
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn integrated design for high-energy, durable zinc–iodine batteries with ultra-high recycling efficiency†
Leiqian
Zhang‡
a,
Han
Ding‡
b,
Haiqi
Gao
b,
Jiaming
Gong
c,
Hele
Guo
d,
Shuoqing
Zhang
e,
Yi
Yu
f,
Guanjie
He
 g,
Tao
Deng
h,
Ivan P.
Parkin
g,
Johan
Hofkens
di,
Xiulin
Fan
*e,
Feili
Lai
*cd and
Tianxi
Liu
*a
g,
Tao
Deng
h,
Ivan P.
Parkin
g,
Johan
Hofkens
di,
Xiulin
Fan
*e,
Feili
Lai
*cd and
Tianxi
Liu
*a
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi, 214122, P. R. China. E-mail: txliu@jiangnan.edu.cn
bState Key Laboratory of Chemistry and Utilization of Carbon Based Energy Resources, Xinjiang University, Urumqi, 830017, Xinjiang, P. R. China
cState Key Laboratory of Metal Matrix Composites, School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai, 200240, P. R. China. E-mail: feililai@sjtu.edu.cn
dDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, Leuven 3001, Belgium. E-mail: feili.lai@kuleuven.be
eState Key Laboratory of Silicon and Advanced Semiconductor Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou, 310027, P. R. China. E-mail: xlfan@zju.edu.cn
fSchool of Chemical Engineering, Zhengzhou University, Zhengzhou, 450001, P. R. China
gDepartment of Chemistry, University College London, London, WC1H 0AJ, UK
hChina-UK Low Carbon College, Shanghai Jiao Tong University, Shanghai, 201306, P. R. China
iDepartment of Molecular Spectroscopy, Max Planck Institute for Polymer Research, Ackermannweg 10, Mainz 55128, Germany
First published on 3rd February 2025
Abstract
Zinc–iodine batteries (ZIBs) have long struggled with the uncontrolled spread of polyiodide in aqueous electrolytes, despite their environmentally friendly, inherently safe, and cost-effective nature. Here, we present an integral redesign of ZIBs that encompasses both the electrolyte and cell structure. The developed self-sieving polyiodide-capable liquid–liquid biphasic electrolyte can achieve an impressive polyiodide extraction efficiency of 99.98%, harnessing a meticulously iodine-containing hydrophobic solvated shell in conjunction with the salt-out effect. This advancement facilitates a membrane-free design with a Coulombic efficiency of ∼100% at 0.1C, alongside an ultra-low self-discharge rate of ∼3.4% per month and capacity retention of 83.1% after 1300 cycles (iodine areal loading: 22.2 mg cm−2). Furthermore, the integrated cell structure, paired with the low-cost electrolyte ($4.6 L−1), enables rapid assembly into A h-level batteries within hours (1.18 A h after 100 cycles with a capacity retention of 86.7%), supports electrolyte regeneration with ∼100% recycling efficiency, and extends to ZIBs with a two-electron iodine conversion reaction. This endeavor establishes a novel paradigm for the development of practical zinc–iodine batteries.
Broader contextIn the contemporary quest for carbon-free and sustainable lifestyles, aqueous zinc-based batteries are shining brightly celebrated for their intrinsic safety, affordability, and environmental friendliness. Especially, zinc–iodine batteries, as a nascent energy storage technology, have recently garnered substantial research attention, distinguished by their remarkable cycle life and rate performance among various zinc-based batteries. Nevertheless, the advancement of zinc–iodine batteries is critically hindered by the inability to fundamentally address the dissolution/diffusion issue of highly water-soluble polyiodide in aqueous electrolytes and iodine-containing active materials recycling. This study, inspired by the extraction concept, proposes a comprehensive redesign of zinc–iodine batteries, encompassing both electrolyte and cell structure, to facilitate the development of A h-grade, cost-effective, shuttle-free, and highly recyclable zinc–iodine batteries. This endeavor presents a versatile research framework for advancing the practical implementation of zinc–iodine batteries. |
Introduction
As the transition from fossil fuel-dependent power generation to carbon-neutral energy sources accelerates, the significance of grid-level electrical energy storage systems (GLEESs) in harnessing intermittent renewable energy is becoming increasingly evident.1–4 In this context, rechargeable lithium-ion batteries (LIBs) have emerged as the preferred choice for GLEESs, owing to their technological maturity and impressive energy density.5 However, the widespread integration of LIBs in durable, secure, and cost-effective grid-level storage solutions faces challenges due to the limited availability and geopolitical risk of raw materials, as well as the inherent flammability.6–8 Consequently, there has been a rapid proliferation of novel rechargeable battery technologies, including sodium/zinc-based batteries and flow batteries, as viable complements or alternatives to LIBs.9–16Aqueous rechargeable zinc–iodine batteries (ARZIBs) represent an innovative battery technology that utilizes the reversible redox process between iodine and zinc metal for energy storage. The theoretical voltage plateaus of ARZIBs are primarily determined by the iodine conversion processes (I3−/I−, E = 0.536 V vs. standard hydrogen electrode (SHE); I2/I−, E = 0.621 V vs. SHE), but typically range around 1.3–1.4 V, effectively mitigating the issues of aqueous electrolyte decomposition.17 Additionally, ARZIBs offer a high theoretical capacity (I2/I−, 211 mA h g−1) and boast attributes such as safety, sustainability, and eco-friendliness.17–19 The liquid-phase conversion mechanism (I−/I3−) in the iodine cathode also endows ARZIBs with excellent rate capability.20–22 Nevertheless, the formation of water-soluble polyiodide poses a significant challenge to the long-term stability of ARZIBs, as these polyiodide can quickly permeate into the electrolytes, causing the depletion of iodine-active material from the cathodes (known as the polyiodide shuttle effect).18,23 Upon diffusing to the zinc anode, the polyiodide promptly engages in a spontaneous reaction with the highly reactive zinc, thus leading to the swift self-discharge of ARZIBs.23,24
To address these challenges, iodine host materials have been conventionally employed to encapsulate iodine species, leveraging their tailored pore structures or active functional groups to confine polyiodide.21,25–27 However, the extensive solubility of iodide species in a wide range of solvents—from low-polarity carbon tetrachloride to high-polarity water—poses a significant obstacle to achieving a definitive solution to polyiodide shuttle effect through this strategy (Fig. 1a).28,29 In an effort to minimize the risks associated with polyiodide shuttling, low iodine loadings (<5 mg cm−2) are typically adopted during cell evaluations, although this deviates from actual operational requirements (Fig. 1b, derived from data in Table S1, ESI†). Moreover, the low Coulombic efficiency (CE) observed at low current densities—typically below 99%,30 alongside the rapid self-discharge rate, which results in over 2% capacity loss per day (Fig. 1c), presents formidable challenges for iodine host materials. Thus, recent advancements have increasingly focused on the design of functional interlayers/separators,31–33 as well as the utilization of high-concentration electrolytes,34,35 to assist iodine host materials in addressing the above issues. For instance, a Ti2O(PO4)2-2H2O/ι-carrageenan interlayer and a 20 m ZnCl2 electrolyte combined with the carbon-based iodine cathodes have achieved iodine loading of 38 and 25.33 mg cm−2, respectively.32,35 Although these strategies partially mitigate the identified challenges, they inherently increase the complexity and cost of the system. Moreover, a long-overlooked practical issue arises regarding the recycling of active materials (Fig. 1d). The conventional sandwich battery structure complicates the separation and reuse of individual active components—such as membranes, interlayers, and iodine cathodes, due to its excessively compact design.36 This challenge is particularly pronounced for customized iodine host materials due to the complexity of their components (involving conductive agents, binders, and active materials) and their strong adsorption of iodine species, which could greatly hinder the separation of iodine species from iodine host materials. Thus, while addressing the shuttle effect is crucial, innovations in electrode and cell design are imperative to enhance the suitability of ARZIBs for GLEESs that prioritize cost-effectiveness (manufacturing costs, recyclability, etc.), safety, and ease of scalability. Notably, the incorporation of a second-phase electrolyte (e.g., acetonitrile) presents a promising solution, as it can mitigate the cross-contamination of polyiodide and offer high scalability.37 However, ARZIBs utilizing such a biphasic electrolyte configuration remain in their infancy and encounter several significant challenges, including low iodine loading (merely 0.4 M I−), limited polyiodide confinement capability (∼95% CE for the scaled-up ARZIB), and a dearth of suitable electrode designs tailored for the biphasic electrolyte structure.
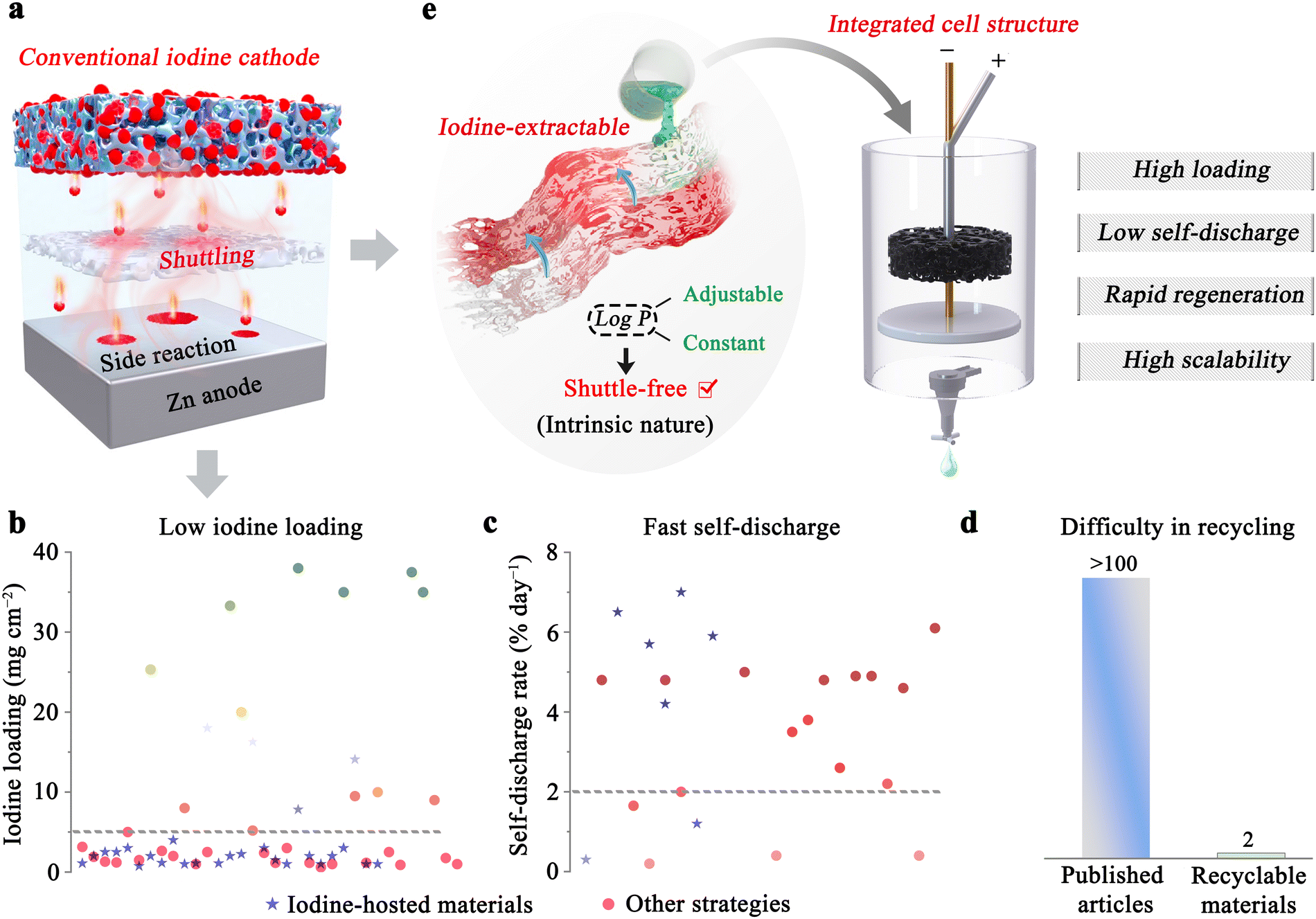 | ||
| Fig. 1 Addressing challenges and design principles for advanced ARZIBs. (a)–(d) The challenges encountered by conventional ARZIBs, including polyiodide shuttling, low loading, fast self-discharge, and recycling difficulties. (e) The benefits of integrated ARZIBs designed based on the extraction principle. | ||
In this study, we engineer a self-sieving polyiodide-capable liquid–liquid biphasic system with a significantly simplified cell structure (Fig. 1e). This system boasts a thermodynamically stable configuration comprising an iodine-rich organic phase atop an aqueous electrolyte phase, complemented by an integrally designed electrode structure, wherein graphite felt serves as the positive current collector and zinc plate functions as both the anode and negative current collector. Specifically, the aqueous phase including significant quantities of sulphates effectively mitigates the crossover between the aqueous and organic phases via the salting-out effect. Concurrently, the organic phase comprises hydrophobic tetrabutylammonium iodide and ionic liquid in acetonitrile solution, endowing the organic phase with a high polyiodide extraction efficiency (99.98%) and low interfacial overpotential (∼62 mV) through the formation of iodine-containing hydrophobic solvated shells and a phase-transfer-type ion transport mechanism. Consequently, the as-developed ARZIB can deliver a reversible specific capacity of 125.1 mA h g−1 after 1300 cycles at 1C, corresponding to 83.1% capacity retention (iodine areal loading of 22.2 mg cm−2). Besides, even under an ultra-high iodine areal loading of 69.8 mg cm−2, the battery showcases an energy density of 110.3 W h kg−1iodine after 200 cycles, with an impressive capacity retention rate of 94.7%. Furthermore, the battery can achieve a remarkable CE of around 100% at 0.1C and maintain a capacity retention of 96.6% in a 30-day self-discharge assessment without ion-exchange membrane. The scalability and practical utility of this battery are underscored by its capacity to accommodate a 6 A h battery stack within hours, reliably store and release solar energy, near-perfect electrolyte cycling efficiency (∼100%), and successful extend to ZIBs featuring a two-electron iodine conversion reaction. This investigation presents a versatile research framework for advancing the practical implementation of zinc–iodine batteries in GLEES.
Results and discussion
Preparation of liquid–liquid biphasic system
Extraction is a widely employed technique for separating compounds by leveraging the differing solubilities of components in immiscible solvents (Fig. 1e). The pivotal factor influencing separation efficiency is the partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), representing the ratio of activity (typically approximated as concentration) of a substance in two immiscible solvents upon achieving equilibrium partitioning.38 Theoretically, the logP value remains constant under specific conditions.39 Thus, it is plausible that if a solvent system is capable of effectively extracting polyiodide from an aqueous electrolyte into its bulk phase, the polyiodide shuttling could not occur in the aqueous electrolyte. This is due to the constant distribution of polyiodide between two phases, an inherent characteristic of this system. To establish such a shuttle-free liquid–liquid biphasic system, the selection of an appropriate solvent is crucial for the efficient extraction of iodine from water. Following the principle of similarity and intermiscibility, iodine exhibits high solubility in non-polar solvents like carbon tetrachloride and carbon disulfide, enabling them to act as effective extractants for polyiodide. However, their low dielectric constants lead to the formation of solutions with typically low ionic conductivity, rendering them unsuitable for application as battery electrolytes. In contrast, acetonitrile (AN), characterized by a high relative permittivity (∼36), is a favoured solvent for numerous inorganic salts due to its low viscosity, minimal toxicity, and high stability, making it a prevalent choice in formulating electrolytes for non-aqueous electrochemical energy storage systems.40–43 Notably, AN also exhibits proficiency in extracting polyiodide from aqueous electrolytes (Fig. 2a). Consequently, AN is a perfectly choice as the solvent to compose the organic phase of the liquid–liquid biphasic system. To endow the organic phase with high ionic conductivity, hydrophobic tetrabutylammonium iodide (TBAI) is introduced to AN. As depicted in Fig. S1 (ESI†), TBAI displays a solubility of up to 1 M in AN (referred to as 1T-AN), resulting in an impressive ionic conductivity reaching 20.4 mS cm−1 that meets the criteria for an exceptional electrolyte solution.44 Especially, the incorporation of TBAI also effectively enhances the solubilization of iodine in AN (up to 2 M), as demonstrated in Fig. S2 (ESI†), which is crucial for augmenting the energy density of the whole system. Besides, the extractability of AN for polyiodide is significantly improved by the robust interaction between TBAI and iodine, as illustrated in Fig. 2a and Fig. S3 (ESI†). Nevertheless, the 1T-AN-based system experiences volatility at room temperature due to the low boiling point of AN (∼82 °C).45 Hence, we propose the incorporation of ionic liquids into 1T-AN to alleviate the volatility of AN. The hydrophobic 1-butyl-3-methylimidazole hexafluorophosphate ([BMIM]PF6) is selected as an additive for 1T-AN owing to its cost-effectiveness and compatibility with AN. As shown in Fig. 2b, the introduction of [BMIM]PF6 not only diminishes AN volatilization but also enhances the ionic conductivity of the electrolyte. However, an excessive amount of [BMIM]PF6 can lead to a gradual decline in the ionic conductivity of electrolytes, coupled with an increase in the fraction of inactive components in the system. Taken together, we opt for a 7:3 (v/v) ratio of 1T-AN to [BMIM]PF6 (noted as 1T-7A3B), where the ionic conductivity of 1T-AN can be elevated from 20.4 to 25.3 mS cm−1, while significantly reducing its volatility.
P), representing the ratio of activity (typically approximated as concentration) of a substance in two immiscible solvents upon achieving equilibrium partitioning.38 Theoretically, the logP value remains constant under specific conditions.39 Thus, it is plausible that if a solvent system is capable of effectively extracting polyiodide from an aqueous electrolyte into its bulk phase, the polyiodide shuttling could not occur in the aqueous electrolyte. This is due to the constant distribution of polyiodide between two phases, an inherent characteristic of this system. To establish such a shuttle-free liquid–liquid biphasic system, the selection of an appropriate solvent is crucial for the efficient extraction of iodine from water. Following the principle of similarity and intermiscibility, iodine exhibits high solubility in non-polar solvents like carbon tetrachloride and carbon disulfide, enabling them to act as effective extractants for polyiodide. However, their low dielectric constants lead to the formation of solutions with typically low ionic conductivity, rendering them unsuitable for application as battery electrolytes. In contrast, acetonitrile (AN), characterized by a high relative permittivity (∼36), is a favoured solvent for numerous inorganic salts due to its low viscosity, minimal toxicity, and high stability, making it a prevalent choice in formulating electrolytes for non-aqueous electrochemical energy storage systems.40–43 Notably, AN also exhibits proficiency in extracting polyiodide from aqueous electrolytes (Fig. 2a). Consequently, AN is a perfectly choice as the solvent to compose the organic phase of the liquid–liquid biphasic system. To endow the organic phase with high ionic conductivity, hydrophobic tetrabutylammonium iodide (TBAI) is introduced to AN. As depicted in Fig. S1 (ESI†), TBAI displays a solubility of up to 1 M in AN (referred to as 1T-AN), resulting in an impressive ionic conductivity reaching 20.4 mS cm−1 that meets the criteria for an exceptional electrolyte solution.44 Especially, the incorporation of TBAI also effectively enhances the solubilization of iodine in AN (up to 2 M), as demonstrated in Fig. S2 (ESI†), which is crucial for augmenting the energy density of the whole system. Besides, the extractability of AN for polyiodide is significantly improved by the robust interaction between TBAI and iodine, as illustrated in Fig. 2a and Fig. S3 (ESI†). Nevertheless, the 1T-AN-based system experiences volatility at room temperature due to the low boiling point of AN (∼82 °C).45 Hence, we propose the incorporation of ionic liquids into 1T-AN to alleviate the volatility of AN. The hydrophobic 1-butyl-3-methylimidazole hexafluorophosphate ([BMIM]PF6) is selected as an additive for 1T-AN owing to its cost-effectiveness and compatibility with AN. As shown in Fig. 2b, the introduction of [BMIM]PF6 not only diminishes AN volatilization but also enhances the ionic conductivity of the electrolyte. However, an excessive amount of [BMIM]PF6 can lead to a gradual decline in the ionic conductivity of electrolytes, coupled with an increase in the fraction of inactive components in the system. Taken together, we opt for a 7:3 (v/v) ratio of 1T-AN to [BMIM]PF6 (noted as 1T-7A3B), where the ionic conductivity of 1T-AN can be elevated from 20.4 to 25.3 mS cm−1, while significantly reducing its volatility.
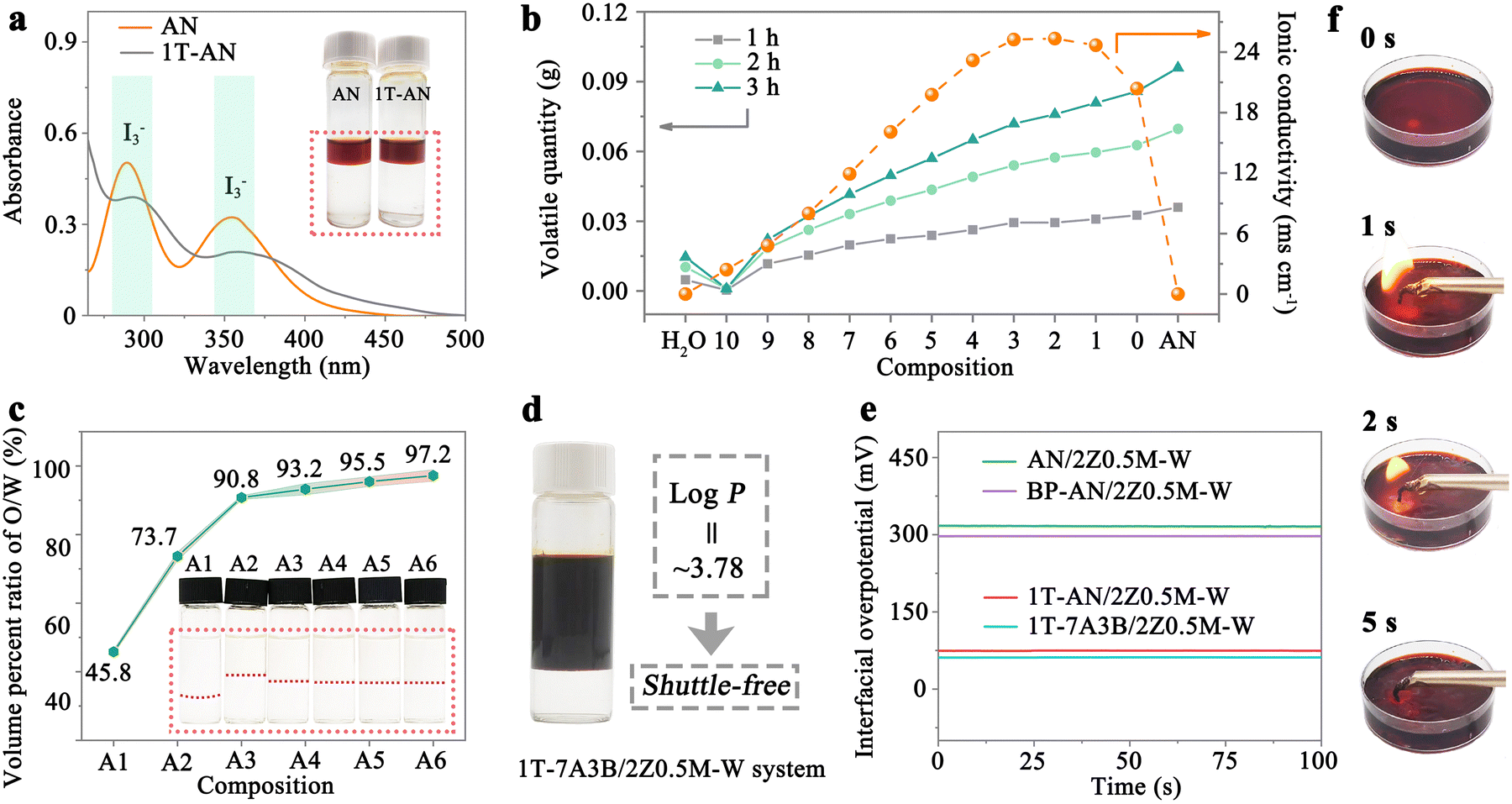 | ||
| Fig. 2 Construction and characterization of liquid–liquid biphasic system. (a) UV-vis spectra of aqueous phase containing polyiodide extracted using AN and 1T-AN. The inset showcases the polyiodide extracted by AN and 1T-AN upon reaching equilibrium. (b) Volatile quantity (left) and ionic conductivity (right) of 1T-AN solutions with varying amounts of [BMIM]PF6. Here, AN and H2O represent pure acetonitrile and water solutions, while 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 correspond to the 1T-AN to [BMIM]PF6 ratios of 10:0, 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, 1:9, and 0:10 (v/v), respectively. (c) Volume percent ratio of the organic (1T-7A3B) to the aqueous phase containing varying salt concentrations (A1: 0 M ZnSO4; A2: 1.0 M ZnSO4; A3: 2.0 M ZnSO4; A4: 2.0 M ZnSO4 + 0.2 M MgSO4; A5: 2.0 M ZnSO4 + 0.5 M MgSO4; A6: 2.0 M ZnSO4 + 1.0 M MgSO4). Here, the inset displays the corresponding degrees of phase separations. (d) 1T-7A3B/2Z0.5M-W system with 0.2 M I2 in the organic phase (top). (e) Interfacial overpotentials of biphasic systems containing different ionic salts in the organic phase. (f) Flammability assessment of 1T-7A3B/2Z0.5M-W system. | ||
To devise the aqueous phase, we adopt a salting-out approach to prevent its intermixing with the organic phase. As illustrated in Fig. 2c, the oil and water phases gradually separate with increasing ZnSO4 concentration. When the concentration of ZnSO4 solution is 2 M, the volume ratio of oil to water reaches 90.8%. Additionally, according to the Hofmeister series, MgSO4 is included due to its low molecular weight and high drainage capacity, which can effectively inhibit the mixing of AN with water at a low dosage. Following the addition of 0.2, 0.5, and 1.0 M MgSO4, the volume percent ratios of oil to water increase to 93.2%, 95.5%, and 97.2%, respectively. After careful consideration of the electrolyte cost and oil–water separation rate, 0.5 M MgSO4 is selected as the optimal choice for the following experiments.
Overall, the organic phase comprises 1T-AN and [BMIM]PF6 (7:3, v/v), while the aqueous phase is constituted of 2 M ZnSO4 and 0.5 M MgSO4 in water. Hence, we denote this liquid–liquid biphasic system as 1T-7A3B/2Z0.5M-W. As depicted in Fig. 2d, there is no discoloration observed in the aqueous phase despite the addition of 0.2 M I2 to the system. The logP value of the system is subsequently calculated from the ultraviolet-visible (UV-vis) standard curve of polyiodide (Fig. S4, ESI†). Impressively, the logP value of polyiodide in the 1T-7A3B/2Z0.5M-W system reaches approximately 3.78, which means that the concentration of polyiodide in the organic phase is 6025 times higher than that in the aqueous phase, corresponding to an extraction efficiency of 99.98%. This substantiates that the biphasic system can considerably solve the challenging issue of polyiodide shuttling, ensuring that polyiodide is consistently retained in the organic phase. The interfacial overpotential, crucial for facilitating rapid ion migration between the two phases, is assessed using the four-electrode method in Fig. 2e. Due to the introduction of TBAI (a phase-transfer catalyst), the interfacial overpotential notably decreases from ∼306 (AN/2Z0.5M-W) to ∼74 mV (1T-AN/2Z0.5M-W). In addition, the incorporation of [BMIM]PF6 ionic liquid (BP-AN/2Z0.5M-W) also contributes to reducing the interfacial overpotential to ∼297 mV. Consequently, the 1T-7A3B/2Z0.5M-W demonstrates a low interfacial overpotential of ∼62 mV, significantly lower than that of most phase-separated systems (>100 mV),46 which is important for realizing high-rate liquid–liquid biphasic-based ARZIBs. Moreover, by leveraging substantial quantities of [BMIM]PF6 ionic liquid and TABI salts, the system boasts low flammability with maximum battery safety (Fig. 2f and Fig. S5, ESI†).
Electrochemical performance assessment of 1T-7A3B/2Z0.5M-W system
To fully leverage the benefits of the liquid–liquid biphasic electrolyte system, we first refined the battery structure. For wires used in iodine cathode, their resistance to iodine-induced corrosion is a critical factor. As shown in Fig. S6 (ESI†), the corrosion resistances of commonly used metals, including copper, aluminum, iron, and titanium, are systematically evaluated. Among these metals, titanium exhibits exceptional resistance to iodine corrosion, outperforming the other metals tested. Therefore, the cathode (housing the iodine-containing organic phase in the top segment) utilizes graphite felt as the current collector and titanium as the wire, while the aqueous phase in the bottom portion employs zinc plate as the anode (also serving as a current collector) and copper as the wire. The device diagram for battery assessment is outlined in Fig. 3a. This integrated top-down configuration obviates the need for supplementary apparatus such as stirrers, pumps, and ion exchange membranes, significantly streamlining the battery manufacturing process and reducing cost. Meanwhile, the utilization of biphasic electrolyte system circumvents the intricate and costly processes associated with traditional battery manufacturing, such as electrode coating, drying, and roller pressing. In this content, the electrochemical characteristics of the 1T-7A3B/2Z0.5M-W system were scrutinized in ARZIBs utilizing this device. Fig. 3b clearly illustrates the remarkable reversibility of the iodine conversion process in the 1T-7A3B/2Z0.5M-W system during the charge and discharge processes. Upon charging, the top segment undergoes a transition from colorless to nearly black, attributed to the oxidation of iodide ions to iodine, whereas after discharging, the organic phase restores its transparency. Notably, the aqueous phase in the bottom section remains colorless throughout the cycling, indicative of the effective mitigation of polyiodide shuttling in the 1T-7A3B/2Z0.5M-W system. The galvanostatic charge–discharge (GCD) curve of the 1T-7A3B/2Z0.5M-W system is depicted in Fig. 3c, with the primary redox process occurring between 1.1 and 1.3 V corresponding to the I−/I3− transition. Additionally, the 1T-7A3B/2Z0.5M-W system exhibits further redox behavior at an elevated plateau of ∼1.5 V, potentially attributed to the I3−/I5− conversion process. These outcomes align with the cyclic voltammetry (CV) curve of the 1T-7A3B/2Z0.5M-W system in Fig. S7 (ESI†), corroborating the iodine conversion pathway in organic electrolytes.47 Additionally, Fig. S8 (ESI†) presents the electrochemical impedance spectroscopy of 1T-7A3B/2Z0.5M-W system, which exhibits a charge transfer resistance of approximately 12 Ω, highlighting the rapid reaction kinetics within the system. | ||
| Fig. 3 Assessment of the reversibility, rate capability, and stability of 1T-7A3B/2Z0.5M-W system. (a) Schematic representation for the structure of the 1T-7A3B/2Z0.5M-W battery system. (b) Visual depiction of the 1T-7A3B/2Z0.5M-W system at different states of charge. (c) Charge/discharge profiles for the 1T-7A3B/2Z0.5M-W system at 0.1C, accompanied by dQ/dV curve. (d) Rate performance evaluation of the 1T-7A3B/2Z0.5M-W system at varying current densities, and (e) the corresponding rate-dependent charge–discharge curves of the 1T-7A3B/2Z0.5M-W system. (f) Cycling stability analysis of the 1T-7A3B/2Z0.5M-W system at 1C. (g) Self-discharge behavior of the 1T-7A3B/2Z0.5M-W system (Inset reveals the self-discharge behavior in ARZIBs derived from polyiodide shuttling). | ||
The rate capability of the 1T-7A3B/2Z0.5M-W system is further elucidated with an iodine loading of 88.8 mg (equivalent to 1 mL of 1T-7A3B), as illustrated in Fig. 3d. Specifically, at a current density of 0.1C (1C = 211 mA g−1 iodine), the 1T-7A3B/2Z0.5M-W system exhibits an initial discharge specific capacity of 169.1 mA h g−1. With the increase of current densities to 0.2, 0.5, 1.0, and 2.0C, the specific capacities of the battery can be preserved at 97.4, 94.2, 91.3, and 59.4%, respectively. Upon returning to current densities of 0.1, 0.2, and 0.5C, the reversible discharge capacities recover to 165.7, 164.2, and 158.7 mA h g−1, respectively, showcasing a recovery rate exceeding 98%. This underscores the exceptional reliability and rate performance of the 1T-7A3B/2Z0.5M-W system. On the other hand, the 1T-7A3B/2Z0.5M-W system also demonstrates nearly 100% Coulombic efficiency (CE) at low current densities of 0.1C, signifying the dramatic elimination of the shuttle effect. The corresponding GCD profiles at various current densities are provided in Fig. 3e. The polarization voltages exhibit gradual increments with current density from 0.1 to 1C, followed by a sharp rise at 2C. Additionally, the energy efficiencies at diverse current densities are computed based on the GCD curves (Fig. S9, ESI†). Despite the energy efficiency being relatively lower at 2C (58.6%), it reaches 95.0%, 90.5%, 83.2%, and 71.9% at current densities of 0.1, 0.2, 0.5, and 1C, respectively, meeting the requisites for grid-scale EESs.48 Notably, the substitution of TBAI with equimolar solid iodine (0.35 M) will result in a rapid decline in battery rate performance, yielding a specific capacity of only 128.6 mA h g−1 at 0.1C (Fig. S10, ESI†). Furthermore, significant fluctuations in battery capacity are observed, with a discharge specific capacity of 84.5 mA h g−1 when the current density increases to 2C. These findings underscore the crucial role of TBAI in achieving high iodine utilization and enhanced rate performance in the 1T-7A3B/2Z0.5M-W system.
The cycling stability of the 1T-7A3B/2Z0.5M-W system is also assessed and presented in Fig. 3f. Despite the substantial iodine loading of 88.8 mg (areal loading: 22.2 mg cm−2), the 1T-7A3B/2Z0.5M-W system demonstrates exceptional stability with a reversible specific capacity of 127.5 mA h g−1 after 700 cycles. It is noteworthy that the declining capacity can be restored through the replenishment of AN. This may arise from the leakage of a small amount of AN during cycling, resulting in the precipitation of active substances (Fig. S11, ESI†). As a result, the 1T-7A3B/2Z0.5M-W system can realize a reversible specific capacity of 125.1 mA h g−1 after 1300 cycles at 1C. This associates to a capacity retention of 83.1% and an impressive average CE of >99.9%, clearly affirming its reliability. The GCD profiles of the 1T-7A3B/2Z0.5M-W system for varying cycle numbers are depicted in Fig. S12 (ESI†), showcasing distinct discharge plateaus (from 1.2 to 1.0 V) across all instances. Even at a current density of 2C, the 1T-7A3B/2Z0.5M-W system exhibits outstanding stability, retaining 82.0% of its specific capacity after 600 cycles (Fig. S13, ESI†). Notably, minimal polyiodide signals can be detected in the aqueous phase post-cycling (Fig. S14, ESI†), validating the efficacy of the 1T-7A3B/2Z0.5M-W system in effectively mitigating polyiodide shuttling, a persistent challenge in ARZIBs. Besides, after resting periods of 0.5, 1, 2, 4, 8, and 30 days, a fully charged 1T-7A3B/2Z0.5M-W system exhibits remarkable capacity retention values of 99.7%, 99.1%, 98.8%, 98.4%, 97.6%, and 96.6%, respectively (Fig. 3g). This exceptional low self-discharge performance surpasses existing literature benchmarks (Table S1, ESI†), as well as commercial Ni-MH (20 to 30% per month) and lead-acid batteries (5 to 20% per month),30 underscoring the unparalleled effectiveness of the 1T-7A3B/2Z0.5M-W system in suppressing polyiodide shuttling. Furthermore, no significant capacity degradation is observed during the forty-five-day self-discharge assessment. Based on these findings, we can assert that the 1T-7A3B/2Z0.5M-W system possesses a high degree of reliability and applicability for GLEESs.
Investigation into the operational mechanism of 1T-7A3B/2Z0.5M-W system
In situ UV-vis spectroscopy was employed to monitor the variations in polyiodide levels within the aqueous phase of the 1T-7A3B/2Z0.5M-W system throughout the charge and discharge processes. The experimental device is shown in Fig. S15a (ESI†). As depicted in Fig. 4a, the absence of detectable polyiodide signals in the aqueous phase throughout battery cycling suggests the completely suppressed shuttle effect in the 1T-7A3B/2Z0.5M-W system. Furthermore, the conversion of iodine species within the organic phase was scrutinized via in situ Raman spectroscopy, with the experimental device illustrated in Fig. S15b (ESI†). During the charging process, the emergence of I3− peaks within the 110–120 cm−1 range, attributed to the oxidation of I−, is observed (Fig. 4b). Subsequently, as the charging voltage nears 1.6 V, signals corresponding to I5− at 155–165 cm−1 are detected, indicative of further oxidation from I3− to I5−.25,47 Upon discharge, the intensities of the I3− and I5− peaks diminish over time, indicating their gradual conversion back to I− species. These findings align with the CV curve of the battery as shown in Fig. S7 (ESI†). Consequently, we can infer that the iodine conversion process within the 1T-7A3B/2Z0.5M-W system follows the reversible pathway of I− ↔ I3− ↔ I5−.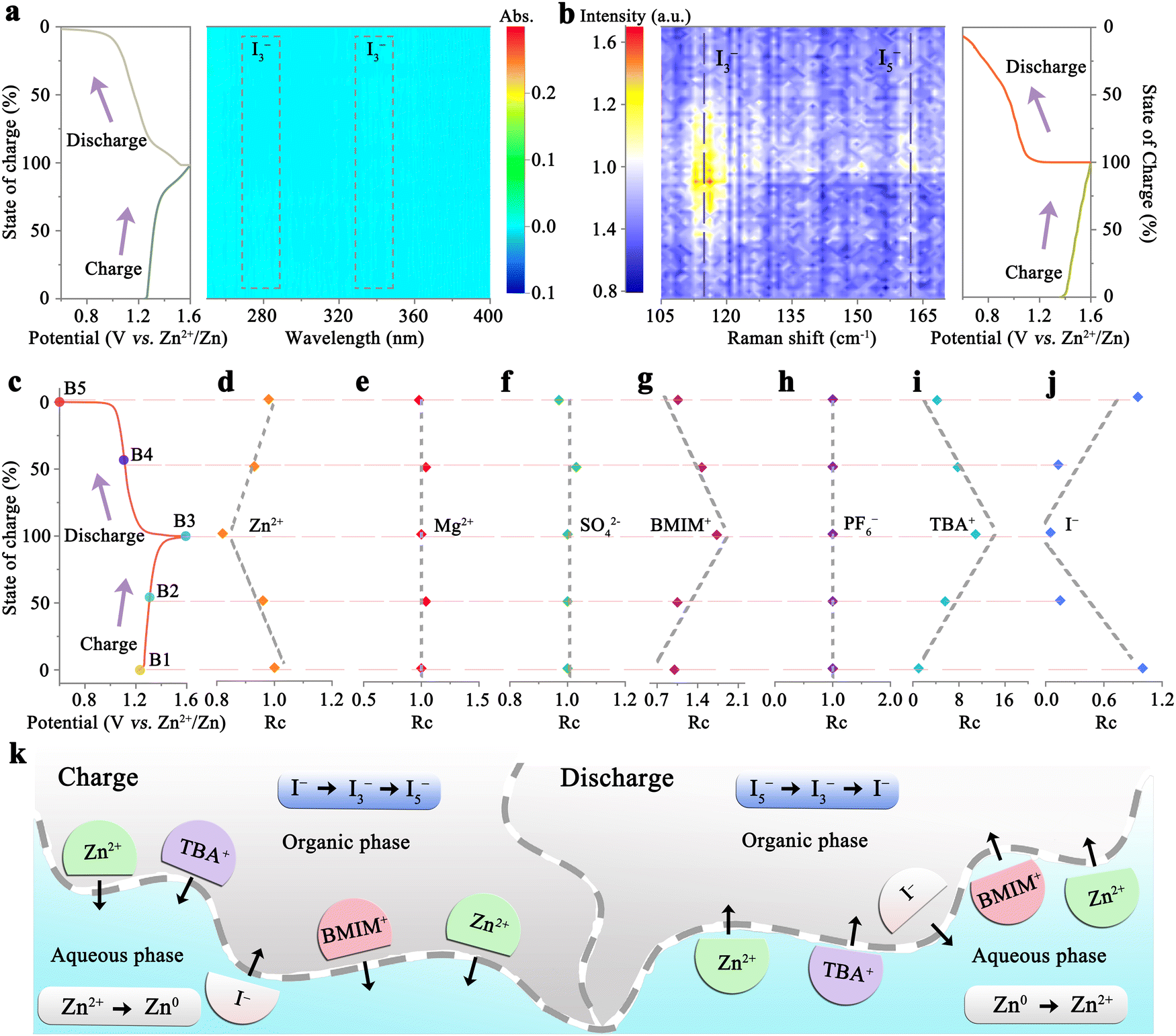 | ||
| Fig. 4 Investigation of energy storage and ion transport mechanisms in the 1T-7A3B/2Z0.5M-W system. (a) In situ UV-vis spectra of the aqueous phase, and (b) in situ Raman spectroscopies of the organic phase along with the corresponding charge–discharge profiles. (c) Charge–discharge profiles illustrating the migration of various ions in the 1T-7A3B/2Z0.5M-W system. The relative concentrations of (d) Zn2+, (e) Mg2+, (f) SO42−, (g) BMIM+, (h) PF6−, (i) TBA+, and (j) I− in the aqueous phase at different SOCs (B1: open-circuit voltage; B2: 1.3 V; B3: 1.6 V; B4: 1.1 V; B5: 0.6 V). (k) Schematic representation elucidating the operational mechanism of the 1T-7A3B/2Z0.5M-W system. | ||
The ion migration dynamics at various states of charge (SOC) between the aqueous and organic phases are comprehensively analyzed in Fig. 4c to j. The concentrations of Zn2+, Mg2+, and SO42− in the aqueous phase were quantified using inductively coupled plasma optical emission spectroscopy. The dissolution-deposition process of Zn2+ ions at the anode leads to a decrease in Zn2+ concentration (initial concentration: ∼1.9 M) during charging and an increase during discharging (Fig. 4d). Notably, a similar migration pattern of Zn2+ (initial concentration: ∼0.1 M) is observed in the organic phase, underscoring their pivotal role in maintaining charge equilibrium between two phases (Fig. S16, ESI†). In contrast, Mg2+ and SO42− predominantly reside in the aqueous phase due to their high hydration energy,49 which are important for preventing miscibility of AN with water (Fig. 4e and f). Besides, BMIM+ (initial concentration: ∼0.02 M) determined by UV-vis spectrum exhibits a reversible mobility process during cycling, i.e., migration to the aqueous phase during charging and back to the organic phase during discharging (Fig. 4g and Fig. S17, ESI†). The alterations in PF6− and TBA+ concentrations were further scrutinized through nuclear magnetic resonance spectroscopy technology (Fig. S18, ESI†). The PF6− (initial concentration: 0 M) consistently remains in the organic phase without SOC-dependent changes, while the migration of TBA+ (initial concentration: ∼0.001 M) displays voltage-dependent behavior (Fig. 4h), transitioning to the aqueous phase during charging and returning to the organic phase during discharging (Fig. 4i). Noteworthy observations include the reversible migration of I− (initial concentration: ∼0.005 M) between two phases, albeit in the opposite direction compared to TBA+ and BMIM+ (Fig. 4j). This observation suggests that the charge difference from the migration of I− can be compensated by these cations. Besides, it is worth noting that even if I− partial diffuses into the aqueous phase (attributable to the dissolution of a fraction of TBAI in water), it can still be effectively utilized through the reversible migration process between two phases. Hence, the CE of the 1T-7A3B/2Z0.5M-W system remains close to 100% even at an ultra-low current density of 0.1C (Fig. 3d).
Collectively, the operational mechanism of the 1T-7A3B/2Z0.5M-W system (Fig. 4k) hinges on the reversible redox reactions in the organic phase (I− ↔ I3− ↔ I5−) and aqueous phase (Zn2+/Zn0), facilitating energy storage and release. On the other hand, the charge discrepancy between the phases resulting from the active substance redox reactions is offset by the reversible phase-transfer process of Zn2+ (dominant), TBA+, and BMIM+ cations, ensuring the continuous and stable operation of the system.
Exploration of extraction mechanisms in the 1T-7A3B/2Z0.5M-W system
The effective suppression of polyiodide shuttling by the 1T-7A3B/2Z0.5M-W system lies in the synergistic interplay among the solvent, ionic salt, and polyiodide. To elucidate the fundamental operational principles of the 1T-7A3B/2Z0.5M-W system, we conducted molecular dynamics (MD) simulation and density functional theory (DFT) calculation. As depicted in Fig. 5a, the polyiodide species within the 1T-7A3B/2Z0.5M-W system remain confined to the organic phase throughout a 100 ns MD simulation, in agreement with previous experimental findings, confirming the absence of polyiodide shuttling phenomenon in the system. The density distribution profiles of ions within the system at this juncture are outlined in Fig. 5b. Despite the slight solubility of AN in water, all constituent substances are distinctly segregated within the system. Particularly noteworthy is the cross-mixing state of ions at the liquid–liquid phase interface, which contributes to the reduction of interfacial overpotential (only ∼62 mV), thereby facilitating the interphase transfer of ions.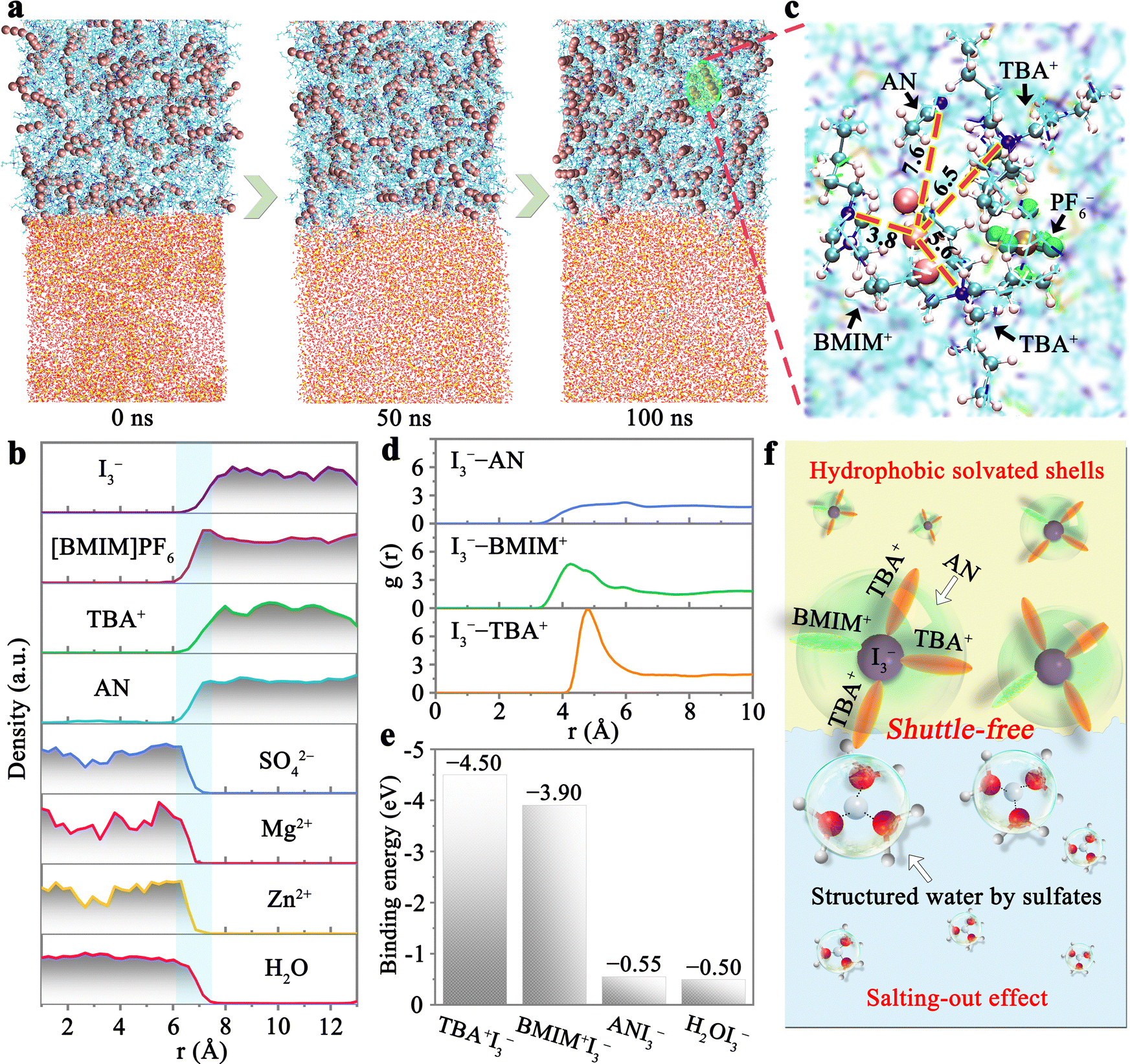 | ||
| Fig. 5 Proposed mechanism for the construction of 1T-7A3B/2Z0.5M-W system by the synergy between iodine-containing hydrophobic solvated shells and salting-out effect. (a) Polyiodide distributions in the 1T-7A3B/2Z0.5M-W system at 0, 50, and 100 ns. (b) Density profiles of ions/molecules within the system at 100 ns. (c) Solvated structure of I3− in the organic phase. (d) RDFs for I3−–AN, I3−–BMIM+, and I3−–TBA+ pairs derived from MD simulation. (e) Binding energy of I3− in TBA+, BMIM+, AN, and H2O. (f) Schematic representation elucidating the mechanism underlying the shuttle-free performance of the 1T-7A3B/2Z0.5M-W. | ||
Further analysis of the solvated structure of polyiodide in the organic phase is presented in Fig. 5c. Each I3− interacts with one AN, one BMIM+, and two TBA+ species, as the pronounced polarity of these ions/molecules enables effective interaction with polyiodide (Fig. S19, ESI†). The radial distribution functions (RDFs) of ions surrounding the I3− species are further elucidated based on the derived models (Fig. 5d). In contrast to the relatively uniform distribution of the I3−–AN pair, the RDF peaks of the I3−–TBA+ and I3−–BMIM+ pairs exhibit higher intensities. Especially, the peak intensity of the I3−–TBA+ pair is approximately 1.9 times compared to that of the I3−–BMIM+ pair. Furthermore, the RDF peak of I3−–TBA+ pair (3.95 Å) appears in a higher position than those of I3−–BMIM+ (3.15 Å) and I3−–AN pair (3.05 Å). These observations indicate that I3− ions are enveloped by a loose BMIM+ inner layer and a dense TBA+ outer layer, with a sparse distribution of AN molecules in the solvated shell structure. This structural arrangement can be attributed to the robust interaction of TBA+ and BMIM+ with polyiodide. As illustrated in Fig. 5e, the binding energies of I3− in TBA+ (−4.50 eV) and BMIM+ (−3.90 eV) are more negative than those in ANI3− (−0.55 eV) and H2OI3− (−0.50 eV). This ensures that I3− ions prefer to be retained inside the hydrophobic shell formed by TBA+ and BMIM+, which is beneficial to eliminating the occurrence of polyiodide shuttle effect. In contrast, the absence of TBA+ and BMIM+ would lead to rapid initiation of the shuttle effect (Fig. S20, ESI†), underscoring the significance of TBA+ and BMIM+ in preventing polyiodide shuttling.
On the other hand, the robust salting-out effect within the aqueous phase plays a pivotal role in upholding the stability of the biphasic system. In instances where the salting-out effect is diminished, the hydrophobic iodine inclusion engendered by TBA+ and BMIM+ will undergo sedimentation due to the diffusion of AN into the aqueous phase (Fig. S21, ESI†). Despite such disruptions in phase equilibrium, significant polyiodide shuttling is averted, as the presence of the hydrophobic shell effectively confines the polyiodide species. Moreover, the aqueous phase with a pronounced salting-out effect serves to curtail the solubilization of polyiodide by restricting free water molecules (Fig. S22, ESI†), thereby bolstering the suppression of the shuttle effect.50 Hence, the shuttle-free characteristic of the 1T-7A3B/2Z0.5M-W system can be attributed to two fundamental factors (Fig. 5f): (1) the hydrophobic solvation shell established by TBA+, BMIM+, and AN adeptly sequesters polyiodide within the organic phase; (2) the robust salting-out effect of the aqueous phase diminishes the cross-diffusion of AN and polyiodide between two phases, safeguarding the rigorous stabilization of the biphasic system.
Evaluation of battery availability performance
The exceptional safety and reliability exhibited by the 1T-7A3B/2Z0.5M-W system have been substantiated in the preceding sections. However, the system may encounter challenges related to insufficient energy density stemming from the low concentration of iodine ions. A notable enhancement in the solubility of iodine in AN has been achieved through the incorporation of TBAI, facilitating the introduction of a substantial quantity of iodine into the organic phase (Fig. S2, ESI†). As illustrated in Fig. S23a (ESI†), the introduction of solid iodine at concentrations of 0.5, 0.75, and 1 M results in iodine ion concentrations in the organic phase reaching up to 1.7 (iodine areal loading: 53.9 mg cm−2), 2.2 (69.8 mg cm−2), and 2.7 M (85.7 mg cm−2), respectively, corresponding to cell capacities of 29.8, 40.1, and 48.8 mA h (1 mL of organic electrolytes) at a current density of 0.1C. The corresponding GCD profiles at 0.1C are depicted in Fig. S23b (ESI†), all of which align with the behavior observed in the 1T-7A3B/2Z0.5M-W system without additional solid iodine, indicative of an I− ↔ I3− ↔ I5− redox process. Upon increase of the current density, batteries containing 1.7 (0.2C: 28.1 mA h; 0.4C: 26.4 mA h; 0.6C: 25.6 mA h; 0.8C: 23.5 mA h) and 2.2 M (0.2C: 38.1 mA h; 0.4C: 35.2 mA h; 0.6C: 29.2 mA h; 0.8C: 21.8 mA h) iodine ions exhibit robust stability. However, poor stability is observed for battery containing 2.7 M iodine ions (0.2C: 38.7 mA h; 0.4C: 22.1 mA h; 0.6C: 14.5 mA h; 0.8C: 9.9 mA h). This may arise from an excess of active material combined with the intrinsically non-conductive nature of iodine, which leaves a substantial portion of the iodine active material unutilized. As a result, the iodine redox process becomes highly susceptible to external perturbations (e.g., temperature), leading to pronounced capacity fluctuations (Fig. S24, ESI†). Nonetheless, the CE remains close to 100% across all scenarios (Fig. S25, ESI†), underscoring the remarkable efficacy of the 1T-7A3B/2Z0.5M-W system in mitigating the shuttle effect. Fig. S26 (ESI†) evaluates the long-term cycling proficiency of batteries containing 1.7 and 2.2 M iodide ions at 0.4C. As expected, both of them have excellent stabilities with capacity retention of 94.3% (1.7 M) and 94.7% (2.2 M) after 200 cycles, corresponding to initial capacities of 26.2 and 35.8 mA h, respectively. Especially, the energy density of the 1T-7A3B/2Z0.5M-W system, containing 2.2 M iodide ions, achieves an impressive 110.3 W h kg−1 (based on the mass of iodine) after 200 cycles, underscoring the potential of this system for significant applications in GLEESs.In light of these findings, we conducted scale-up experiments utilizing the 1T-7A3B/2Z0.5M-W system incorporating 2.2 M iodide ions within the organic phase. Benefiting from an integrated battery structure, the 1T-7A3B/2Z0.5M-W system facilitates rapid scaling to A h levels within hours, achieved through a streamlined four-step protocol: electrolyte configuration, electrode fabrication, electrolyte filling, and cell encapsulation. As illustrated in Fig. 6a, employing an iodine loading of approximately 4185 mg (equivalent to 15 mL of organic electrolyte), the as-fabricated battery exhibits an initial capacity of 387.3 mA h at 0.3C. Subsequent activation of the iodine cathode leads to a gradual increase in iodine utilization, culminating in a capacity of 453.5 mA h after stabilizing 16 cycles. Following 160 cycles, the battery demonstrates a commendable reversible capacity of 422.6 mA h, translating to an impressive capacity retention rate of 93.2%. The A h-level 1T-7A3B/2Z0.5M-W system also demonstrates exceptional stability, retaining 86.7% of its capacity over 100 cycles with an initial discharge capacity of 1.36 A h (Fig. S27, ESI†). Remarkably, even upon the introduction of 50 g of iodine active substance (approximately 180 mL of organic electrolyte), the battery sustains high efficiency, showcasing stable capacities of around 6 A h (equivalent to 33.3 A h L−1) and 3 A h (equivalent to 16.7 A h L−1) at 0.1C and 0.2C, respectively (Fig. 6b).
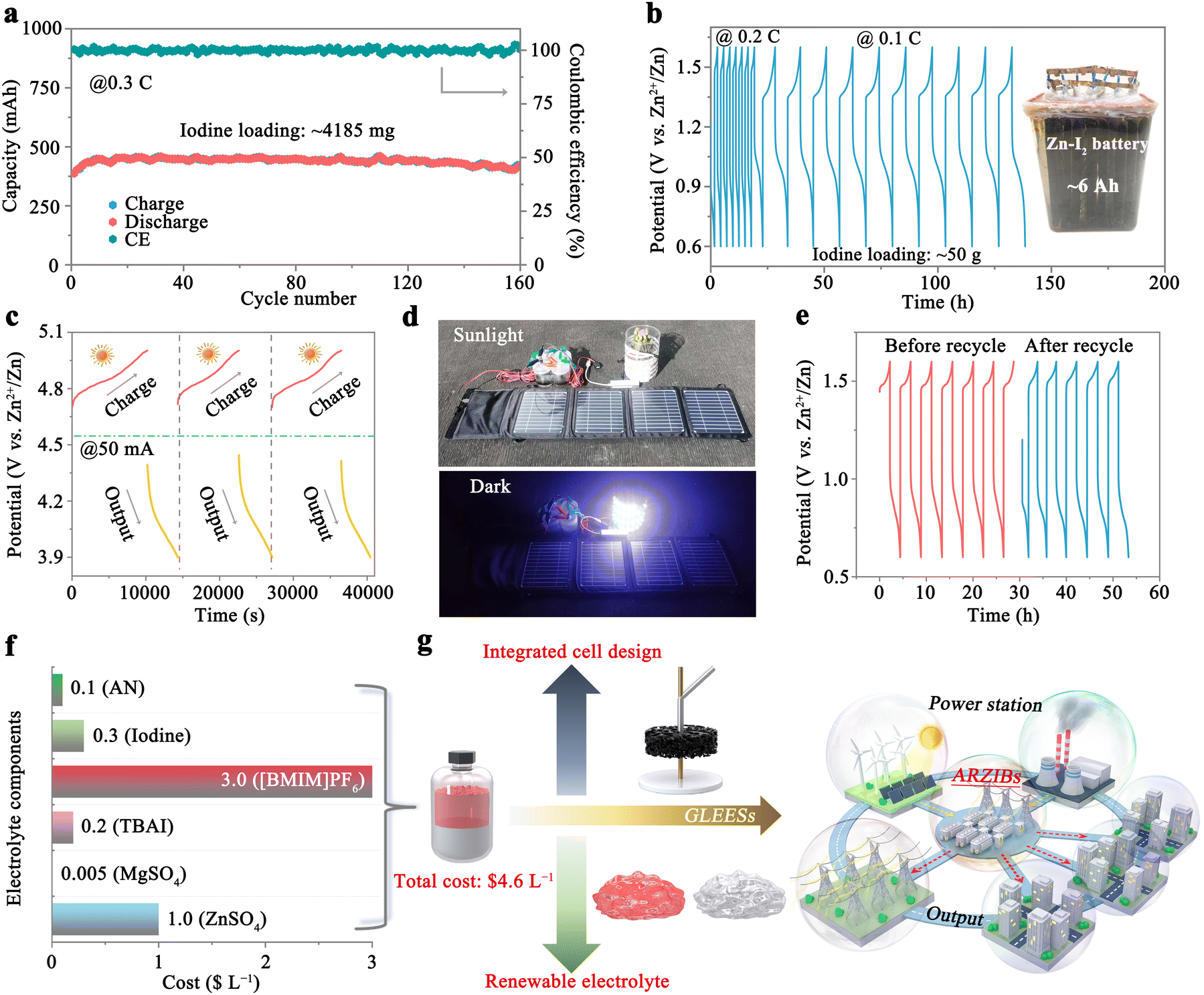 | ||
| Fig. 6 Scaled-up 1T-7A3B/2Z0.5M-W system for practical energy storage applications. (a) Evaluation of cycling performance for the 1T-7A3B/2Z0.5M-W system with an iodine loading of approximately 4185 mg. (b) Charge–discharge profiles of a 6 A h 1T-7A3B/2Z0.5M-W system, with an inset showcasing the digital image of the 6 A h battery. (c) Charge/discharge profile of the 1T-7A3B/2Z0.5M-W system utilizing a solar panel as the power source. (d) Operation of a solar-powered battery energy storage system during the daylight and nighttime. (e) Charge–discharge profiles pre- and post-electrolyte recycling for the 1T-7A3B/2Z0.5M-W system at 0.2 C. (f) Cost of each electrolyte component within the proposed 1T-7A3B/2Z0.5M-W system. (g) Advantages of the 1T-7A3B/2Z0.5M-W system for application in GLEESs. | ||
In a proof-of-concept demonstration, we assembled four advanced ARZIBs interconnected in series, leveraging the 1T-7A3B/2Z0.5M-W system as the foundational framework to establish an energy storage module. By harnessing solar energy during daylight, we charge the battery to 5 V using a solar cell (30 W), while discharging it to 3.9 V at 50 mA in the nighttime. Notably, the charge–discharge profiles of the batteries exhibit negligible alterations over the course of the three-day testing period (Fig. 6c). Fig. 6d clearly illustrates the actual energy storage process facilitated by the 1T-7A3B/2Z0.5M-W system, wherein electricity converted from solar energy during the daylight is stored and subsequently released at night to power electrical devices such as LEDs, smartphone (Fig. S28a, ESI†), and electric fan (Fig. S28b, ESI†). Notably, the battery also demonstrates excellent stability over 10 cycles at 0.15C within the voltage range of 3.2–6 V (Fig. S29, ESI†). These results exemplify the unparalleled reliability of the 1T-7A3B/2Z0.5M-W system.
The recyclability of electrolytes plays a pivotal role in significantly reducing the cost associated with energy storage in batteries. As an example, a 1T-7A3B/2Z0.5M-W system comprising 8 mL of organic electrolyte (2.2 M I−) was employed to demonstrate electrolyte recovery. As depicted in Fig. S30 (ESI†), following a cycling period, the battery exhibits a charging capacity of 253.4 mA h. Subsequently, the battery is disassembled, while the electrolyte is retrieved through a series of liquid separation, washing, and concentration procedures (refer to the Experimental section of the ESI† for detailed protocols). Consequently, the battery utilizing the recycled electrolyte showcases an initial discharge capacity of 155.4 mA h, swiftly recovering to performance levels observed prior to electrolyte recycling during subsequent cycling, suggesting an active material recycling efficiency approaching ∼100%. Fig. 6e presents the charge/discharge profiles of the battery utilizing fresh and recycled electrolytes, demonstrating a remarkable level of consistency throughout cycling, thereby substantiating the superior availability of recycled electrolytes.
Ultimately, the cost of the electrolyte in the proposed 1T-7A3B/2Z0.5M-W system is evaluated in Fig. 6f (refer to Table S2 for further information, ESI†). With all electrolyte components being commercially available, the cost for one-liter (comprising an organic to aqueous phase ratio of 1 to 5 by volume) amounts to a mere $4.6 L−1. This indicates that the battery can be manufactured at a comparatively low cost during further scale-up. In parallel, this system also exhibits remarkable advantages in stability (≥1300 cycles), CE (∼100%), and rate performance (2C ≈ 9.4 mA cm−2) compared to similar biphasic battery systems, which typically demonstrate stability of less than 100 cycles, a CE below 90%, and rate performance under 2 mA cm−2.46 Furthermore, this cocktail electrolyte structure supports an integrated electrode design, allowing for the seamless incorporation of graphite felt and zinc collectors into the current conductor in a top-to-bottom configuration, thereby streamlining the production process. Consequently, this design enables the rapid scale-up of the 1T-7A3B/2Z0.5M-W system to A h level within hours. Besides, the remarkable regenerative capacity of the electrolyte significantly contributes to the reduction of overall battery costs. Given these advantages, we can consider that the 1T-7A3B/2Z0.5M-W system holds exceptional promise for integration into GLEESs, facilitating the storage and reutilization of intermittent power (Fig. 6g). Importantly, it remains considerable potential for future enhancements, such as optimizing energy density through modifications to the iodine reaction pathway (as depicted in Fig. S31, ESI†) and the reduction of aqueous phase proportion (Table S3, ESI†), and accelerating the charge–discharge rate via the incorporation of catalysts.25,51 Besides, the design concept of this system is highly versatile, as demonstrated by its adaptation to a zinc–bromine battery (Fig. S32, ESI†), where replacing TBAI with tetrabutylammonium bromide yields modest cycling stability and high CE, attributed to the effective suppression of polybromide shuttling. All of these will effectively bolster the competitiveness of the 1T-7A3B/2Z0.5M-W system for GLEES.
Conclusions
In summary, we have successfully engineered a shuttle-free and highly scalable zinc–iodine battery system, characterized by a self-sieving polyiodide-capable liquid–liquid biphasic electrolyte and an integrated cell structure. The fundamental pillars of the biphasic electrolyte encompass three critical elements: (1) an iodine-containing hydrophobic solvated shell, which effectively constrain the polyiodide within the organic phase, (2) a salting-out effect, which ensures the robust stabilization of the biphasic system, and (3) a reversible phase-transfer process of cations (BMIM+, TBA+, Zn2+) and anions (I−), which offsets the charge difference resulting from redox reactions at the cathode (I− ↔ I3− ↔ I5−) and anode (Zn2+/Zn0), thus ensuring continuous and stable operation. Consequently, the battery system exhibits remarkable stability, exemplified by a capacity retention of 83.1% after 1300 cycles (iodine areal loading: 22.2 mg cm−2), alongside an ultra-low self-discharge rate (3.4% per month). In conjunction with the integrated cell structure, the system further presents remarkable advantages for rapid scale-up, underscored by its low-cost biphasic electrolyte ($4.6 L−1), renewable nature (∼100% recycling efficiency of active materials), and an exceedingly simplified cell structure. This groundbreaking research presents a promising sustainable power source for large-scale energy storage and a versatile strategy toward constructing a high-performance, intrinsically safe, and low-cost aqueous zinc–iodine battery.Author contributions
T. Deng, X. Fan, F. Lai, and T. Liu proposed and designed the idea. L. Zhang performed the experiments, electrochemical tests and wrote the paper. H. Ding, H. Gao, and Y. Yu performed the theoretical calculations. J. Gong, H. Guo, and S. Zhang assisted with the data analysis and characterization. G. He, I. P. Parkin, and J. Hofkens contributed to the discussion and electrochemical measurements. All authors analyzed the results and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely acknowledge financial support from the Shanghai Scientific and Technological Innovation Project (22520710100). J. H. acknowledges financial support from the Research Foundation-Flanders (FWO, Grant No. G983.19N and ZW15_09-G0H6316N), the Flemish government through long-term structural funding Methusalem (CASAS2, Meth/15/04) and the Moonshot cSBO project P2C (HBC.2019.0108), Interne Fondsen KU Leuven through project C3/20/067 and the MPI as MPI fellow. We thank the central laboratory, School of Chemical and Materials Engineering, Jiangnan University for their help in characterization, and the Supercomputing Center of Zhengzhou University for the support in DFT calculations.Notes and references
- J. Hao, S. Zhang, H. Wu, L. Yuan, K. Davey and S. Z. Qiao, Chem. Soc. Rev., 2024, 53, 4312–4332 RSC.
- M. Ferrara, Y.-M. Chiang and J. M. Deutch, Joule, 2019, 3, 2585–2588 CrossRef.
- M. S. Ziegler, J. M. Mueller, G. D. Pereira, J. Song, M. Ferrara, Y.-M. Chiang and J. E. Trancik, Joule, 2019, 3, 2134–2153 CrossRef.
- P. Li, X. Wang, S. Yang, K. Chu, H. Zhang, D. Chen and Q. Li, Mater. Today Chem., 2024, 41, 102274 CrossRef CAS.
- E. Fan, L. Li, Z. Wang, J. Lin, Y. Huang, Y. Yao, R. Chen and F. Wu, Chem. Rev., 2020, 120, 7020–7063 CrossRef CAS PubMed.
- C. Grosjean, P. H. Miranda, M. Perrin and P. Poggi, Renewable Sustainable Energy Rev., 2012, 16, 1735–1744 CrossRef.
- H. Jia, K. Liu, Y. Lam, B. Tawiah, J. H. Xin, W. Nie and S.-X. Jiang, Adv. Fiber Mater., 2022, 5, 36–58 CrossRef.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- H. Jiang, G. Qian, R. Liu, W.-D. Liu, Y. Chen and W. Hu, Sci. China Mater., 2023, 66, 4542–4549 CrossRef CAS.
- G. L. Soloveichik, Chem. Rev., 2015, 115, 11533–11558 CrossRef CAS PubMed.
- R. Usiskin, Y. Lu, J. Popovic, M. Law, P. Balaya, Y.-S. Hu and J. Maier, Nat. Rev. Mater., 2021, 6, 1020–1035 CrossRef CAS.
- J. Hao, L. Yuan, B. Johannessen, Y. Zhu, Y. Jiao, C. Ye, F. Xie and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 25114–25121 CrossRef CAS PubMed.
- J. Chen, J. Ma, B. Liu, Z. Li, X. Zhang, S. Sun, K. Lu, J. Yin, S. Chen, X. Zu, Z. Zhang, X. Qiu, Y. Qin and W. Zhang, Compos. Commun., 2023, 38, 101524 CrossRef.
- W. Wan, D. Sun, W. Ma, Y. Cao and H. Chai, Compos. Commun., 2024, 48, 101910 CrossRef.
- C. Liu, C. Qian, H. Dong, Z. Lu, Q. Huang and Y. Chen, Compos. Commun., 2023, 40, 101610 CrossRef.
- D. Lin and Y. Li, Adv. Mater., 2022, 34, e2108856 CrossRef PubMed.
- L. Zhang, H. Guo, W. Zong, Y. Huang, J. Huang, G. He, T. Liu, J. Hofkens and F. Lai, Energy Environ. Sci., 2023, 16, 4872–4925 RSC.
- W. Qu, C. Wen, B. Chen, Y. Cai and M. Zhang, Sci. China Mater., 2024, 67, 2889–2897 CrossRef CAS.
- J. Ma, M. Liu, Y. He and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 12636–12647 CrossRef CAS PubMed.
- L. Zhang, M. Zhang, H. Guo, Z. Tian, L. Ge, G. He, J. Huang, J. Wang, T. Liu and I. P. Parkin, Adv. Sci., 2022, 9, 2105598 CrossRef CAS PubMed.
- Z. Pei, Z. Zhu, D. Sun, J. Cai, A. Mosallanezhad, M. Chen and G. Wang, Mater. Res. Bull., 2021, 141, 111347 CrossRef CAS.
- L. Zhang, J. Huang, H. Guo, L. Ge, Z. Tian, M. Zhang, J. Wang, G. He, T. Liu and J. Hofkens, Adv. Energy Mater., 2023, 13, 2203790 CrossRef CAS.
- H. Yang, Y. Qiao, Z. Chang, H. Deng, P. He and H. Zhou, Adv. Mater., 2020, 32, e2004240 CrossRef PubMed.
- M. Liu, Q. Chen, X. Cao, D. Tan, J. Ma and J. Zhang, J. Am. Chem. Soc., 2022, 144, 21683–21691 CrossRef CAS PubMed.
- S. J. Zhang, J. Hao, H. Li, P. F. Zhang, Z. W. Yin, Y. Y. Li, B. Zhang, Z. Lin and S. Z. Qiao, Adv. Mater., 2022, 34, e2201716 CrossRef PubMed.
- X. Guo, H. Xu, Y. Tang, Z. Yang, F. Dou, W. Li, Q. Li and H. Pang, Adv. Mater., 2024, 36, 2408317 CrossRef CAS PubMed.
- Y. Zhao, L. Wang and H. R. Byon, Nat. Commun., 2013, 4, 1896 CrossRef PubMed.
- J. H. Hildebrand and C. A. Jenks, J. Am. Chem. Soc., 1920, 42, 2180–2189 CrossRef CAS.
- H. Wu, J. Hao, S. Zhang, Y. Jiang, Y. Zhu, J. Liu, K. Davey and S. Z. Qiao, J. Am. Chem. Soc., 2024, 146, 16601 CrossRef CAS PubMed.
- Y. Kang, G. Chen, H. Hua, M. Zhang, J. Yang, P. Lin, H. Yang, Z. Lv, Q. Wu, J. Zhao and Y. Yang, Angew. Chem., Int. Ed., 2023, 62, e202300418 CrossRef CAS PubMed.
- Y. Xu, M. Zhang, R. Tang, S. Li, C. Sun, Z. Lv, W. Yang, Z. Wen, C. Li, X. Li and Y. Yang, Energy Environ. Sci., 2024, 17, 6656–6665 RSC.
- Q. Chen, J. Hao, Y. Zhu, S. J. Zhang, P. Zuo, X. Zhao, M. Jaroniec and S. Qiao, Angew. Chem., Int. Ed., 2024, 64, e202413703 CrossRef PubMed.
- W. Zong, J. Li, C. Zhang, Y. Dai, Y. Ouyang, L. Zhang, J. Li, W. Zhang, R. Chen, H. Dong, X. Gao, J. Zhu, I. P. Parkin, P. R. Shearing, F. Lai, K. Amine, T. Liu and G. He, J. Am. Chem. Soc., 2024, 146, 21377–21388 CrossRef PubMed.
- Y. Ji, J. Xie, Z. Shen, Y. Liu, Z. Wen, L. Luo and G. Hong, Adv. Funct. Mater., 2023, 33, 2210043 CrossRef CAS.
- J. Mao, C. Ye, S. Zhang, F. Xie, R. Zeng, K. Davey, Z. Guo and S. Qiao, Energy Environ. Sci., 2022, 15, 2732–2752 RSC.
- X. Hu, Z. Zhao, Y. Yang, H. Zhang, G. Lai, B. Lu, P. Zhou, L. Chen and J. Zhou, InfoMat, 2024, 6, e12620 CrossRef CAS.
- J. C. Dearden and G. M. Bresnen, Quant. Struct.-Act. Relat., 1988, 7, 133–144 CrossRef CAS.
- D. C. Scott and J. W. Clymer, Pharm. Technol., 2002, 26, 30–40 CAS.
- L. Zhang, K. Tsay, C. Bock and J. Zhang, J. Power Sources, 2016, 324, 615–624 CrossRef CAS.
- W.-w Liu, X.-b Yan, J.-w Lang, J.-b Pu and Q.-j Xue, New J. Chem., 2013, 37, 2186–2195 RSC.
- J. Krummacher, C. Schütter, L. H. Hess and A. Balducci, Curr. Opin. Electrochem., 2018, 9, 64–69 CrossRef CAS.
- J. F. Côté, D. Brouillette, J. E. Desnoyers, J.-F. Rouleau, J.-M. StArnaud and G. Perron, J. Solution Chem., 1996, 25, 1163–1173 CrossRef.
- J. Meng, Q. Tang, L. Zhou, C. Zhao, M. Chen, Y. Shen, J. Zhou, G. Feng, Y. Shen and Y. Huang, Joule, 2020, 4, 953–966 CrossRef CAS.
- A. W. Francis, J. Chem. Eng. Data, 1965, 10, 145–151 CrossRef CAS.
- X. Li, Z. Qin, Y. Deng, Z. Wu and W. Hu, Adv. Sci., 2022, 9, e2105468 CrossRef PubMed.
- Z. Cheng, H. Pan, F. Li, C. Duan, H. Liu, H. Zhong, C. Sheng, G. Hou, P. He and H. Zhou, Nat. Commun., 2022, 13, 125 CrossRef CAS PubMed.
- K. Mongird et al. , 2020 Grid Energy Storage Technology Cost and Performance Assessment, 2020, vol. 117 Search PubMed.
- M. Li, B. Zhuang, Y. Lu, L. An and Z. G. Wang, J. Am. Chem. Soc., 2021, 143, 773–784 CrossRef CAS PubMed.
- Y. Yang, S. Liang, B. Lu and J. Zhou, Energy Environ. Sci., 2022, 15, 1192–1200 RSC.
- B. Li, J. Liu, Z. Nie, W. Wang, D. Reed, J. Liu, P. McGrail and V. Sprenkle, Nano Lett., 2016, 16, 4335–4340 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee05873a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |