
Comprehensive crystallization retardation of inorganic perovskites for high performance inverted solar cells†
Zezhang
Wang
a,
Tianfei
Xu
a,
Nan
Li
a,
Zhen
Chang
a,
Jing
Shan
a,
Yong
Wang
a,
Minfang
Wu
a,
Fengwei
Xiao
a,
Shengzhong
Liu
 *bcd and
Wanchun
Xiang
*a
*bcd and
Wanchun
Xiang
*a
aKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, Shaanxi Key Laboratory for Advanced Energy Devices, Shaanxi Engineering Lab for Advanced Energy Technology, School of Materials Science and Engineering, Shaanxi Normal University, Xi’an, 710119, China. E-mail: wanchun.xiang@snnu.edu.cn
bDalian National Laboratory for Clean Energy, iChEM, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: szliu@dicp.ac.cn
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing, 100049, P. R. China
dCNNP Optoelectronics Technology, 2828 Canghai Road, Lingang, Shanghai, China
First published on 21st March 2025
Abstract
Inverted inorganic perovskite solar cells (PSCs) are ideal top cells for tandem configurations due to their ideal bandgap and excellent thermal stability. However, water-induced rapid crystallization during inorganic perovskite film processing in ambient air is difficult to control. Here, we report a crystallization retardation method to prepare inorganic perovskite films by incorporating an acrylonitrile-methyl acrylate copolymer (AMAC) in perovskite precursor solution. Firstly, the strong interaction between the AMAC and the precursor solution yields increased colloidal size, delays dimethyl sulfoxide (DMSO) volatilization during annealing and postpones the phase transition. Secondly, the interaction between AMAC and dimethylamine (DMA+) slows down ion exchange with Cs+. These interactions retard perovskite crystallization, increase pack-crystal grain size and reduce residual stress. Combined with the functional groups in AMAC, the incorporation of the AMAC reduces defects in perovskite films, modulates interfacial energy levels, and prolongs charge lifetimes, inhibiting the migration of iodide ions. Ultimately, the power conversion efficiency (PCE) of the AMAC-incorporated inverted (p–i–n) and conventional (n–i–p) PSCs reaches 21.7% and 21.8%, respectively, while the unencapsulated devices show only 8% degradation over 2500 h of maximum power point tracking and continuous operation.
Broader contextInorganic perovskite materials outperform their organic–inorganic hybrid counterparts in terms of excellent thermal stability and ideal bandgap for tandem solar cells, and thus have attracted great attention from the photovoltaic community. To realize high performance perovskite solar cells prepared in air, high quality perovskite films with low defects are required. However, water-induced rapid crystallization during the preparation of inorganic perovskite films in ambient air is difficult to control, which exerts challenges in maximizing power conversion efficiency and operational stability of devices. To overcome this issue, we adopted an acrylonitrile–methyl acrylate copolymer (AMAC) as an additive in the perovskite precursor to retard the crystallization of inorganic perovskites. The extended time span for grain growth and released lattice strain is attributed to the interaction between the AMAC and precursors to slow down ion exchange and solvent evaporation. Additionally benefitting from further reduced defects, the AMAC incorporation considerably increases the device efficiency from 19.4% to 21.7% under 100 mW cm−2 irradiation, the highest value for inverted inorganic PSCs, along with improved operational stability. |
Introduction
In recent years, the photovoltaic (PV) research community has witnessed perovskite solar cells (PSCs) with a series of breakthroughs in power conversion efficiencies (PCE).1–3 Further surpassing the Shockley–Queisser (S–Q) limit of single junction solar cells relies on the multi-junction tandem structure.4–7 To ensure high performance of tandem devices, the top perovskite subcells with an inverted structure are required to be highly stable with high voltage output.8,9 In this regard, metal halide inorganic perovskites outperform their organic–inorganic hybrid counterparts in two aspects: (1) inorganic perovskites without a significantly altered halide ratio exhibit ideal bandgaps for the tandem structure10,11 and (2) inorganic perovskites demonstrate superior thermal stability.12,13 Despite these attractive features, the development of inorganic PSCs with an inverted structure is still lagging behind that of the conventional structure.14,15The high performance of inorganic PSCs is strictly linked to high quality perovskite films with low defects.16–18 Therefore, regulating crystallization of perovskite during film processing is of great importance.19,20 It has been found that a trace amount of water can assist the formation of inorganic perovskites by accelerating nucleation and crystallization processes.21,22 This allows fabrication of inorganic perovskites in air without a N2 atmosphere. However, the water-induced fast crystallization process is difficult to control, which severely affects the reproducibility of the film.14,23,24 This is because the preparation of inorganic perovskites generally requires intermediate phase engineering with organic salts (such as dimethylamine iodide, DMAI) to regulate the crystallization.25–27 In this process, a DMAPbI3 hybrid perovskite is initially formed, followed by ion exchange between DMA+ and Cs+ with preserved lattice geometry.26,28,29 Water-induced fast crystallization will disturb the desired nucleation speed and efficient ion exchange between DMA+ and Cs+, which may result in inhomogeneous and low-crystalline films, due to the multiple pathways of crystal nucleation originating from various intermediate phases in the film-forming process.30–32 The low quality perovskite film contains numerous defects, which leads to nonradiative recombination and fast degradation of perovskites.32,33 Therefore, modulating crystallization for high quality and stable inorganic perovskite films in air with low defect density is essential for low nonradiative energy loss and long-term stability of PSCs, which is beneficial for mass production.14,34
It has been reported that the integration of a polymer as an additive in precursors can facilitate perovskite grain growth with a preferred orientation by increasing the activation energy of nucleation, which can retard perovskite crystal growth.26,35,36 The effective chemical bonding between polymers and perovskite materials can suppress trap states and prevent ion migration.37–39 Furthermore, long-chain polymers are dispersed at grain boundaries, providing a strong barrier against perovskite film degradation.26,40 These ultimately contribute to the overall improvement in the efficiency and operational stability of PSCs. However, the PV performance of the corresponding devices with polymer additives in precursor solutions is generally lower than those of devices modified with small molecule additives. The underlying reason could probably be the single type of functional groups in the polymer and the weak interaction between the polymer and the perovskite precursors, which may result in inefficient crystallization regulation. Consequently, the design of novel polymers with multiple functionalities for efficiently regulating the crystallization of inorganic perovskite films is of great significance for high performance devices.
In this work, an acrylonitrile–methyl acrylate copolymer (AMAC) is introduced into CsPbI3−xBrx perovskite precursor solution to prepare inverted inorganic PSCs. AMAC is obtained from copolymerization of acrylonitrile and methyl acrylate, with multiple functional groups such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C
O and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, which can potentially form coordination and hydrogen bond interactions simultaneously with perovskite precursors and solvent and as a result modulate film formation. In addition, we found that the strong hydrogen bonding between AMAC and precursor solvent contributes to the retarded crystallization process of the CsPbIxBr3−x film. These interactions result in a remarkable increase of grain size, with much reduced grain boundaries and defects. The improved quality of inorganic perovskite films displays a noticeable decrease of nonradiative recombination and prolonged charge lifetime. Benefiting from these improvements, the AMAC-incorporated inverted inorganic PSCs achieved a champion PCE of 21.7%, with an open circuit voltage (VOC) loss as low as 0.457 V, which is the highest PCE of inverted inorganic PSCs so far (Fig. S1 and Table S1, ESI†). Meanwhile, the conventional n–i–p device with AMAC incorporation also achieves a 21.8% PCE. In terms of stability, the addition of AMAC not only reduces the residual strain within the perovskite, but also influences the chemical environment of the I− ions and effectively inhibits I− ion migration, which contributes to a significant improvement in the operational stability of the device. With maximum power point (MPP) tracking and continuous standard irradiation, the AMAC device decays by only 8% at 2500 h, while the reference-device has decayed to 28% at 750 h.
