
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electricity-to-ammonia interconversion in protonic ceramic cells: advances, challenges and perspectives
Mingzhuang
Liang†
a,
Jinwook
Kim†
 b,
Xiaomin
Xu
c,
Hainan
Sun
d,
Yufei
Song
e,
SungHyun
Jeon
a,
Tae Ho
Shin
*f,
Zongping
Shao
*c and
WooChul
Jung
*ag
b,
Xiaomin
Xu
c,
Hainan
Sun
d,
Yufei
Song
e,
SungHyun
Jeon
a,
Tae Ho
Shin
*f,
Zongping
Shao
*c and
WooChul
Jung
*ag
aResearch Institute of Advanced Materials, Seoul National University (SNU), Seoul, 08826, Republic of Korea. E-mail: wcjung@snu.ac.kr
bDepartment of Materials Science and Engineering, Northwestern University, Evanston, Illinois 60208, USA
cWA School of Mines: Minerals, Energy and Chemical Engineering (WASM-MECE), Curtin University, Perth, WA 6845, Australia. E-mail: zongping.shao@curtin.edu.au
dSchool of Chemistry and Chemical Engineering, Nantong University, Nantong, 226019, P. R. China
eState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemical Engineering, Nanjing Tech University, Nanjing, 210009, P. R. China
fDivision for Low Carbon Energy & Materials DX, Korea Institute of Ceramic Engineering and Technology (KICET), Jinju-si 52851, Republic of Korea. E-mail: ths@kicet.re.kr
gDepartment of Materials Science and Engineering, Seoul National University, Seoul, 08826, Republic of Korea
First published on 10th March 2025
Abstract
NH3 is an attractive alternative fuel to hydrogen and methane, offering advantages such as easy compression at room temperature, straightforward storage and transportation, high volumetric energy density, and carbon-free nature. However, conventional NH3 synthesis requires high temperatures and pressures, resulting in substantial energy consumption and increased equipment and maintenance costs. Protonic ceramic cells (PCCs), as a cutting-edge energy conversion technology, can realize NH3 synthesis at moderate pressures and low-to-intermediate temperatures by utilizing surplus renewable electricity generated by wind and solar power. Additionally, PCCs can be employed to convert NH3 into electricity to meet instantaneous demand, providing a means to address the seasonal and intermittent nature of renewable energy sources. Despite their potential, the commercial application of electricity-to-NH3 interconversion in PCCs faces several challenges, primarily related to insufficient performance and durability. This review systematically explores the mechanisms and challenges of electricity-to-NH3 interconversion in PCCs, highlights recent advancements in NH3 synthesis using PCCs and direct NH3-fueled proton ceramic fuel cells (DA-PCFCs), and discusses perspectives for realizing high-efficiency electricity-to-NH3 interconversion. This review aims to establish a scientific foundation for efficient electricity-to-NH3 interconversion via PCCs and provides critical insights for designing high-performance and durable PCC components.
 Mingzhuang Liang | Mingzhuang Liang obtained his PhD in Chemical Engineering from Nanjing Tech University, China, under the supervision of Prof. Zongping Shao. He is currently a Postdoctoral Fellow at Seoul National University. His research interests are mainly focused on the development of key materials for applications in solid oxide cell. |
 Jinwook Kim | Jinwook Kim is currently a Postdoctoral Fellow at the School of Materials Science and Engineering, at Northwestern University, USA. He received his PhD in Materials Science and Engineering from Korea Advanced Institute of Science and Technology, Korea, in 2023. His research interest focuses on the fabrication and characterization of novel solid oxide fuel cell electrodes for efficient energy storage and conversion. |
 Tae Ho Shin | Tae Ho Shin is a Director and Principal researcher for Division of Carbon Neutrality & Materials Digitalization at Korea Institute of Ceramic Engineering and Technology (KICET). He received his PhD at Kyushu University, Japan. He worked as Research Fellow in the School of Chemistry at the University of St Andrews, and he then joined KICET, Korea (2015). His research involves developing new oxide electrodes for electrochemical devices such as solid oxide fuel cells and electrolysis cells. An area of particular interest is crystal structure analysis for understanding and predicting the design of materials with targeted electrochemical properties. |
 Zongping Shao | Zongping Shao is a John Curtin Distinguished Professor at Curtin University, Australia. He obtained his PhD from Dalian Institute of Chemical Physics, China, in 2000. He worked as a visiting scholar at the Institute Researches Sur La Catalyse, CNRS, France, and then a postdoctoral fellow at California Institute of Technology, USA, from 2000 to 2005. His research interests include mixed conducting membranes, solid oxide electrochemical cells, electrocatalysis, advanced energy storage devices including lithium/sodium-(ion) batteries, metal–air batteries, supercapacitors, and solar cells. He has been recognized as a Highly Cited Researcher by Clarivate Analytics since 2017. |
 WooChul Jung | WooChul Jung is an Associate Professor at Seoul National University, Korea. He received his PhD degree at MIT and served as a postdoctoral fellow at Caltech. The main goal of his research activities is to understand the reactions that occur at the interfaces between ionic solids (oxides in particular) and gases and thereby to improve the reaction kinetics for applications in chemical and electrochemical catalysis, such as, solid oxide fuel cells, electrolyzers, and hydrocarbon reformers. He has been developing model oxide structures with well-defined interface geometries and analyzing true surface properties and reaction characteristics. |
Broader contextThe conventional Haber–Bosch process for NH3 synthesis necessitates energy-intensive high-temperature and high-pressure conditions, leading to substantial energy consumption and significant operational costs. Proton ceramic cells (PCCs) offer a compelling solution by enabling the electrochemical synthesis of NH3 from renewable electricity at intermediate temperatures and pressures. Furthermore, PCCs can efficiently convert the synthesized NH3 back into electricity to meet instant demand, addressing the intermittency inherent in renewable energy sources. While PCCs exhibit immense potential, achieving efficient bidirectional electrochemical interconversion between electricity and NH3 remains a critical challenge for their practical application. This review comprehensively examines the mechanisms and advantages of this electrochemical interconversion in PCCs, with a particular focus on recent advancements and critical challenges in NH3 synthesis utilizing PCCs and direct NH3-fueled proton ceramic fuel cells (DA-PCFCs). The review concludes with a discussion of the future prospects and critical research directions towards realizing efficient electrochemical interconversion between electricity and NH3 using PCC technology. This review aims to establish a scientific foundation for efficient electricity-to-NH3 interconversion via PCCs and provides critical insights for designing high-performance and durable PCC components. |
1. Introduction
The rapid expansion of the global economy has intensified the use of traditional fossil fuels, leading to significant environmental pollution and heightened greenhouse gas emissions. To ensure environmental protection and sustainable development, there is an urgent need to identify clean energy alternatives to fossil fuels and to advance energy conversion technologies. Recent decades have seen extensive exploration of renewable energy sources, including wind, solar, and tidal energy. However, the intermittent nature of these sources poses challenges in meeting immediate energy demands.1–3 Hydrogen energy, an efficient and clean secondary energy source, shows promise as a key component of future energy systems due to its sole combustion byproduct being water.4–7 Despite its advantages, H2 presents challenges for storage and transportation due to its small molecular size, propensity to leak, and the difficulty of liquefaction under normal temperatures and pressures, all of which contribute to increased costs.8–10 Consequently, it is crucial to develop effective H2 storage and transport methods. Among the various options, NH3 has emerged as a promising H2 carrier, offering a potential solution to these challenges.11,12NH3 is a nitrogenous compound comprising one nitrogen atom and three hydrogen atoms. It can be readily liquefied under ambient conditions, offering significant economic and safety benefits for storage and transportation. With an exceptional volumetric energy density of 12.9 MJ L−1, NH3 significantly outperforms liquid H2 (8.6 MJ L−1), making it an attractive option for energy storage.13,14 Moreover, global NH3 production exceeds 170 million tons annually, supported by a well-established, cost-effective production and supply chain. The mature technology for NH3 synthesis, combined with its ideal combustion products being primarily N2 and water without CO2 emissions, underscores its potential to mitigate global climate change. These characteristics position NH3 as a promising clean fuel and H2 carrier. The Haber–Bosch process, the current dominant method for NH3 synthesis, requires high temperatures (400–500 °C) and pressures (150–350 atm) to drive the reaction between N2 and H2 over an iron-based catalyst.15,16 However, this process has significant drawbacks, including substantial energy consumption (1–2% of global energy consumption from steam-methane reforming), considerable CO2 emissions (∼1.8% of global CO2 emissions), and the high equipment and maintenance costs associated with such extreme operating conditions. Moreover, the H2 required for this process is typically derived from methane, further leading to substantial carbon emissions.17,18 Consequently, alternative NH3 synthesis methods, including electrochemical approaches, plasma techniques, and solid nitride cycling, have attracted considerable interest.18–20 Among these, electrochemical approaches, with their potential for seamless integration with renewable energy systems, are the most promising technologies for sustainable NH3 production. Based on the operating temperature, electrochemical NH3 synthesis devices can be categorized into low-temperature (<100 °C), intermediate-temperature (200–600 °C), and high-temperature (>700 °C) systems.21 Compared to other low- and intermediate-temperature devices such as alkaline water electrolyzers and proton exchange membrane electrolyzers, solid oxide electrolysis cells (SOECs) typically require fewer noble metal catalysts, exhibit faster reaction rates, and achieve higher energy efficiency.22–24
Solid oxide cells (SOCs), encompassing both SOECs and solid oxide fuel cells (SOFCs), are an emerging technology for energy conversion and storage.25–27 These cells offer the versatility to switch between electrolysis cell and fuel cell modes, facilitating the efficient interconversion between electrical and chemical energy.28–30 This reversible-mode capability positions SOCs as a promising technology for enhancing the flexibility, stability, and sustainability of energy systems, marking them a key focus for future energy research. SOCs are categorized based on the type of charge carrier in their electrolytes: oxygen ion conducting SOCs (O-SOCs) and protonic ceramic cells (PCCs).31,32 PCCs typically operate at intermediate to low temperatures (<600 °C) due to the relatively low activation energy required for proton transport compared to oxygen ion transport.33 PCCs can function in two modes: protonic ceramic electrolysis cells (PCECs) and protonic ceramic fuel cells (PCFCs). In PCEC mode, excess renewable electricity can be utilized for NH3 synthesis and storage.34,35 Conversely, PCFCs can convert stored NH3 back into electricity to meet immediate energy demands.36,37 Despite the potential of PCCs, challenges remain in the electricity-to-NH3 interconversion, particularly related to catalyst stability and efficiency, material and manufacturing costs, and the optimization of reaction conditions.38–41 Overcoming these challenges is crucial for advancing the practical application of PCCs in future energy systems.
Numerous studies have previously been published, focusing on the development of electrolytes, electrode catalysts, and operating conditions for NH3 synthesis in PCCs and direct NH3-fueled proton ceramic fuel cells (DA-PCFCs). This review begins by outlining the fundamental mechanisms involved in electricity-to-NH3 interconversion in PCCs, followed by a detailed analysis of the challenges associated with achieving such conversions. Subsequently, the advancements and achievements in electricity-to-NH3 interconversion using PCCs over the past few decades are summarized. Finally, the review concludes with a forward-looking perspective on the future applications of PCCs in electricity-to-NH3 interconversion, highlighting the critical role they could play in shaping next-generation sustainable energy systems. By providing an in-depth examination of the state of the art, the objective of this review is to serve as a comprehensive guide for researchers engaged in the advancement of proton-conducting ceramic technologies, addressing both the current obstacles and potential breakthroughs that could drive innovation in clean energy conversion, which is critical for accelerating the transition to a sustainable and carbon-neutral global energy landscape.
2. Mechanisms of the electricity-to-NH3 interconversion in PCCs
2.1. NH3 synthesis in PCECs
PCECs have been employed for NH3 synthesis through various electrochemical processes, with the goal of achieving reactors that offer high energy efficiency and minimal greenhouse gas emissions.38,42,43 Currently, three main types of PCEC reactors are utilized for NH3 synthesis, each based on different reactions: the hydrogen oxidation reaction (HOR), methane-steam reforming reaction (MSRR), and water oxidation reaction (WOR), as illustrated in Fig. 1. In the following sections, we will examine the operating mechanisms of these reactors and discuss the challenges in NH3 synthesis by PCECs. | ||
| Fig. 1 The PCC reactors for NH3 synthesis via (a) HOR, (b) MSRR and (c) WOR. (d)–(f) The potential mechanisms of the NRR. | ||
In Reactor 1, H2 is supplied to the anode, while N2 is introduced at the cathode (Fig. 1a). Under an applied voltage, H2 molecules dissociate into protons and electrons (H2 → 2H+ + 2e−). The generated protons then migrate across the electrolyte to the cathode and combine with adsorbed nitrogen to produce NH3 (6H+ + N2 + 6e− → 2NH3).44,45 This approach offers significant advantages over the traditional high-pressure NH3 synthesis methods, as it operates under moderate conditions and directly converts electrical energy into chemical energy. However, a primary challenge lies in the need for a reliable H2 source. Consequently, Reactor 1 must be coupled with H2 production systems, such as methane reforming or water electrolysis, which significantly increases the overall system cost.
Reactor 2, a more integrated approach, utilizes renewable electricity to reform methane and water steam into H2 and CO2 on the anode side (Fig. 1b).46,47 The protons from H2 dissociation then migrate across the electrolyte to the cathode, combining with adsorbed nitrogen to form NH3. Unlike Reactor 1, Reactor 2 eliminates the need for additional H2 production equipment, greatly simplifying the overall system. By combining the endothermic reforming reaction with the exothermic nitrogen reduction, Reactor 2 achieves a thermoneutral state, enhancing energy efficiency. Additionally, the generated CO2 can be further reformed with methane to produce CO, reducing carbon emissions. However, the anode's exposure to methane, steam, and CO2 necessitates a stable and highly active electrode material to ensure optimal performance and longevity. Fortunately, numerous methane steam reforming anode catalysts developed for SOFCs can be adapted for use in Reactor 2.48–50
Recent advancements in PCECs have enabled efficient H2 production through water electrolysis. Therefore, researchers have increasingly explored PCECs for a more environmentally friendly approach to NH3 synthesis (Fig. 1c).51,52 In Reactor 3, the WOR at the anode is driven by renewable electricity, leading to the generation of protons and oxygen. These protons migrate through the electrolyte to the cathode, where they react with absorbed nitrogen to form NH3.52,53 This process, utilizing only water and N2, offers a carbon-free pathway for NH3 production. To ensure optimal performance and longevity in the high-steam environment required for Reactor 3, the development of advanced anode materials is crucial. Fortunately, some anode materials already used in PCEC water electrolysis for H2 production, such as PrBa0.5Ca0.5Co2O5+δ (PBCC), Ce0.2Ba0.2Sr0.2La0.2Ca0.2CoO3−δ (CBSLCC), and BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY), are also well-suited for this NH3 production process.54–56
In all the three pioneering NH3 synthesis reactors, despite varying anode reactions, the cathode, where the nitrogen reduction reaction (NRR) takes place, shares a striking resemblance. Currently, there are three main NRR mechanisms, as illustrated in Fig. 1d–f. In Fig. 1d, the NRR within the PCEC reactor proceeds via a dissociative mechanism. Initially, N2 molecules are adsorbed onto the catalyst surface, undergoing subsequent activation. Subsequently, hydrogen protons from the anode and electrons combine with the activated nitrogen in a hydrogenation process to form NH3. Finally, the NH3 molecules desorb from the catalyst surface.57,58 Owing to the substantial energy barrier for N2 dissociation on the catalyst surface, this step is generally regarded as the rate-determining step (RDS) in NH3 synthesis. Consequently, promoting N2 dissociation can effectively enhance NH3 production rate.59,60Fig. 1e and f depict the NRR associative mechanism, further categorized into alternating and distal hydrogenation pathways based on distinct hydrogenation sequences.61 In Fig. 1e, a N2 molecule initially adsorbs onto the catalyst surface. Hydrogenation occurs preferentially at the distal nitrogen atom of the adsorbed N2 molecule. This stepwise hydrogenation process culminates in the formation of an NH3 molecule, which then desorbs from the catalyst surface. Subsequent hydrogenation of the remaining nitrogen atom completes the catalytic cycle, yielding a second NH3 molecule that desorbs from the catalyst surface.21 Conversely, in Fig. 1f, both N atoms of the adsorbed N2 molecule experience sequential hydrogenation. When the distal nitrogen atom reaches the NH3 state and desorbs, the proximal nitrogen atom continues to be hydrogenated until it also forms NH3 and desorbs.62 Consequently, for the associative mechanism, enhancing adsorbed N2 activation significantly augments NH3 synthesis rates via NRR.63,64 Unlike ambient temperature processes, the elevated operating temperature of PCECs (300–600 °C) promotes the dissociation of the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond, a critical step in NH3 synthesis. To elevate the electricity-to-NH3 conversion rate, developing cathodes with exceptional NRR activity is paramount. The PCEC reactor's cathode typically consists of metal nanoparticles supported on an oxide, which is conducive to the NRR process.38,65,66 The catalytic activity of catalysts for the NRR in PCECs is generally influenced by their electronic structure, crystal structure, and surface morphology.67,68 Moreover, different preparation methods can significantly affect the catalyst's particle size, dispersion, and specific surface area, thus impacting its catalytic performance.
N bond, a critical step in NH3 synthesis. To elevate the electricity-to-NH3 conversion rate, developing cathodes with exceptional NRR activity is paramount. The PCEC reactor's cathode typically consists of metal nanoparticles supported on an oxide, which is conducive to the NRR process.38,65,66 The catalytic activity of catalysts for the NRR in PCECs is generally influenced by their electronic structure, crystal structure, and surface morphology.67,68 Moreover, different preparation methods can significantly affect the catalyst's particle size, dispersion, and specific surface area, thus impacting its catalytic performance.
2.2. Direct NH3 utilization in PCFCs
PCFCs exhibit excellent fuel flexibility, enabling the utilization of electrochemically produced NH3 for power generation. In DA-PCFCs, water steam is generated at the cathode, preventing dilution of the fuel gas at the anode, thus enhancing fuel utilization.69–71 Additionally, the electrolyte effectively separates oxygen ions from NH3, preventing the formation of NOx.72,73In DA-PCFCs, the NH3 molecule is initially adsorbed onto the anode surface and dissociates into H2 and N2. Subsequently, the electrochemical process involves the oxidation of H2 at the anode, generating protons. These protons are conducted through the electrolyte to the cathode, where they participate in the reduction of oxygen to form H2O (Fig. 2a).74 An effective anode catalyst enhances NH3 decomposition by providing a greater number of active sites for adsorption and subsequent decomposition.75,76 An ideal anode catalyst should possess high NH3 adsorption capacity, low N–H bond dissociation energy, and excellent resistance to poisoning.77,78 To date, a diverse array of anode catalysts has been developed, encompassing noble metal-based catalysts (Pt, Pd, Ru, etc.), transition metal oxide catalysts (Ni, Co, Fe, etc.), and metal-ceramic catalysts (Ni–BZCY, Ni–BZCYYb, Sr2CoMo0.8Ni0.2O6−δetc.).79–82 The catalytic activity of these catalysts is influenced by a complex interplay of factors, including their electronic structure, surface properties, and the nature of their interactions with the support material.
 | ||
| Fig. 2 (a) Schematic illustration of the operating mechanism of the DA-PCFC. (b) The mechanism of NH3 decomposition on the anode side. | ||
NH3 decomposition at the anode surface is a complex process involving the following steps:83,84
| NH3 (g) + * ↔ NH3* |
| NH3* + * → NH2* + H* |
| NH2* + * → NH* + H* |
| NH* + * → N* + H* |
| 2H* → H2 (g) + 2* |
| 2N* → N2 (g) + 2* |
3. Challenges of electricity-to-NH3 interconversion in PCCs
3.1. NH3 synthesis in PCECs
Research and development of PCEC technology for NH3 synthesis is currently in its nascent stages, with significant potential for future advancements. However, achieving high faradaic efficiency (FE) for NH3 synthesis remains a critical challenge for the advancement of PCEC technology, primarily due to the competitive relationship between the hydrogen evolution reaction (HER) and the NRR.88 This competition is influenced by various factors, including the applied potential, local reactant availability (H+/N2), and the characteristics of the PCEC cathode, which determine the binding of reactants and electron transfer.89 Although higher temperatures can promote N2 dissociation and adsorption, they can also induce NH3 decomposition, leading to a decrease in FE. Thus, precise control of the reaction temperature is essential.67,68 In the reactor depicted in Fig. 1a, the NH3 yield can be significantly enhanced by adjusting the partial pressures of H2 and N2.90,91 Additionally, numerous strategies have been reported to modify the electronic structure and increase the active sites of NRR catalysts, including tailoring the size and morphology, elemental doping, and introducing defects, which can enhance the FE of NH3 production in PCECs.22,92,93 Although reported NH3 synthesis rates (10−13 to 10−8 mol cm−2 s−1) surpass those of low-temperature electrochemical methods (<100 °C), the low FE remains a hurdle for practical implementation. Therefore, rational design of both the cathode and reaction conditions is imperative for advancing PCEC NH3 synthesis technology.3.2. Direct NH3 utilization in PCFCs
Despite the significant advantages of DA-PCFCs, their practical implementation is currently hindered by insufficient stability and suboptimal low-temperature performance.4. Recent advances
4.1. NH3 synthesis in PCECs
Recent advances in PCEC NH3 synthesis research can be grouped into three main approaches: hydrogen-based, methane-based, and water-based synthesis. Each approach presents unique advantages and challenges, and the focus of research varies based on the materials used. Hydrogen-based synthesis has garnered the most attention due to its simple system and relatively established pathways.109 Researchers are mainly focusing on optimizing the NRR catalyst to enhance overall efficiency and NH3 selectivity. Methane-based synthesis is less explored but presents a unique advantage in reducing the complexity of the reactor system.110 Since methane can be directly reformed to produce H2, it eliminates the need for separate reformers or other processing units, which can reduce both the size of the overall system and streamline the synthesis process by decreasing the number of required steps. This streamlined process has the potential to lower costs and simplify industrial-scale implementation, making it a promising alternative for future research. However, not only the NRR but also the methane-steam reforming reaction (MSRR) should be considered, and the selection of materials are limited due to carbon coking.48,111,112 Water-based synthesis represents the ultimate goal for a fully sustainable NH3 production method.113 This approach offers a sustainable pathway for NH3 synthesis by utilizing water as the source of protons and nitrogen from the air, eliminating carbon emissions and relying solely on abundant resources. However, water-based synthesis currently faces significant challenges, particularly in terms of energy consumption and reaction efficiency. Despite these obstacles, the potential environmental benefits have spurred considerable research activity, and advancements in catalyst and cell design are steadily addressing the technical hurdles.As PCEC research progressed, the development of highly active triple-conducting oxides (TCO) significantly advanced the field.5,98 These materials, which can conduct protons, oxygen ions, and electrons or holes, have been widely adopted to increase cell performance from wide active reaction sites. Therefore, several studies were conducted by using TCO material as the PCEC electrode for NH3 production.115 However, in the case of NH3 production, simply achieving high current densities is not sufficient; NH3 selectivity is equally critical. To improve selectivity for NH3 synthesis, many studies have introduced catalysts like Ru, which is known for its high NRR activity and selectivity.65,116–119 Ru-based catalysts, especially when used in exsolution techniques, allow for efficient catalyst utilization even in small quantities. However, the high cost of precious metals such as Ru presents a challenge, even when used in small amounts, as it significantly raises the overall cost of the electrode. In an effort to overcome this limitation, researchers have explored the use of alternative materials, including transition metals like Fe, Co, and Ni.38,39,44,66,68,115,120,121 These metals have shown promise as catalysts for NH3 synthesis, offering a cost-effective solution while maintaining reasonable activity.
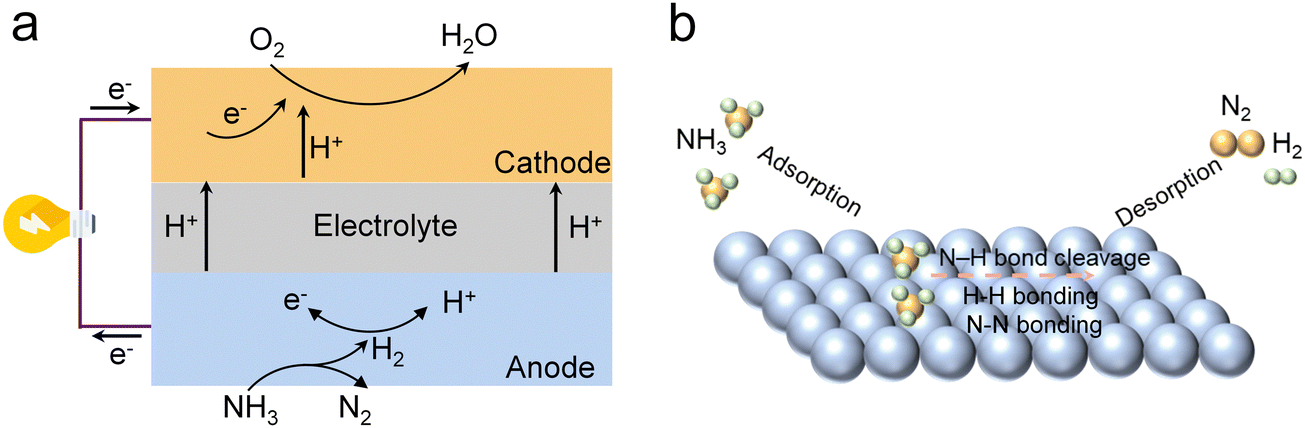
More recently, perovskite electrodes have garnered attention for their potential in NH3 synthesis.22,67,68 The La0.9Bi0.1FeO3−δ (LBiF) perovskite was prepared by Chen et al. and was used as a cathode in a PCEC to realize NH3 synthesis (Fig. 3a and b).67 The introduction of Bi dopants resulted in the formation of new Bi3+/Bi2+/Bi0 redox electron pairs, which facilitated electron transfer and thereby improved the electrical conductivity of the material (Fig. 3c). Furthermore, Bi significantly boosted the NRR activity and effectively suppressed the HER. When LBiF was used as the NRR electrocatalyst, the faradaic efficiency (FE) for NH3 synthesis reached a maximum value of 2.03% at 650 °C and 0.4 V, with an NH3 production rate of 4.47 × 10−9 mol cm−2 s−1, surpassing that of LaFeO3−δ (LF), La0.9FeO3−δ (L0.9F), and LBi (Fig. 3d and e). In their other research, an NRR electrocatalyst, LaCu0.1Fe0.9O3−δ (LCuF), for NH3 synthesis in a PCEC was fabricated by Cu doping (Fig. 3f).68 The synergistic effect of Cu and Fe facilitates electron transfer through the Fe–O2−δ–Cu pathway, enriching Fe sites with Fe4+ and Cu sites with Cu+. This enhanced electron transfer kinetics boosts NRR performance. At an operating temperature of 650 °C and a cell voltage of 0.4 V, the PCEC with the LCuF electrode achieved a FE of 2.8% and an NH3 production rate of 5.12 × 10−9 mol cm−2 s−1 (Fig. 3g and h). Furthermore, they prepared Sr0.9Ti0.6Fe0.4O3−δ (S0.9TF) via A-site defect engineering. The abundant oxygen vacancies, Ti3+ species, and exsolved Fe active particles enhanced N2 adsorption and activation, improving NRR activity. At 650 °C and 0.6 V, PCEC with S0.9TF exhibited a maximum NH3 production rate of 6.84 × 10−9 mol cm−2 s−1 and a FE of 2.79%. Through a simple B-doping method, they synthesized a synergistic mixed catalyst, Sr(Ti0.6Fe0.4)0.8B0.2O3−δ, composed of Sr3B2O6 (SB) and Sr1−yTi0.6Fe0.4O3−δ (S1−yTF).122 SB acts as a proton acceptor, effectively suppressing the HER and promoting proton-coupled electron transfer for NH3 synthesis. Furthermore, the grain boundaries between S1−yTF and SB introduce more defects, significantly enhancing the NH3 production rate and FE. In these reports, the NH3 content was quantified using the indophenol blue method, which detects NH3 by forming a blue-colored complex with phenol and hypochlorite under alkaline conditions, measured spectrophotometrically to determine the NH3 concentration.
 | ||
| Fig. 3 (a) Schematic of NH3 synthesis in PCEC with the LBiF electrode. (b) Rietveld refined XRD pattern of LBiF. (c) Electrical conductivities of LF and LBiF in 10% H2 + 90% N2. (d) The FE values and (e) NH3 production rate in PCEC with the LBiF electrode. Reproduced with permission.67 Copyright 2024, Elsevier. (f) Schematic diagram of the NH3 synthesis device. (g) The FE values and (h) NH3 production rate in PCEC with the LCuF electrode. Reproduced with permission.68 Copyright 2024, Royal Society of Chemistry. | ||
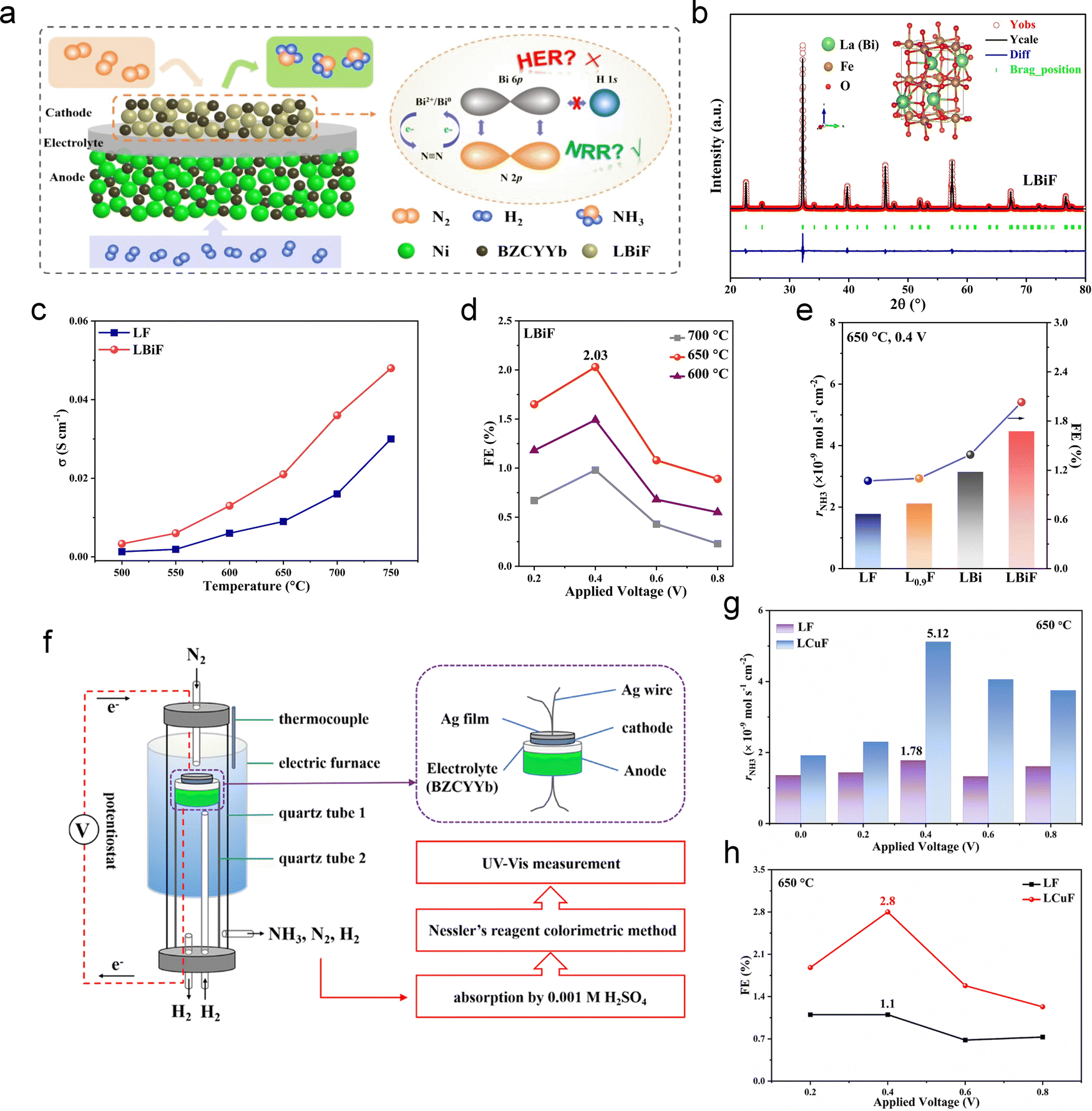
While most research has focused on NRR electrodes and catalysts, recent studies have also investigated the role of electrolytes in enhancing NH3 synthesis. An ideal PCEC electrolyte should exhibit high proton conductivity. In the NH3 synthesis process, H2 is supplied electrochemically in the form of protons.123 Therefore, the H2 supply rate is limited by the maximum proton flux. In the early stages of PCEC NH3 production research, Ma et al. synthesized cubic perovskite BaCe0.85Y0.15O3−δ (BCY15) samples via a microemulsion method, exhibiting nearly pure proton conductivity in wet H2. Cavity ring-down spectroscopy was employed to quantify NH3 production in a symmetric Ag–Pd electrode cell with a BCY15 electrolyte. By measuring the ring-down time of a laser beam in an optical cavity, shortened by NH3 absorption, this technique enables the detection of exceedingly low NH3 concentrations. Under an applied current of 0.75 mA and a temperature of 500 °C, a maximum NH3 synthesis rate of 2.1 × 10−9 mol s−1 cm−2 was achieved.124 Other Ba-based electrolytes, such as BaZr0.8Y0.2O3−δ, BaCe0.65Zr0.2Er0.15O3−δ, BaCe0.2Zr0.7Y0.1O3−δ, and BaZr0.1Ce0.7Y0.1Yb0.1O3−δ, have been extensively studied.109,123,125,126 More recently, Wang et al. developed cells using La5.5WO11.25−δ (LWO) electrolyte instead of the traditional Ba-rich materials (Fig. 4a).45 LWO electrolyte possesses high stability under CO2 conditions and unique conductivity properties.127,128 This high proton conduction ensures that activated protons can efficiently participate in the NRR, leading to enhanced NH3 production without compromising the FE. Consequently, the LWO electrochemical membrane reactor demonstrated superior performance across a wide temperature range. Quantitative analysis of NH3 production was conducted using the indophenol blue method. Its FE and NH3 production rate surpassed those of most other electrochemical NH3 synthesis systems, including those operating at room temperature with Ru-based catalysts and those at elevated temperatures above 500 °C (Fig. 4b). Furthermore, the reactor demonstrated excellent stability, maintaining a consistent NH3 production rate and FE over long-term testing at 350 °C (Fig. 4c). This durability, along with its enhanced performance, makes LWO a highly promising material for use in PCEC NH3 synthesis.
 | ||
| Fig. 4 (a) Cross-section SEM image of the LWO membrane reactor. (b) Comparison NH3 production rate and FE of the LWO membrane reactor with other reference data. (c) Long-term stability of the LWO membrane reactor at 2500 μA cm−2 and 350 °C. Reproduced with permission.45 Copyright 2023, Cell Press. | ||
The study by Stoukides et al. presents an approach that combines methane reforming reaction and NH3 synthesis in a protonic ceramic membrane reactor.129Fig. 5a shows the integrated cell used for NH3 synthesis via methane reforming reaction. The system combines the vanadium nitride–iron (VN–Fe) cathode and Ni–BaZr0.7Ce0.2Y0.1O3−δ (Ni–BZCY72) anode with a BaZr0.8Ce0.1Y0.1O3−δ (BZCY81) electrolyte, facilitating both NH3 synthesis and methane reforming reactions. This integration simplifies the process by combining methane reforming and NH3 synthesis in a single reactor unit. Fig. 5b–g present the results of the study, displaying CH4 conversion, CO2 selectivity, H2 production rate, NH3 production rate, FE for NH3 production, and current–voltage characteristics at various operating temperature (550 °C, 600 °C, 650 °C). A maximum NH3 synthesis rate of 68 mmol NH3 m−2 h−1 with a FE of 5.5% was achieved at 600 °C, as determined by the indophenol blue method.
 | ||
| Fig. 5 (a) Schematic and SEM images of the VN–Fe (cathode)/BZCY81 (electrolyte)/Ni–BZCY72 (anode) cell for NH3 synthesis using methane and steam. Experimental results on (b) CH4 conversion, (c) CO2 selectivity, (d) H2 generated rate, (e) NH3 synthesis rate, (f) FE to NH3, and (g) cell voltage versus open-circuit voltage as a function of current. Reproduced with permission.129 Copyright 2020, Cell Press. | ||
Ding et al. developed Ru/La0.25Ce0.75O2−x catalysts via hydrothermal treatment, introducing hydroxyl groups and inducing electronic restructuring for PCEC NH3 synthesis. In situ generated Ce3+–OH/Ru sites facilitated NN bond cleavage and N–H bond formation, significantly enhancing the NRR process.110Fig. 6a illustrates the catalyst synthesis method using a hydrothermal process, which integrates Ru into the La0.25Ce0.75O2−δ (LDCRu) support to create a catalyst with optimized particle size and surface hydroxyl groups. These modifications enable better NRR activity by creating multiple active sites for NH3 production. Fig. 6b and c provide cross-sectional images of the constructed PCEC using different materials for each purpose. Fig. 6b shows the Ni–BZCYYb based anode support cell used to facilitate the electrochemical reactions with H2 fuel, while Fig. 6c displays an electrolyte support configuration. Both setups integrate LDCRu catalysts in the cathode to drive the NH3 synthesis reactions. Fig. 6d shows the current density as a function of applied bias voltage for different cathodes: PrBa0.5Sr0.5Co1.5Fe0.5O3−δ/Ru (P/Ru), P/LDCRu-D (dry), and P/LDCRu-W (wet). It is evident that the wet-prepared P/LDCRu-W catalyst outperforms the others in terms of current density, indicating the importance of surface hydroxyl groups in facilitating proton transport and NH3 production. Fig. 6e presents the NH3 production rate, which was determined using the Nessler reagent colorimetric method. The P/LDCRu-W loaded cell significantly enhances the production rate compared to the P/LDCRu-D loaded cell and the conventional Ru catalyst loaded cell. This finding indicates that the surface hydroxyl groups, which are likely formed during the wet preparation process, play a critical role in facilitating the NRR and enhancing its efficiency. Fig. 6f demonstrates the operational stability of the Ni–BZCYYb/P/LDCRu-W system, where the current density remained stable over 550 h at 400 °C. The P/LDCRu-W cathode demonstrated excellent performance, achieving the highest NH3 production rate of 0.452 mol m−2 h−1 at an applied voltage of 1.5 V and the highest FE of 36.5% at a lower voltage of 0.6 V (Fig. 6g). They also conducted a similar experiment using C2H6 (ethane) as the hydrogen source at the anode. In this case, the highest NH3 production rate was 0.840 mol h−1 m−2 at 1.2 V, while the highest FE of 35.0% was achieved at 0.6 V (Fig. 6h). Moreover, the cell with the P/LDCRu-W cathode showed stable FE for NH3 production under different hydrocarbon gas feeds and operating voltages (Fig. 6i). The results show that the system maintains stable NH3 production across different conditions, confirming the effectiveness of the Ru/LDC catalyst.
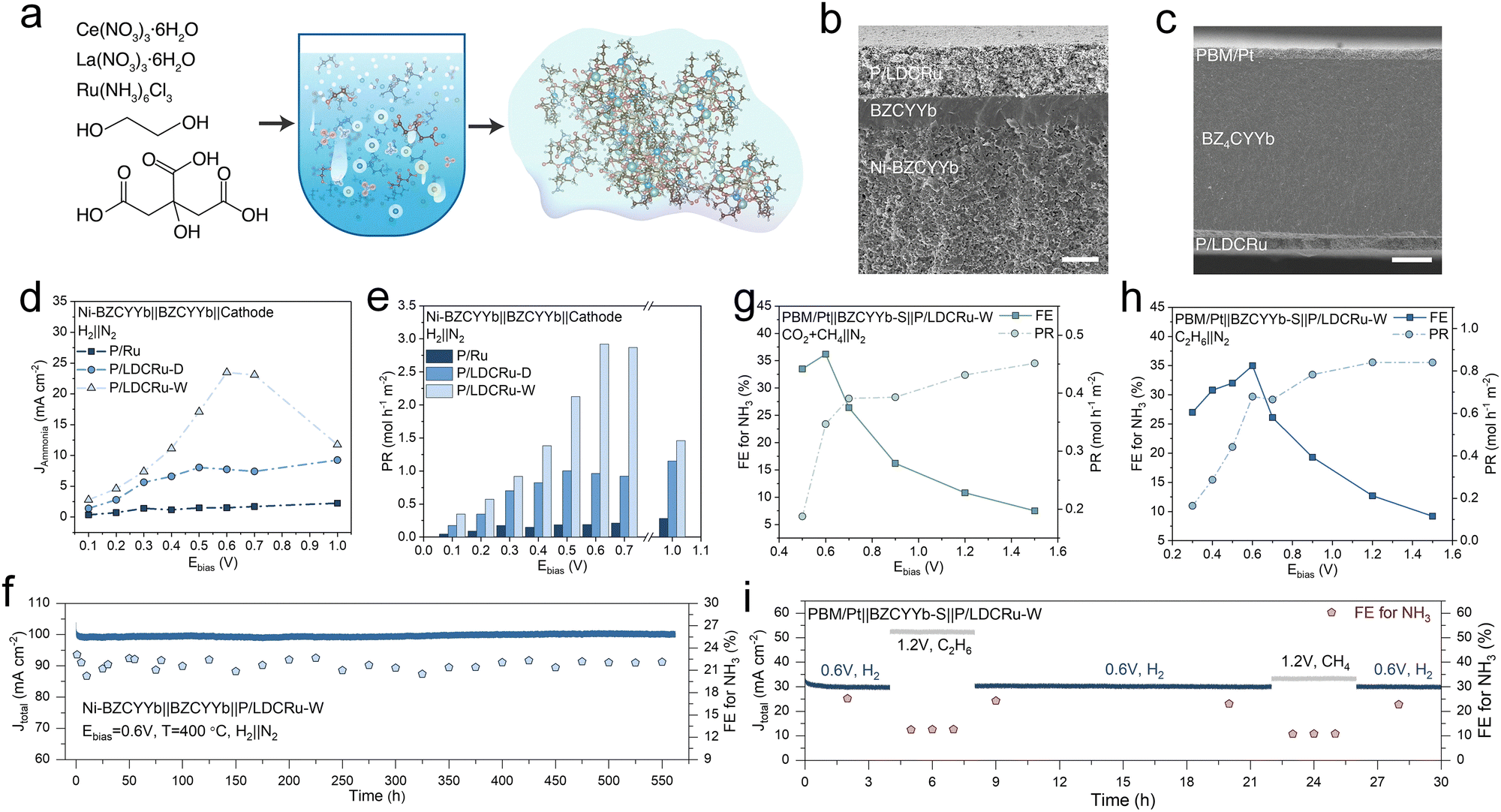 | ||
| Fig. 6 (a) Schematic of the gel preparation process for the Ru/LDC catalyst. (b) and (c) SEM of two types of PCC for NH3 production. (d)–(f) Current density, NH3 production rate, and long term stability of anode support type cells fueled with H2. (g) FE and production rate of NH3 with electrolyte support cells with a mixture of CO2 and CH4 as fuel. (h) FE and production rate of NH3 with electrolyte support cells with C2H6 as the fuel. (i) Long-term stability of electrolyte support type cell with various fuels. Reproduced with permission.110 Copyright 2022, Elsevier. | ||
Overall, methane reforming coupled with optimized catalysts constitutes a significant advancement towards efficient and scalable NH3 synthesis via PCEC. While challenges remain, such as improving long-term stability and addressing carbon coking, the advancements highlighted in these studies pave the way for future breakthroughs in NH3 production.
One promising approach is the use of external catalysts integrated into the system to improve reaction kinetics and stability. Sullivan et al., introduced an external Ru-based catalyst to enhance NH3 production in a PCEC.113Fig. 7a–c show a schematic of the PCC with the external catalyst layer integrated into the system. The Ru-based catalyst with high dispersion and uniformity is positioned to facilitate the NH3 synthesis process. In Fig. 7d, the FE of the cell shows nearly 100% for H2 between 500 and 1000 mA cm−2. In Fig. 7e, as the driving current density increases from 2000 to 5000 mA cm−2, the NH3 synthesis rate normalized to the mass of Ru was approximately 2.5 × 10−8 mol cm−2 s−1 under a current of 5000 mA cm−2. The NH3 generation rate was quantified using a Drager tube method, enabling direct gas-phase estimation of NH3 concentration, albeit with lower precision compared to spectroscopic techniques. Despite these improvements, the resulting NH3 production rates, when normalized to the mass of Ru catalyst, are still comparatively lower than those achieved with other Ru-based catalysts reported in the literature. To address this limitation, the researchers pressurized the NH3 synthesis reactor up to 12.5 bar by introducing additional hydrogen. This pressurization led to a dramatic increase in NH3 synthesis rates, reaching as high as 2.1 × 10−6 mol cm−2 s−1, which is 100 times higher than the rates achieved under ambient pressure (Fig. 7f). The increased pressure not only enhances reaction kinetics but also shifts the equilibrium to favor NH3 synthesis, demonstrating the potential of high-pressure conditions for scaling up this technology. A unique aspect of Sullivan et al.'s hybrid approach is its capability for cyclic operation, directly enabling transitions between using NH3 to generate power and using power to synthesize NH3. Fig. 7g showcased the system's versatility in switching seamlessly between energy generation and NH3 production modes, making it a promising candidate for practical applications in sustainable energy systems.
 | ||
| Fig. 7 (a) Schematic for NH3 production and the power generation process using the Ru–B2CA catalyst. (b) SEM and (c) TEM image of the Ru–B2CA catalyst. (d) FE and voltage as a function of the current density of the cell. (e) NH3 synthesis rate as a function of the current density of the cell. (f) NH3 synthesis rate as a function of operation pressure. (g) Reversible NH3 synthesis/NH3 fuel cell operation result. Reproduced with permission.113 Copyright 2021, Springer Nature. | ||
Another innovative approach is plasma-assisted NH3 synthesis. Plasma technology provides a powerful method to activate N2 by breaking its triple bond, which is one of the most energy-intensive steps in the NRR. In a study by Sanden et al., a radio frequency (RF) plasma source was applied externally to the PCEC system to aid in N2 dissociation.17 The plasma pre-activated N2 molecules before they reached the cathode, reducing the activation energy required for the NRR. Fig. 8a presents a schematic of the plasma-electrocatalysis setup, showing how the RF plasma is applied to activate nitrogen molecules prior to the electrochemical reaction. The generated NH3 was diluted with He and then quantified by mass spectrometry. Fig. 8b shows the resulting NH3 concentration and production rate as a function of the current density, demonstrating the significantly increased NH3 production rates with plasma activation. Fig. 8c highlights the FE, illustrating that the plasma-assisted process effectively lowers the activation energy for the NRR and improves selectivity. At lower current densities, the plasma-assisted system achieves a FE of up to 88%, though this efficiency decreases as current density increases due to the competing HER reaction. These additional techniques offer new pathways for overcoming the inherent limitations of steam-based NH3 synthesis via the WOR. By addressing the challenges of electrode performance, nitrogen activation, and overall system efficiency, these innovative approaches bring NH3 synthesis in PCECs closer to being a competitive alternative to traditional methods.
 | ||
| Fig. 8 (a) Schematic representation of the hybrid plasma electrochemical reactor setup. (b) NH3 production rate as a function of current density and (c) faradaic efficiency of NH3 production with/without 80 W plasma power. Reproduced with permission.17 Copyright 2020, American Chemical Society. | ||
4.2. Direct NH3 utilization in PCFCs
Unlike NH3 synthesis, NH3 decomposition is an endothermic reaction (2NH3 → 3H2 + N2, ΔH = +45.9 kJ mol−1). By reducing the flow rate of the reactant gas mixture, the residence time of NH3 molecules on the catalyst surface is extended, allowing for more opportunities for reaction. Consequently, elevated operating temperatures and decreased flow rates can promote NH3 decomposition.102,131 The overall performance of DA-PCFCs is strongly dependent on the rate at which NH3 is decomposed at the anode, highlighting the crucial role of anode catalysis in these systems. The NH3 decomposition rate increases with NH3 partial pressure and decreases with H2 partial pressure, which can be described by the Temkin–Pyzhev equation:132,133 | (1) |
Additionally, the materials and thicknesses of the anode, electrolyte, and cathode significantly impact the overall performance of DA-PCFCs.4,134,135 Therefore, the recent research on DA-PCFCs will be described from the aspects of anodes, electrolytes, and cathodes.
Among single-metal catalysts, Ru-based catalysts exhibit the high activity and stability for NH3 decomposition, as depicted in Fig. 9a.138,139 Masel et al. identified N2 desorption as the rate-determining step (RDS) in NH3 decomposition over Fe, Co, and Ni surfaces, while the cleavage of N–H bonds is the RDS on Rh, Ir, Pd, Pt, and Cu surfaces. They also experimentally determined the following activity order: Ru > Ni > Rh > Co > Ir > Fe ≫ Pt > Cr > Pd > Cu ≫ Te, Se, Pb.140 Although Ru offers superior catalytic activity to Ni, its high cost and scarcity have hindered its widespread application. Therefore, Ni was extensively investigated as a potential catalyst for NH3 decomposition. Furthermore, the excellent electronic conductivity of Ni is beneficial for enhancing the NH3 decomposition process (Fig. 9b).141 Therefore, Ni has been the focus of extensive research as an NH3 decomposition catalyst.
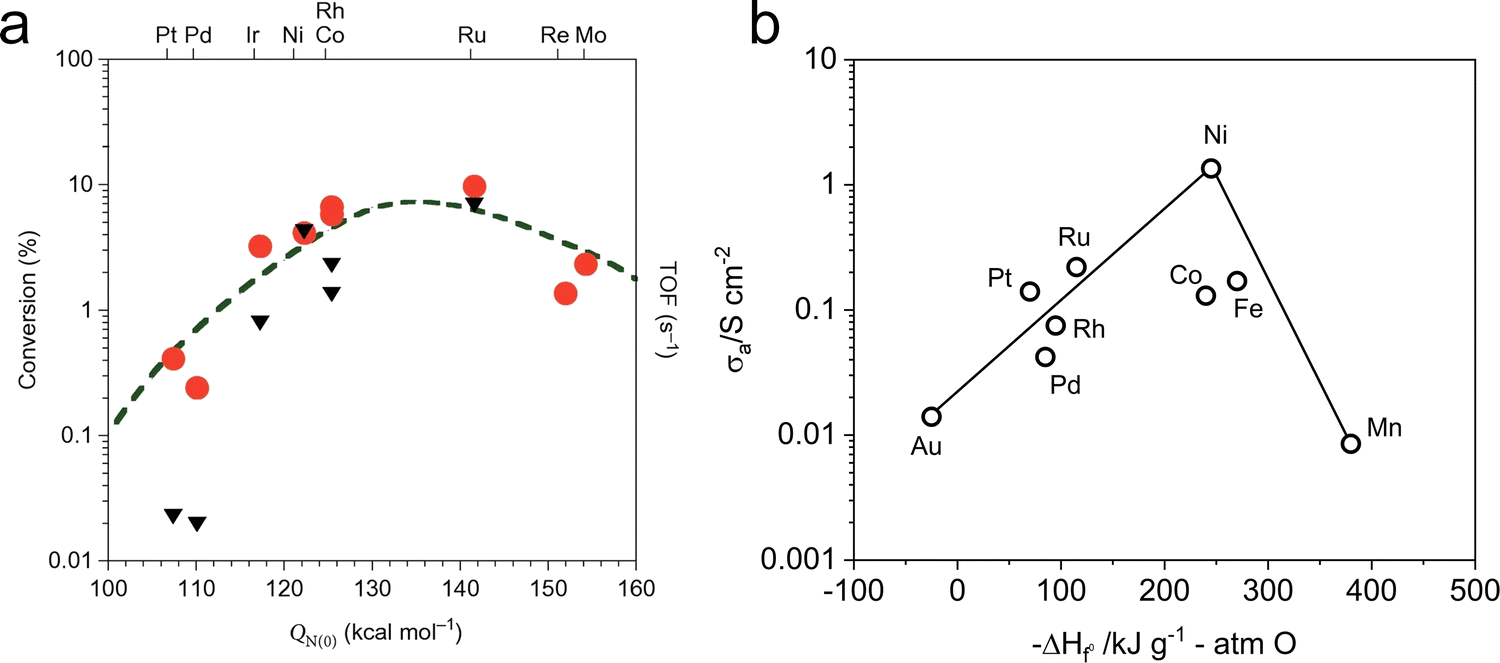 | ||
| Fig. 9 (a) Relationship of theoretical NH3 decomposition volcano curve and experimental turnover frequencies (TOFs) of various transition-metal catalysts at 850 K obtained by microkinetic modeling and the nitrogen binding energies (QN(0)). Reproduced with permission.138 Copyright 2021, American Chemical Society. (b) The relationship diagram between anodic polarization conductivity of various metals at 1000 °C and heat of oxide generation function. Reproduced with permission.141 Copyright 2024, Wiley-VCH. | ||
In 2015, Yang et al. introduced Ni–BaCe0.75Y0.25O3−δ (Ni–BCY25) as a promising anode for DA-PCFCs.142 Notably, the NH3 decomposition rate of Ni–BCY25 at temperatures ranging from 400 to 700 °C significantly surpassed those of Ni–8mol% Y2O3–ZrO2 and Ni–Ce0.90Gd0.10O1.95 (Fig. 10a). They further investigated the impact of steam content on NH3 decomposition over the Ni–BCY25 anode, as depicted in Fig. 10b. Introducing 0.8% steam led to a significant decline in the NH3 decomposition rate. This reduction primarily stems from steam adsorption onto the BCY25 surface. The resulting species, including hydroxide groups and protons, can potentially occupy active reaction sites at the interface between the nickel catalyst and the BCY25 support, resulting in a significant water poisoning effect.143,144 When operating on NH3 fuel, a promising peak power density (PPD) of 216 mW cm−2 was attained at 650 °C in a single cell designated as Ni–BCY25|BaCe0.9Y0.1O3−δ|Sm0.5Sr0.5CoO3−δ (Fig. 10c). Miyazaki et al. assessed the suitability of Ni–Ba(Zr,Y)O3−δ (Ni–BZY) as DA-PCFC anodes by comparing the NH3 decomposition activity of Ni–BZY10, Ni–BZY20, Ni–BCY10, and Ni–YSZ.145 As depicted in Fig. 10d, Ni–BZY10 and Ni–BZY20 catalysts exhibited superior NH3 decomposition performance compared to Ni–YSZ, suggesting that proton conductors play a more significant role in the decomposition process. Among the catalysts evaluated for NH3 decomposition under varying H2 partial pressures, Ni–BZY20 exhibited the highest NH3 conversion activity and resistance to hydrogen inhibition. Additionally, a DA-PCFC comprising a Ni–BZY20 anode, a BZY20 electrolyte, and a Pt cathode achieved a PPD of 125 mW cm−2 at 700 °C. The study by Haile et al. using Ni–BZCYYb4411 as the anode in a DA-PCFC revealed a strong correlation between NH3 conversion rate and open circuit voltage.146 While NH3 conversion rates above 85% were achieved at temperatures above 550 °C (Fig. 10e), a substantial decrease to 40% was observed at 450 °C, likely explaining the sharp OCV reduction at this temperature (Fig. 10f). A further observation was the rapid decrease in OCV with increasing NH3 flow rate, suggesting that higher flow rates hinder NH3 decomposition and consequently affect the cell performance. The Ni component within the Ni-based ceramic anode of DA-PCFCs is susceptible to the formation of Ni3N when exposed to NH3. However, Ni3N can be readily reduced by hydrogen, leading to structural degradation of the anode and compromising the long-term stability of DA-PCFCs.11,105,147 The strong chemisorption of nitrogen atoms onto Ni-based catalysts, while promoting NH3 decomposition, can also hinder N2 desorption, resulting in a poisoning effect and limiting the overall NH3 decomposition rate.141,148
 | ||
| Fig. 10 (a) NH3 conversion on several Ni-based anodes. (b) NH3 conversion on Ni–BCY25 in dry and wet gases. (c) I–V–P curves of a cell with the structure of Ni–BCY25|BCY10|SSC. Reproduced with permission.142 Copyright 2015, American Chemical Society. (d) NH3 decomposition rate as a function of partial pressure of hydrogen on several Ni-based anodes at 500 °C. Reproduced with permission.145 Copyright 2017, Elsevier. (e) NH3 conversion rate of the Ni–BCZYYb4411 anode on a PCFC. (f) OCV as functions of NH3 flow rate from 450 to 600 °C. Reproduced with permission.146 Copyright 2024, American Chemical Society. | ||
The catalytic activity of Ni-based ceramic anodes for NH3 decomposition can be enhanced by the in situ exsolution of B-site cations. Shao et al. proposed a Pd-doped perovskite BZCYYb (BZCYYbPd) and evaluated its NH3 decomposition activity as a DA-PCFC anode (Fig. 11a–e).149 As indicated in Fig. 11b and c, Pd metal nanoparticles exsolved from the BZCYYbPd lattice, creating B-site deficient structures that facilitate proton conduction.150Fig. 10d demonstrates that Ni–BZCYYbPd exhibits superior NH3 decomposition activity compared with Ni–BZCYYb, primarily attributed to the beneficial effects of Pd nanoparticles and B-site defect structures. Therefore, the Ni–BZCYYbPd anode enabled the DA-PCFC to achieve a PPD of 724 mW cm−2 at 650 °C (Fig. 11e). They also developed a Ru and Fe co-doped PCFC anode material, Ni–Ba(Zr0.1Ce0.7Y0.1Yb0.1)0.94Ru0.03Fe0.03O3−δ.135 Secondary oxidation–reduction treatment led to the formation of more RuFe nanoparticles on the surface, enhancing its catalytic activity for NH3 decomposition. Consequently, the electrochemical performance of the DA-PCFC with this anode was significantly improved. Recently, Ciucci et al. developed a novel DA-PCFC anode denoted as Ni–Ba(Zr0.1Ce0.7Y0.1Yb0.1)0.95Ni0.05O3−δ (BZCYYbNi), which significantly improved the electrochemical performance of DA-PCFCs.151 Ni nanoparticles exsolved from the BZCYYbNi perovskite structure create numerous active sites for NH3 decomposition, significantly boosting the catalytic activity of the Ni–BZCYYbNi anode (Fig. 11f). A DA-PCFC employing the Ni–BZCYYbNi ceramic anode achieved a PPD of 523 mW cm−2 at 650 °C, as illustrated in Fig. 11g. The Ni–BZCYYbNi anode offers a more cost-effective alternative to Pd-based anodes for DA-PCFCs due to the lower cost of Ni.
 | ||
| Fig. 11 (a) The operating mechanism of DA-PCFCs with the Ni–BZCYYbPd anode and NH3 decomposition pathway on the Ni–BZCYYbPd anode. (b) XRD patterns and (c) HR-TEM images of BZCYYbPd powder before and after H2 reduction at 650 °C. (d) NH3 conversion rates of Ni–BZCYYb and Ni–BZCYYbPd. (e) I–V–P curves of the DA-PCFC. Reproduced with permission.149 Copyright 2021, Wiley-VCH. (f) Schematic of DA-PCFCs: (left) Ni–BZCYYb anode and BZCYYb electrolyte; (right) Ni–BZCYYbNi anode and BZCYYbNi electrolyte. (g) I–V–P curves of a DA-PCFC with the Ni–BZCYYbNi anode. Reproduced with permission.151 Copyright 2022, Wiley-VCH. | ||
Incorporating high catalytic activity species (Ru, Fe, etc.) into Ni-based anodes can significantly enhance DA-PCFCs’ performance and operating stability. Chen et al. successfully developed high-performance DA-PCFCs by decorating Ni–BZCYYb anodes with Fe, achieving a PPD of 1.257 W cm−2 at 650 °C.152 Fe modification alters the adsorption strength of NH3 and the barrier for N2 associative desorption, contributing to improved performance and stability. In their another study, they infiltrated Ru0.95Cu0.05 into the Ni–BZCYYb anode, resulting in the formation of Ru0.95Cu0.05Nix (RCN) catalysts through an in situ reaction between Ru0.95Cu0.05 nanoparticles and Ni grains (Fig. 12a and b).99 As shown in Fig. 12c, the RCN–BZCYYb anode exhibited the highest NH3 decomposition rate among the evaluated anodes (∼98% at 550 °C). Consequently, DA-PCFCs with RCN-catalyzed Ni–BZCYYb anodes demonstrated a high PPD of 732 mW cm−2 and enhanced stability. Recently, Shim et al. treated a Ni–BZCYYb anode with a Pd catalyst using atomic layer deposition (ALD).153 This process formed a Pd catalytic layer on the Ni–BZCYYb surface and introduced some Pd atoms into the anode's interior (Fig. 12d). This approach effectively improved NH3 decomposition rates, inhibited Ni3N formation, and enhanced DA-PCFC performance and durability (Fig. 12e).
 | ||
| Fig. 12 (a) Schematic illustration of NH3 decomposition on the RCN nanoparticle-modified Ni–BZCYYb anode. (b) STEM-EDX mapping results of the RCN–Ni–BZCYYb anode. (c) NH3 decomposition rates of Ni–BZCYYb and RuxCu1−x–Ni/BZCYYb (x = 0–1) anodes. Reproduced with permission.152 Copyright 2022, Royal Society of Chemistry. (d) Bright-field, BZCYYb, NiO, and Pd TEM-EDS mapping images. (e) Schematic illustration of the nitridation of Ni in bare Ni–BZCYYb (left) and Pd-treated Ni–BZCYYb (right) anodes of DA-PCFCs. Reproduced with permission.153 Copyright 2023, Wiley-VCH. | ||
 | ||
| Fig. 13 (a) The ultrasonic spray coating process for preparing thin-film electrolytes and SEM images of the single cell. Reproduced with permission.165 Copyright 2023, Nature Portfolio. (b) Fabrication process of the single cell using a tape casting technique. Reproduced with permission.152 Copyright 2022, Royal Society of Chemistry. | ||
O’Hayre et al. demonstrated a high-performance PCFC cathode by infiltrating the triple conductor BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY) into a BaZr0.3Ce0.6Y0.1O3−δ (BZCY63) framework (Fig. 14a).97 The high oxygen vacancy concentration and proton conductivity of BCFZY contribute to its superior catalytic activity,166 enabled the corresponding PCFC to achieve a PPD of 0.648 W cm−2 at 600 °C (Fig. 14b) and maintain stable operation for over 1100 h at 500 °C (Fig. 14c). Some high-performance BCFZY derivatives were further developed, such as Ba0.9Co0.4Fe0.4Zr0.1Y0.1O3−δ,167 BaCo0.7Fe0.1Zr0.1Y0.1O3−δ,168 Ba(Co0.4Fe0.4Zr0.1Y0.1)0.95Ni0.05O3−δ,169 BCFZY–NiO,170 Ba(Co0.4Fe0.4Zr0.1Y0.1)0.95Mg0.05O3−δ32 and Ba0.95(Co0.4Fe0.4Zr0.1Y0.1)0.95Ni0.05O3−δ,171 which can also be applied to DA-PCFCs.
 | ||
| Fig. 14 (a) Process for preparing BCFZY infiltrated BCZY63 composite. (b) I–V–P and (c) stability of the cells under H2/air. Reproduced with permission.97 Copyright 2015, American Association for the Advancement of Science. | ||
Shao et al. developed a PCFC triple-conducting nanocomposite cathode BaCo0.7(Ce0.8Y0.2)0.3O3 (BCCY), comprising BaCe0.8Y0.2O3 (P-BCCY) as a proton conductor, BaCo0.9(Ce0.8Y0.2)0.1O3 (M-BCCY) as an oxygen-ion conductor, and BaCoO3−δ (BC) with high electronic conductivity (Fig. 15a–c).172 This synergistic combination endowed BCCY with excellent catalytic activity. When employed as a PCFC cathode, BCCY achieved a high PPD of 508 mW cm−2 at 550 °C and demonstrated an operational stability exceeding 800 h (Fig. 15d and e).
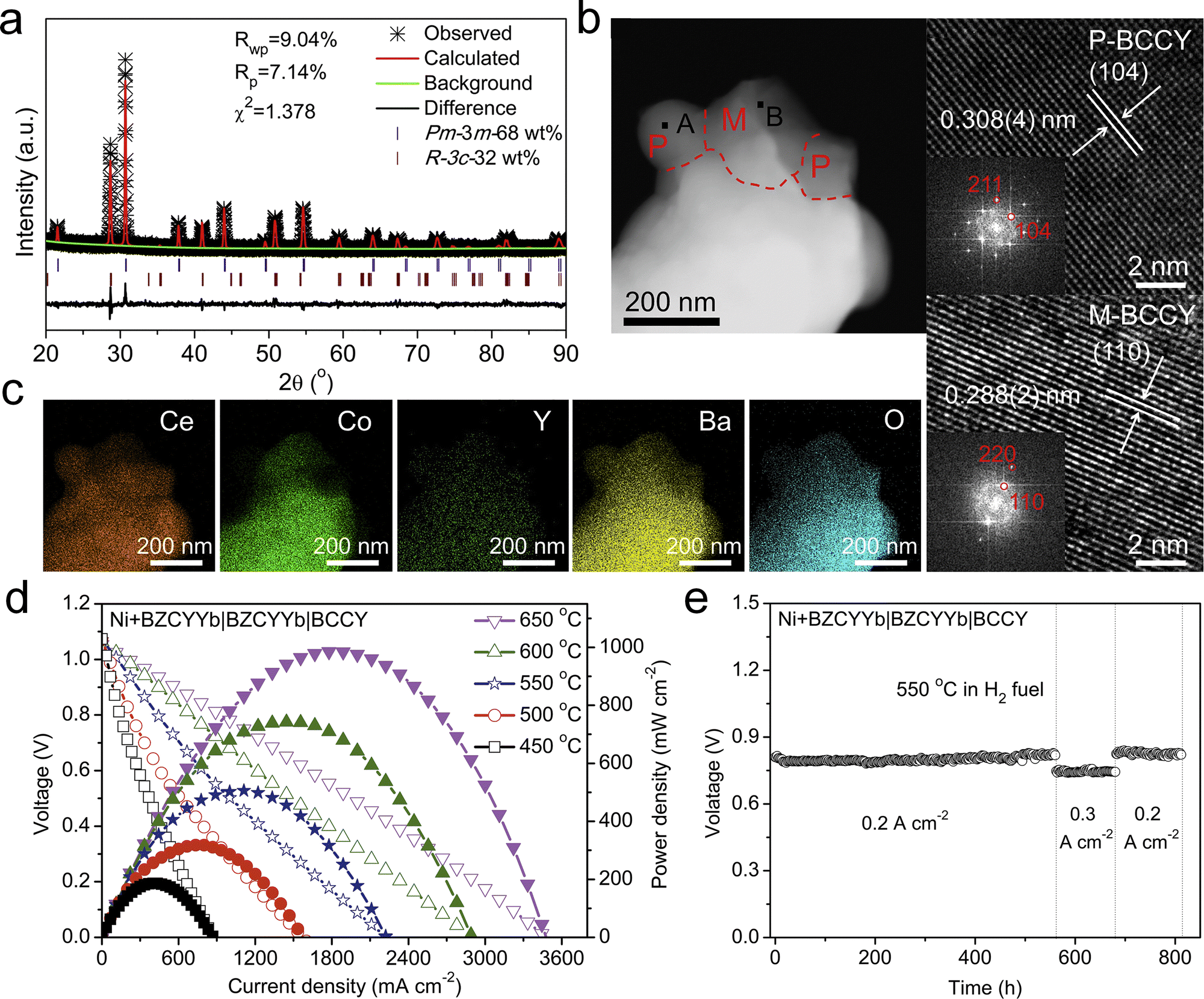 | ||
| Fig. 15 (a) Refined XRD profiles of the BCCY sample. (b) STEM image and (c) STEM-EDX result of BCCY. (d) I–V–P curves and (e) durability of PCFCs with the BCCY cathode. Reproduced with permission.172 Copyright 2019, Cell Press. | ||
Incorporating an anode catalytic layer (ACL) can also effectively enhance both the performance and durability of DA-PCFCs. Liu et al. fabricated tubular DA-PCFCs featuring an iron catalytic layer on the Ni–BZCYYb anode (Fig. 16a).173 These DA-PCFCs, utilizing a highly active Fe catalyst for NH3 decomposition, achieved a record PPD of 1.078 W cm−2 at 700 °C for tubular DA-PCFCs. By decomposing most of the NH3 into H2 and N2, the Fe catalytic layer minimizes direct contact between NH3 and the Ni–BZCYYb anode, thus enhancing the stability of the tubular DA-PCFCs. The high catalytic activity of Fe for NH3 decomposition has also been demonstrated in other studies.106,174 Chen et al. prepared a material designated as Fe–CeOx (Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ce = 6:4) via a wet chemical method and incorporated it as a catalytic layer on the anode side of DA-PCFCs (Fig. 16b).175 This composite material not only maintains a high NH3 decomposition rate but also suppresses the formation of iron nitride, ensuring the long-term operational stability of DA-PCFCs.
:Ce = 6:4) via a wet chemical method and incorporated it as a catalytic layer on the anode side of DA-PCFCs (Fig. 16b).175 This composite material not only maintains a high NH3 decomposition rate but also suppresses the formation of iron nitride, ensuring the long-term operational stability of DA-PCFCs.
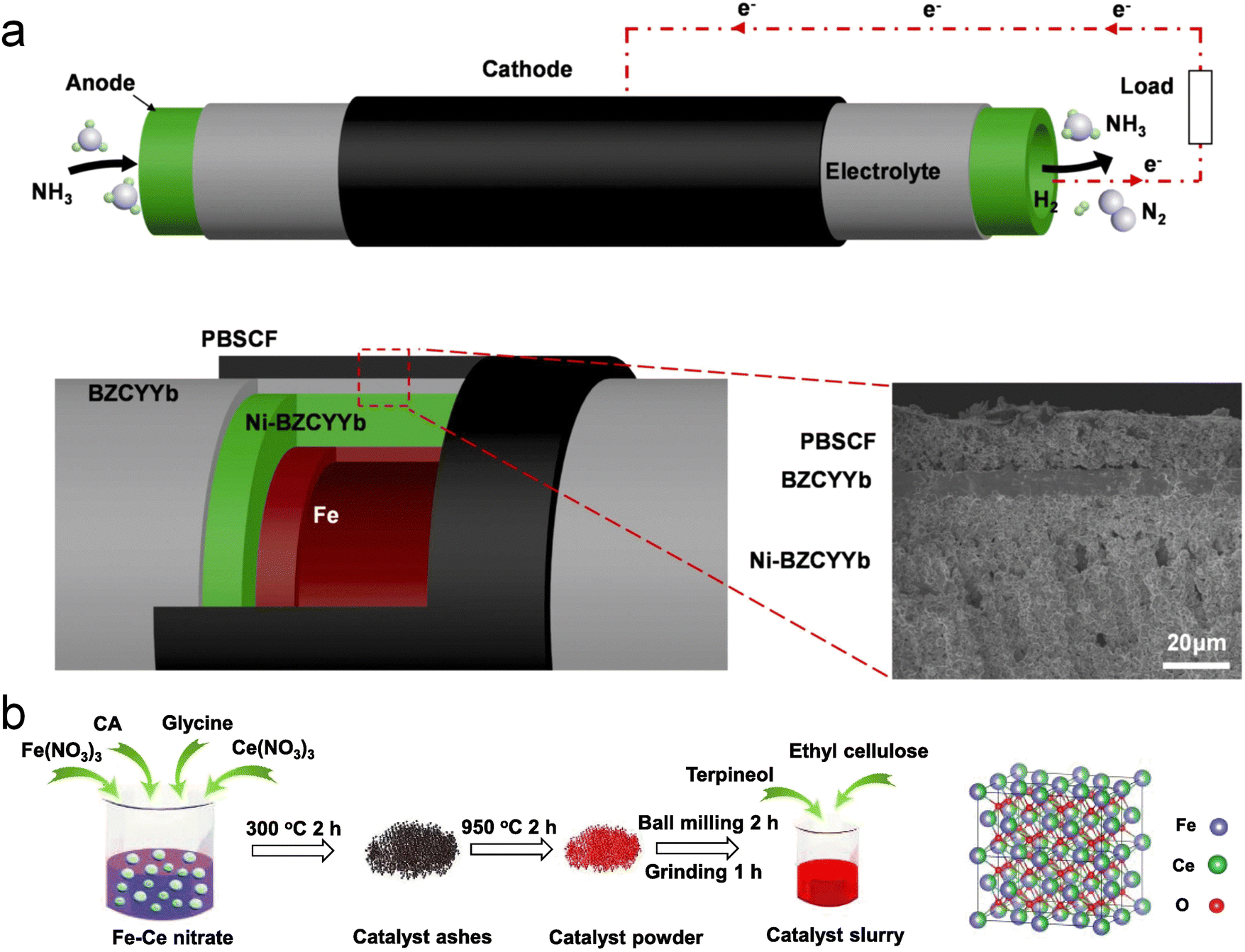 | ||
| Fig. 16 (a) Schematic representation of a tubular cell utilizing NH3 as the fuel.173 (b) Schematic diagram of the synthesis process and the crystal structure of the Fe–CeOx (Fe:Ce = 6:4) catalyst. Reproduced with permission.175 Copyright 2022, Elsevier. | ||
Beyond simple metal catalytic layers, perovskite oxides present an alternative for the DA-PCFC catalytic layer, enhancing cell performance and stability. In 2023, Chen et al. developed the reduced Sr2Fe1.35Mo0.45Cu0.2O6−δ (r-SFMC) anode catalytic layer, which demonstrates high NH3 adsorption and decomposition capabilities (Fig. 17a and b).176 The tubular Ni–BZCYYb anode-supported DA-PCFCs equipped with an r-SFMC ACL achieved a remarkable PPD of 1.02 W cm−2 and exhibited outstanding stability over 200 h at 650 °C, marking a significant advancement in DA-PCFC technology (Fig. 17c). More recently, Shao et al. substituted 5, 10, 15 and 20% Ru into Pr0.6Sr0.4Co0.2Fe0.8O3−δ (PSCF), respectively, and found reduced-Pr0.6Sr0.4(Co0.2Fe0.8)0.85Ru0.15O3−δ (r-PSCFR15) exhibits the optimal NH3 decomposition activity among these catalysts (Fig. 17d).100 During the reduction process, CoFe (CF) and CoFeRu (CFR) alloys form on the PSCF and PSCFR15 surfaces, respectively. The CFR alloy promotes the N2 desorption process in the NH3 decomposition reaction, thereby boosting NH3 decomposition efficiency (Fig. 17e and f). Under the influence of the r-PSCFR15 catalytic layer, the DA-PCFC exhibited a significant improvement in cell performance, reaching 625 mW cm−2 at 650 °C. Additionally, the DA-PCFC demonstrated robust durability, maintaining stable performance over 340 h (Fig. 17g). After the operational stability test, the Ni particles in the anode of the PCFC with r-PSCFR15 ACL were noticeably smaller compared to those on the anodes without ACL (Fig. 17h and i), which indicates that the r-PSCFR15 effectively prevented the Ni sintering in the Ni-based anode.
 | ||
| Fig. 17 (a) NH3 temperature-programmed desorption profiles of r-SFMC and Ni–BZCYYb. (b) NH3 catalytic activities of r-SFMC, Ni–BZCYYb, and their mixture. (c) PPDs of DA-PCFCs with and without r-SFMC ACL fueled by wet H2 and dry NH3. Reproduced with permission.176 Copyright 2023, Wiley-VCH. (d) NH3 decomposition rates of r-PSCF, r-PSCFR05, r-PSCFR10, r-PSCFR15, r-PSCFR20, Ni–BZCYYb, and Ni–BZCYYb|r-PSCFR15. ENH3 and EN2 calculated for (e) CF(011) and (f) CFR(011). (g) The operational stability of DA-PCFCs under NH3 fuel conditions. The SEM images of Ni–BZCYYb anodes of DA-PCFCs (h) without ACL and (i) with r-PSCFR15 ACL after the stability test. Reproduced with permission.100 Copyright 2024, Wiley-VCH. | ||
5. Perspectives for electricity-to-NH3 interconversion in PCCs
5.1. NH3 synthesis in PCECs
The low FE and NH3 production rate in PCEC NH3 synthesis are primarily attributed to the competitive HER.88 To enhance these metrics, future research should prioritize the development of catalysts with high NRR activity. Optimizing the electronic structure, crystal structure, and surface morphology of catalysts can effectively suppress the HER and promote the NRR. Exploring novel catalytic materials, such as nitride catalysts, is also crucial. DFT calculations and advanced in situ characterization techniques can aid in identifying the active sites for the HER and NRR, significantly benefiting the development of NH3 synthesis catalysts. Moreover, optimizing electrolyte design and NH3 synthesis operating conditions is of paramount importance.5.2. Direct NH3 utilization in PCFCs
6. Conclusions
PCCs represent a promising energy conversion technology, enabling both the storage of excessive renewable electricity via NH3 synthesis and on-demand electricity generation from NH3. This review comprehensively introduces the reaction mechanisms of electricity-to-NH3 interconversion in PCCs, analyzes the challenges of this technology and summarizes the research advancements in this field. Moreover, this review emphasizes the need for researchers to explore not only novel catalytic electrodes and electrolyte materials, but also to optimize operating conditions and identify active sites for the NRR and HER in order to achieve efficient NH3 production in PCECs. For DA-PCFCs, the development of anode electrodes or anode catalytic layers with high catalytic activity for NH3, as well as the fabrication of DA-PCFC stacks, should be the primary focus of future research. This review aims to encapsulate the progress in PCC-based electricity-to-NH3 interconversion technology and provide insights for future research in this field.Author contributions
Mingzhuang Liang: conceptualization, visualization, and writing – original draft. Jinwook Kim: investigation and writing – review & editing. Xiaomin Xu: writing – review & editing. Hainan Sun: writing – review & editing. Yufei Song: writing – review & editing. SungHyun Jeon: writing – review & editing. Tae Ho Shin: supervision and writing – review & editing. Zongping Shao: supervision and writing – review & editing. WooChul Jung: supervision, writing – review & editing, and project administration.Data availability
The data supporting the findings of this study are available in the manuscript, and additional data are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Ceramic Strategic Technology R&D program through the Korea Institute of Ceramic Engineering & Technology (KICET) (grant NTIS no. 240002182). This work was also supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2021M3H4A3A02086497). We appreciate the support from Research Institute of Advanced Materials.Notes and references
- H. Ding, W. Wu, C. Jiang, Y. Ding, W. Bian, B. Hu, P. Singh, C. J. Orme, L. Wang, Y. Zhang and D. Ding, Nat. Commun., 2020, 11, 1907 CAS.
- F. Zhu, Z. Du, K. Xu, F. He, Y. Xu, Y. Liao and Y. Chen, Adv. Energy Mater., 2024, 14, 2401048 CrossRef CAS.
- D. Kim, I. Jeong, S. Ahn, S. Oh, H. N. Im, H. Bae, S. J. Song, C. W. Lee, W. Jung and K. T. Lee, Adv. Energy Mater., 2024, 14, 2304059 CrossRef CAS.
- Y. Wang, Y. Ling, B. Wang, G. Zhai, G. Yang, Z. Shao, R. Xiao and T. Li, Energy Environ. Sci., 2023, 16, 5721–5770 RSC.
- S. Choi, T. C. Davenport and S. M. Haile, Energy Environ. Sci., 2019, 12, 206–215 RSC.
- J. H. Kim, J. Hong, D.-K. Lim, S. Ahn, J. Kim, J. K. Kim, D. Oh, S. Jeon, S.-J. Song and W. Jung, Energy Environ. Sci., 2022, 15, 1097–1105 RSC.
- J. H. Kim, K. Jang, D.-K. Lim, S. Ahn, D. Oh, H. Kim, J. Seo, P.-P. Choi and W. Jung, J. Mater. Chem. A, 2022, 10, 2496–2508 RSC.
- F. Jiao and B. Xu, Adv. Mater., 2019, 31, 1805173 CrossRef PubMed.
- N. Tsvetkov, D. Kim, I. Jeong, J. H. Kim, S. Ahn, K. T. Lee and W. Jung, Adv. Mater. Technol., 2023, 8, 2201075 CAS.
- S. H. Oh, S.-Y. Park, S. Kim, K. J. Yoon, H. C. Shin, K. T. Lim and J.-H. Lee, J. Korean Ceram. Soc., 2024, 61, 34–43 CAS.
- Z. Wan, Y. Tao, J. Shao, Y. Zhang and H. You, Energy Convers. Manage., 2021, 228, 113729 CAS.
- A. Afif, N. Radenahmad, Q. Cheok, S. Shams, J. H. Kim and A. K. Azad, Renewable Sustainable Energy Rev., 2016, 60, 822–835 CAS.
- D. S. Dhawale, G. Kaur and S. Giddey, Inorg. Chem. Front., 2023, 10, 6176–6192 CAS.
- A. C. Chien, W. Y. Chen and M. S. Zheng, J. Electrochem. Soc., 2023, 170, 044505 CAS.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 CAS.
- R. Li, T. Li, X. Liu, C. Xie, Q. Zhen, S. Bashir and J. L. Liu, Energy Sci. Eng., 2023, 11, 2293–2301 CAS.
- R. K. Sharma, H. Patel, U. Mushtaq, V. Kyriakou, G. Zafeiropoulos, F. Peeters, S. Welzel, M. C. Van De Sanden and M. N. Tsampas, ACS Energy Lett., 2020, 6, 313–319 Search PubMed.
- Y.-i Kwon, S. K. Kim, Y. B. Kim, S. J. Son, G. D. Nam, H. J. Park, W.-C. Cho, H. C. Yoon and J. H. Joo, ACS Energy Lett., 2021, 6, 4165–4172 CAS.
- I. Goyal, N. C. Kani, S. A. Olusegun, S. Chinnabattigalla, R. R. Bhawnani, K. D. Glusac, A. R. Singh, J. A. Gauthier and M. R. Singh, ACS Energy Lett., 2024, 9, 4188–4195 CAS.
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2020, 142, 14374–14383 CAS.
- H. M. Vieri, M.-C. Kim, A. Badakhsh and S. H. Choi, Energies, 2024, 17, 441 CrossRef CAS.
- K. Wang, H. Chen, S.-D. Li and Z. Shao, J. Mater. Chem. A, 2022, 10, 24813–24823 RSC.
- J. Yu, L. Liu, Y. Du, Y. Li, D. Zhang, B. Li, X. Liu, L. Cheng, X. Zhang and Y. Zhang, Energy Technol., 2024, 12, 2401169 CrossRef CAS.
- B. Lee, D. Lim, H. Lee and H. Lim, Renewable Sustainable Energy Rev., 2021, 143, 110963 CAS.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CAS.
- Y. Song, X. Zhang, K. Xie, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1902033 CrossRef CAS PubMed.
- S. Park, E.-I. Kim, B. Singh and S.-J. Song, J. Korean Ceram. Soc., 2024, 61, 419–428 CrossRef CAS.
- C. Zhao, Y. Li, W. Zhang, Y. Zheng, X. Lou, B. Yu, J. Chen, Y. Chen, M. Liu and J. Wang, Energy Environ. Sci., 2020, 13, 53–85 Search PubMed.
- A. Hauch, R. Küngas, P. Blennow, A. B. Hansen, J. B. Hansen, B. V. Mathiesen and M. B. Mogensen, Science, 2020, 370, eaba6118 CAS.
- J.-h Myung, D. Neagu, D. N. Miller and J. T. Irvine, Nature, 2016, 537, 528–531 CAS.
- M. Liang, Y. Wang, Y. Song, D. Guan, J. Wu, P. Chen, A. Maradesa, M. Xu, G. Yang, W. Zhou, W. Wang, R. Ran, F. Ciucci and Z. Shao, Appl. Catal., B, 2023, 331, 122682 CAS.
- M. Liang, Y. Song, D. Liu, L. Xu, M. Xu, G. Yang, W. Wang, W. Zhou, R. Ran and Z. Shao, Appl. Catal., B, 2022, 318, 121868 CAS.
- Y. Song, J. Liu, Y. Wang, D. Guan, A. Seong, M. Liang, M. J. Robson, X. Xiong, Z. Zhang, G. Kim, Z. Shao and F. Ciucci, Adv. Energy Mater., 2021, 11, 2101899 CAS.
- B. Wang, T. Li, F. Gong, M. H. D. Othman and R. Xiao, Fuel Process. Technol., 2022, 235, 107380 CAS.
- F. Liu, D. Ding and C. Duan, Adv. Sci., 2023, 10, 2206478 CAS.
- M. Ni, M. K. Leung and D. Y. Leung, Int. J. Energy Res., 2009, 33, 943–959 CAS.
- Z. Li, C. Wang, I. T. Bello, M. Guo, N. Yu, M. Zhu and M. Ni, J. Power Sources, 2023, 556, 232505 CAS.
- M. Okazaki and J. Otomo, Solid State Ionics, 2024, 414, 116649 CrossRef CAS.
- M. Okazaki and J. Otomo, ACS Omega, 2023, 8, 40299–40308 CrossRef CAS PubMed.
- K. Miyazaki, H. Muroyama, T. Matsui and K. Eguchi, Sustainable Energy Fuels, 2020, 4, 5238–5246 RSC.
- Y. Han, W. Gao and Y. Qin, Energy, 2024, 297, 131287 CrossRef CAS.
- Y. Du, X. Su, X. Wang, L. Ye and K. Xie, New J. Chem., 2024, 48, 10060–10066 CAS.
- J. T. Irvine, S. Wilson, S. Amnuaypanich, G. J. Irvine, M. C. Verbraeken, K. Nowicki and G. M. Carins, Faraday Discuss., 2023, 243, 296–306 RSC.
- S. Klinsrisuk and J. T. Irvine, Catal. Today, 2017, 286, 41–50 CrossRef CAS.
- G. Weng, S. Lei, R. Wang, K. Ouyang, J. Dong, X. Lin, J. Xue, L.-X. Ding and H. Wang, Joule, 2023, 7, 1333–1346 CrossRef CAS.
- H. Malerød-Fjeld, D. Clark, I. Yuste-Tirados, R. Zanón, D. Catalán-Martinez, D. Beeaff, S. H. Morejudo, P. K. Vestre, T. Norby, R. Haugsrud, J. M. Serra and C. Kjølseth, Nat. Energy, 2017, 2, 923–931 CrossRef.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef CAS.
- F. Liu, H. Deng, Z. Wang, A. M. Hussain, N. Dale, Y. Furuya, Y. Miura, Y. Fukuyama, H. Ding, B. Liu and C. Duan, J. Am. Chem. Soc., 2024, 146, 4704–4715 CAS.
- K. Hong, M. Choi, Y. Bae, J. Min, J. Lee, D. Kim, S. Bang, H.-K. Lee, W. Lee and J. Hong, Nat. Commun., 2023, 14, 7485 CAS.
- Y. Guo, S. Wang, R. Li, J. Yu, X. Zhang, M. Li, X. Zheng, J. Zhu, Y. Song, G. Wang and X. Bao, Joule, 2024, 8, 2016–2032 CAS.
- F. Kosaka, T. Nakamura, A. Oikawa and J. Otomo, ACS Sustainable Chem. Eng., 2017, 5, 10439–10446 CrossRef CAS.
- C.-Y. Yoo, J. H. Park, K. Kim, J.-I. Han, E.-Y. Jeong, C.-H. Jeong, H. C. Yoon and J.-N. Kim, ACS Sustainable Chem. Eng., 2017, 5, 7972–7978 CAS.
- R. Lan, J. T. Irvine and S. Tao, Sci. Rep., 2013, 3, 1145 CrossRef PubMed.
- Y. Zhou, E. Liu, Y. Chen, Y. Liu, L. Zhang, W. Zhang, Z. Luo, N. Kane, B. Zhao and L. Soule, ACS Energy Lett., 2021, 6, 1511–1520 CAS.
- F. He, M. Hou, D. Liu, Y. Ding, K. Sasaki, Y. Choi, S. Guo, D. Han, Y. Liu, M. Liu and Y. Chen, Energy Environ. Sci., 2024, 17, 3898–3907 CAS.
- C. Duan, R. Kee, H. Zhu, N. Sullivan, L. Zhu, L. Bian, D. Jennings and R. O’Hayre, Nat. Energy, 2019, 4, 230–240 CrossRef CAS.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 Search PubMed.
- S. Gunduz, D. J. Deka and U. S. Ozkan, J. Catal., 2020, 387, 207–216 Search PubMed.
- Q. Hu, C. Tian, D. Bao, H. Zhong and X. Zhang, Next Energy, 2024, 4, 100144 Search PubMed.
- A. L. Garden and E. Skulason, J. Phys. Chem. C, 2015, 119, 26554–26559 Search PubMed.
- E. Skulason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 CAS.
- J.-C. Liu, X.-L. Ma, Y. Li, Y.-G. Wang, H. Xiao and J. Li, Nat. Commun., 2018, 9, 1610 Search PubMed.
- A. U. Shetty and R. Sankannavar, J. Energy Chem., 2024, 92, 681–697 CrossRef CAS.
- W. Q. Li, M. Xu, J. S. Chen and T. N. Ye, Adv. Mater., 2024, 36, 2408434 CrossRef CAS PubMed.
- H. Kim, Y. S. Chung, T. Kim, H. Yoon, J. G. Sung, H. K. Jung, W. B. Kim, L. B. Sammes and J. S. Chung, Solid State Ionics, 2019, 339, 115010 CrossRef CAS.
- J. Humphreys, R. Lan, D. Du, W. Xu and S. Tao, Int. J. Hydrogen Energy, 2018, 43, 17726–17736 CAS.
- W. Guo, Y. Li, S.-D. Li, Z. Shao and H. Chen, Chem. Eng. J., 2024, 498, 155124 CAS.
- W. Guo, Y. Li, S.-D. Li, Z. Shao and H. Chen, J. Mater. Chem. A, 2024, 12, 1200–1210 CAS.
- K. Pei, Y. Zhou, K. Xu, H. Zhang, Y. Ding, B. Zhao, W. Yuan, K. Sasaki, Y. Choi, Y. Chen and M. Liu, Nat. Commun., 2022, 13, 2207 CAS.
- Y. Huang, F. He, K. Xu, H. Gao, X. Zhang, Y. Xu, Z. Du, F. Zhu, W. Gong, C. Jian and Y. Chen, Adv. Funct. Mater., 2024, 34, 2409598 CAS.
- Z. Liu, Z. Tang, Y. Song, G. Yang, W. Qian, M. Yang, Y. Zhu, R. Ran, W. Wang, W. Zhou and Z. Shao, Nano-Micro Lett., 2022, 14, 217 CAS.
- H. Zhu, C. Karakaya and R. J. Kee, Int. J. Green Energy, 2022, 19, 1568–1582 CAS.
- M. M. Rahman, A. M. Abdalla, L. A. Omeiza, V. Raj, S. Afroze, M. S. Reza, M. R. Somalu and A. K. Azad, Processes, 2023, 11, 2728 CAS.
- M. Ni, D. Y. Leung and M. K. Leung, J. Power Sources, 2008, 183, 687–692 CAS.
- K. Xu, F. Zhu, M. Hou, C. Li, H. Zhang and Y. Chen, Nano Res., 2023, 16, 2454–2462 CAS.
- S. Appari, V. M. Janardhanan, S. Jayanti, L. Maier, S. Tischer and O. Deutschmann, Chem. Eng. Sci., 2011, 66, 5184–5191 CAS.
- Y. Song, H. Li, M. Xu, G. Yang, W. Wang, R. Ran, W. Zhou and Z. Shao, Small, 2020, 16, 2001859 CAS.
- S. Sorcar, H. Zinowits, E. P. Komarala, N. Moshe, I. Agranovich and B. A. Rosen, J. Mater. Chem. A, 2022, 10, 24115–24126 CAS.
- N. Khatun, C.-Y. Chiu, C.-J. Lin, J.-Y. Lin, S.-F. Wang and T. C.-K. Yang, J. Power Sources, 2024, 600, 234252 CAS.
- F. Zhong, X. Zhao, H. Fang, Y. Luo, S. Wang, C. Chen and L. Jiang, Appl. Catal., B, 2024, 360, 124522 Search PubMed.
- X. Xiong, J. Yu, X. Huang, D. Zou, Y. Song, M. Xu, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Mater. Sci. Technol., 2022, 125, 51–58 CrossRef CAS.
- Y. Yi, J. Chen, M. Xu, G. Yang, R. Ran, W. Zhou, W. Wang and Z. Shao, Catalysts, 2023, 13, 996 CrossRef CAS.
- S. Mukherjee, S. V. Devaguptapu, A. Sviripa, C. R. Lund and G. Wu, Appl. Catal., B, 2018, 226, 162–181 CrossRef CAS.
- P. Xie, Y. Yao, Z. Huang, Z. Liu, J. Zhang, T. Li, G. Wang, R. Shahbazian-Yassar, L. Hu and C. Wang, Nat. Commun., 2019, 10, 4011 CrossRef PubMed.
- S. Sun, Q. Jiang, D. Zhao, T. Cao, H. Sha, C. Zhang, H. Song and Z. Da, Renewable Sustainable Energy Rev., 2022, 169, 112918 CrossRef CAS.
- B. Lu, L. Li, M. Ren, Y. Liu, Y. Zhang, X. Xu, X. Wang and H. Qiu, Appl. Catal., B, 2022, 314, 121475 CAS.
- A. Takahashi and T. Fujitani, J. Chem. Eng. Jpn., 2016, 49, 22–28 CAS.
- C. A. Fernandez, N. M. Hortance, Y.-H. Liu, J. Lim, K. B. Hatzell and M. C. Hatzell, J. Mater. Chem. A, 2020, 8, 15591–15606 CAS.
- L. Hu, Z. Xing and X. Feng, ACS Energy Lett., 2020, 5, 430–436 CAS.
- A. Skodra and M. Stoukides, Solid State Ionics, 2009, 180, 1332–1336 CAS.
- M. Ouzounidou, A. Skodra, C. Kokkofitis and M. Stoukides, Solid State Ionics, 2007, 178, 153–159 CAS.
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chem, 2021, 7, 1708–1754 CAS.
- R. Zhao, H. Xie, L. Chang, X. Zhang, X. Zhu, X. Tong, T. Wang, Y. Luo, P. Wei and Z. Wang, EnergyChem, 2019, 1, 100011 Search PubMed.
- S. H. Jeon, W. G. Jung, H. Bae, S. Ahn, B. Koo, W. J. Yu, S. Kim, D. H. Oh, U. Kim, S. A. Barnett, J. Seo, B.-J. Kim and W. C. Jung, Adv. Mater., 2024, 36, 2404103 CAS.
- H. Zhang, K. Xu, F. He, Y. Zhou, K. Sasaki, B. Zhao, Y. Choi, M. Liu and Y. Chen, Adv. Energy Mater., 2022, 12, 2200761 CAS.
- M. Choi, D. Kim, T. K. Lee, J. Lee, H. S. Yoo and W. Lee, Adv. Energy Mater., 2025, 15, 2400124 CAS.
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O’Hayre, Science, 2015, 349, 1321–1326 CAS.
- J. H. Kim, D. Kim, S. Ahn, K. J. Kim, S. Jeon, D.-K. Lim, J. K. Kim, U. Kim, H.-N. Im, B. Koo, K. T. Lee and W. C. Jung, Energy Environ. Sci., 2023, 16, 3803–3814 CAS.
- H. Zhang, K. Xu, Y. Xu, F. He, F. Zhu, K. Sasaki, Y. Choi and Y. Chen, Energy Environ. Sci., 2024, 17, 3433–3442 CAS.
- M. Liang, Y. Song, B. Xiong, D. Liu, D. Xue, L. Shen, K. Shi, Y. Song, J. Li, Q. Niu, M. G. Xu, F. Ciucci, W. Zhou and Z. Shao, Adv. Funct. Mater., 2024, 34, 2408756 CAS.
- B. Stoeckl, V. Subotić, M. Preininger, M. Schwaiger, N. Evic, H. Schroettner and C. Hochenauer, Electrochim. Acta, 2019, 298, 874–883 CAS.
- J. Yang, A. F. S. Molouk, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, ACS Appl. Mater. Interfaces, 2015, 7, 28701–28707 CrossRef CAS PubMed.
- B. Miao, Z. Deng, P. Han, N. Yan, Z. Pan and S. H. Chan, Chem. Eng. J., 2024, 159062 Search PubMed.
- N. Jantakananuruk, J. R. Page, C. D. Armstrong, J. Persky, R. Datta and A. R. Teixeira, J. Power Sources, 2022, 548, 231999 CrossRef CAS.
- L. Chen, H. Zhang, K. Xu, Y. Xu, X. Zhang, F. Zhu, F. He and Y. Chen, Mater. Today Catal., 2024, 7, 100072 CrossRef.
- H. Lan, J. Chu, X. Chen, Q. Zhou, W. Jin, Y. Zhang and J. Zhou, J. Power Sources, 2024, 593, 233987 CrossRef CAS.
- Y. Wang, Y. Gu, H. Zhang, J. Yang, J. Wang, W. Guan, J. Chen, B. Chi, L. Jia, H. Muroyama, T. Matsui, K. Eguchi and Z. Zhong, Appl. Energy, 2020, 270, 115185 CrossRef CAS.
- Y. Zhang, B. Chen, D. Guan, M. Xu, R. Ran, M. Ni, W. Zhou, R. O’Hayre and Z. Shao, Nature, 2021, 591, 246–251 CrossRef CAS PubMed.
- J. Chen, W. Gao, L. Zhu, H. Tao, S. Feng, H. Cao, J. Guo, Y. Chen and P. Chen, J. Mater. Chem. A, 2024, 12, 26667–26677 RSC.
- M. Li, B. Hua, W. Wu, L.-C. Wang, Y. Ding, M. M. Welander, R. A. Walker and D. Ding, Mater. Today, 2022, 60, 31–40 CrossRef CAS.
- L. Cheng, Y. Zhou, L. Luo, L. Wang, X. Xu, D. Guan, W.-H. Huang, C.-W. Pao, Z. Hu, J. Zhou, S. Wang and Z. Shao, Chem. Eng. J., 2025, 505, 159587 CrossRef CAS.
- S. Zhai, R. Zhao, H. Liao, L. Fu, S. Hao, J. Cai, Y. Wu, J. Wang, Y. Jiang, J. Xiao, T. Liu and H. Xie, J. Energy Chem., 2024, 96, 39–48 CrossRef CAS.
- L. Zhu, C. Cadigan, C. Duan, J. Huang, L. Bian, L. Le, C. H. Hernandez, V. Avance, R. O’Hayre and N. P. Sullivan, Commun. Chem., 2021, 4, 121 CrossRef CAS PubMed.
- G. Marnellos and M. Stoukides, Science, 1998, 282, 98–100 CrossRef CAS PubMed.
- W. Wang, X. Cao, W. Gao, F. Zhang, H. Wang and G. Ma, J. Membr. Sci., 2010, 360, 397–403 CrossRef CAS.
- J. Otomo, N. Noda and F. Kosaka, ECS Trans., 2015, 68, 2663 CrossRef CAS.
- F. Kosaka, N. Noda, T. Nakamura and J. Otomo, J. Mater. Sci., 2017, 52, 2825–2835 CrossRef CAS.
- F. Kosaka, T. Nakamura and J. Otomo, J. Electrochem. Soc., 2017, 164, F1323 CrossRef CAS.
- Y. Kobayashi, N. Shimoda, Y. Kimura and S. Satokawa, ECS Trans., 2017, 75, 43 CrossRef CAS.
- M. Okazaki and J. Otomo, ECS Trans., 2022, 109, 3 CrossRef CAS.
- E. Vasileiou, V. Kyriakou, I. Garagounis, A. Vourros, A. Manerbino, W. Coors and M. Stoukides, Solid State Ionics, 2016, 288, 357–362 CAS.
- K. Wang, W. Zan, Y. Li, S. D. Li, Z. Shao and H. Chen, Adv. Funct. Mater., 2024, 2418404 Search PubMed.
- E. Vasileiou, V. Kyriakou, I. Garagounis, A. Vourros and M. Stoukides, Solid State Ionics, 2015, 275, 110–116 CrossRef CAS.
- Y. Guo, B. Liu, Q. Yang, C. Chen, W. Wang and G. Ma, Electrochem. Commun., 2009, 11, 153–156 CrossRef CAS.
- J. Yin, X. Wang, J. Xu, H. Wang, F. Zhang and G. Ma, Solid State Ionics, 2011, 185, 6–10 CAS.
- D. S. Yun, J. H. Joo, J. H. Yu, H. C. Yoon, J.-N. Kim and C.-Y. Yoo, J. Power Sources, 2015, 284, 245–251 CAS.
- C. Solís, L. Navarrete, M. Balaguer and J. M. Serra, J. Power Sources, 2014, 258, 98–107 Search PubMed.
- C. Solis, L. Navarrete, S. Roitsch and J. M. Serra, J. Mater. Chem., 2012, 22, 16051–16059 CAS.
- V. Kyriakou, I. Garagounis, A. Vourros, E. Vasileiou and M. Stoukides, Joule, 2020, 4, 142–158 CAS.
- H. Zhu, S. Ricote, C. Duan, R. P. O’Hayre and R. J. Kee, J. Electrochem. Soc., 2018, 165, F845 CAS.
- J. Yang, T. Akagi, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, Fuel Cells, 2015, 15, 390–397 CAS.
- M. Kishimoto, N. Furukawa, T. Kume, H. Iwai and H. Yoshida, Int. J. Hydrogen Energy, 2017, 42, 2370–2380 CAS.
- Y. Luo, Y. Shi, S. Liao, C. Chen, Y. Zhan, C.-T. Au and L. Jiang, J. Power Sources, 2019, 423, 125–136 CAS.
- J. Cao, Y. Ji and Z. Shao, Energy Environ. Sci., 2022, 15, 2200–2232 CAS.
- Z. Liu, H. Di, D. Liu, G. Yang, Y. Zhu, Z. Luo, R. Ran, W. Wang, W. Zhou and Z. Shao, Adv. Funct. Mater., 2024, 2420214 CrossRef.
- S. Oh, M. J. Oh, J. Hong, K. J. Yoon, H.-I. Ji, J.-H. Lee, H. Kang, J.-W. Son and S. Yang, iscience, 2022, 25, 105009 CrossRef CAS PubMed.
- G. Jeerh, M. Zhang and S. Tao, J. Mater. Chem. A, 2021, 9, 727–752 RSC.
- C. Chen, K. Wu, H. Ren, C. Zhou, Y. Luo, L. Lin, C. Au and L. Jiang, Energy Fuels, 2021, 35, 11693–11706 CrossRef CAS.
- A. M. Mehdi, A. Hussain, M. Z. Khan, M. B. Hanif, R.-H. Song, W. W. Kazmi, M. M. Ali, S. Rauf, Y. Zhang and M. M. Baig, Russ. Chem. Rev., 2023, 92, RCR5098 CrossRef.
- J. C. Ganley, F. Thomas, E. Seebauer and R. I. Masel, Catal. Lett., 2004, 96, 117–122 CAS.
- H. Zhang, K. Xu, F. He, F. Zhu, Y. Zhou, W. Yuan, Y. Liu, M. Liu, Y. Choi and Y. Chen, Adv. Mater., 2024, 36, 2313966 CAS.
- J. Yang, A. F. S. Molouk, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, ACS Appl. Mater. Interfaces, 2015, 7, 7406–7412 CAS.
- A. Kruth and J. T. Irvine, Solid State Ionics, 2003, 162, 83–91 Search PubMed.
- A. Kruth, R. Davies, M. Islam and J. T. S. Irvine, Chem. Mater., 2007, 19, 1239–1248 CrossRef CAS.
- K. Miyazaki, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, J. Power Sources, 2017, 365, 148–154 CAS.
- J. Yun, G. Xiong, S. Kim, D. Bardgett, S. Choi and S. M. Haile, ACS Energy Lett., 2024, 9, 5520–5528 CAS.
- O. B. Rizvandi, A. Nemati, M. Chen and H. L. Frandsen, Int. J. Hydrogen Energy, 2024, 50, 961–976 Search PubMed.
- T. Su, B. Guan, J. Zhou, C. Zheng, J. Guo, J. Chen, Y. Zhang, Y. Yuan, W. Xie and N. Zhou, Energy Fuels, 2023, 37, 8099–8127 CAS.
- F. He, Q. Gao, Z. Liu, M. Yang, R. Ran, G. Yang, W. Wang, W. Zhou and Z. Shao, Adv. Energy Mater., 2021, 11, 2003916 Search PubMed.
- F. He, Z. Teng, G. Yang, C. Zhou, D. Guan, S. Chen, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Power Sources, 2020, 460, 228105 CAS.
- Y. Song, J. Chen, M. Yang, M. Xu, D. Liu, M. Liang, Y. Wang, R. Ran, W. Wang, F. Ciucci and Z. Shao, Small, 2022, 18, 2200450 CrossRef CAS PubMed.
- H. Zhang, Y. Zhou, K. Pei, Y. Pan, K. Xu, Y. Ding, B. Zhao, K. Sasaki, Y. Choi, Y. Chen and M. Liu, Energy Environ. Sci., 2022, 15, 287–295 RSC.
- H. J. Jeong, W. Chang, B. G. Seo, Y. S. Choi, K. H. Kim, D. H. Kim and J. H. Shim, Small, 2023, 19, 2208149 CrossRef CAS PubMed.
- Q. Ma, R. Peng, Y. Lin, J. Gao and G. Meng, J. Power Sources, 2006, 161, 95–98 CrossRef CAS.
- L. Zhang and W. Yang, J. Power Sources, 2008, 179, 92–95 CrossRef CAS.
- K. Xie, Q. Ma, B. Lin, Y. Jiang, J. Gao, X. Liu and G. Meng, J. Power Sources, 2007, 170, 38–41 CrossRef CAS.
- N. Maffei, L. Pelletier, J. Charland and A. McFarlan, J. Power Sources, 2005, 140, 264–267 CrossRef CAS.
- Y. Yoo, M. Tuck, N. Lim, A. McFarlan and N. Maffei, ECS Trans., 2007, 7, 2305 CrossRef CAS.
- Y. Aoki, S. Kobayashi, E. Tsuji and H. Habazaki, ECS Trans., 2015, 68, 2735 CrossRef CAS.
- K. Xie, R. Yan, G. Meng and X. Liu, Ionics, 2009, 15, 115–119 CAS.
- K. Xie, R. Yan, D. Dong, S. Wang, X. Chen, T. Jiang, B. Lin, M. Wei, X. Liu and G. Meng, J. Power Sources, 2008, 179, 576–583 CAS.
- Y. Yoo, N. Lim, M. Phongaksorn, A. McFarlan and N. Maffei, ECS Trans., 2008, 12, 691 CAS.
- Y. Aoki, T. Yamaguchi, S. Kobayashi, C. Zhu and H. Habazaki, ECS Trans., 2017, 78, 1511 CAS.
- Y. Aoki, T. Yamaguchi, S. Kobayashi, D. Kowalski, C. Zhu and H. Habazaki, Glob. Chall., 2018, 2, 1700088 Search PubMed.
- F. Liu, H. Deng, D. Diercks, P. Kumar, M. H. A. Jabbar, C. Gumeci, Y. Furuya, N. Dale, T. Oku, M. Usuda, P. Kazempoor, L. Fang, D. Chen, B. Liu and C. Duan, Nat. Energy, 2023, 8, 1145–1157 CrossRef CAS.
- C. Zhou, J. Sunarso, J. Dai, R. Ran, Y. Song, F. He, W. Zhou and Z. Shao, J. Membr. Sci., 2020, 596, 117709 CrossRef CAS.
- R. Ren, Z. Wang, C. Xu, W. Sun, J. Qiao, D. W. Rooney and K. Sun, J. Mater. Chem. A, 2019, 7, 18365–18372 RSC.
- Y. Shin, Y.-d Kim, M. Sanders, S. P. Harvey, M. Walker and R. O'Hayre, J. Mater. Chem. A, 2022, 10, 24839–24853 RSC.
- M. Liang, F. He, C. Zhou, Y. Chen, R. Ran, G. Yang, W. Zhou and Z. Shao, Chem. Eng. J., 2021, 420, 127717 CrossRef CAS.
- H. Lee, H. Jung, C. Kim, S. Kim, I. Jang, H. Yoon, U. Paik and T. Song, ACS Appl. Energy Mater., 2021, 4, 11564–11573 CrossRef CAS.
- M. Liang, Y. Zhu, Y. Song, D. Guan, Z. Luo, G. Yang, S. P. Jiang, W. Zhou, R. Ran and Z. Shao, Adv. Mater., 2022, 34, 2106379 CrossRef CAS PubMed.
- Y. Song, Y. Chen, W. Wang, C. Zhou, Y. Zhong, G. Yang, W. Zhou, M. Liu and Z. Shao, Joule, 2019, 3, 2842–2853 CrossRef CAS.
- Y. Pan, H. Zhang, K. Xu, Y. Zhou, B. Zhao, W. Yuan, K. Sasaki, Y. Choi, Y. Chen and M. Liu, Appl. Catal., B, 2022, 306, 121071 Search PubMed.
- Z. Huang, T. Chen, X. Zhang, K. Liu, T. Li, S. Duo, H. Zhang, Y. Ling and S. Wang, Ceram. Int., 2024, 50, 10551–10559 CrossRef.
- M. Hou, Y. Pan and Y. Chen, Sep. Purif. Technol., 2022, 297, 121483 Search PubMed.
- F. He, M. Hou, Z. Du, F. Zhu, X. Cao, Y. Ding, Y. Zhou, M. Liu and Y. Chen, Adv. Mater., 2023, 35, 2304957 Search PubMed.
- S. Wang, F. Gong, Q. Zhou, Y. Xie, H. Li, M. Li, E. Fu, P. Yang, Y. Jing and R. Xiao, Appl. Catal., B, 2023, 339, 123134 Search PubMed.
- A. W. Tricker, K. L. Hebisch, M. Buchmann, Y.-H. Liu, M. Rose, E. Stavitski, A. J. Medford, M. C. Hatzell and C. Sievers, ACS Energy Lett., 2020, 5, 3362–3367 CrossRef CAS.
- S. Zhou, X. Yang, X. Xu, S. X. Dou, Y. Du and J. Zhao, J. Am. Chem. Soc., 2019, 142, 308–317 CrossRef PubMed.
- V. C. Graça, L. I. Holz, A. J. Araújo, F. J. Loureiro and D. P. Fagg, J. Energy Storage, 2023, 68, 107769 CrossRef.
- I. Valov, B. Luerssen, E. Mutoro, L. Gregoratti, R. A. De Souza, T. Bredow, S. Günther, A. Barinov, P. Dudin, M. Martin and J. Janek, Phys. Chem. Chem. Phys., 2011, 13, 3394–3410 RSC.
- D. Yao, C. Tang, L. Li, B. Xia, A. Vasileff, H. Jin, Y. Zhang and S. Z. Qiao, Adv. Energy Mater., 2020, 10, 2001289 CrossRef CAS.
- F. Chang, H. Wu, R. V. D. Pluijm, J. Guo, P. Ngene and P. E. De Jongh, J. Phys. Chem. C, 2019, 123, 21487–21496 CrossRef CAS PubMed.
- K. Yamazaki, M. Matsumoto, M. Ishikawa and A. Sato, Appl. Catal., B, 2023, 325, 122352 CrossRef CAS.
- K. Xu, Y. Y. Zhang, W. W. Wang, M. Peng, J. C. Liu, C. Ma, Y. W. Zhang, C. J. Jia, D. Ma and C. H. Yan, Angew. Chem., 2025, 137, e202416195 CrossRef.
- Y. Li, Q. Guan, G. Huang, D. Yuan, F. Xie, K. Li, Z. Zhang, X. San and J. Ye, Adv. Energy Mater., 2022, 12, 2202459 CAS.
- M. Kishimoto, H. Muroyama, S. Suzuki, M. Saito, T. Koide, Y. Takahashi, T. Horiuchi, H. Yamasaki, S. Matsumoto, H. Kubo, N. Takahashi, A. Okabe, S. Ueguchi, M. Jun, A. Tateno, T. Matsuo, T. Matsui, H. Iwai, H. Yoshida and K. Eguchi, Fuel Cells, 2020, 20, 80–88 CAS.
- S. Kim, H. Lee, C. Kim, I. Jang, K. Lee, S. Sun, D. Lee, J. Kim, K. Park, G. Lee, H. Jeong, H. Yoon, U. Paik and T. Song, J. Power Sources, 2022, 548, 232082 CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |