic cations with appropriate spatial conformation. From the XPS spectrum of N 1s (Fig. 2j), the APCl-modulated perovskite film has the largest displacement (from 400.7 eV to 398.2 eV), indicating a strong interaction between N and Pb2+ in AP+. Although N also exists in TFACl, its bonding ability with Pb2+ is weakened due to inappropriate steric hindrance. Specifically, the displacement of N 1s of the TFACl and phFACl modulated perovskite thin films is only from 400.7 eV to 399.9 eV and 399.8 eV, respectively. This effect further confirms the necessity of appropriate spatial conformation for achieving ideal dual-site passivation.
Investigation of crystal phase after organic salt modulation
From the perspective of crystallinity and crystal structure, Fig. 3a–d display the grazing-incidence wide-angle X-ray scattering (GIWAXS) of the perovskite films without and with organic salts post-modulation. Compared with the control perovskite films, no new diffraction ring was found in the APCl-modulated perovskite film. However, we observed a new diffraction ring at q = 0.52 after phFACl modulation and at q = 0.68 after TFACl modulation, which is attributed to the formation of low-dimensional perovskites. Regardless of precursor solvents, the XRD results in Fig. 3e and f further confirmed the generation of low-dimensional perovskites after phFACl and TFACl modulation whereas no low-dimensional perovskite was formed upon APCl modulation. With increasing concentrations of APCl, there is still no appearance of low-dimensional components (Fig. 3g), which further consolidated our findings—APCl modulation tends not to produce low-dimensional perovskite. For phFACl and TFACl, it is easy to form low-dimensional perovskites by single active site interaction. In contrast, it is difficult to form low-dimensional perovskite for APCl because it interacts with perovskite via strong dual-site chemical anchoring due to the suitable distance between two interaction sites and spatial conformation, which was validated by previous theoretical results. | ||
| Fig. 3 Investigation of crystal phase after organic salt modulation. GIWAXS of the (a) control, (b) phFACl, (c) APCl, and (d) TFACl-modulated perovskite films. (e) XRD patterns of the control and phFACl, APCl, and TFACl-modulated perovskite films based on mixed solvents of DMF/NMP = 7/1. (f) XRD patterns of the control and phFACl, APCl, and TFACl modulated perovskite films based on mixed solvents of DMF/DMSO = 4/1. (g) XRD patterns of the perovskite films modulated with different concentrations of APCl. | ||
Characterizations of perovskite film quality
The effects of surface modulation by three salts on the morphology and crystallinity of perovskite films were studied by scanning electron microscopy (SEM), atomic force microscopy (AFM), and light-beam-induced current (LBIC). After three organic salt modulations, the grain size was maintained but the surface morphology was changed and the grain boundaries were blurred (Fig. S2, ESI†). From the AFM image in Fig. 4a, the APCl-modulated perovskite film exhibited the flattest surface morphology, which was evidenced by its lowest roughness. The flat and uniform perovskite film will promote electron extraction at the perovskite/C60 interface and reduce nonradiative recombination losses. To verify the positive effect on defect passivation and photocurrent uniformity, we first performed LBIC measurement involving a laser beam of 520 nm light to obtain a photoresponse mapping. The brightness of LBIC results represents higher photon electron conversion efficiency. Fig. 4b shows that the phFACl, APCl, and TFACl-modulated PSC devices had better photocurrent output uniformity than control devices. The external quantum efficiency of the control device decreased significantly after ageing for 7 days, while the changes in LBIC mapping for the three salts-modulated perovskite films were not significant. The LBIC results suggest that phFACl, APCl, and TFACl modulation can inhibit ion migration in perovskite and thereby hinder the chemical reaction between I− and Ag. It was revealed that APCl-passivated devices exhibited optimal uniformity and photocurrent output intensity. | ||