N, which can potentially form coordination and hydrogen bond interactions simultaneously with perovskite precursors and solvent and as a result modulate film formation. In addition, we found that the strong hydrogen bonding between AMAC and precursor solvent contributes to the retarded crystallization process of the CsPbIxBr3−x film. These interactions result in a remarkable increase of grain size, with much reduced grain boundaries and defects. The improved quality of inorganic perovskite films displays a noticeable decrease of nonradiative recombination and prolonged charge lifetime. Benefiting from these improvements, the AMAC-incorporated inverted inorganic PSCs achieved a champion PCE of 21.7%, with an open circuit voltage (VOC) loss as low as 0.457 V, which is the highest PCE of inverted inorganic PSCs so far (Fig. S1 and Table S1, ESI†). Meanwhile, the conventional n–i–p device with AMAC incorporation also achieves a 21.8% PCE. In terms of stability, the addition of AMAC not only reduces the residual strain within the perovskite, but also influences the chemical environment of the I− ions and effectively inhibits I− ion migration, which contributes to a significant improvement in the operational stability of the device. With maximum power point (MPP) tracking and continuous standard irradiation, the AMAC device decays by only 8% at 2500 h, while the reference-device has decayed to 28% at 750 h.
Results and discussion
Firstly, we explored the effect of AMAC on the crystallization process and the morphology of CsPbI3−xBrx thin films through a series of tests. We used a one-step spin-coating method to prepare inorganic perovskite films and performed post-annealing at 210 °C in air with a relative humidity (RH) of 10–20%. Fig. S2 (ESI†) recorded the effect of AMAC on the crystallization process of the CsPbI3−xBrx film at different times during annealing. Both as-prepared films were colorless. The reference film started to turn black in 10 s, which was significantly postponed to 40 s for the AMAC-film. We also monitored the phase transition of the perovskite with and without AMAC incorporation using in situ X-ray diffraction (XRD, Fig. 1a). Both as-prepared wet films showed a diffraction peak at 11.9° ascribed to DMAPbI3. This DMAPbI3 phase gradually transformed into the perovskite phase at 14.6° during film annealing. However, the appearance of the perovskite signal was remarkably delayed for the AMAC-film compared with the reference film, along with the prolonged dwelling time of the DMAPbI3 phase, indicating delayed onset of phase transition with AMAC incorporation. | ||
| Fig. 1 (a) In situ X-ray diffraction tracking of films with and without AMAC incorporation annealed at 210 °C. (b) In situ PL measurements of the perovskite films during the thermal annealing process. (c) Evolution of PL intensity at 680 nm and PL peak position for perovskite films during the annealing process. (d) Calculation of binding energy between AMAC and DMSO, DMA+. (e) Average viscosity of DMSO solutions containing different concentrations of AMAC (inset: viscosity over time). (f) Particle size in solution–solvent complexes of perovskite precursors with different concentrations of AMAC (inset: Particle size distribution). | ||
Additionally, we tracked the optical variation of perovskite films with and without AMAC incorporation by in situ photoluminescence (PL) spectroscopy (Fig. 1b). Prior to nucleation, the PL signal of the as-prepared film is negligible. A broad PL signal can be detected in the wavelength range of 600–700 nm once nucleation occurs. As the nucleus grows, the PL peaks appear red-shifted due to the increase in the crystal size and the occurrence of ion-exchange reactions between precursor components. We observed a significant delay in the appearance of the PL signal and prolonged time to reach the maximum PL intensity with AMAC incorporation (Fig. 1c). This suggests that the nucleation and growth stages of the inorganic perovskite have been retarded, which is consistent with the in situ XRD results described above.
We assume that AMAC has interactions (e.g. hydrogen binding) with components in the perovskite precursor solution, such as DMA+ and dimethyl sulfoxide (DMSO), which slows down the ion exchange reaction between DMA+ and Cs+, and the upcoming crystallization process. To prove it, we performed FTIR measurements (Fig. S3, ESI†). The molecular interaction between AMAC and DMSO can be well recognized by observing SO and CO peak shifts. This is similar to those between AMAC and DMA+. DFT calculation results in Fig. 1d further reveal that the average binding energy of AMAC is stronger with DMSO than that with DMA+. This implies that the addition of AMAC may lead to the retardation of DMSO evaporation during film annealing as a result of sufficient time window for crystal growth and phase transition. This result is also solidified by viscosity measurement, whose results are recorded in Fig. 1e. The average viscosity of precursor solutions is gradually increased from 2.08 to 3.45 with increasing AMAC concentration, indicating that interactions exist.
We also turned to look at the colloidal chemistry in perovskite precursors with AMAC incorporation, using the dynamic light scattering (DLS) technique. As shown in Fig. 1f, the diameter of the perovskite–solvent complex becomes larger and the distribution narrows with the increase of AMAC incorporation. This result indicates that the incorporation of AMAC has a significant effect on the nucleation kinetics of perovskite grains, which may be due to the strong interaction between the AMAC and the perovskite precursor. It is noteworthy that the colloidal size becomes smaller and the distribution widens when the AMAC incorporation is increased to 0.6 mg mL−1. This suggests that excess AMAC will make the diameter distribution of the perovskite–solvent complexes inhomogeneous, thus affecting the film quality. Combined with previous results, we speculate that the incorporation of a small amount of AMAC reduces the number of nucleation sites, thus decreasing the nucleation rate during perovskite nucleus formation. The perovskite film with a larger grain size is therefore realized with improved uniformity during the subsequent crystallization process.41
The strong interactions between AMAC and precursors will inevitably lead to the modification of the perovskite film morphology, as is evaluated by top-view scanning electron microscopy (SEM, Fig. 2a and Fig. S4, ESI†). All films display condensed coverage without pinholes. The grains become smoother and the average grain size increases with AMAC incorporation, which is conducive to suppressing the generation of defects during the growth of perovskite grains. The changes in perovskite grains after AMAC incorporation are consistent with the DLS results. Through atomic force microscopy (AFM) characterization, it is found that the AMAC-films have much smaller root-mean-square (RMS) roughness than the reference film (Fig. 2b). The reduced RMS is beneficial to the improvement of the layer-to-layer contact, which should consequently reduce the series resistance at the interface.42,43
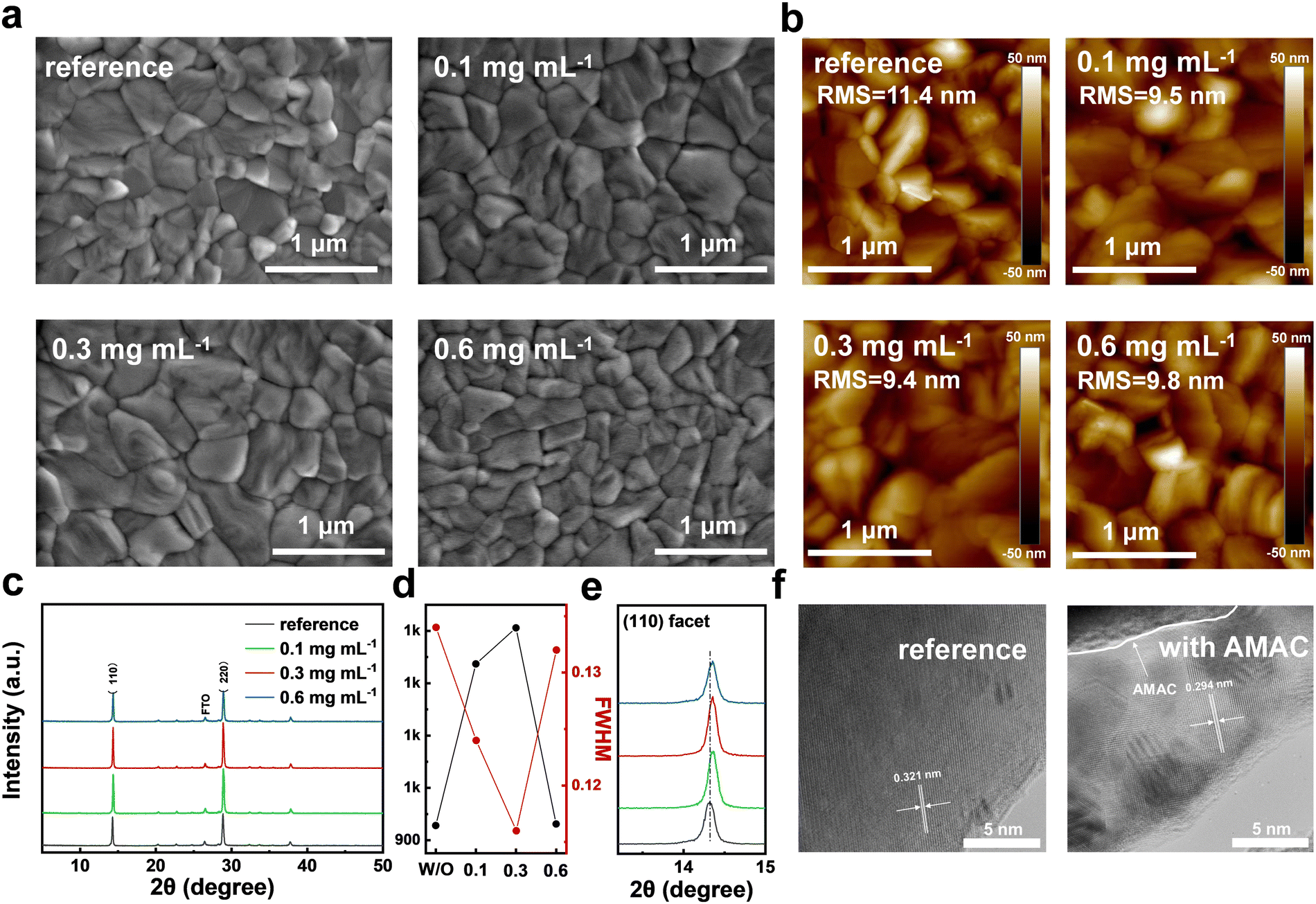 | ||
| Fig. 2 (a) Top-view SEM images with different concentrations of AMAC incorporation. (b) AFM images of perovskite films with different concentrations of AMAC incorporation. (c) XRD patterns of inorganic perovskite films with different concentrations of AMAC incorporation. (d) Peak intensity and FWHM derived from XRD patterns. (e) XRD patterns of the perovskite (110) facet. (f) TEM images of inorganic perovskite films. | ||
The variation of the perovskite structure and crystallinity with and without AMAC incorporation was then characterized by XRD. As shown in Fig. 2c, all films exhibited characteristic perovskite peaks at 14.4° and 28.9°, corresponding to (110) and (220) facets. As AMAC concentration increases, the peak intensity shows a tendency to grow stronger, followed by a decrease. In particular, the 0.3 mg mL−1 AMAC-film exhibits peaks with the highest intensity and the narrowest full-width at half-maximum (FWHM), indicating the highest crystallinity (Fig. 2d). In addition, we found that the incorporation of AMAC shifts the perovskite diffraction peaks slightly to a large angle, which implies lattice contraction (Fig. 2e). Transmission electron microscopy (TEM) images confirm this observation, with the lattice spacing decreasing from 0.321 nm to 0.294 nm with AMAC incorporation (Fig. 2f). The lattice contraction may be due to a complete volatilization of DMA+ as a result of favorable interaction with AMAC.26,44,45
The electrostatic surface potential (ESP) of the AMAC monomer in Fig. S5 (ESI†) shows that the CO and CN functional groups exhibit higher electron density, which is expected to couple with the undercoordinated lead from the perovskite. Experimentally, X-ray photoelectron spectroscopy (XPS) was employed to investigate the molecular interaction between the AMAC and the perovskite. As shown in Fig. 3a, the Pb 4f peaks of the reference film are 137.8 eV and 142.7 eV, which shift to 137.3 eV and 142.2 eV with AMAC incorporation. The N 1s and O 1s peaks existing at 399.7 eV and 531.0 eV in pure AMAC shift to 399.9 eV and 532.2 eV, respectively, after AMAC was incorporated into perovskite (Fig. S6, ESI†). We postulate that the N 1s and O 1s peaks shifting towards higher binding energy and Pb 4f peaks shifting towards lower binding energy is due to the synergistic bonding effect of CN⋯Pb2+ and CO⋯Pb2+. We also observed a slight shift of I 3d peaks with AMAC incorporation, which may be owing to the intervention of AMAC to change the coupling of the Pb–I bond, or the interaction of AMAC with I ions (Fig. 3b). We further conducted Fourier transform infrared (FTIR) measurements on pure AMAC and AMAC–PbI2 complex to probe the interaction of AMAC with the perovskite (Fig. S7, ESI†). The CO signal from AMAC shifts from 1732 cm−1 to 1722 cm−1, and the signal assigned to CN shifts from 2243 cm−1 to 2246 cm−1 (Fig. 3c), indicating interactions between the two. Based on the above analysis, we depicted the schematic diagram of crystallization pathways of CsPbI3−xBrx with AMAC incorporation in Fig. 3d.
 | ||
| Fig. 3 (a) and (b) XPS spectra of Pb 4f and I 3d from perovskite films with and without AMAC incorporation. (c) FTIR spectra of AMAC and AMAC–PbI2 films. (d) Schematic diagram of crystallization pathways of the perovskite with AMAC incorporation. | ||
We then fabricated inverted inorganic PSCs with and without AMAC incorporation in an air environment. The inverted device structure is F-doped tin oxide (FTO)/NiOx/[2-(3,6-dimethoxy-9H-carbazol-9-yl)ethyl] phosphonic acid (MeO-2PACz)/perovskite/[6,6]-phenyl-C61-butyric acid methyl ester (PCBM)/bathocuproine (BCP)/Ag. The current–density–voltage (J–V) performance of the devices with different amounts of AMAC was evaluated under 100 mw cm−2 illumination. The reference PSCs exhibit a VOC of 1.172 V, a JSC of 20.30 mA cm−2, an FF of 81.4% and a PCE of 19.4%. At an AMAC concentration of 0.3 mg mL−1, the PSCs exhibit a VOC of 1.273 V, a JSC of 20.30 mA cm−2, and an FF of 83.9%, yielding a champion PCE of 21.7%, with a VOC loss of only 0.457 V, marking the highest reported PCE for inverted inorganic PSCs (Fig. 4a and Table 1). Fig. 4b shows the statistical parameter distributions without and with 0.3 mg mL−1 AMAC-incorporated PSCs from 30 devices each. The hysteresis index (HI) in Fig. 4c decreases from 8.2% to 4.1% with AMAC incorporation (detailed J–V parameters shown in Table S2, ESI†), implying enhanced charge diffusion and transfer. The external quantum efficiency (EQE) spectra are shown in Fig. 4d. The integrated JSC values are 20.22 mA cm−2 and 20.24 mA cm−2 for the reference and AMAC devices, respectively, which are in agreement with the J–V performance. Fig. 4e shows the stabilized output power (SPO) of the PSCs detected under MPP. The PCEs of the reference and the AMAC device show stabilized values of 19.0% and 21.4% under one sun illumination for 600 s. We also fabricated conventional n–i–p CsPbI3−xBrx PSCs with a structure of FTO/TiO2/perovskite/2,2',7,7'-tetrakis [N,N-di(4-methoxyphenyl)amino]-9,9'-spirobifluorene (spiro-OMeTAD)/Au. The resultant J–V performance in Fig. 4f shows a PCE improvement from 19.6% to 21.8%, with a VOC of 1.265 V, a JSC of 20.82 mA cm−2, an FF of 82.7%. These results highlight the universality of AMAC incorporation for inorganic PSCs with different structures.
| Device | V OC (V) | PCE (%) | FF (%) | J SC (mA cm−2) |
|---|---|---|---|---|
| Reference | 1.172 | 19.4 | 81.4 | 20.30 |
| (1.169 ± 0.004) | (19.1 ± 0.1) | (80.6 ± 0.5) | (20.25 ± 0.03) | |
| 0.1 mg mL−1 | 1.254 | 21.2 | 83.3 | 20.29 |
| (1.251 ± 0.005) | (21.0 ± 0.1) | (82.9 ± 0.3) | (20.27 ± 0.03) | |
| 0.3 mg mL−1 | 1.273 | 21.7 | 83.9 | 20.30 |
| (1.267 ± 0.004) | (21.5 ± 0.1) | (83.5 ± 0.2) | (20.26 ± 0.03) | |
| 0.5 mg mL−1 | 1.235 | 20.9 | 83.2 | 20.30 |
| (1.229 ± 0.004) | (20.6 ± 0.1) | (82.7 ± 0.3) | (20.26 ± 0.03) |
 | ||
| Fig. 4 (a) J–V characterization of p–i–n CsPbI3−xBrx PSCs incorporated with different amounts of AMAC under 100 mW cm−2 illumination (the active area is 0.09 cm2). (b) Statistical box charts of PV parameters. (c) Hysteresis with and without AMAC-incorporated PSCs. (d) EQE and integrated JSC. (e) SPO at MPP tracking. (f) J–V characterization of n–i–p CsPbI3−xBrx PSCs with and without AMAC incorporation. | ||
To investigate the effect of AMAC incorporation on defect control in inorganic perovskite thin films, we performed steady-state photoluminescence (PL) and time-resolved photoluminescence (TRPL) measurements. The PL spectra in Fig. 5a display a greatly enhanced peak intensity by more than a factor of three with AMAC incorporation, with a slight blue shift, which indicates that defects in the perovskite films have been passivated. The TRPL spectra of both films in Fig. 5b show double exponential decay, where the fitted average charge lifetimes are 66.07 ns and 118.02 ns for reference and incorporated films, respectively (Table S3, ESI†). The PL-mapping measurements also display longer average lifetime in a monitored area of 50 μm2 (Fig. S8, ESI†). These results suggest that AMAC incorporation improves carrier lifetimes in the perovskite film. We additionally illuminated FTO/NiOx/perovskite (with or without AMAC)/PCBM laminated samples in different excitation directions to see the interfacial charge transfer behavior. Fig. S9 (ESI†) shows that the AMAC-sample exhibits greater quenching than the reference one, suggesting that the incorporation of AMAC facilitates interfacial charge extraction. The space-charge-limited currents (SCLC) were measured using the hole-only devices structured FTO/NiOx/perovskite/spiro-OMeTAD/Au. The trap-filled limit voltage (VTFL) extracted from the J–V curve is used to estimate the defect density, which in the reference device is 6.24 × 1015 cm−3, and is reduced to 4.30 × 1015 cm−3 with AMAC incorporation (Fig. 5c and d). In addition, femtosecond transient absorption spectroscopy (TAS) was utilized to further investigate carrier complexation in perovskite films with and without AMAC incorporation. Fig. S10 (ESI†) shows the visual photoinduced absorption at delay time and wavelength. Upon excitation, the negative ground state bleaching band is centered at 701 nm, and the reference film and AMAC film decay kinetics follow double exponential decay with average time constants of 3.8 ns and 7.4 ns, respectively (Fig. 5e). In addition, we performed electrochemical impedance spectroscopy (EIS) studies on PSCs with and without AMAC incorporation in a dark environment (Fig. S11, ESI†). The values of the recombination resistance (Rrec) based on the fitted Nyquist plots are shown in Table S3 (ESI†). The Rrec of the AMAC device is 6551 Ω, which is much larger than that of the reference device (Rrec = 2827 Ω), suggesting that AMAC incorporation resulted in a reduction of charge recombination in the device. The dark J–V characterizations in Fig. S12 (ESI†) show that the current output of the AMAC-incorporated device is lower than that of the reference device. Since the rate of carrier production in dark environments is proportional to the trap density,46 AMAC incorporation can effectively passivate defects in the perovskite. Fig. 5f shows the variation of Voc at different light intensities for PSCs with and without AMAC incorporation. The ideal factor of the AMAC device (n = 1.117) is much lower than that of the reference device (n = 1.873), indicating that the incorporation of AMAC effectively suppresses the defect-assisted recombination.
 | ||
| Fig. 5 (a) Steady-state PL spectra of perovskite films with and without AMAC incorporation. (b) TRPL spectra of perovskite films with and without AMAC incorporation. (c) and (d) SCLC measurements without and with AMAC incorporation, devices structure: FTO/NiOx/perovskite/Spiro-OMeTAD/Au. (e) Dynamic evolution of the bleaching recovery from measuring F-TAS. (f) VOC variation with light intensity. | ||
In addition to defects, the energy level mismatch between the perovskite and the neighboring transporting layer will lead to interfacial charge accumulation, which is unfavorable for charge collection. In order to investigate the effect of AMAC incorporation on the energy levels of perovskite, ultraviolet-visible absorption spectroscopy (UV-vis) and ultraviolet photoelectron spectroscopy (UPS) were carried out to obtain the energy level information of reference and AMAC-films (Fig. 6a and b). The corresponding energy diagram is depicted in Fig. 6c. The reference perovskite has a Fermi level of −4.22 eV, a conduction band (CB) of −3.88 eV, and a valence band (VB) of −5.61 eV. The AMAC-incorporated film has an n-type nature with a shallower Fermi level of −3.94 eV, a CB of −3.75 eV, and a VB of −5.48 eV. This energy level arrangement with AMAC incorporation is more favorable for efficient charge separation and transfer, thus increasing both VOC and FF. This conclusion is further corroborated by Kelvin probe force microscopy (KPFM) measurements. Fig. 6d and e shows the surface potential in a range of 2 μm. The surface potential of the reference film is 65 mV, which rises to 183 mV with AMAC incorporation, along with increased Fermi level. Interestingly, the surface potential of the AMAC film is more uniform, which should be attributed to effective defect control. The Mott–Schottky analysis was carried out to measure the built-in potential (Vbi) induced by photogenerated carrier separation in perovskite films. The C–V curves in Fig. 6f show that the Vbi value of the AMAC-incorporated device (1.22 V) is 90 mV higher than that of the reference device (1.13 V). A higher Vbi facilitates the separation and transfer of the effective charge to the respective charge transport layer, thus increasing the VOC.
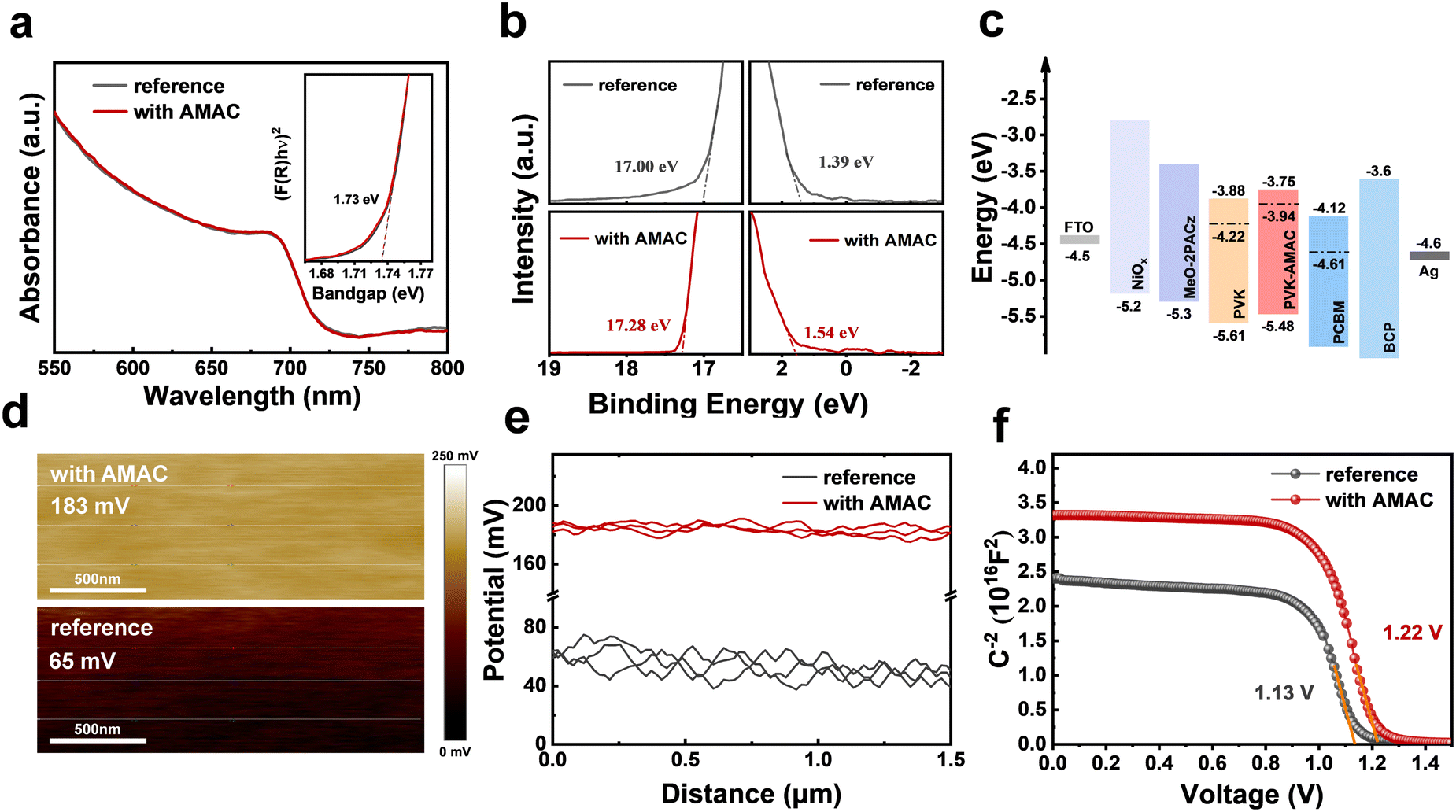 | ||
| Fig. 6 (a) UV-vis spectra of films with and without AMAC incorporation (inset: Tauc plot). (b) UPS patterns of perovskite films with and without AMAC incorporation. (c) Energy diagram (vs. vacuum). (d) and (e) KPFM images and corresponding surface potential. (f) C–V characteristic plots of devices. | ||
Apart from appreciably enhanced device PV performance, we also anticipate that the AMAC incorporation can contribute to stability improvement of both inorganic perovskite films and devices. Fig. S13 (ESI†) shows the photographs of perovskite films with and without AMAC incorporation stored under an atmosphere with an RH value of 20–30%. Both fresh films exhibited black perovskite phase upon preparation. The reference film started to show small yellow dots in 120 h and turned completely yellow in 216 h. In contrast, the AMAC film remained completely black in 408 h. We further stored the films with and without AMAC incorporation at an RH value of ∼ 40% with a fixed temperature of 180 °C and collected the photographs of the films at different time points. The corresponding XRD patterns were also collected to monitor the structural evolution (Fig. S14, ESI†). The reference film started to decompose at 360 s with the appearance of the δ-phase. In 600 s, the black phase has been completely transformed into the δ-phase along with the appearance of PbI2 phase. However, the diffraction intensity of the black phase of the AMAC-film shows a negligible decrease at 600 s without phase transformation. These results indicate that AMAC incorporation leads to substantially enhanced stability of inorganic perovskite films, not only under ambient but also under heat and damp conditions.
As for the device stability, we evaluated the ambient stability of the unencapsulated PSCs by storing them in an RH of 20–30%. As can be seen in Fig. 7a, after aging under ambient conditions for 2000 h, the PCEs with AMAC incorporation maintained 92% of the initial value, whereas the reference device has decayed to half of initial value in 1000 h. Further evaluation of the device thermal stability was conducted under continuous heating at 65 °C in a nitrogen environment. After 300 h of continuous heating, the devices without and with AMAC incorporation maintained 41.5% and 84.2% of their original PCEs (Fig. S15, ESI†). In addition, we tested the long-term stability of the unencapsulated devices under the MPP tracking mode in RH ∼ 10% (Fig. 7b). After 2500 h of continuous illumination under one sun, the AMAC device maintained 92% of the initial PCE. However, the reference device dropped to half of the initial PCE in 650 h. These results indicate that AMAC incorporation remarkably improves the long-term environmental and operational stabilities of PSCs.
 | ||
| Fig. 7 (a) Device stability stored at RH 20–30%. (b) Stabilized MPP tracking. (35 °C, RH ∼ 10%) encapsulated devices exposed to continuous one sun illumination for 2500 h. (c) and (d) GIXRD spectra of perovskite films without and with AMAC at different slant angles. (e) linear fit of 2θ-sin2φ for perovskite films with and without AMAC. (f) and (g) ToF-SIMS spectra of PSCs without and with AMAC incorporation before and after MPP aging. (h) Ion migration activation energy of perovskite films without and with AMAC incorporation. | ||
Since AMAC incorporation can adjust the perovskite grain growth, we expect that residue strain on the perovskite surface can be regulated as well, which may contribute to the enhanced stability.11,47 Therefore, grazing incidence X-ray diffraction (GIXRD) was utilized to investigate the residual strain on the perovskite surface. To ensure the reliability of the structural information, the (110) surface with a diffraction angle of 14.4° was tested and calculated using equation 2θ = sin2φ (Fig. 7c and d).48,49 With tilt angle (φ) increasing from 0.3° to 1.5°, the diffraction peaks of the AMAC film are almost unchanged, while that of the reference film is gradually shifted to small angles. This peak shift implies that the perovskite lattice spacing is extended along the in-plane direction. Fig. 7e shows the linear relationship between 2θ and sin2φ, where the slopes of the fitted lines reflect the different degrees of residual strain within the perovskite crystal.50–52 The slope increases from −0.033 to −0.005 with AMAC incorporation, suggesting that AMAC can effectively release the residual strain in the perovskite. According to Bragg's Law and Hooke's Law, the residual strain (σ) in different films can be further quantified by the following equation:47
 | (1) |
Furthermore, ion migration is known to be one of the origins of device performance degradation. It can induce phase separation and undesirable chemical or physical processes in perovskite films.58,59 It has been found that the migration of I− ions with the lowest mobility energy tends to be more prevalent in the perovskite and illumination accelerates the I− ion migration process.60–62 We have shown in XPS results that AMAC incorporation affects the chemical environment of I− ions, and the AMAC-film exhibits greatly enlarged perovskite grain size and reduced grain boundaries. Therefore, we believe that the migration of I− ions should be inhibited. To prove it, we performed the time-of-flight secondary-ion mass spectrometry (ToF-SIMS) measurements on devices with and without AMAC after aging in the MPP mode used in Fig. 7b. As can be seen in Fig. 7f and g, after aging, the signal of I− ions is increased in the adjacent PCBM layer for the reference device, while the AMAC device shows an almost unchanged profile of I− ions. This means that the incorporation of AMAC has suppressed the migration of I− ions under operational conditions. Interestingly, we also noticed that the Ag signals in PCBM and perovskite layers are increased significantly for the reference device, in contrast to the negligible change of the Ag signal in the AMAC device. It has been reported in the literature that the penetration of the Ag metal into the perovskite is detrimental as it can react with iodide, causing severe short circuit and perovskite degradation.63–65 The successful prohibition of Ag migration provides additional credit for the enhanced device stability.
We further performed the measurement of the ionic conductivity upon varied temperatures with a device structure of FTO/perovskite (with or without AMAC)/Au, a widely adopted method for characterizing ion migration in the perovskite film.66–68 The activation energy (Ea) for ion migration can be extracted by a linear fitting of the data based on the Nernst–Einstein equation, σ(T) = σ0T exp(−EakBT), where σ(T) is the conductivity as a function of the temperature T, σ0 is a constant and kB is the Boltzmann constant. The fitted Ea value for the AMAC-film in Fig. 7h is 111 meV, which is appreciably higher than that of the reference film (75 meV), implying that AMAC incorporation can effectively immobilize ions and inhibit ion migration, thus contributing to the enhanced device operational stability.
Conclusions
In this work, we incorporated copolymer AMAC into the perovskite precursor solution to fabricate efficient and stable inverted inorganic CsPbI3−xBrx PSCs in an air environment. We found that the incorporation of AMAC retarded rapid crystallization of the perovskite in air and improved film quality. Specifically, the stronger interaction between AMAC and the precursor solvent prevents DMSO evaporation during perovskite annealing, delaying the onset of the phase transition. Additionally, the interaction between AMAC and DMA+ reduces the exchange rate between Cs+ and DMA+ ions during phase transition. The comprehensive retardation crystallization results in inorganic perovskite films with an enlarged grain size and reduced defect density. With the optimized AMAC concentration, the device obtained a PCE of 21.7%, much higher than the reference device (19.4%), with a high VOC value of 1.273 V. Encouragingly, with MPP tracking and continuous standard irradiation, the AMAC-incorporated device can maintain an initial efficiency of 92% for 2500 h, far better than the reference device's initial efficiency of only 28% for less than 750 h. The improved operational stability is mainly because the AMAC incorporation not only reduces the residual stress within the perovskite and minimizes defects, but also increases energy barrier for iodide ion migration. We extended this method to conventional structured inorganic PSCs with a high efficiency of 21.8%, highlighting the importance of comprehensive retardation crystallization by simultaneously regulating solvent volatilization and ion exchange during inorganic perovskite film preparation in air for high quality inorganic PSCs.Data availability
Further information and requests for resources should be directed to and will be fulfilled by the corresponding author.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work is supported by the Shaanxi Provincial Science Fund for Distinguished Young Scholars (2023-JC-JQ34), the National Natural Science Foundation of China (52211540391 and 52350710208), the Chinese National 1000-Talent-Plan Program (1110010341), the Fundamental Research Funds for the Central Universities (GK202309025), and the Young Scientist Innovation Project of School of Materials Science and Engineering at Shaanxi Normal University (SNNU) (2023YSIP-MSE-SNNU006).References
- Best Research-Cell Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency.html, (accessed: 2024).
- C. Wang, Y. Jiang, H. Xu, N. Zheng, G. Bai, Y. Zha, H. Qi, Z. Bian, X. Zhan and Z. Liu, eScience, 2023, 3, 100113 CrossRef.
- P. Zhu, D. Wang, Y. Zhang, Z. Liang, J. Li, J. Zeng, J. Zhang, Y. Xu, S. Wu, Z. Liu, X. Zhou, B. Hu, F. He, L. Zhang, X. Pan, X. Wang, N.-G. Park and B. Xu, Science, 2024, 383, 524–531 CrossRef CAS PubMed.
- J. Liu, Y. He, L. Ding, H. Zhang, Q. Li, L. Jia, J. Yu, T. W. Lau, M. Li, Y. Qin, X. Gu, F. Zhang, Q. Li, Y. Yang, S. Zhao, X. Wu, J. Liu, T. Liu, Y. Gao, Y. Wang, X. Dong, H. Chen, P. Li, T. Zhou, M. Yang, X. Ru, F. Peng, S. Yin, M. Qu, D. Zhao, Z. Zhao, M. Li, P. Guo, H. Yan, C. Xiao, P. Xiao, J. Yin, X. Zhang, Z. Li, B. He and X. Xu, Nature, 2024, 635, 596–603 Search PubMed.
- F. Xu, E. Aydin, J. Liu, E. Ugur, G. T. Harrison, L. Xu, B. Vishal, B. K. Yildirim, M. Wang, R. Ali, A. S. Subbiah, A. Yazmaciyan, S. Zhumagali, W. Yan, Y. Gao, Z. Song, C. Li, S. Fu, B. Chen, A. ur Rehman, M. Babics, A. Razzaq, M. De Bastiani, T. G. Allen, U. Schwingenschlögl, Y. Yan, F. Laquai, E. H. Sargent and S. De Wolf, Joule, 2024, 8, 224–240 Search PubMed.
- S. Liu, J. Li, W. Xiao, R. Chen, Z. Sun, Y. Zhang, X. Lei, S. Hu, M. Kober-Czerny, J. Wang, F. Ren, Q. Zhou, H. Raza, Y. Gao, Y. Ji, S. Li, H. Li, L. Qiu, W. Huang, Y. Zhao, B. Xu, Z. Liu, H. J. Snaith, N.-G. Park and W. Chen, Nature, 2024, 632, 536–542 CrossRef PubMed.
- T. Li, J. Xu, R. Lin, S. Teale, H. Li, Z. Liu, C. Duan, Q. Zhao, K. Xiao, P. Wu, B. Chen, S. Jiang, S. Xiong, H. Luo, S. Wan, L. Li, Q. Bao, Y. Tian, X. Gao, J. Xie, E. H. Sargent and H. Tan, Nat. Energy, 2023, 8, 610–620 CrossRef CAS.
- K. O. Brinkmann, P. Wang, F. Lang, W. Li, X. Guo, F. Zimmermann, S. Olthof, D. Neher, Y. Hou, M. Stolterfoht, T. Wang, A. B. Djurišić and T. Riedl, Nat. Rev. Mater., 2024, 9, 202–217 CrossRef CAS.
- Q. Jiang and K. Zhu, Nat. Rev. Mater., 2024, 9, 399–419 CrossRef CAS.
- S. Wang, S. Qi, H. Sun, P. Wang, Y. Zhao and X. Zhang, Adv. Mater., 2024, 63, e202400018 Search PubMed.
- Z. Wang, T. Xu, N. Li, Y. Liu, K. Li, Z. Fan, J. Tan, D. Chen, S. Liu and W. Xiang, Energy Environ. Sci., 2024, 17, 7271–7280 RSC.
- W. Xiang, S. Liu and W. Tress, Energy Environ. Sci., 2021, 14, 2090–2113 RSC.
- J. Qiu, X. Mei, M. Zhang, G. Wang, S. Zou, L. Wen, J. Huang, Y. Hua and X. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202401751 CrossRef CAS PubMed.
- C. Shen, T. Ye, P. Yang and G. Chen, Adv. Mater., 2024, 36, 2401498 Search PubMed.
- T. Xu, W. Xiang, X. Ru, Z. Wang, Y. Liu, N. Li, H. Xu and S. Liu, Adv. Mater., 2024, 36, 2312237 CrossRef CAS PubMed.
- H. Liu, Z. Zhang, J. Xu, H. Han, C. Zhao, Y. Fu, K. Lang, F. Shen, P. Zou, X. Liu, R. Shi, Z. Su, X. Gao, S. F. Liu and J. Yao, Adv. Energy Mater., 2024, 2402142 CrossRef CAS.
- S. Fu, J. Le, X. Guo, N. Sun, W. Zhang, W. Song and J. Fang, Adv. Mater., 2022, 34, 2205066 CrossRef CAS PubMed.
- W. Zhang, H. Yuan, X. Li, X. Guo, C. Lu, A. Liu, H. Yang, L. Xu, X. Shi, Z. Fang, H. Yang, Y. Cheng and J. Fang, Adv. Energy Mater., 2023, 35, 2303674 CrossRef CAS.
- Y.-M. Xie, X. Qifan and H.-L. Yip, Adv. Energy Mater., 2021, 11, 2100784 CrossRef CAS.
- Z. Shao, Z. Wang, Z. Li, Y. Fan, H. Meng, R. Liu, Y. Wang, A. Hagfeldt, G. Cui and S. Pang, Angew. Chem., Int. Ed., 2019, 58, 5587–5591 CrossRef CAS PubMed.
- K. Liu, Y. Luo, Y. Jin, T. Liu, Y. Liang, L. Yang, P. Song, Z. Liu, C. Tian, L. Xie and Z. Wei, Nat. Commun., 2022, 13, 4891 CrossRef CAS.
- R. Guo, Q. Xiong, A. Ulatowski, S. Li, Z. Ding, T. Xiao, S. Liang, J. E. Heger, T. Guan, X. Jiang, K. Sun, L. K. Reb, M. A. Reus, A. Chumakov, M. Schwartzkopf, M. Yuan, Y. Hou, S. V. Roth, L. M. Herz, P. Gao and P. Müller-Buschbaum, Adv. Mater., 2024, 36, 2310237 CrossRef CAS PubMed.
- Y. Du, Q. Tian, X. Chang, J. Fang, X. Gu, X. He, X. Ren, K. Zhao and S. Liu, Adv. Mater., 2022, 34, 2106750 CrossRef CAS.
- Y. Li, Y. Yan, Y. Fu, W. Jiang, M. Liu, M. Chen, X. Huang, G. Lu, X. Lu, J. Yin, S. Wu and A. K.-Y. Jen, Angew. Chem., Int. Ed., 2024, 63, e202412515 CrossRef CAS.
- Y. Jiang, T.-F. Xu, H.-Q. Du, M. U. Rothmann, Z.-W. Yin, Y. Yuan, W.-C. Xiang, Z.-Y. Hu, G.-J. Liang, S.-Z. Liu, M. K. Nazeeruddin, Y.-B. Cheng and W. Li, Joule, 2023, 7, 2905–2922 CrossRef CAS.
- M.-H. Li, S. Wang, X. Ma, R. Long, J. Wu, M. Xiao, J. Fu, Z. Jiang, G. Chen, Y. Jiang and J.-S. Hu, Joule, 2023, 7, 2595–2608 CrossRef CAS.
- N. Sun, S. Fu, Y. Li, L. Chen, J. Chung, M. M. Saeed, K. Dolia, A. Rahimi, C. Li, Z. Song and Y. Yan, Adv. Funct. Mater., 2024, 34, 2309894 CrossRef CAS.
- W. Ke, I. Spanopoulos, C. C. Stoumpos and M. G. Kanatzidis, Nat. Commun., 2018, 9, 4785 CrossRef.
- K. Wang, Z. Jin, L. Liang, H. Bian, D. Bai, H. Wang, J. Zhang, Q. Wang and S. Liu, Nat. Commun., 2018, 9, 4544 Search PubMed.
- T. Li, W. Li, K. Wang, Y. Tong, H. Wang, Y. Chen, H. Qi, Z. Kang and H. Wang, Angew. Chem., Int. Ed., 2024, 63, e202407508 Search PubMed.
- W. Chen, X. Li, Y. Li and Y. Li, Energy Environ. Sci., 2020, 13, 1971–1996 RSC.
- J. Huang, H. Wang, C. Jia, Y. Tang, H. Yang, C. Chen, K. Gou, Y. Zhou, D. Zhang and S. Liu, Prog. Mater. Sci., 2024, 141, 101223 CrossRef CAS.
- J. Wang, L. Bi, Q. Fu and A. K.-Y. Jen, Adv. Energy Mater., 2024, 14, 2401414 CrossRef CAS.
- Z. Wu, S. Sang, J. Zheng, Q. Gao, B. Huang, F. Li, K. Sun and S. Chen, Angew. Chem., Int. Ed., 2024, 63, e202319170 CrossRef CAS PubMed.
- C. Chen, X. Wang, Z. Li, X. Du, Z. Shao, X. Sun, D. Liu, C. Gao, L. Hao, Q. Zhao, B. Zhang, G. Cui and S. Pang, Angew. Chem., Int. Ed., 2022, 61, e202113932 CrossRef CAS.
- D. Bi, C. Yi, J. Luo, J.-D. Décoppet, F. Zhang, Shaik M. Zakeeruddin, X. Li, A. Hagfeldt and M. Grätzel, Nat. Energy, 2016, 1, 16142 CrossRef CAS.
- L. Zuo, H. Guo, D. W. de Quilettes, S. Jariwala, N. De Marco, S. Dong, R. DeBlock, D. S. Ginger, B. Dunn, M. Wang and Y. Yang, Sci. Adv., 2017, 3, e1700106 CrossRef PubMed.
- L. Liu, B. Farhadi, J. Li, S. Liu, L. Lu, H. Wang, M. Du, L. Yang, S. Bao, X. Jiang, X. Dong, Q. Miao, D. Li, K. Wang and S. F. Liu, Angew. Chem., Int. Ed., 2024, 63, e202317972 CrossRef CAS PubMed.
- H. Dong, G. Shen, H. Fang, Y. Li, X. Gao, Q. Song, X. Xu, Y. Wang, C. Mu and D. Xu, Adv. Funct. Mater., 2024, 34, 2402394 CrossRef CAS.
- T. Wang, Z. Wan, X. Min, R. Chen, Y. Li, J. Yang, X. Pu, H. Chen, X. He, Q. Cao, G. Feng, X. Chen, Z. Ma, L. Jiang, Z. Liu, Z. Li, W. Chen and X. Li, Adv. Energy Mater., 2024, 14, 2302552 CrossRef CAS.
- N. Li, S. Tao, Y. Chen, X. Niu, C. K. Onwudinanti, C. Hu, Z. Qiu, Z. Xu, G. Zheng, L. Wang, Y. Zhang, L. Li, H. Liu, Y. Lun, J. Hong, X. Wang, Y. Liu, H. Xie, Y. Gao, Y. Bai, S. Yang, G. Brocks, Q. Chen and H. Zhou, Nat. Energy, 2019, 4, 408–415 CrossRef CAS.
- J. Ge, R. Chen, Y. Ma, Y. Wang, Y. Hu, L. Zhang, F. Li, X. Ma, S.-W. Tsang, J. You, A. K. Y. Jen and S. F. Liu, Angew. Chem., Int. Ed., 2024, 63, e202319282 CrossRef CAS.
- Z. Chang, D. Zheng, S. Zhao, L. Wang, S. Wu, L. Liu, Z. Li, L. Zhang, Q. Dong, H. Wang, S. Wang, K. Wang and S. Liu, Adv. Funct. Mater., 2023, 33, 2214983 CrossRef CAS.
- R. Prasanna, A. Gold-Parker, T. Leijtens, B. Conings, A. Babayigit, H.-G. Boyen, M. F. Toney and M. D. McGehee, J. Am. Chem. Soc., 2017, 139, 11117–11124 CrossRef CAS PubMed.
- J. Tong, J. Gong, M. Hu, S. K. Yadavalli, Z. Dai, F. Zhang, C. Xiao, J. Hao, M. Yang, M. A. Anderson, E. L. Ratcliff, J. J. Berry, N. P. Padture, Y. Zhou and K. Zhu, Matter, 2021, 4, 1365–1376 CrossRef.
- C. Li, Z. Song, D. Zhao, C. Xiao, B. Subedi, N. Shrestha, M. M. Junda, C. Wang, C.-S. Jiang, M. Al-Jassim, R. J. Ellingson, N. J. Podraza, K. Zhu and Y. Yan, Adv. Energy Mater., 2019, 9, 1803135 CrossRef.
- H. Wang, C. Zhu, L. Liu, S. Ma, P. Liu, J. Wu, C. Shi, Q. Du, Y. Hao, S. Xiang, H. Chen, P. Chen, Y. Bai, H. Zhou, Y. Li and Q. Chen, Adv. Mater., 2019, 31, 1904408 CrossRef PubMed.
- G. Yuan, W. Xie, Q. Song, S. Ma, Y. Ma, C. Shi, M. Xiao, F. Pei, X. Niu, Y. Zhang, J. Dou, C. Zhu, Y. Bai, Y. Wu, H. Wang, Q. Fan and Q. Chen, Adv. Funct. Mater., 2023, 35, 2211257 Search PubMed.
- C. Shi, Q. Song, H. Wang, S. Ma, C. Wang, X. Zhang, J. Dou, T. Song, P. Chen, H. Zhou, Y. Chen, C. Zhu, Y. Bai and Q. Chen, Nano-Micro Lett., 2022, 32, 2201193 CrossRef.
- R. Cao, K. Sun, C. Liu, Y. Mao, W. Guo, P. Ouyang, Y. Meng, R. Tian, L. Xie, X. Lü and Z. Ge, Nano-Micro Lett., 2024, 16, 178 CrossRef CAS PubMed.
- Y. Li, Y. Duan, Z. Liu, L. Yang, H. Li, Q. Fan, H. Zhou, Y. Sun, M. Wu, X. Ren, N. Yuan, J. Ding, S. Yang and S. Liu, Adv. Mater., 2024, 36, 2310711 CrossRef CAS.
- G. Abadias, E. Chason, J. Keckes, M. Sebastiani, G. B. Thompson, E. Barthel, G. L. Doll, C. E. Murray, C. H. Stoessel and L. Martinu, J. Vac. Sci. Technol., A, 2018, 36, 020801 CrossRef.
- J. Wu, S.-C. Liu, Z. Li, S. Wang, D.-J. Xue, Y. Lin and J.-S. Hu, Natl. Sci. Rev., 2021, 8(8), nwab047 CrossRef CAS PubMed.
- W. Meng, K. Zhang, A. Osvet, J. Zhang, W. Gruber, K. Forberich, B. Meyer, W. Heiss, T. Unruh, N. Li and C. J. Brabec, Joule, 2022, 6, 458–475 CrossRef CAS.
- Y. Zhao, I. Yavuz, M. Wang, M. H. Weber, M. Xu, J.-H. Lee, S. Tan, T. Huang, D. Meng, R. Wang, J. Xue, S.-J. Lee, S.-H. Bae, A. Zhang, S.-G. Choi, Y. Yin, J. Liu, T.-H. Han, Y. Shi, H. Ma, W. Yang, Q. Xing, Y. Zhou, P. Shi, S. Wang, E. Zhang, J. Bian, X. Pan, N.-G. Park, J.-W. Lee and Y. Yang, Nat. Mater., 2022, 21, 1396–1402 CrossRef CAS PubMed.
- J. T.-W. Wang, Z. Wang, S. Pathak, W. Zhang, D. W. de Quilettes, F. Wisnivesky-Rocca-Rivarola, J. Huang, P. K. Nayak, J. B. Patel, H. A. Mohd Yusof, Y. Vaynzof, R. Zhu, I. Ramirez, J. Zhang, C. Ducati, C. Grovenor, M. B. Johnston, D. S. Ginger, R. J. Nicholas and H. J. Snaith, Energy Environ. Sci., 2016, 9, 2892–2901 RSC.
- Y. Wang, D. Zheng, K. Wang, Q. Yang, J. Qian, J. Zhou, S. Liu and D. Yang, Angew. Chem., Int. Ed., 2024, 63, e202405878 CrossRef CAS PubMed.
- L. A. Muscarella and B. Ehrler, Joule, 2022, 6, 2016–2031 CrossRef CAS.
- Z. Fang, T. Nie, S. Liu and J. Ding, Adv. Func. Mater., 2024, 34, 2404402 CrossRef CAS.
- H. Zai, Y. Ma, Q. Chen and H. Zhou, J. Energy Chem., 2021, 63, 528–549 CrossRef CAS.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 RSC.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O’Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 7497 CrossRef CAS.
- M. Ghasemi, B. Guo, K. Darabi, T. Wang, K. Wang, C.-W. Huang, B. M. Lefler, L. Taussig, M. Chauhan, G. Baucom, T. Kim, E. D. Gomez, J. M. Atkin, S. Priya and A. Amassian, Nat. Mater., 2023, 22, 329–337 CrossRef CAS PubMed.
- W.-Q. Wu, P. N. Rudd, Z. Ni, C. H. Van Brackle, H. Wei, Q. Wang, B. R. Ecker, Y. Gao and J. Huang, J. Am. Chem. Soc., 2020, 142, 3989–3996 CrossRef CAS PubMed.
- A. Liu, X. Li, W. Zhang, H. Yang, X. Guo, C. Lu, H. Yuan, W. Ou-Yang and J. Fang, Adv. Funct. Mater., 2024, 34, 2307310 CrossRef CAS.
- Y. Ma, X. Du, R. Chen, L. Zhang, Z. An, A. K.-Y. Jen, J. You and S. Liu, Adv. Mater., 2023, 35, 2306947 CrossRef CAS PubMed.
- Y. Wang, J. Chen, Y. Zhang, W. L. Tan, Z. Ku, Y. Yuan, Q. Chen, W. Huang, C. R. McNeill, Y.-B. Cheng and J. Lu, Adv. Mater., 2024, 36, 2401416 CrossRef CAS PubMed.
- J. Cheng, H. Cao, S. Zhang, J. Shao, W. Yan, C. Peng, F. Yue and Z. Zhou, Adv. Mater., 2024, 36, 2410298 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00149h |
| This journal is © The Royal Society of Chemistry 2025 |