
Crystalline nitrogen-doped-carbon anchored well-dispersed Fe3O4 nanoparticles for real-time scalable neutral H2O2 electrosynthesis†
Hao
Yin
a,
Jili
Yuan
 *a,
Jun
Wang
*b,
Shiwei
Hu
*c,
Pingshan
Wang
b and
Haibo
Xie
*a
*a,
Jun
Wang
*b,
Shiwei
Hu
*c,
Pingshan
Wang
b and
Haibo
Xie
*a
aDepartment of Polymer Materials and Engineering, College of Materials & Metallurgy, Guizhou University, Huaxi District, Guiyang 550025, P. R. China. E-mail: jlyuan@gzu.edu.cn; hbxie@gzu.edu.cn
bEnvironmental Research at Greater Bay Area, Key Laboratory for Water Quality and Conservation of the Pearl River Delta, Ministry of Education, Guangzhou University, Guangzhou 510006, China. E-mail: wangjun@gzhu.edu.cn; chemwps@csu.edu.cn
cInstitute of Mechanics, Chinese Academy of Sciences, Beijing, China. E-mail: hushiwei@imech.ac.cn
First published on 22nd January 2025
Abstract
Salt-free neutral H2O2 electrosynthesis via a 2-electron oxygen reduction reaction (2e−-ORR) remains challenging owing to the absence of efficient electrocatalysts and well-matched practical processes. Herein, we report an important progress and understanding of neutral H2O2 electrosynthesis of 2e−-ORR at a scalable rate using crystalline nitrogen-doped-carbon anchored Fe3O4 nanoparticles (NPs, Fe3O4@TNC) as efficient electrocatalysts, which were derived from the pyrolysis of a mixture of g-C3N4 and Fe@Tpy, achieving a salt-free, real-time and continuous H2O2 production process. Based on rotating ring-disk electrodes, Fe3O4@TNC achieved nearly 100% selectivity from 0 to 0.75 V vs. RHE and a limiting diffusion current density up to 5.2 mA cm−2 at 0 V vs. RHE. It was revealed that the exposed (220) facet of Fe3O4 NPs obtained a thermodynamically optimal binding of *OOH and rapid *OOH-mediated kinetic pathway. The integration of Fe3O4@TNC into scalable cells exhibited superior performance and techno-economic potential for neutral H2O2 electrosynthesis as industrially relevant current densities were achieved with remarkable real-time continuous production while maintaining relatively large faradaic efficiency. This work provides in-depth mechanistic insights into neutral H2O2 electrosynthesis and offers an advanced and economical process for integrating efficient electrocatalysts and scalable electrolyzer for industrially relevant neutral H2O2 production.
Broader contextNeutral H2O2 electrosynthesis via oxygen reduction reaction is an attractive route for sustainable and on-site application. However, it remains challenging to design efficient electrocatalysts at industrially relevant and affordable production rates. Herein, crystalline nitrogen-doped-carbon-anchored well-dispersed Fe3O4 nanoparticles (NPs, Fe3O4@TNC) were constructed by pyrolyzing a mixture of g-C3N4 and Fe2+-terpyridine. Fe3O4@TNC showed superior performance for neutral H2O2 production, achieving nearly 100% selectivity from 0 to 0.75 V vs. RHE and a limiting diffusion current density up to 5.2 mA cm−2 at 0 V vs. RHE. It was revealed that the mainly exposed (220) facet of Fe3O4 NPs facilitated thermodynamically optimal binding of *OOH and rapid *OOH-mediated kinetic pathway. Moreover, integrating Fe3O4@TNC into scalable cells led to a record production rate of 8.89 mol h−1 gcat.−1 and 19.23 mM for real-time continuous salt-free neutral H2O2 production, showing a techno-economical H2O2 production rate. |
Introduction
Hydrogen peroxide (H2O2) is an important green oxidant in large-scale industrial processes and smaller on-site activities,1 such as industrial bleaching,2 chemical synthesis,3 disinfection,4 and fuel cell technologies.5 Compared to the current process for industrial H2O2 production,6 H2O2 electrosynthesis via a 2-electron oxygen reduction reaction (2e−-ORR) is a promising alternative approach that is applicable on different scales for H2O2 production in a green and sustainable manner.1,7–9 The current main challenge is achieving high efficiency at industrially relevant current densities for a positive techno-economic value of H2O2 electrosynthesis.9,10 Although a desired system of neutral H2O2 electrosynthesis was presented,10–15 it is not yet satisfactory for large-scale productivity at an affordable cost owing to the competitive reduction reaction of the accumulated H2O2, extra separation purity from the salt-containing electrolyte, and further concentration costs. Therefore, it is highly promising to develop a salt-free and scalable neutral H2O2 electrosynthesis system, particularly including efficient electrocatalyst and a well-matched process.To date, although many efficient electrocatalysts have been developed for neutral 2e−-ORR electrocatalysis,13,15–18 they rarely show versatile production potential for neutral H2O2 electrosynthesis owing to their relatively large overpotential, low current density, and lack of a process for continuous neutral H2O2 electrosynthesis.19–21 In particular, this kind of neutral electrolytes or salt-free aqueous solution usually has high ohmic loss and low ionization, resulting in sluggish ORR kinetics and low binding of OOH*.22 For example, although porous carbon-supported Pd nanoparticles (NPs) achieved a high selectivity of neutral 2e−-ORR of about 95%, its ORR current density is still lower than the alkaline activity of this kind of catalysts. Similarly, transition metal-based or metal-free carbon electrocatalysts also present a similar trend of selectivity for neutral 2e−-ORR, and their overpotential and current density are also far less than those under alkaline conditions.23,24 Besides, even though these efficient electrocatalysts in a flow cell contribute a considerable H2O2 yield, their neutral sluggish kinetics for 2e−-ORR also limit the continuous accumulation of H2O2 due to a thermodynamic decomposition potential of H2O2 of about 1.766 V.19 Therefore, it is highly challenging to design efficient electrocatalysts and rational neutral H2O2 electrosynthesis systems to realize superior and scalable neutral H2O2 production.
Compared with various types electrocatalysts, carbon-based metal electrocatalysts, benefiting from their low-cost, easily adjustable structure of active sites, optimizable number of active sites, and high conductivity, show more positive onset potential and relatively large current density of neutral 2e−-ORR.25–27 However, owing to water as the proton supply, the kinetics of *OOH formation over carbon-based single-atom metal electrocatalysts is slow, requiring a more negative potential for H2O2 electrosynthesis.26 Fortunately, carbon-supported metal NPs can achieve higher current density of 2e−-ORR at a more positive potential due to the available active sites accelerating the proton coupling electron transfer process to hydrogenate the adsorbed O2, thus forming *OOH.28,29 On the other hand, pure metal NPs as active site for ORR thermodynamically favor cleavage of the O–O bond because of the favorable adsorption of O2 in the side-on manner.30 In contrast, carbon-anchored metal compound NPs, owing to the well-dispersed metal atom, show great potentials for theoretically higher efficient neutral 2e−-ORR at more positive potential ranges.26 However, neutral H2O2 electrosynthesis still has not achieved a practical rate and industrially relevant current densities owing to the rapid consumption of proton in water, which causes alkalization of aqueous solution.19 Therefore, it is significantly meaningful to integrate efficient carbon-anchored metal compound NPs electrocatalysts and scalable electrolyzers for developing an advanced neutral H2O2 electrosynthesis process.
Herein, we report crystalline nitrogen-doped-carbon anchored well-dispersed Fe3O4 nanoparticles (NPs, Fe3O4@TNC) as efficient electrocatalysts toward real-time continuous and scalable neutral H2O2 production (Scheme 1). Experimental observations and theoretical calculations indicated that Fe3O4@TNC with a main exposure (220) facet achieved a high onset potential of about 0.75 V vs. RHE, an exclusive neutral H2O2 selectivity at a record current density, attributed to the main exposed Fe3O4 (220) facet with an optimal binding of *OOH and rapid *OOH-mediated kinetic pathway. The integration of Fe3O4@TNC into scalable cells exhibited scalable neutral H2O2 electrosynthesis with a positive techno-economic potential.
 | ||
| Scheme 1 Schematic of the synthesis procedure of Fe3O4@TNC and the integration of Fe3O4@TNC into solid-state electrolysis (SE) cell for scalable neutral H2O2 electrosynthesis via O2 reduction. | ||
Results and discussion
Synthesis and structural characterization of catalysts
The formation mechanism of Fe3O4@TNC electrocatalyst was first revealed by X-ray photoelectron spectroscopy (XPS), thermogravimetry (TG), and Fourier transform infrared (FTIR) spectroscopy to monitor the pyrolysis process of precursors. The XPS spectra in Fig. S1 (ESI†) suggest that both g-C3N4 and Tpy as precursors mainly contain two kinds of carbon and nitrogen elements. Moreover, their high-resolution XPS C 1s and N 1s spectra were deconvoluted into a series of peaks that correspond to the typical position of g-C3N4 and Tpy.31,32 The HR-XPS O 1s spectra for g-C3N4 and Tpy can also be deconvoluted into H2O adsorption of two peaks (Fig. S1, ESI†). Fe@Tpy as the precursor includes C, N, Fe, Cl, and O five kinds of elements (Fig. S1, ESI†), which are ascribed to FeCl2 and methanol used for the assembly of Fe@Tpy.32 In contrast to Tpy, a new peak appears in the HR-XPS C 1s and O 1s spectrum, assigned to the C–O bond possibly due to the existence of methanol. TG-FTIR was further used to investigate the pyrolysis process of the mixture of both Fe@Tpy and g-C3N4 (Fig. 1(a) and Fig. S2, S3, ESI†). As the pyrolysis temperature increases, the peak at 1293 cm−1 appears and gradually increases for pure Fe@Tpy and the mixture, assigned to the vibration of C–O bond,33 indicating that methanol was partially encapsulated in Fe@Tpy.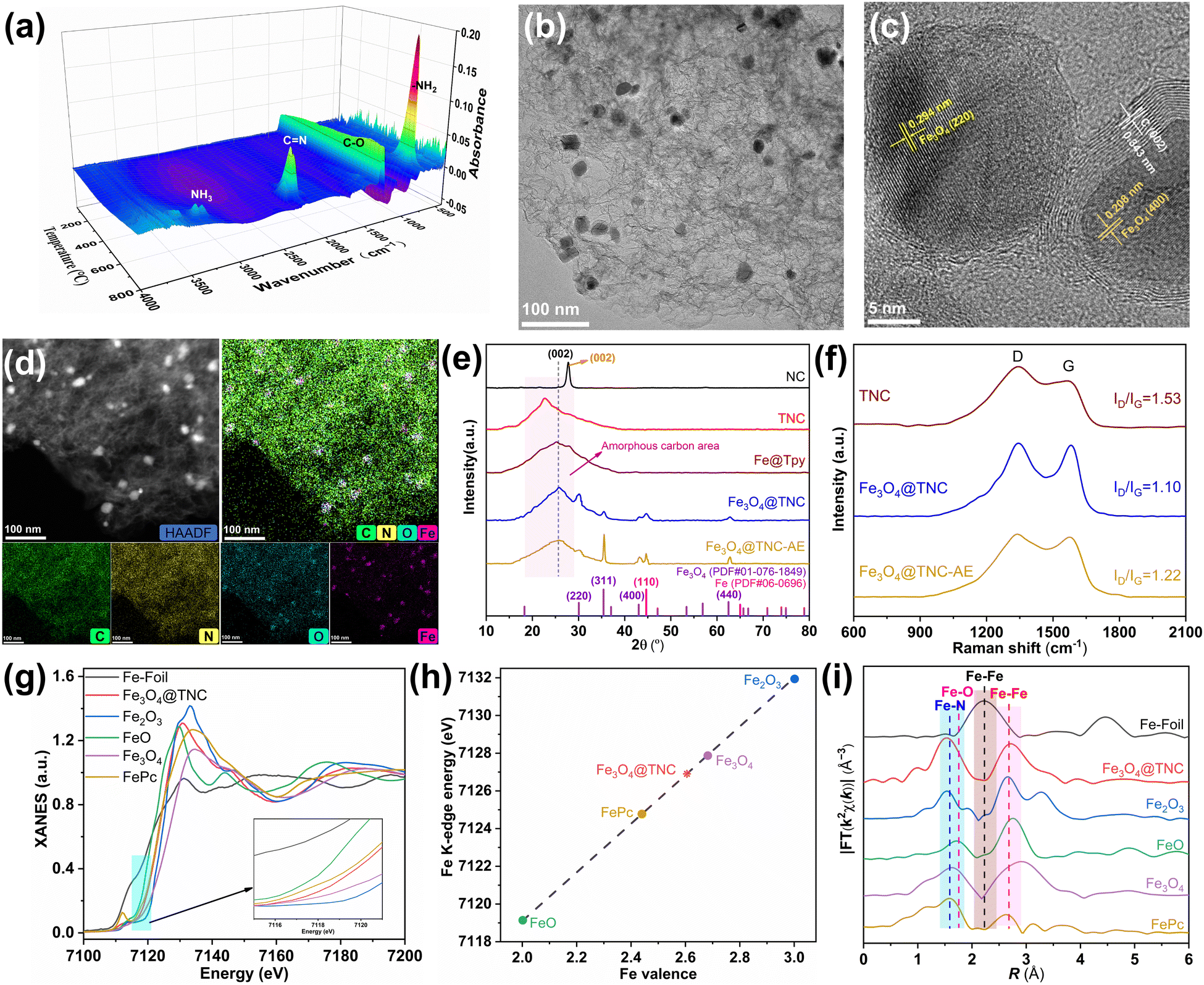 | ||
| Fig. 1 (a) 3D colormap surface of FTIR for the escaping gases for carbonizing the mixture of g-C3N4 and Fe@Tpy. (b) TEM image and (c) HRTEM image of Fe3O4@TNC. (d) HAADF-STEM and EDS mapping (C, N, O, and Fe) of Fe3O4@TNC. (e) X-ray diffraction pattern of NC, TNC, Fe@Tpy, and Fe3O4@TNC. (f) Raman spectra of TNC, Fe3O4@TNC, and Fe3O4@TNC-AE. (g) Fe K-edge XANES spectra of Fe3O4@TNC, Fe foil, FeO, Fe2O3, Fe3O4, and FePc. (h) Linear fit of Fe valence versus Fe K-edge energy position, (i) FT k2-weighted χ(k)-functions of the EXAFS spectra of the Fe K-edge for Fe3O4@TNC and the reference samples. | ||
Moreover, with the decomposition of g-C3N4 at the beginning of 620 °C (Fig. 1(a) and Fig. S2, ESI†), it could be observed that the peak of C–O for the mixture also decreased significantly at about 700 °C (Fig. 1(a)). The peak of C–O keeps increasing from about 500 to 800 °C during the pyrolysis of Fe@Tpy (Fig. S2 and S3a, ESI†). These results suggest that methanol should be an oxygen source for Fe3O4@TNC. Compared with the XPS of C, N, and O 1s for TNC derived from carbonizing the mixture of g-C3N4 and Tpy, Fe3O4@TNC presents new peaks for C–O (287.0 eV), Fe–N (399.5 eV), and Fe–O (530.1 eV)34 (Fig. S4 and Tables S1, S2, ESI†). Moreover, in contrast to the Fe 2p HR-XPS of Fe@Tpy (Fig. S1j, ESI†), Fe3O4@TNC presents a low-intensity peak for Fe of zero value (Fig. S4g, ESI†), suggesting the formation of a little bit of metallic Fe. Besides, the intensity of the Fe2+ and Fe3+ peaks dominate in the Fe 2p HR-XPS of Fe3O4@TNC (Fig. S4g, ESI†), confirming the main existence of Fe metal in the zero oxidation state. In contrast to the porous layer structure of NC, which originated from the pyrolysis of pure g-C3N4, TNC displays a typical lamellar stack, and Fe3O4@TNC shows an interconnected and stacked structure consisting of stacked linear and layer structure (Fig. S5, ESI†) derived from the carbon framework of the linear polymer of Fe@Tpy and g-C3N4, indicating that iron species were confined by the nitrogen-doped carbon matrix.
Transmission electron microscopy (TEM) images further confirm the presence of nanoparticles of about 25 nm size that are well-distributed on the carbon matrix (Fig. 1(b) and Fig. S6, ESI†). High-resolution TEM (HRTEM) images further show distinct lattice fringes with interlayer spacings of 0.294, 0.208, and 0.343 nm (Fig. 1(c)), which are assigned to the (220) and (400) crystal planes of cubic Fe3O4 and the (002) crystal plane of graphitized nitrogen-doped carbon matrix,35 respectively. Besides, it could be observed in Fig. 1(c) and Fig. S6a (ESI†) that some Fe3O4 NPs were encapsulated by the nitrogen-doped carbon matrix, demonstrating nitrogen-doped carbon support with a good crystallinity. The selected-area electron-diffraction (SAED) pattern displays a ring-like pattern and the exposed planes of Fe3O4 NPs, further confirming the graphitized nature of the nitrogen-doped carbon matrix with a good crystallinity and the facet species of Fe3O4 NPs over Fe3O4@TNC (Fig. S7, ESI†). The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image further confirms the mainly uniform monodispersed bright NPs (Fig. 1(d)). The energy-dispersive X-ray spectroscopy (EDS) elemental mapping verifies the elemental composition of Fe3O4@TNC with uniform distribution of C, N, O, and Fe elements. Particularly, the bright region of oxygen and iron elements is well overlapped (Fig. 1(d)), further confirming the well-dispersed Fe3O4 NPs anchored on the nitrogen-doped carbon (donated as Fe3O4@TNC). The crystalline structure of Fe3O4 NPs and nitrogen-doped carbon in Fe3O4@TNC was further confirmed by the X-ray diffraction (XRD) pattern (Fig. 1(e)). The diffraction peak at about 26.5° is attributed to the (002) plane of graphitic carbon, suggesting a nitrogen-doped carbon support with a good crystallinity, while other diffraction peaks of Fe3O4@TNC, including (220), (311), (440), and (400), correspond well to cubic phase Fe3O4 (card no. 01-076-1849).34,36 Particularly, the (220) peak of Fe3O4 over Fe3O4@TNC with strong intensity relative to other peaks indicates the main facet over Fe3O4@TNC. Besides, Fe (110) in Fe3O4@TNC is also observed in Fig. 1(e) (card no. 06-0696),34 which agrees with the Fe 2p XPS spectra (Fig. S4g, ESI†). Therefore, it could be demonstrated that the obtained Fe-based components in Fe3O4@TNC include Fe3O4 and metallic Fe components, with the Fe3O4 component dominating particularly.34,37
To further confirm the exposed Fe-based components over Fe3O4@TNC, Fe3O4@TNC was etched by acid washing (denoted as Fe3O4@TNC-AE), the XRD pattern of Fe3O4@TNC-AE shows a remarkable enhancement of the intensity of Fe3O4 (311), while the intensity of Fe (110) and Fe3O4 (440) and the (400) peak is stronger than that of Fe3O4@TNC (Fig. S8, ESI†), indicating that these Fe-components were encapsulated by crystalline nitrogen-doped-carbon supports. More importantly, in contrast to Fe3O4@TNC, the intensity of the Fe3O4 (220) peak decreased obviously after acid etching (Fig. 1(e)), further demonstrating the (220) peak of Fe3O4, which mainly stays in the exposed state rather than the encapsulated situation relative to the other peaks. The ID/IG ratio in the Raman spectrum also increases from 1.10 to 1.22 after acid etching and is far lower than that of TNC (Fig. 1(f)), confirming that the carbon lattice fringes are etched.38 This result suggests that the formation of Fe3O4 or Fe NPs over Fe3O4@TNC favors the formation of the crystalline nitrogen-doped-carbon. Besides, N 1s, O 1s, and Fe 2p HR-XPS of Fe3O4@TNC-AE also show peaks for Fe–N, Fe–O, and Fe of zero value, respectively (Fig. S8, ESI†). In particular, the content of O and Fe in Fe3O4@TNC-AE is lower than that in Fe3O4@TNC, suggesting that the crystalline carbon matrix was destroyed and simultaneously the encapsulated Fe-component was further exposed. Therefore, the above results suggest that Fe3O4 (220) dominates in the exposed plane of Fe3O4@TNC, where the Fe (110) and Fe3O4 (440), (400), (311) are mainly encapsulated by the carbon matrix, which is because this strategy for acid etching enables to etch the carbon matrix of the encapsulated Fe-based components.39 After the acid etching of Fe3O4@TNC, the specific surface areas decreased sharply from 537.68 to 251.80 m2 g−1, and the distribution of the pore size also decreased from 22.18 to 6.12 nm (Fig. S9 and Table S3, ESI†). The content of Fe decreased significantly (Fig. S10 and Table S1, ESI†), further verifying that the carbon matrix and exposed Fe3O4 NPs in Fe3O4@TNC were etched, leading to the formation of a smaller porous structure. Moreover, it could be concluded that the encapsulated Fe-based components in Fe3O4@TNC enables to support the porous layer structure of the crytalline nitrogen-doped-carbon matrix, consequently realizing the mainly exposed (220) facet of Fe3O4 NPs with a well-dispersed.
To further reveal the chemical state and coordination environment for Fe-based components in Fe3O4@TNC, the Fe K-edge X-ray absorption near-edge structure (XANES) spectrum in Fig. 1(g) suggests that Fe3O4@TNC is different from those of Fe foil and iron phthalocyanine (FePc) but is very close to that of FeO, Fe2O3, and Fe3O4. The valence state (+2.61) of Fe in Fe3O4@TNC is higher than that of FePc (+2.44) and somewhat lower than that of Fe3O4 (+2.67) (Fig. 1(h)), suggesting that Fe3O4 dominates in Fe-based components of Fe3O4@TNC and electron transfer occurs from the coordination network to Fe sites in Fe3O4@TNC.40 Moreover, k2-weighted Fourier transform (FT) EXFAS spectra demonstrate that Fe3O4@TNC displays two kinds of peaks at 1.52 and 2.72 Å (Fig. 1(i)), corresponding to the Fe–N and Fe–O scattering path and Fe–Fe scattering path, respectively, which is becuase the peak at 1.52 Å is close to the Fe–N path of FePc (1.58 Å) and the Fe–O path of Fe2O3 (1.54 Å) and Fe3O4 (1.61 Å) but far away from FeO (1.72 Å), and the peak of the Fe–Fe path at 2.72 Å is positioned between FeO (2.76 Å) and Fe2O3 (2.67 Å). The wavelet transforms of K-edge EXAFS (WT-EXAFS) in Fe3O4@TNC and the reference samples are shown in Fig. S11 (ESI†); the two intensities maximum of Fe3O4@TNC are attributed to the contribution of the backscattering of Fe–O–Fe, Fe–O, Fe–N, and Fe–Fe bonds, which agrees with the XPS spectra (Fig. S4 and Table S2, ESI†). Particularly, the backscattering center of Fe–O–Fe and Fe–O bonds enables them to nearly align to the two intensity maxima of the reference Fe3O4. This further suggests that Fe-based components in Fe3O4@TNC are mainly Fe3O4.41 Besides, the intensity of Fe3O4@TNC-AE in the K-edge energy position around 7116 eV of XANES spectra is higher than Fe3O4@TNC and the reference Fe3O4 and FePc (Fig. S12a, ESI†). FT-EXAFS of Fe3O4@TNC-AE exhibits a main intensity maximum, possibly ascribed to the backscattering of Fe–Fe bonds (Fig. S12b, ESI†). The contribution of the backscattering of Fe–O–Fe, Fe–O, and Fe–N bonds is also presented in Fe3O4@TNC-AE (Fig. S12c, ESI†), further confirming that Fe metal was exposed in Fe3O4@TNC-AE and also included partial Fe3O4, which agrees with the XRD pattern (Fig. S8, ESI†). Based on the above results, it could be concluded that the Fe-based components of Fe3O4@TNC include Fe3O4 NPs with the main exposed Fe3O4 (220) facet and the main encapsulated Fe3O4 (440), (400), and (311) facets, Fe NPs with the main encapsulated (110) facet, and these Fe-based components are anchored in crystalline nitrogen-doped-carbon support.
Electrocatalytic performance evaluations of the catalysts
The electrochemical ORR performance was first evaluated using a rotating ring-disk electrode (RRDE) at room temperature in an O2-saturated neutral electrolyte of 0.1 M LiClO4. All potentials were iR-compensated (80%) and converted to the reversible hydrogen electrode (RHE) scale. As shown in Fig. 2(a), Fe3O4@TNC achieved the largest disk current density (jdisk) of 4.69 mA cm−2 and ring current of 0.48 mA (Iring) at 0.3 V and a positive onset potential of 0.75 V compared with NC, Fe@Tpy, TNC, and Fe3O4@TNC-AE. The faradaic efficiency (FE) of Fe3O4@TNC for H2O2 selectivity was nearly 100% at the potential region from 0.60 to 0 V and also about 96% at 0.70 V (Fig. 2(b)). The electron transferred number (n) was calculated to be 2.05 at 0.60 V and 2.46 at 0.70 V (Fig. 2(c)). The rotating speeds of the RRDE have little influence on the H2O2 selectivity of Fe3O4@TNC (Fig. S13, ESI†). These results revealed Fe3O4@TNC shows superior 2e−-ORR performance in both activity and selectivity toward neutral H2O2 electrosynthesis. In comparison, Fe3O4@TNC-AE exhibits a wide fluctuation of electron transfer ranging between 2.5 and 3.69 (Fig. 2(c)), suggesting that the exposed Fe3O4 (440), (400), and (311) facets and Fe (110) over Fe3O4@TNC-AE are not conducive to 2e−-ORR. The structural difference between Fe3O4@TNC and Fe3O4@TNC-AE can reveal that the exposed Fe3O4 (220) in Fe3O4@TNC should be the main active site for H2O2 electrosynthesis via 2e−-ORR. | ||
| Fig. 2 (a) Polarization curves recorded at 1600 rpm with a scan rate of 5 mV s−1 in an O2-saturated electrolyte containing 0.1 M LiClO4 and the ring electrode at a constant potential of 1.2 V, including disk current density (jdisk) and ring current (Iring). (b) H2O2 faradaic efficiency. (c) Electron transfer number (n). (d) Tafel plots. (e) TOF values of Fe3O4@TNC and Fe3O4@TNC-AE. (f) Capacitance current densities measured at 1.0 V using 40 μF cm−2 as the specific capacitance standard. (g) Mass activity for H2O2 production at 0.3 V. (h) Stability test of Fe3O4@TNC at a potential of 0.5 V. (i) Comparison of H2O2 selectivity of Fe3O4@TNC and previously reported catalysts (Table S5, ESI†) at 2 mA cm−2 of jdisk. | ||
To better understand the role of each component in Fe3O4@TNC in the process of neutral H2O2 electrosynthesis, the study explored the effect of different factors such as the type of Fe-based components, crystalline degree of graphitic carbon matrix, and nitrogen-doped contents and types in Fe3O4@TNC. These factors were adjusted by varying the pyrolysis temperature during the fabrication process. Under a low carbonization temperature, the as-prepared sample (Fe3O4@TNC-700) shows a strong intensity for the typical (002) peak of g-C3N4, but no Fe-based component-relevant diffraction peak appears42 (Fig. S14, ESI†), while showing a high content of N residual to form pyridinic-N, pyrrolic-N, graphitic-N and Fe–N bond (Fig. S8 and Table S1, ESI†). In addition, a low intensity of Fe0 and Fe–O peaks also appeared34 (Fig. S15, ESI†). These structural changes result in the as-prepared sample with a low graphitic carbon degree (ID/IG, 1.56), low specific surface area (201.42 m2 g−1), and larger pore size of about 19.66 nm (Fig. S16, S17 and Table S3, ESI†), and also lead to an ultralow activity and selectivity for H2O2 electrosynthesis (Fig. S18, ESI†), further confirming that both the formation of Fe3O4 NPs and graphitic degree of the carbon matrix are critical for neutral H2O2 electrosynthesis performance. After the pyrolysis temperature increases, the as-prepared sample (Fe3O4@TNC-900) displays three peaks with slightly strong intensity, assigned to graphitic carbon (002), cubic phase Fe3O4 (311), and Fe (110),36 (Fig. S14, ESI†). However, the cubic phase Fe3O4 (220) in Fe3O4@TNC-900 almost disappears, further demonstrating that a higher pyrolysis temperature is unfavorable for the formation of Fe3O4 (220). In addition, the Fe0 content sharply increases due to the reduction of Fe2+ under high temperatures (Fig. S15, ESI†), the crystalline degree of graphitic carbon increases due to the ID/IG ratio decreasing from 1.65 to 1.05 (Fig. S16, ESI†), and the specific surface area and pore size decreased to 478.31 m2 g−1 and 16.0 nm, respectively (Fig. S17 and Table S3, ESI†). This kind of structural change also causes low selectivity of H2O2 electrosynthesis and decrease of the ORR activity (Fig. S18, ESI†), further confirming that the Fe3O4 (311) and (400), and Fe (110) planes in Fe3O4@TNC are unfavorable for 2e−-ORR. Moreover, the different usage of g-C3N4 for the preparation of Fe3O4@TNC has a certain effect on the onset potential and activity of 2e−-ORR (Fig. S19, ESI†), demonstrating that the crystalline degree of nitrogen-doped-carbon also affects the 2e−-ORR performance over the exposed (220) facet of Fe3O4 NPs. Furthermore, it can be concluded that proper pyrolysis temperature and usage of g-C3N4 are beneficial for the formation of the mainly exposed Fe3O4 (220) and a proper crystalline nitrogen-doped-carbon.
The relationship between Fe-based components in Fe3O4@TNC and its superior performance was revealed by electrochemical evaluation. The Tafel slope of 147 mV dec−1 for Fe3O4@TNC is smaller than that for Fe3O4@TNC-AE (210 mV dec−1), TNC (167 mV dec−1), Fe@Tpy (292 mV dec−1), and NC (445 mV dec−1) (Fig. 2(d)), demonstrating more favorable reaction kinetics of H2O2 electrosynthesis over the main exposed (220) facet of Fe3O4 NPs in Fe3O4@TNC.8 Moreover, the turnover frequency (TOF) values of Fe3O4@TNC are higher than those for Fe3O4@TNC-AE at potentials of 0.2, 0.4, and 0.6 V; particularly, a TOF value of 18.5 s−1 was achieved for Fe3O4@TNC at 0.6 V, which is about 11-fold for Fe3O4@TNC-AE (Fig. 2(e)), further confirming the exposed Fe3O4 NPs (220) plane in Fe3O4@TNC showing a superior intrinsic performance of neutral H2O2 production per unit time, but the exposed Fe3O4 (311) plane in Fe3O4@TNC-AE shows less selectivity toward 2e−-ORR. Electrochemically active surface area (ECSA, Fig. 2(f), Fig. S20 and Table S4) with a double layer capacitance of 11.1 mF cm−2 was observed for Fe3O4@TNC, which is also higher than that of TNC (4.6 mF cm−2) and Fe3O4@TNC-AE (6.7 mF cm−2), indicating superior electron transfer between the active sites of Fe3O4@TNC and electrolytes and more active sites available for oxygen activation.43 As indicated in Fig. 2(g), Fe3O4@TNC exhibited a mass current density of 93.1 A g−1 at 0.3 V under O2-saturated electrolyte, which is approximately 1.9-fold, 7.6-fold, 133-fold, and 465.5-fold higher than that of Fe3O4@TNC-AE, TNC, Fe@Tpy, and NC, respectively, further indicating that the exposed Fe3O4 (220) plane in Fe3O4@TNC should be the active sites for 2e−-ORR. However, the mass current density of Fe3O4@TNC is about 12.6 A g−1 at 0.3 V using air as the feeding gas (Fig. 2(g)), which is far lower 93.1 A g−1, suggesting that the limited mass transportation of O2 has a large influence on the activity of neutral H2O2 electrosynthesis. Thus, it can be further concluded that Fe3O4@TNC with a large specific surface area (537.68 m2 g−1) and proper pore width (22.18 nm) presents a huge potential for an instantaneously large yield rate of H2O2 under satisfactory O2 supply.
The long-term stability of Fe3O4@TNC is revealed in Fig. 2(h). After continuous electrocatalysis for 12 hours, there is no obvious decay in the current density at 400 ppm of the rotating rate or static condition. Post-analysis of TEM images confirmed that obvious changes in Fe3O4@TNC before and after the long-term electrolysis (Fig. S21, ESI†). The potential of 0.55 V and H2O2 selectivity (nearly 100%) at the current density of 2 mA cm−2 observed for Fe3O4@TNC are among the high values for all previously reported benchmarking H2O2 electrosynthesis catalysts under neutral electrolyte for ORR (Fig. 2(i) and Table S5, ESI†), further suggesting the main exposed Fe3O4 (220) plane over Fe3O4@TNC with a huge potential for realizing the desired neutral H2O2 electrosynthesis with low overpotential under large current density.
Theoretical calculations and in situ characterization
The above comparable experiments have demonstrated that the exposed Fe3O4 (220) anchored on a porous nitrogen-doped carbon matrix is critically important for the superior performance of neutral H2O2 electrosynthesis via 2e−-ORR. Density functional theory (DFT) calculations were performed to unveil the catalytic mechanisms of H2O2 synthesis. According to the structural characterization, both Fe-based components- and carbon matrix-relevant models in Fe3O4@TNC were constructed, including nitrogen-doped carbon with vacancy (CNV), Fe3O4- and Fe-based relevant configurations, and their heterojunction configurations (Fig. S22, ESI†). First, O2 adsorption behavior was investigated on the surface Fe atom of the above models (Fig. S23, ESI†). Both CNV and Fe3O4(400) displayed a positive value, and the rest of the models all presented a negative value (Fig. 3(a)), suggesting that the adsorption of O2 on these models is spontaneous, other than CNV and Fe3O4(400). Moreover, in comparison with Fe3O4-based facets, the structural heterojunction between CNV and various Fe3O4-based facets all present a different change trend for the adsorption of O2. For instance, in contrast to −2.34 eV of Fe3O4(220) and −2.14 eV of Fe3O4(311), the heterojunctions of both Fe3O4(220)–CNV and Fe3O4(311)–CNV are at about −1.37 eV and −2.56 eV, respectively (Fig. 3(a)). In addition, compared with Fe(110) (−0.81 eV), Fe(110)–CNV has an O2 adsorption energy of −2.16 eV. These results suggest that there is a strong interaction between the carbon-based matrix and Fe3O4, which affects the adsorption strength of O2, thereby affecting the adsorption of *OOH.33Fig. 3(b) shows the free energy profile of H2O2 formation at an equilibrium potential of 0.7 V on the above models. DFT predicts that the formation of *OOH on Fe3O4(220)–CNV is an endothermic process and has the lowest barrier energy of 0.07 eV compared to CNV, Fe(110), Fe(110)–CNV, Fe3O4(400), Fe3O4(400)–CNV, Fe3O4(220), Fe3O4(311), and Fe3O4(311)–CNV, suggesting that the exposed Fe3O4(220) in Fe3O4@TNC is the most favorable for 2e−-ORR for H2O2 electrosynthesis. The ORR pathway is further investigated by the tendency of O–O bond dissociation (ΔG*OOH). Fig. 3(c) shows a volcano plot for oxygen reduction via the 2e− pathway as a function of ΔG*OOH. Compared with 3.83 eV of pure Fe3O4(220), the free energy of *OOH (ΔG*OOH) on the Fe atom of Fe3O4(220)–CNV is 4.19 eV, closer to the ideal value of 4.22 eV. However, Fe3O4(311)–CNV has ΔG*OOH of 2.25 eV compared to 3.48 eV for Fe3O4(311), further confirming a strong interaction at the inside of the heterojunction between CNV and Fe3O4 NPs. Besides, both Fe3O4(400)–CNV and Fe(110)–CNV of ΔG*OOH are 4.55 and 3.63 eV, respectively, which is relatively closer to 4.22 eV. The charge density difference in Fe3O4(311)–CNV and Fe3O4(220)–CNV for the adsorption of O2 and the OOH intermediate further reveal the effect of the heterojunction between CNV and Fe3O4 NPs (Fig. 3(d)). When O2 and OOH intermediate adsorb on the Fe atom of Fe3O4(220)–CNV, the adsorbed O2 and OOH achieve 0.69 e− and 0.54 e− electrons, respectively, but the adsorbed O2 and OOH on the Fe atom of Fe3O4(311)–CNV accumulate the electrons of 0.73 e− and 0.56 e−, respectively. Therefore, the nitrogen-doped carbon matrix as support could modify the surface electron density of the exposed Fe3O4 facet, thereby adjusting the number of electron transfers from the Fe atom in Fe3O4 to the adsorbed OOH with a desired binding strength. | ||
| Fig. 3 (a) Adsorption energy of O2, (b) free energy diagram at U = 0.7 V for the reduction of O2 to H2O2, (c) computed activity volcano plots on the possible relevant crystalline facet of Fe3O4@TNC. (d) Charge density difference for both O2 and *OOH adsorbed on Fe3O4 (220)–CNV and Fe3O4 (311)–CNV. (e) In situ ATR-FTIR spectra collected on Fe3O4@TNC catalyst in O2-saturated 0.1 M LiClO4 electrolyte at different potentials from OCP, 1.0 to −0.3 V. (f) In situ electrochemical Raman spectra of the ORR process on Fe3O4@TNC at various potentials. | ||
Combining the structural characterization of Fe3O4@TNC and Fe3O4@TNC-AE with their electrocatalytic performance, although both contain various Fe-based components, such as Fe(110), Fe3O4(400), Fe3O4(220), and Fe3O4(311), the exposed Fe-based components in Fe3O4@TNC should be the Fe3O4(220) facet with an excellent performance of 2e−-ORR, while the mainly exposed Fe3O4(311) in Fe3O4@TNC-AE exhibited a low selectivity for H2O2 electrosynthesis (Fig. 2(b) and Fig. S8, ESI†). Therefore, the agreement between both theoretical calculations and experimental observations suggests that the exposed Fe3O4 facet in Fe3O4@TNC should be the (220) facet and the other Fe-based NPs should be mainly encapsulated in the carbon matrix. In situ attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectroscopy was used to monitor the interactions between oxygen intermediates and Fe3O4@TNC in O2-saturated 0.1 M LiClO4 electrolyte (Fig. S24a and b, ESI†). As illustrated in Fig. 3(e), the intensity of the peak at 1054 cm−1 presents a gradually increasing trend as the potential, associated with the adsorbed ClO4− in the electrolyte.8 Moreover, the bands at 1450 cm−1, typically assigned to the adsorbed *O2 species,44 present an obvious intensity from the potential of 0.4 to −0.1 V, indicating the hydrogenation of the adsorbed *O2 with a fast kinetics. Besides, the adsorption bands at about 890, 1240, and 1396 cm−1 appear on applying a potential of 0.8 V are assigned to the M–O and O–O stretching vibration of the adsorbed *OOH and the adsorbed *HOOH species,13,38 respectively. It is observed that the peak intensity of both adsorbed *OOH and *HOOH species is close, further demonstrating this hydrogenation step from *OOH to *HOOH with a rapid kinetics process.
Furthermore, the bands of the adsorbed *OOH and *HOOH species are enhanced from the potential of 0.8 to 0.4 V and slightly rise by further decreasing the potential during neutral H2O2 electrosynthesis, attributed to the limited O2 supply, which follows the trend for the potential at about 0.4 V of the limited current in polarization curve of Fe3O4@TNC (Fig. 2(a)). Overall, these results demonstrate the rapid kinetic process of O2 hydrogenation via *OOH-mediated two-electron ORR pathway on the Fe3O4@TNC catalyst, which agrees with the thermodynamically spontaneous process from the DFT results (Fig. 3(b)). The strong electromagnetic field of Fe3O4 NPs in Fe3O4@TNC could effectively enhance the Raman signal of the species on the exposed Fe3O4 NP surface, which allows us to detect trace intermediates during the ORR process. In situ electrochemical Raman measurements were performed in 0.1 M LiClO4 electrolyte using a customized cell (Fig. S24c and d, ESI†). Both D-band and G-band Raman scattering of Fe3O4@TNC were observed at about 1350 and 1640 cm−1 under open circuit potential (OCP) or N2-saturated electrolyte condition, respectively, which agreed with the Raman spectrum of Fe3O4@TNC (Fig. 1(f)). During ORR electrocatalysis, as observed in Fig. 3(f), the intensity of the broad Raman band at about 1093 cm−1 is slightly enhanced as the applied potential increases from the onset potential of 0.7 V for ORR, corresponding to the absorbed O–O stretching vibration of superoxide species (*O2−).45 Moreover, a new band at about 1528 cm−1 appears at a higher potential than 0.7 V, assigned to the absorbed *OOH species,21 and the intensity of *OOH slightly increases as the potential rises. Besides, the intensity of *O2− and *OOH becomes obvious under a sufficient potential due to the fast consumption rate, further suggesting the neutral 2e−-ORR on Fe3O4@TNC via the *OOH-mediated pathway with a fast kinetic process.
Scalable neutral production of H2O2 electrosynthesis
To evaluate the potential for scalable neutral H2O2 electrosynthesis, Fe3O4@TNC was first investigated at a large potential range from 0.75 to −0.7 V in O2-saturated neutral electrolyte of 0.1 M LiClO4 using RRDE (Fig. S25, ESI†). Fe3O4@TNC with the mainly exposed Fe3O4 (220) plane displays a large rising trend for current density in the potential range from 0.75 to −0.7 V, with nearly 100% selectivity for H2O2 production, further confirming its great application potential for industrially relevant yield rate of neutral H2O2 production under satisfactory O2 supply and large current density. In addition, Fe3O4@TNC was deposited on a hydrophobic gas-diffusion electrode as a cathode for enhancing O2 supply and Pt gauze as anode. As illustrated in Fig. 4(a), the two-electrode system was assembled in a solid-state electrolyte (SE) cell with a sandwiched double proton exchange membranes (PEM) configuration containing a porous polymer ion conductor in the SE layer to eliminate the iR-drop between the cathode and anode.12,14,46 The optimized conditions, including the mass loading of the catalyst and water flow rate for the selectivity of H2O2 production, are summarized in Fig. S26 (ESI†) under enough O2 supply with a flow rate of 100 sccm. Increasing the water flow rate within 5–40 mL min−1 is favorable for bringing out the generated H2O2 and minimizing its further electroreduction (Fig. S27, ESI†), particularly, the current density of SE cells up to 60 mA cm−2. A higher mass loading of Fe3O4@TNC requires a larger flow rate of DI water, which obtains a relatively high H2O2 FE of at least 40 mL min−1 due to the high resistance of O2 gas and generated H2O2 mass transport.15 Therefore, the optimal mass loading of Fe3O4@TNC and DI water flow were respectively determined to be 0.1 mg cm−2 and 30 mL min−1, which ensure not only the proton resources supply but also efficiently brings out the produced H2O2. The I–V curve of the 4-cm2 three-chamber double-layer-PEM SE cell is plotted in Fig. 4(b); as the overall current density increased from 2.5 to 60 mA cm−2, the cell voltage of the SE reactor gradually increased from 1.9 to 2.3 V, and the trend for FE of H2O2 decreased gradually from 98% to 83% at the cathode side (Fig. 4(b)). Besides, it can be observed in Fig. 4(b) that a small amount of partially generated H2O2 could get through the PEM into the middle layer, corresponding to about 1–4% H2O2 FE. The maximum total H2O2 FE in the SE cell is nearly 100% in the current density range from 2.5 to 20 mA cm−2 (Fig. 4(c)). Subsequently, H2O2 FE decreased to 87% under a high current density of 60 mA cm−2, suggesting that a small amount of the partially generated H2O2 is further reduced to H2O at the cathode side. Even then, the yield rate of H2O2 increased sharply as the applied current density increased. Particularly, 8.57 and 8.89 mol h−1 gcat.−1 were obtained at a current density of 50 and 60 mA cm−2, respectively. | ||
| Fig. 4 (a) Schematic of H2O2 electrosynthesis via ORR in the SE cell with double-PEM configuration. (b) The I–V curve and corresponding FEs for generating H2O2 in the middle chamber and cathode, (c) the corresponding total yield rate of H2O2 and FEs. (d) Chronopotentiometry stability test by directly flowing 1 M Na2SO4 solution in the middle chamber at 50 mA cm−2 current density. (e) Chronopotentiometry test using 300 mA cm−2 for H2O2 production by varying H2O flow rate from 1 to 12 mL min−1. (f) Techno-economic evaluation of neutral H2O2 electrosynthesis with two different conditions in the SE cell. | ||
The stability of H2O2 electrosynthesis was further investigated by holding a cell current density of 50 mA cm−2 (200 mA total current). As shown in Fig. 4(d), the continuous generation of H2O2 solution on the cathode side can stably run for over 60 h without decline with about 91% of H2O2 FE, suggesting Fe3O4@TNC with an excellent stability for H2O2 electrosynthesis under scalable H2O2 production. The structural and morphological stability of Fe3O4@TNC after 60 h of testing is reflected in the XRD and TEM images (Fig. S28, S29 and Table S6, ESI†), with minimal change in the (002) crystal plane. To further investigate the scalable H2O2 production of SE cell using Fe3O4@TNC as a catalyst, air atmosphere was used to replace pure O2 gas and the optimized operating condition of SE cell was continuously used. ORR activity is observed under low voltage of SE cell due to the close current density (Fig. S30a, ESI†), and H2O2 FE is nearly 100% within the current density of 2.5–20 mA cm−2. Subsequently, the H2O2 FE sharply decreases from 30 to 50 mA cm−2 as the current density of SE cell increases (Fig. S30b, ESI†). This result is mainly due to the limited O2 supply responding to the diffusion-limited current density (Fig. S31, ESI†). Moreover, continuous H2O2 generation at the cathode side occurred stably for 40 h, maintaining about 90% selectivity of H2O2, and the voltage of the SE cell increases slightly (Fig. S30c, ESI†), further confirming engaging Fe3O4@TNC into the SE cell showing a huge application potential for real-time continuous H2O2 production with air and pure water. Furthermore, to explore the industrially relevant and economical potential for neutral H2O2 electrosynthesis over Fe3O4@TNC in the SE cell, different flow rates of DI water were adopted to regulate the real-time H2O2 concentration produced from the cathode under a current density of 300 mA cm−2. As displayed in Fig. 4(e), as the flow rate of DI water decreases from 12 to 1 mL min−1, the concentration of real-time generated H2O2 at the cathode side presents a rising trend from 5.01 to 19.23 mM, showing a large region of H2O2 concentration, indicating a real-time H2O2 supply potential through the SE cell with the Fe3O4@TNC catalyst. Moreover, techno-economic evaluation, derived from SE electrolyzer results under current densities of 50 and 300 mA cm−2, estimated the plant-gate levelized costs for H2O2 production using Fe3O4@TNC at a market price of about 73% (US$586) and 40% (US$316) (Fig. 4(f) and Fig. S32, S33, ESI†), respectively, compared with the market price of about US$800 tonne−1 for electron-grade 30% H2O2 (ref. 9,10). Moreover, the industrially relevant current density of 300 mA cm−2 for neutral H2O2 electrosynthesis achieves larger room for benefits with respect to 50 mA cm−2 (Fig. S34, ESI†), demonstrating that this process of H2O2 electrosynthesis via the integration of efficient electrocatalysts into the SE cell presents a huge potential for economical and scalable H2O2 production in pure water.
Conclusion
In summary, a Fe3O4@TNC catalyst was developed by pyrolyzing the mixture of both g-C3N4 and Fe@Tpy, which demonstrated a scalable and economical rate of H2O2 production in the SE cell. This strategy employed crystalline nitrogen-doped-carbon supported exposed and encapsulated Fe3O4 NPs as the electrocatalysts. We first found that the exposed Fe3O4 NPs with the (220) facet on TNC is the most active site, which facilitates the thermodynamically lowest barrier energy process for 2e−-ORR via *OOH mediated with rapid kinetics for neutral H2O2 electrosynthesis due to the interaction between the exposed (220) facet of Fe3O4 NPs and crystalline nitrogen-doped-carbon. Under the RRDE test conditions, the Fe3O4@TNC catalyst exhibits a high onset potential of 0.75 V, a nearly 100% selectivity toward H2O2 production at a broad potential range from 0.75 to −0.6 V, and a current density of 2 mA cm−2 at 0.5 V vs. RHE in the neutral media, which is superior to that of the previously reported electrocatalysts. The scalable current densities up to 60 mA cm−2 (240 mA) with 87% FE were obtained for continuous H2O2 production at a record rate of 8.89 mol h−1 gcat.−1 in the SE cell. Particularly, a real-time continuous 19.23 mM of H2O2 production was achieved at a current density of 300 mA cm−2, showing a huge techno-economic potential for industrially relevant H2O2 production in pure water.Experimental section
Synthesis of Tpy
A mixture of terephthalaldehyde (5.0 g, 37.3 mmol), 2-acetylpyridine (21.67 g, 179.1 mmol), anhydrous ethanol (300 mL), and powdered NaOH (8.95 g, 223.8 mmol) was added to a 1000 mL round-bottom flask. The mixture was stirred at room temperature for 12 h, 200 mL of NH3 H2O (25–28%) was added and heated at 85 °C for 48 h to get a brown suspension solution. After being cooled to room temperature, the brown precipitate was filtered and then recrystallized twice under reflux with a mixed solvent of CHCl3/CH3OH (v/v, 2/1) to obtain the target compound (1.4 g, 1.96 mmol) as a grey solid (donated as Tpy) in 35% yield.Synthesis of Fe@Tpy
Typically, the obtained Tpy (1.85 mmol) was dissolved in 100 mL CH2Cl2 at 85 °C (1.85 × 10−2 mol L−1), and FeCl2·4H2O was used as the metal source to form a metal ion methanol solution (1.85 × 10−2 mol L−1). The metal ions with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of mass of Tpy were easily dropped into the Tpy solution, reacted at 85 °C for 16 h, and the target product was separated at the end of the reaction, labeled as Fe@Tpy.
:1 ratio of mass of Tpy were easily dropped into the Tpy solution, reacted at 85 °C for 16 h, and the target product was separated at the end of the reaction, labeled as Fe@Tpy.
Synthesis of TNC, Fe3O4@TNC, and Fe3O4@TNC-AE
The synthesis of g-C3N4 was through the thermal polymerization of melamine in a tube furnace at 550 °C for 3 h, with a heating rate of 5 °C min−1 under an Ar atmosphere. The mixture of Fe@Tpy and g-C3N4 was carbonized under different temperatures and Ar atmosphere (700, 800, and 900 °C) using a heating rate of 5 °C min−1 for 2 h. The final products were obtained after cooling and denoted as Fe3O4@TNC-700, Fe3O4@TNC-800, and Fe3O4@TNC-900. Additionally, pyrolyzing the mixture for the different ratios of g-C3N4 to Fe@Tpy, a series of powders was prepared after the pyrolysis of these mixtures at 800 °C for 2 h using a heating rate of 5 °C min−1, which were labeled as Fe3O4@TNC-20, Fe3O4@TNC-40, Fe3O4@TNC-60, and Fe3O4@TNC-80. Generally, TNC is prepared by directly pyrolyzing the mixture of Tpy and g-C3N4 under the same preparation conditions of Fe3O4@TNC-800, particularly, the usage ratio between Tpy and g-C3N4, and the pyrolysis temperature. Typically, Fe3O4@TNC-AE was achieved by the etching of Fe3O4@TNC-800. First, Fe3O4@TNC was dispersed into a 2 M HNO3 solution and sonicated for 1 h. The resultant ink solution was further stirred at room temperature for 24 hours and then washed in ultrapure water. The final product was obtained after drying, known as Fe3O4@TNC-AE.Computational details
Spin-polarized DFT calculations were performed using the Vienna ab initio simulation package.47 The generalized gradient approximation toward the Perdew–Burke–Ernzerhof level was adopted in the projector-augmented wave potentials.48 The Monkhorst–Pack type of k-point sample was shifted to the gamma center to adapt for all the structures in this work. The Gaussian smearing of 0.05 eV was adopted to satisfy the self-consistence and convergence of the electron density. All the atomic positions were allowed to relax until the forces were less than 0.005 eV Å−1, and the electron convergence energy was set to 10−5 eV. The vacuum was set to 15 Å for all the interfacial structures to avoid interactions between its periodic images. The plane wave cutoff was set to 520 eV. The DFT-D3 scheme was adopted to correct the van der Waals interaction.49 Here, we used the computational hydrogen electrode (CHE) model proposed by Nørskov et al. to calculate the free energy levels of all intermediates:50,51 ΔGads = ΔEads + ΔZPE − TΔS + eU, where ΔEads is the binding energy of adsorption species *O2 and *OOH. ΔZPE, ΔS, U, and e− are the ZPE changes, entropy changes applied potential at the electrode, and charge transferred, respectively. The contributions of each component for ΔGads were obtained from literature.51 As the ground state of the O2 molecule is poorly described in DFT calculations, we used gas-phase H2O and H2 as reference states as they are readily treated in the DFT calculations. In our simulations, solvation effects were not considered.Neutral H2O2 production in a solid-state electrolyte cell with double-PEM configuration
A solid electrolyte (SE) cell with a sandwiched double-PEM design was used for continuous H2O2 electrosynthesis. The cell structures and production setup are shown in Fig. 4(a). The cathode was supplied with a mixture of O2 gas (100 sccm) and DI water (30 mL min−1), with the gas flow regulated by a mass flow controller (MFC) and the water flow controlled by a peristaltic pump. The cathode and anode were positioned on opposite sides of 0.5 cm thick PEEK plates, each containing 2 cm × 2 cm channels. The catalyst layers faced the flowing liquid electrolyte, providing a geometric surface area of 4 cm−2. A measuring cylinder calibrated the flow rate of H2O2 produced at the outlet. The rapid flow of water in the gas/liquid mixture via the cathode chamber is good for removing the produced H2O2 molecules and reducing additional H2O2 electroreduction. The SE in the middle chamber consisted of Dowex 50W X8 hydrogen form (Sigma-Aldrich), a styrene–divinylbenzene sulfonated copolymer cation conductor. A syringe pump was used to introduce a solution containing H2SO4 and/or Na2SO4 into the SE layer. At the anode side, a 0.1 M H2SO4 solution was circulated at 3 mL min−1, while a 1 M Na2SO4 solution flowed through the SE layer at the same rate in a single pass. Potentials were measured using a two-electrode setup and manually adjusted for 100% accuracy.Solid-state electrolyte cell
The cathode side was continuously supplied with a mixture of O2 gas and water flow for 2e−-ORR, while the anode side was circulated with 0.1 M H2SO4 solution for water oxidation and proton supply. In the middle chamber, 1 M Na2SO4 solution flowed through in the SE layer to introduce the cation effects, maintaining ion conduction and a neutral environment at the catalyst/PEM interface, thus promoting H2O2 generation. The H2O2 molecules produced at the cathode were efficiently carried away by the O2 and DI water flow. Meanwhile, the generated protons from water oxidation at the anode moved into the SE layer to compensate for the charge.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (22466010), Guizhou Provincial Basic Research Program (Natural Science) ZK[2023]47 and Key Program ZK[2025]075, Innovation and Entrepreneurship Project for overseas Talents in Guizhou Province [2022]02, Specific Natural Science Foundation of Guizhou University (X202207), and Science and Technology Department of Guizhou Province (grant no. Platform & Talents [2019]5607). The authors would like to thank Shiyanjia Lab (https://www.shiyanjia.com) for the XPS and TEM analysis. The authors also would like to SCI-GO (https://www.sci-go.com) for the HRTEM analysis and the computing support of Chengdu DiYiYuanLi Technology Co., Ltd and the State Key Laboratory of Public Big Data at Guizhou University.Notes and references
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS.
- R. Hage and A. Lienke, Angew. Chem., Int. Ed., 2006, 45, 206–222 CrossRef CAS PubMed.
- B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2473 CrossRef CAS PubMed.
- S. Gao, Y. Jin, K. Ge, Z. Li, H. Liu, X. Dai, Y. Zhang, S. Chen, X. Liang and J. Zhang, Adv. Sci., 2019, 6, 1902137 CrossRef CAS PubMed.
- J. Ma, N. A. Choudhury and Y. Sahai, Renewable Sustainable Energy Rev., 2010, 14, 183–199 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed.
- J. Gao, H. bin Yang, X. Huang, S.-F. Hung, W. Cai, C. Jia, S. Miao, H. M. Chen, X. Yang and Y. Huang, Chem, 2020, 6, 658–674 CAS.
- B.-H. Lee, H. Shin, A. S. Rasouli, H. Choubisa, P. Ou, R. Dorakhan, I. Grigioni, G. Lee, E. Shirzadi and R. K. Miao, Nat. Catal., 2023, 6, 234–243 CrossRef CAS.
- J. Qi, Y. Du, Q. Yang, N. Jiang, J. Li, Y. Ma, Y. Ma, X. Zhao and J. Qiu, Nat. Commun., 2023, 14, 6263 CrossRef CAS PubMed.
- T. H. Jeon, B. Kim, C. Kim, C. Xia, H. Wang, P. J. J. Alvarez and W. Choi, Energy Environ. Sci., 2021, 14, 3110–3119 RSC.
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef CAS PubMed.
- P. Cao, X. Quan, X. Nie, K. Zhao, Y. Liu, S. Chen, H. Yu and J. G. Chen, Nat. Commun., 2023, 14, 172 CrossRef CAS PubMed.
- X. Zhang, X. Zhao, P. Zhu, Z. Adler, Z.-Y. Wu, Y. Liu and H. Wang, Nat. Commun., 2022, 13, 2880 CrossRef CAS PubMed.
- H. Li, P. Wen, D. S. Itanze, Z. D. Hood, S. Adhikari, C. Lu, X. Ma, C. Dun, L. Jiang and D. L. Carroll, Nat. Commun., 2020, 11, 3928 CrossRef CAS PubMed.
- R. D. Ross, H. Sheng, Y. Ding, A. N. Janes, D. Feng, J. Schmidt, C. U. Segre and S. Jin, J. Am. Chem. Soc., 2022, 144, 15845–15854 CrossRef CAS PubMed.
- S. Siahrostami, S. J. Villegas, A. H. Bagherzadeh Mostaghimi, S. Back, A. B. Farimani, H. Wang, K. A. Persson and J. Montoya, ACS Catal., 2020, 10, 7495–7511 CrossRef CAS.
- S. Ding, B. Xia, M. Li, F. Lou, C. Cheng, T. Gao, Y. Zhang, K. Yang, L. Jiang, Z. Nie, H. Guan, J. Duan and S. Chen, Energy Environ. Sci., 2023, 16, 3363–3372 RSC.
- Y. Tian, D. Deng, L. Xu, M. Li, H. Chen, Z. Wu and S. Zhang, Nano-Micro Lett., 2023, 15, 122 CrossRef CAS PubMed.
- X. Huang, M. Song, J. Zhang, T. Shen, G. Luo and D. Wang, Nano-Micro Lett., 2023, 15, 86 CrossRef CAS PubMed.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS PubMed.
- Y. Zhao, H. Wang, J. Li, Y. Fang, Y. Kang, T. Zhao and C. Zhao, Adv. Funct. Mater., 2023, 33, 2305268 CrossRef CAS.
- L. Jing, W. Wang, Q. Tian, Y. Kong, X. Ye, H. Yang, Q. Hu and C. He, Angew. Chem., Int. Ed., 2024, 63, e202403023 CrossRef CAS PubMed.
- N. Wang, Y.-S. Zhang, D.-D. Wei, H.-M. Duan, L.-M. Mo and H.-Y. Wang, J. Mater. Chem. A, 2023, 11, 4162–4169 RSC.
- X. Sun, J. Yang, X. Zeng, L. Guo, C. Bie, Z. Wang, K. Sun, A. K. Sahu, M. Tebyetekerwa, T. E. Rufford and X. Zhang, Angew. Chem., Int. Ed., 2024, e202414417 CAS.
- M. Yan, Z. Wei, Z. Gong, B. Johannessen, G. Ye, G. He, J. Liu, S. Zhao, C. Cui and H. Fei, Nat. Commun., 2023, 14, 368 CrossRef CAS PubMed.
- L. Liu, L. Kang, J. Feng, D. G. Hopkinson, C. S. Allen, Y. Tan, H. Gu, I. Mikulska, V. Celorrio and D. Gianolio, Nat. Commun., 2024, 15, 4079 CrossRef CAS PubMed.
- S. Li, L. Chen, J. Wang, T. Liu, D. Li, Z. Yang, X. Xiao, C. Chu and B. Chen, Environ. Sci. Technol., 2024, 58, 10072–10083 CrossRef CAS PubMed.
- P. Guo, B. Liu, F. Tu, Y. Dai, Z. Zhang, Y. Xia, M. Ma, Y. Zhang, L. Zhao and Z. Wang, Energy Environ. Sci., 2024, 17, 3077–3087 RSC.
- X.-K. Gu, J. S. A. Carneiro, S. Samira, A. Das, N. M. Ariyasingha and E. Nikolla, J. Am. Chem. Soc., 2018, 140, 8128–8137 CrossRef CAS PubMed.
- J. Yuan, X. Liu, Y. Tang, Y. Zeng, L. Wang, S. Zhang, T. Cai, Y. Liu, S. Luo and Y. Pei, Appl. Catal., B, 2018, 237, 24–31 CrossRef CAS.
- M. Eguchi, M. Momotake, F. Inoue, T. Oshima, K. Maeda and M. Higuchi, ACS Appl. Mater. Interfaces, 2017, 9, 35498–35503 CrossRef CAS PubMed.
- X. Li, C. Li, Y. Xu, Q. Liu, M. Bahri, L. Zhang, N. D. Browning, A. J. Cowan and J. Tang, Nat. Energy, 2023, 8, 1013–1022 CrossRef CAS.
- J. Geng, S. Ji, M. Jin, C. Zhang, M. Xu, G. Wang, C. Liang and H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202210958 CrossRef CAS PubMed.
- C. Santoro, P. Bollella, B. Erable, P. Atanassov and D. Pant, Nat. Catal., 2022, 5, 473–484 CrossRef CAS.
- X. Yang, Y. Tian, S. Mukherjee, K. Li, X. Chen, J. Lv, S. Liang, L. K. Yan, G. Wu and H. Y. Zang, Angew. Chem., Int. Ed., 2023, 62, e202304797 CrossRef CAS PubMed.
- G. Ye, S. Liu, K. Huang, S. Wang, K. Zhao, W. Zhu, Y. Su, J. Wang and Z. He, Adv. Funct. Mater., 2022, 32, 2111396 CrossRef CAS.
- S. Chen, T. Luo, X. Li, K. Chen, J. Fu, K. Liu, C. Cai, Q. Wang, H. Li and Y. Chen, J. Am. Chem. Soc., 2022, 144, 14505–14516 CrossRef CAS PubMed.
- Y. Wang, Y. Zhou, Y. Feng and X. Y. Yu, Adv. Funct. Mater., 2022, 32, 2110734 CrossRef CAS.
- X. Lei, Q. Tang, Y. Zheng, P. Kidkhunthod, X. Zhou, B. Ji and Y. Tang, Nat. Sustain., 2023, 6, 816–826 CrossRef.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng and J. Cheng, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- R. D. Ross, H. Sheng, A. Parihar, J. Huang and S. Jin, ACS Catal., 2021, 11, 12643–12650 CrossRef CAS.
- Y. Li, J. Chen, Y. Ji, Z. Zhao, W. Cui, X. Sang, Y. Cheng, B. Yang, Z. Li, Q. Zhang, L. Lei, Z. Wen, L. Dai and Y. Hou, Angew. Chem., Int. Ed., 2023, 62, e202306491 CrossRef CAS PubMed.
- S. Nayak, I. J. McPherson and K. A. Vincent, Angew. Chem., Int. Ed., 2018, 130, 13037–13040 CrossRef.
- J. Wei, D. Xia, Y. Wei, X. Zhu, J. Li and L. Gan, ACS Catal., 2022, 12, 7811–7820 CrossRef CAS.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitski, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Ehrlich, J. Moellmann, W. Reckien, T. Bredow and S. Grimme, ChemPhysChem, 2011, 12, 3414–3420 CrossRef CAS PubMed.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee05796a |
| This journal is © The Royal Society of Chemistry 2025 |