| Fig. 4 Characterizations of perovskite film quality. (a) AFM and (b) LBIC mapping of the control, phFACl, APCl, and TFACl-modulated PSC devices. (c) Steady-state PL and (d) TRPL spectra of the control, phFACl, APCl and TFACl-modulated perovskite films on a bare glass substrate. (e) TAS plots of the control and modulated PSCs. (f) I–V curves of the electron-only devices based on ITO/SnO2/perovskite (phFACl, APCl and TFACl)/PC61BM/Ag structures and (g) the hole-only devices based on ITO/NiOx/perovskite (phFACl, APCl, and TFACl)/spiro-OMeTAD/Ag structures. (h) Statistical chart of the defect densities of the perovskite films. | ||
The experimental results confirmed the chemical interaction and defect passivation ability of three organic salts. Subsequently, we qualitatively and quantitatively analyzed the defects using spectroscopic and electrical methods. Fig. 4c and d present the steady-state and time-resolved photoluminescence (PL and TRPL) of perovskite films on bare glass. As illustrated in Fig. 4c, the perovskite films modulated with APCl exhibited the highest PL intensity among all perovskite films, indicating the inhibition of nonradiative recombination and the reduction of defect density. The lifetime of the perovskite films was investigated by TRPL characterizations. The TRPL spectra in Fig. 4d were fitted by the biexponential function.49,50 The fitting carrier lifetime results are shown in Table S1 (ESI†). The average lifetime (τavg) of the phFACl, APCl, and TFACl modulated perovskite films was calculated to be 1.93 μs, 2.05 μs, 1.76 μs, respectively, which are longer than that of the control film (0.99 μs). The longest carrier lifetime of the APCl-modulated perovskite film is due to dual-site anchoring and passivation of AP+.51
A reduced trap density in the perovskite films was also verified by the thermal admittance spectroscopy (TAS) measurement results in Fig. 4e. After post-modulation, all modulated perovskite films exhibited reduced defect density compared with the pristine film. The APCl-modulated perovskite film had the lowest defect density in the energy range of 0.3–0.4 eV, which is ascribed to the most excellent passivation of APCl for the surface defects of perovskite films. To further evaluate the passivation effect of different modulation molecules from a quantitative perspective, we calculated the trap state density of perovskite films using the space charge limited current (SCLC) method. Fig. 4f and g show dark current–voltage (I–V) curves of the electron-only devices (ITO/SnO2/perovskite/PC61BM/Ag) and the hole-only devices (ITO/NiOX/perovskite/spiro-OMeTAD/Ag), respectively.49 As for the electron-transporting only devices, the trap-filled limit voltage (VTFL) of 0.15 V for the APCl-modulated device was lower than 0.19 V, 0.17 V, and 0.18 V for the control, phFACl and TFACl-modulated devices. Regarding hole-transporting only devices, the VTFL was estimated to be 0.08 V, 0.04 V and 0.10 V for the phFACl, APCl and TFACl modulated devices, which is lower than 0.18 V for the control device (Fig. 4h). The defect density of states (Nt) of the device could be obtained according to the following eqn (1):
 | (1) |
Interfacial energy level alignment modulation
Kelvin probe force microscopy (KPFM) measurement was carried out to gain insights into the surface potentials of the perovskite films and the changes in the energy level structure inside the PSC device. As shown in Fig. 5a and b, the average surface potential of the control, phFACl, APCl and TFACl-modulated perovskite films was −225 mV, −450 mV, −500 mV and −350 mV, respectively. Clearly, the perovskite film modulated with APCl exhibited the greatest increase in surface potential, which correlates with the observed enhancement in VOC for the APCl-modulated devices. From the ultraviolet photoelectron spectroscopy (UPS) (Fig. 5c) and corresponding bandgap structure schematic diagram (Fig. 5d), we can conclude that the Fermi energy levels of the phFACl, APCl, and TFACl-modulated perovskite films shifted upward compared with the control perovskite film. In particular, the APCl-modulated perovskite film exhibited a most obvious Fermi energy shift, indicating that APCl modulation induced more n-type characteristics. In addition, we obtained the energy level alignment of the film from Fig. 5d and Fig. S3 (ESI†). Meanwhile, Fig. S4 (ESI†) provides a more complete energy level diagram. It is revealed that the APCl-modulated film has a smaller conduction band offset of 0.16 eV with C60, which facilitates electron extraction and reduces charge carrier recombination at the interface. This is related to the reason that APCl passivated the electronic defects on the surface of the perovskite and formed a back surface field with the bulk phase of the perovskite. The formed back surface field is in the same direction as the built-in electric field of the device, thus making the stronger built-in potential (Vbi) of 0.93 V, which is higher than 0.80 V for control devices from the Mott–Schottky plots (Fig. S5, ESI†), thus promoting electron transport and extraction. Actually, organic cation-induced n-type doping and back electric field at the perovskite/ETL interface have been reported to improve the PCE and stability of inverted PSCs.53 | ||
| Fig. 5 Interfacial energy level alignment modulation. (a) KPFM images, (b) the corresponding cross-sectional surface potential curves, (c) UPS spectra and (d) energy level diagram of the control, phFACl, APCl and TFACl-modulated perovskite films. | ||
Photovoltaic performance
We prepared inverted PSCs with the device structure of ITO/NiOx/Me-4PACz/perovskite (phFACl, APCl, or TFACl modulation)/C60/BCP/Cu (Fig. 6a) by employing a vacuum flash evaporation process. Unless otherwise stated, the composition for fabricating the perovskite films and devices was Cs0.05FA0.95PbI3. The effect of the concentrations of three organic salts on the photovoltaic performance was systematically studied (Fig. S6–S8, ESI†). From the photovoltaic parameters (PCE, VOC, JSC, and FF) of the PSCs summarized in Fig. 6b, the phFACl, APCl, and TFACl-modulated PSCs exhibited an average PCE value of 25.54%, 26.53% and 25.08%, respectively, higher than 24.49% for the control devices. It is worth noting that the APCl-modulated devices exhibited the most noticeable enhancement in VOC and FF, leading to the highest PCE. The dual-site passivation of AP+ should be responsible for the distinguished photovoltaic performance. The amplitude of the PSC photovoltaic performance improvement follows the sequence of the passivation ability of trap defects: APCl > phFACl > TFACl. | ||
| Fig. 6 Photovoltaic performance and stability of PSCs. (a) The configuration of PSCs. (b) Statistics of VOC, FF, PCE and JSC of 10 individual devices based on the control, phFACl, APCl, and TFACl-modulated perovskite films. (c) J–V curves of the PSCs based on the control, phFACl, APCl, and TFACl-modulated perovskite films. (d) EQE curves of the PSCs based on the control, phFACl, APCl, and TFACl-modulated perovskite films. (e) J–V curves of the wide-bandgap PSCs based on the control and APCl-modulated perovskite films. (f) J–V curves and (g) photograph of the APCl-modulated perovskite module with an aperture area of 40.1 cm2. (h) Normalized PCE for the control and phFACl, APCl and TFACl-modulated PSC devices over a long period in an environment with a relative humidity of 30% ± 5% and without encapsulation. (i) MPPT stability curves of the encapsulated PSCs without and with APCl. The inset picture shows a batch of PSC device samples used for stability tests. | ||
Fig. 6c presents the champion J–V curves of the control and modulated PSC devices in reverse scan (RS) and forward scan (FS). The control, phFACl, APCl and TFACl-modulated PSCs delivered champion PCEs of 25.02% (a JSC of 26.04 mA cm−2, a VOC of 1.165 V, and an FF of 82.45%), 25.68% (a JSC of 26.01 mA cm−2, a VOC of 1.191 V, and an FF of 82.90%), 26.83% (a JSC of 26.36 mA cm−2, a VOC of 1.194 V, and an FF of 85.27%) and 25.31% (a JSC of 26.10 mA cm−2, a VOC of 1.178 V, and an FF of 82.34%), respectively. The APCl-modified PSCs exhibit a steady-state PCE of 26.32% (Fig. S9–S14, ESI†). To the best of our knowledge, our obtained steady-state PCE of 26.32% is the best PCE ever reported for inverted PSCs based on the vacuum flash method in ambient conditions for perovskite deposition. In addition, the hysteresis index (HI) calculated according to the formula of HI = (PCEReverse − PCEForwad)/PCEReverse was 4.0%, 2.1%, 0.5% and 3.0% for the control, phFACl, APCl, and TFACl-modulated PSCs, respectively (Table S2, ESI†). The hysteresis was mitigated for all modulated devices, and the smallest hysteresis was found for the APCl-modulated PSCs, which resulted from reduced defect density and facilitated interfacial electron extraction and thus suppressed interfacial charge accumulation. The integrated current density from the EQE spectra of the control, phFACl, APCl, and TFACl-modulated PSCs was 25.22 mA cm−2, 25.73 mA cm−2, 25.80 mA cm−2 and 25.70 mA cm−2, respectively, which matches well with the values from J–V measurements (Fig. 6d). Furthermore, APCl modulation helped to achieve an enhanced PCE from 20.40% to 22.16% in the wide-bandgap 1.66 eV Cs0.05MA0.15FA0.8Pb(I0.76Br0.24)3 based PSCs (Fig. 6e). Our perovskite film fabrication procedure based on vacuum flash and ambient conditions is compatible with large-area modules. To further confirm the effectiveness of our APCl passivation method, we fabricated an APCl-modulated large-area module by the vacuum flash method, where 11 subcells were connected in series (Fig. 6f and g). The champion module with an aperture area (including dead area) of 40.1 cm2 achieved an appealing PCE of 19.83%. The efficiency gap between modules and small active area-PSCs may be because upscaling to large-area modules compromises film uniformity and crystallization control, causing edge-to-center thickness variations, bulk defects and non-uniform coverage.54,55 Additionally, series connection constrains module current to its lowest-performing subcell, lowering the overall efficiency. The above results certify that our passivation technique is very effective in large-area and small-area PSCs.56
To effectively corroborate the intrinsic reasons for the performance improvement of the device, we conducted a series of electrical characterizations to analyze the charge transfer and recombination processes. As presented in Fig. S15 (ESI†), the ideality factor m (1.11) of the APCl-modulated device is closer to 1 than those of the control (1.29), phFACl (1.15), and TFACl (1.17) modulated devices, indicating that APCl is the most effective passivator in suppressing interfacial nonradiative recombination. The interfacial charge extraction and recombination were further evaluated by performing transient photocurrent (TPC) and transient photovoltage decay (TPV) tests. In Fig. S16 (ESI†), the shortest photocurrent decay lifetime (τrec) was found for the APCl-modulated device (0.34 μs) among all devices, which demonstrated its most efficient charge transfer and extraction due to reduced interface defects and favourable band alignment. From TPV in Fig. S17 (ESI†), the APCl-modulated PSC devices exhibited the longest charge carrier recombination lifetime (2.77 ms), which is indicative of the smallest nonradiative recombination losses, explaining why it has the highest VOC and FF. To understand the sensitivity and response speed of the APCl-modulated devices, we also perform −3 dB measurements to characterize the response speed to light, thereby reflecting the reduced carrier trapping effects of trap defects. In Fig. S18 (ESI†), the −3 dB bandwidth (f−3dB) for the phFACl, APCl, and TFACl-modulated photovoltaic devices is 0.265, 0.294, and 0.223 MHz, respectively, while it is 0.186 MHz for the control device. This demonstrates the reduced density of surface trap defects and faster charge carrier extraction capability in the APCl-modulated devices.
Long-term stability
The long-term stability of devices without and with APCl modulation was explored. Firstly, we tracked the long-term stability of the unencapsulated devices by placing them in the air with a relative humidity of 30 ± 5% (temperature of 25 ± 5 °C) in the dark (Fig. 6h). After 1200 h of aging, the efficiency of the APCl-modulated devices remained above 90%, while the control devices could only maintain over 60% of their initial efficiency. We assessed the operational stability of 1.53 eV-Cs0.05MA0.05FA0.9PbI3-based PSCs without and with APCl modulation. The operational light stability of the device was investigated by cyclic J–V tests under continuous LED illumination (Fig. S19, ESI†). Encapsulation using a capping glass front with UV-curable adhesive encapsulant for sealing was applied. The APCl-modulated PSC device retained 73.7% of the initial PCE over 1500 h. In contrast, the control device dropped to 72.1% of the initial PCE after only 300 h. We further conducted the typical MPPT of the control and APCl-modulated PSC devices under continuous 100 mW cm−2 3A+ white LED light irradiation at room temperature of 25 ± 5 °C and stored in a 99.99% nitrogen environment. As shown in Fig. 6i, after 2000 h of continuous light exposure, the APCl-modulated device could retain 95.8% of the initial PCE, while the control device dropped to 63.7% after 1600 h. This outstanding MPPT stability is at the forefront of current reported research work. We attribute the improved light stability to the reduced interfacial trap defects by the dual-site passivation of anionic and cationic defects. The contact angles of the control, phFACl, APCl, and TFACl-modified perovskite films are 31.9°, 43.7°, 49.5°, and 35.8°, respectively (Fig. S20, ESI†). This indicates that APCl-modified perovskite film has the highest hydrophobicity, which typically enhances the environmental humidity stability of the device.Discussion
We realized dual-site passivation of different charged defects in perovskite through heterocycle functionalized amidinium cations, which enabled highly efficient and stable inverted PSCs and modules. Dual-site passivation was achieved by modulating the distance between two active sites and spatial conformation. The optimized APCl demonstrated outstanding defect passivation ability due to horizontal binding and dual-site anchoring induced by the appropriate distance between two binding functional groups. The APCl-modulated inverted PSCs delivered a PCE of 26.83%, which is the best PCE ever reported for PSCs fabricated based on vacuum flash and in ambient air without anti-solvents. Moreover, APCl passivation also resulted in a high-efficiency large-area module with a PCE of 19.83% (aperture area of 40.1 cm2). The APCl modulation also resulted in excellent device light stability, maintaining 95.8% of their initial performance after 2000 h of continuous MPP tracking. This work provides an effective avenue to maximize the potential of organic cations and salts to ameliorate the PCE and operational stability of PSCs, which paves the way for the commercial deployment of perovskite photovoltaic technology.Author contributions
M. L., J. C. and C. C. conceived the idea. J. C. and C. C. supervised this project. M. L., J. D., T. P. and Q. M. fabricated devices and samples for various characterizations. M. L., Z. Z. and C. C. conducted the light stability measurement. J. Z. and B. Z. conducted the DFT calculation. Y. W. facilitated large-area modules. M. L. and C. C. wrote the first draft of the manuscript. J. C., J. T., and C. C. wrote the final version of the manuscript. All authors discussed the results and contributed to the revisions of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (U21A2076, 62274018, 52462031), the Science and Technology Development Fund, Macau SAR (No. 0009/2022/AGJ), the Central Guidance on Local Science and Technology Development Fund of Hebei Province (226Z4305G), the Hebei Province Higher Education Science and Technology Research Project (JZX2024030), the Shijiazhuang Basic Research Project at Hebei-based Universities (241790847A), the Natural Science Foundation of Hebei Province (E2024202086), the Xinjiang Construction Corps Key Areas of Science and Technology Research Project (2023AB029), the Anhui Provincial Natural Science Foundation (2408085MA001), the Natural Science Research Project of Anhui Educational Committee (2024AH051450), and the S&T Program of Hebei (24464401D). A portion of this work is based on the data obtained at BSRF-1W1A. The authors gratefully acknowledge the cooperation of the beamline scientists at the BSRF-1W1A beamline.References
- H. Chen, C. Liu, J. Xu, A. Maxwell, W. Zhou, Y. Yang, Q. Zhou, A. S. R. Bati, H. Wan, Z. Wang, L. Zeng, J. Wang, P. Serles, Y. Liu, S. Teale, Y. Liu, M. I. Saidaminov, M. Li, N. Rolston, S. Hoogland, T. Filleter, M. G. Kanatzidis, B. Chen, Z. Ning and E. H. Sargent, Science, 2024, 384, 189–193 CrossRef CAS PubMed.
- Y. Zhao, F. Ma, Z. Qu, S. Yu, T. Shen, H.-X. Deng, X. Chu, X. Peng, Y. Yuan, X. Zhang and J. You, Science, 2022, 377, 531–534 CrossRef CAS PubMed.
- M. Tao, Y. Wang, K. Zhang, Z. Song, Y. Lan, H. Guo, L. Guo, X. Zhang, J. Wei, D. Cao and Y. Song, Joule, 2024, 8(11), 3142–3152 CrossRef CAS.
- X. Zhao, Z. Zhang, Y. Zhu, F. Meng, M. Li, C. Wang, W. Gao, Y. Feng, R. Li, D. He, J. Chen and C. Chen, Nano Lett., 2023, 23, 11184–11192 CrossRef CAS PubMed.
- S. Liu, J. Li, W. Xiao, R. Chen, Z. Sun, Y. Zhang, X. Lei, S. Hu, M. Kober-Czerny, J. Wang, F. Ren, Q. Zhou, H. Raza, Y. Gao, Y. Ji, S. Li, H. Li, L. Qiu, W. Huang, Y. Zhao, B. Xu, Z. Liu, H. J. Snaith, N.-G. Park and W. Chen, Nature, 2024, 632, 536–542 CrossRef PubMed.
- C. Ma, F. T. Eickemeyer, S.-H. Lee, D.-H. Kang, S. J. Kwon, M. Grätzel and N.-G. Park, Science, 2023, 379, 173–178 CrossRef CAS PubMed.
- J. Zhu, Y. Xu, Y. Luo, J. Luo, R. He, C. Wang, Y. Wang, K. Wei, Z. Yi, Z. Gao, J. Wang, J. You, Z. Zhang, H. Lai, S. Ren, X. Liu, C. Xiao, C. Chen, J. Zhang, F. Fu and D. Zhao, Sci. Adv., 2024, 10, eadl2063 CrossRef CAS PubMed.
- Y. Gao, Q. Cai, Y. He, D. Zhang, Q. Cao, M. Zhu, Z. Ma, B. Zhao, H. He, D. Di, Z. Ye and X. Dai, Sci. Adv., 2024, 10, eado5645 CrossRef CAS PubMed.
- Y. Zheng, Y. Li, R. Zhuang, X. Wu, C. Tian, A. Sun, C. Chen, Y. Guo, Y. Hua, K. Meng, K. Wu and C.-C. Chen, Energy Environ. Sci., 2024, 17, 1153–1162 RSC.
- X. Li, S. Gao, X. Wu, Q. Liu, L. Zhu, C. Wang, Y. Wang, Z. Liu, W. Chen, X. Li, P. Xiao, Q. Huang, T. Chen, Z. Li, X. Gao, Z. Xiao, Y. Lu, X. Zeng, S. Xiao, Z. Zhu and S. Yang, Joule, 2024, 8(11), 3169–3185 CrossRef CAS.
- Y. Yang, H. Chen, C. Liu, J. Xu, C. Huang, C. D. Malliakas, H. Wan, A. S. R. Bati, Z. Wang, R. P. Reynolds, I. W. Gilley, S. Kitade, T. E. Wiggins, S. Zeiske, S. Suragtkhuu, M. Batmunkh, L. X. Chen, B. Chen, M. G. Kanatzidis and E. H. Sargent, Science, 2024, 386, 898–902 CrossRef CAS PubMed.
- D. Gao, R. Li, X. Chen, C. Chen, C. Wang, B. Zhang, M. Li, X. Shang, X. Yu, S. Gong, T. Pauporté, H. Yang, L. Ding, J. Tang and J. Chen, Adv. Mater., 2023, 35, 2301028 CrossRef CAS PubMed.
- W. Gao, J. Ding, Q. Ma, H. Zhang, J. Zhang, Z. Zhang, M. Li, Y. Wang, B. Zhang, T. Pauporté, J.-X. Tang, J. Chen and C. Chen, Adv. Mater., 2024, 2413304 Search PubMed.
- D. He, D. Ma, R. Li, B. Liu, Q. Zhou, H. Yang, S. Lu, Z. Zhang, C. Li, X. Li, L. Ding, J. Feng, J. Yi and J. Chen, ACS Energy Lett., 2024, 9, 2615–2625 CrossRef CAS.
- B. Liu, Q. Zhou, Y. Li, Y. Chen, D. He, D. Ma, X. Han, R. Li, K. Yang, Y. Yang, S. Lu, X. Ren, Z. Zhang, L. Ding, J. Feng, J. Yi and J. Chen, Angew. Chem., Int. Ed., 2024, 63, e202317185 CrossRef CAS PubMed.
- H. Li, C. Zhang, C. Gong, D. Zhang, H. Zhang, Q. Zhuang, X. Yu, S. Gong, X. Chen, J. Yang, X. Li, R. Li, J. Li, J. Zhou, H. Yang, Q. Lin, J. Chu, M. Grätzel, J. Chen and Z. Zang, Nat. Energy, 2023, 8, 946–955 CrossRef CAS.
- C. Gong, X. Chen, J. Zeng, H. Wang, H. Li, Q. Qian, C. Zhang, Q. Zhuang, X. Yu, S. Gong, H. Yang, B. Xu, J. Chen and Z. Zang, Adv. Mater., 2024, 36, 2307422 CrossRef CAS PubMed.
- N. Li, X. Niu, Q. Chen and H. Zhou, Chem. Soc. Rev., 2020, 49, 8235–8286 RSC.
- H. Zhu, S. Teale, M. N. Lintangpradipto, S. Mahesh, B. Chen, M. D. McGehee, E. H. Sargent and O. M. Bakr, Nat. Rev. Mater., 2023, 8, 569–586 CrossRef.
- J. Chen and N.-G. Park, Adv. Mater., 2019, 31, 1803019 CrossRef CAS PubMed.
- J. Chen and N.-G. Park, ACS Energy Lett., 2020, 5, 2742–2786 CrossRef CAS.
- B. Chen, P. N. Rudd, S. Yang, Y. Yuan and J. Huang, Chem. Soc. Rev., 2019, 48, 3842–3867 RSC.
- B. Liu, H. Bi, D. He, L. Bai, W. Wang, H. Yuan, Q. Song, P. Su, Z. Zang, T. Zhou and J. Chen, ACS Energy Lett., 2021, 6, 2526–2538 CrossRef CAS.
- Z. Ni, C. Bao, Y. Liu, Q. Jiang, W.-Q. Wu, S. Chen, X. Dai, B. Chen, B. Hartweg, Z. Yu, Z. Holman and J. Huang, Science, 2020, 367, 1352–1358 CrossRef CAS PubMed.
- Y. Yuan and J. Huang, Acc. Chem. Res., 2016, 49, 286–293 CrossRef CAS PubMed.
- H. Zai, Y. Ma, Q. Chen and H. Zhou, J. Energy Chem., 2021, 63, 528–549 CrossRef CAS.
- Z. Zhang, M. Li, R. Li, X. Zhuang, C. Wang, X. Shang, D. He, J. Chen and C. Chen, Adv. Mater., 2024, 36, 2313860 CrossRef CAS PubMed.
- J.-W. Lee, H.-S. Kim and N.-G. Park, Acc. Chem. Res., 2016, 49, 311–319 CrossRef CAS PubMed.
- C. Li, X. Wang, E. Bi, F. Jiang, S. M. Park, Y. Li, L. Chen, Z. Wang, L. Zeng, H. Chen, Y. Liu, C. R. Grice, A. Abudulimu, J. Chung, Y. Xian, T. Zhu, H. Lai, B. Chen, R. J. Ellingson, F. Fu, D. S. Ginger, Z. Song, E. H. Sargent and Y. Yan, Science, 2023, 379, 690–694 CrossRef CAS PubMed.
- X. Zhao, Z. Zhang, Y. Zhu, F. Meng, M. Li, C. Wang, W. Gao, Y. Feng, R. Li, D. He, J. Chen and C. Chen, Nano Lett., 2023, 23, 11184–11192 CrossRef CAS PubMed.
- X. Li, W. Zhang, X. Guo, C. Lu, J. Wei and J. Fang, Science, 2022, 375, 434–437 CrossRef CAS PubMed.
- S. Yang, S. Chen, E. Mosconi, Y. Fang, X. Xiao, C. Wang, Y. Zhou, Z. Yu, J. Zhao, Y. Gao, F. De Angelis and J. Huang, Science, 2019, 365, 473–478 CrossRef CAS PubMed.
- J. Hu, C. Wang, S. Qiu, Y. Zhao, E. Gu, L. Zeng, Y. Yang, C. Li, X. Liu, K. Forberich, C. J. Brabec, M. K. Nazeeruddin, Y. Mai and F. Guo, Adv. Energy Mater., 2020, 2000173 CrossRef CAS.
- D. Gao, R. Li, X. Chen, C. Chen, C. Wang, B. Zhang, M. Li, X. Shang, X. Yu, S. Gong, T. Pauporté, H. Yang, L. Ding, J.-X. Tang and J. Chen, Adv. Mater., 2023, 35, 2301028 CrossRef CAS PubMed.
- Y. Zhu, Z. Zhang, X. Zhao, M. Li, Y. Feng, S. Zhang, W. Gao, J. Chen, J.-X. Tang and C. Chen, Adv. Energy Mater., 2024, 14, 2303946 CrossRef CAS.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 460–466, DOI:10.1038/s41566-019-0398-2.
- C. Zhang, H. Li, C. Gong, Q. Zhuang, J. Chen and Z. Zang, Energy Environ. Sci., 2023, 16, 3825–3836 RSC.
- C. Liu, Y. Yang, H. Chen, J. Xu, A. Liu, A. S. R. Bati, H. Zhu, L. Grater, S. S. Hadke, C. Huang, V. K. Sangwan, T. Cai, D. Shin, L. X. Chen, M. C. Hersam, C. A. Mirkin, B. Chen, M. G. Kanatzidis and E. H. Sargent, Science, 2023, 382, 810–815 CrossRef CAS PubMed.
- H. Tan, A. Jain, O. Voznyy, X. Lan, F. P. García de Arquer, J. Z. Fan, R. Quintero-Bermudez, M. Yuan, B. Zhang, Y. Zhao, F. Fan, P. Li, L. N. Quan, Y. Zhao, Z.-H. Lu, Z. Yang, S. Hoogland and E. H. Sargent, Science, 2017, 355, 722–726 CrossRef CAS PubMed.
- J. Chen, X. Zhao, S.-G. Kim and N.-G. Park, Adv. Mater., 2019, 31, 1902902 CrossRef PubMed.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- J. Xue, R. Wang, X. Chen, C. Yao, X. Jin, K.-L. Wang, W. Huang, T. Huang, Y. Zhao, Y. Zhai, D. Meng, S. Tan, R. Liu, Z.-K. Wang, C. Zhu, K. Zhu, M. C. Beard, Y. Yan and Y. Yang, Science, 2021, 371, 636–640 CrossRef CAS PubMed.
- L. Yan, H. Huang, P. Cui, S. Du, Z. Lan, Y. Yang, S. Qu, X. Wang, Q. Zhang, B. Liu, X. Yue, X. Zhao, Y. Li, H. Li, J. Ji and M. Li, Nat. Energy, 2023, 8, 1158–1167 CrossRef CAS.
- S. Heo, G. Seo, Y. Lee, M. Seol, S. H. Kim, D.-J. Yun, Y. Kim, K. Kim, J. Lee, J. Lee, W. S. Jeon, J. K. Shin, J. Park, D. Lee and M. K. Nazeeruddin, Adv. Mater., 2019, 31, 1805438 CrossRef PubMed.
- L. Yang, Z. Fang, Y. Jin, H. Feng, B. Deng, L. Zheng, P. Xu, J. Chen, X. Chen, Y. Zhou, C. Shi, W. Gao, J. Yang, X. Xu, C. Tian, L. Xie and Z. Wei, Adv. Mater., 2024, 36, 2311923 CrossRef CAS PubMed.
- S. Wang, C. Yao, L. Li, T. Huang, S. Tan, P. Shi, S. Jin, C. Zhu, Y. Yang, B. Zhu, J. Xue and R. Wang, Joule, 2024, 8, 1105–1119 CrossRef CAS.
- L. Wang, L. Fu, B. Li, H. Li, L. Pan, B. Chang and L. Yin, Sol. RRL, 2021, 5, 2000720 CrossRef CAS.
- T. Liu, J. Guo, D. Lu, Z. Xu, Q. Fu, N. Zheng, Z. Xie, X. Wan, X. Zhang, Y. Liu and Y. Chen, ACS Nano, 2021, 15, 7811–7820 CrossRef CAS PubMed.
- W. Gao, J. Ding, Q. Ma, H. Zhang, J. Zhang, Z. Zhang, M. Li, Y. Wang, B. Zhang, T. Pauporté, J.-X. Tang, J. Chen and C. Chen, Adv. Mater., 2024, 2413304, DOI:10.1002/adma.202413304.
- Z. Zhang, M. Li, R. Li, X. Zhuang, C. Wang, X. Shang, D. He, J. Chen and C. Chen, Adv. Mater., 2024, 36, 2313860 CrossRef CAS PubMed.
- S. Hu, K. Otsuka, R. Murdey, T. Nakamura, M. Truong, T. Yamada, T. Handa, K. Matsuda, K. Nakano, A. Sato, K. Marumoto, K. Tajima, Y. Kanemitsu and A. Wakamiya, Energy Environ. Sci., 2022, 15, 2096 RSC.
- Q. Lin, A. Armin, R. C. R. Nagiri, P. L. Burn and P. Meredith, Nat. Photonics, 2015, 9, 106–112 CrossRef CAS.
- M. Li, Z. Zhu, Z. Wang, W. Pan, X. Cao, G. Wu and R. Chen, Adv. Mater., 2024, 36, 2309428 CrossRef CAS PubMed.
- S. Strohm, F. Machui, S. Langner, P. Kubis, N. Gasparini, M. Salvador, I. McCulloch, H. J. Egelhaaf and C. J. Brabec, Energy Environ. Sci., 2018 10.1039/C8EE01150H.
- Y. Yun, Q. Chang, J. Yan, Y. Tian, S. Jiang, W. Wei, S. Li, Y. Guo, J. Yin, J. Li, M. Chen, K. Huang, C. Li and R. Zhang, Sci. Adv., 2025, 11, eadp3112 CrossRef CAS PubMed.
- Z. Zhang, Y. Feng, J. Ding, Q. Ma, H. Zhang, J. Zhang, M. Li, T. Geng, W. Gao, Y. Wang, B. Zhang, T. Pauporté, J. Tang, H. Chen, J. Chen and C. Chen, Nat. Commun., 2025, 16, 753 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00524h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |