
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNylon electrolyte chemistry in high-energy Li-metal batteries†
Zhiming
Zhao
 *ab,
Georgian
Melinte
c,
Dong
Guo
ab,
Yongjiu
Lei
ab,
Mohamed N.
Hedhili
c,
Xianrong
Guo
c,
Zixiong
Shi
ab,
Yizhou
Wang
ab,
Jehad K.
El-Demellawi
d,
Wenli
Zhao
ab and
Husam N.
Alshareef
*ab
*ab,
Georgian
Melinte
c,
Dong
Guo
ab,
Yongjiu
Lei
ab,
Mohamed N.
Hedhili
c,
Xianrong
Guo
c,
Zixiong
Shi
ab,
Yizhou
Wang
ab,
Jehad K.
El-Demellawi
d,
Wenli
Zhao
ab and
Husam N.
Alshareef
*ab
aCenter for Renewable Energy and Storage Technologies (CREST), King Abdullah University of Science and Technology (KAUST), Thuwal 239955-6900, Saudi Arabia. E-mail: zhiming.zhao@utexas.edu; husam.alshareef@kaust.edu.sa
bMaterials Science and Engineering, Physical Science and Engineering (PSE) Division, King Abdullah University of Science and Technology (KAUST), Thuwal 239955-6900, Saudi Arabia
cCore Labs, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
dKAUST Upstream Research Center (KURC), EXPEC-ARC, Saudi Aramco, Thuwal 23955-6900, Saudi Arabia
First published on 13th February 2025
Abstract
The practical options for polymer electrolytes (PEs) are predominantly confined to polyether matrices, which unfortunately falter at voltages exceeding 4 V. Herein, we introduce a pioneering approach using polyamide (PA, Nylon), a classical polymer with oxidation-resistant amide linkages, reengineered as high-voltage PEs compatible with Li-metal batteries (LMBs). Despite its resistance to most solvents, we observed that PA is susceptible to dissolution in dilute Li+ electrolytes. Upon dissolution, Li+ ions engage with the carbonyl moieties of PA, while anions establish H-bonds with their amido groups, thereby disrupting intrinsic H-bonds and fracturing the semi-crystalline framework of pure PA. These interactions facilitate Li+ transport and immobilize anions within the PA networks, resulting in (semi-)solid electrolytes with outstanding ionic conductivity (1.23 × 10−3 S cm−1) and a high Li+ transference number (0.71). Notably, the nitrogen-rich nature of PA fosters the formation of a nitrided interphase on Li-metal, securing a completely dendrite-free Li chemistry. Consequently, the Li‖LiNi0.8Co0.1Mn0.1O2 (NCM811) battery exhibits a prolonged cycling life surpassing 800 cycles under practical conditions. Additionally, PA within the electrolyte can be easily regenerated using water-based solvents. This innovation not only establishes a high-voltage PE for high-energy LMBs but also introduces a sustainable new chemistry for utilizing and regenerating PA via benign Li+ solutions.
Broader contextPolymer electrolytes represent a vital avenue for engineering high-energy LMBs because they can potentially confer a dendrite-free characteristic to Li-metal anodes, thereby providing safety merits. To date, poly(ethylene oxide) (PEO) is the only mature polymer matrix known to exhibit compatibility with Li-metal. However, the EO groups are prone to oxidation, limiting the operational voltage of the battery to 4 V. In parallel to poly(ether) electrolytes, poly(carbonate) electrolytes, characterized by the strong electron-withdrawing ability of carbonyl groups that can inherently withstand higher voltages, has been unfortunately overlooked. In this study, we describe, for the first time, polyamide (PA, nylon 6) electrolytes, classified within the poly(carbonate) category, as high-voltage polymer electrolytes compatible with LMBs. As a semicrystalline polymer, PA 6 is traditionally soluble only in corrosive acids; however, we found that PA 6 can be readily dissolved in Li+ carbonate electrolytes. The resulting semisolid PA electrolytes exhibit a high voltage tolerance exceeding 5.1 V, high ionic conductivity, and a high Li+ transference number. LMBs paired with high-voltage layered oxide cathodes demonstrate a prolonged cycling life surpassing 800 cycles. This work introduces a new polymer option for high-energy LMBs and a novel mechanism for the dissolution and recycling of PA. |
Introduction
Li metal is celebrated as the ultimate anode material for future Li batteries, offering a high theoretical capacity (3860 mA h g−1) and the lowest redox potential (−3.04 V versus the standard hydrogen electrode) among alternatives. Yet, its exceedingly low electrochemical potential concurrently engenders a great challenge, that is, the incompatibility with most solvents typically used in today's Li-ion batteries. While interphase layers at the Li-metal and electrolyte boundary can offer temporary respite, the underlying thermodynamic discrepancies foster ongoing parasitic reactions and dangerous growth of Li dendrites, which cast shadows over the safety and the practical adoption of LMBs.1In the quest for stability and safety, solid-state electrolytes (SSEs) have emerged as beacons of new hope. Solid inorganic electrolytes, notable for their robust thermal stability and ionic conductivities rivaling those of liquid counterparts, are distinguished as one of the most promising candidates for LMBs. As an example, Li10GeP2S12 features an impressive ionic conductivity of 12 mS cm−1 at ambient temperatures, coupled with a high Young's modulus (∼30 Gpa) and Li+ transference number (∼0.9).2 These properties significantly fortify the electrolyte against Li dendrites and thus enhance safety. Yet, the narrative of inorganic SSEs is tinged with the challenges of brittleness and stiffness, which can readily lead to cracking and high interfacial resistance with electrodes. Moreover, the complexities and costs involved in their production and integration weave a tapestry of hurdles that must be surmounted for their practical deployment.3 Not to be overshadowed, PEs stand as another pillar in the realm of SSEs, cherished for their flexibility and ease of molding into thin films. As a classical model, the exploration of poly(ethylene oxide) (PEO)-based SSEs dates back to the late 1960s when Lundberg,4 Armand, and their colleagues first identified the ion transport phenomenon within PEO matrices.5 The subsequent decades have seen a flowering of efforts to enhance their ionic conductivity, Li+ transference numbers, and mechanical properties. Emerging strategies include employing salts with bulk anions such as bis(trifluoromethanesulfonyl)imide (TFSI−) to plasticize the polymer matrix and curb the migration of anions,6,7 introducing nano-sized inorganic fillers to prevent the crystallization of ethylene oxide (EO) chains,8,9 and pioneering in situ polymerizations with functional groups.10,11 Despite these advances, a fundamental limitation persists: EO groups are susceptible to oxidation at voltages exceeding 4 V versus Li/Li+, restricting their compatibility with high-voltage cathodes like LiNixMnyCo1−x−yO2, and thus curtailing their potential application in high-energy battery systems (Fig. 1a).12,13
 | ||
| Fig. 1 (a) Comparison of anodic stability and ionic conductivity between polyether and nylon electrolytes (PAE reported in this work), with corresponding operational voltages of different cathodes shown (LFP: LiFePO4; and S: sulfur cathode). (b) The dissolution ability of PA 6 in carbonate (DEC), ether (dimethoxyethane, DME), and aqueous solutions with different salts at 60 °C. Red bars indicate that PA 6 can be dissolved in the corresponding solution; longer lines indicate higher concentrations, and scale bars are provided for reference. Green bars indicate that PA 6 cannot be dissolved in the corresponding solution. (c) Schematic illustration depicting Nylon chemistry (left) and the failure mechanisms of conventional carbonate electrolytes (right) in high-energy Li-metal batteries (LMBs). | ||
Exploring parallel paths, PEs featuring carbonyl groups deserve attention comparable to that given to polyether, akin to the attention shared between carbonate and ether electrolytes in the liquid electrolyte domain. However, these PEs have not been as widely studied or adopted, primarily due to challenges in processing them effectively for ion conduction, except for certain in situ polymerized forms which exhibit an inferior polymerization degree and low ionic conductivity.14–16 Nonetheless, the inherent oxidation stability provided by carbonyl groups in PEs promises their suitability for high-voltage applications. PA, a classical synthetic macromolecular polymer enriched with carbonyl groups in their amide linkages and boasting a history of nearly 90 years, has revolutionized materials science since its discovery.17–19 PA has found widespread applications across industries such as textiles, automotive, electronics, and packaging. However, its utilization in batteries has been limited, primarily to roles as separators, with rare examples as functional ion-conducting electrolytes.20–22 The propensity of PA to form ordered lamellar crystal structures through dense interchain H-bonds between carbonyl and amido groups lends them high crystallinity, combining strength, toughness, and resistance to solvents. To date, only a few acids such as formic acid have proven capable of dissolving PA, limiting its potential modification and incorporation with charge carriers.23 The question of how low-cost and sustainable PA can be effectively utilized as high-voltage electrolytes in LMBs remains an enticing challenge for current PE design. Recent advancements have been achieved with a poly(amide)-based quasi-solid electrolyte, developed through in situ polymerization of precursors, for use in Li‖NCM622 batteries.24 However, the focus of this research was not on the macromolecule polymer material itself.
Upon detailed observation of PA, it becomes evident that its amide linkages (–CONH–), are akin to the peptide bond in proteins, illustrating how human innovation has drawn inspiration from natural molecular structures in creating new materials. To manipulate the behavior of proteins in solution environments, Franz Hofmeister proposed the Hofmeister series in 1888, which elucidates how various salts influence the behavior of proteins in solution.25 This series classifies ions into “chaotropic”, which disrupts the protein structure, and “kosmotropic”, which stabilizes it. Chaotropic ions tend to disrupt H-bonds among molecules, reducing structural stability and increasing solubility. Building on this principle, despite PA being challenging to dissolve in aprotic solvents, it holds great potential for dissolution in chaotropic salt solutions.
Our investigations here reveal that macromolecular PA can be specifically solubilized in Li+-based ether/carbonate solutions due to the higher Lewis acidity of Li+ compared to other alkali metal ions (Fig. 1b and Fig. S1–S3, ESI†). The reason PA cannot dissolve in aqueous solutions is likely due to the strong solvation ability of water for Li+, which hinders the exertion of Lewis acidity of Li+. As a model, the dissolution behavior of PA 6 ([–HN(CH2)5CO–]n, hereafter abbreviated as PA) in a widely used carbonate electrolyte, 1 M LiPF6 in a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) with 5 wt% fluoroethylene carbonate (FEC) (1 M LiPF6 in EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC (1:1 by vol%) + 5 wt% FEC, henceforth designated as BE), was thoroughly investigated. Concentrations of PA in BE can reach up to 10 wt% at 60 °C. Detailed analysis shows that upon introducing PA into BE, Lewis-acidic Li+ cations interact with the carbonyl groups (C
:DEC (1:1 by vol%) + 5 wt% FEC, henceforth designated as BE), was thoroughly investigated. Concentrations of PA in BE can reach up to 10 wt% at 60 °C. Detailed analysis shows that upon introducing PA into BE, Lewis-acidic Li+ cations interact with the carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) on the PA chains, whereas PF6− anions form H-bonds with the amido groups (N–H). These interactions effectively “unzip” the original PA chains bonded by intrinsic H-bonds. At a PA concentration of 10 wt% within BE, the electrolyte transitions to a semisolid GPE at room temperature (referred to as PAE hereafter), characterized by pronounced mechanical properties (elevated viscoelasticity and modulus), rapid ion transport kinetics (ionic conductivity: 1.23 × 10−3 S cm−1 at 20 °C; Li+ transference number: 0.71), and a high oxidation voltage threshold exceeding 5.1 V. Importantly, a highly nitrided SEI is formed on the Li-metal anode, bestowing a completely dendrite-free Li anode morphology and high reversibility. Consequently, the Li‖NCM811 cell with a practical N/P (negative capacity/positive capacity) ratio of 3.5 demonstrates an ultra-long cycling life exceeding 800 cycles with over 70% capacity retention and a high energy density near 400 W h kg−1. These novel chemistries of Nylon in high-energy LMBs are vividly depicted in Fig. 1c and will be detailed in the subsequent content.
O) on the PA chains, whereas PF6− anions form H-bonds with the amido groups (N–H). These interactions effectively “unzip” the original PA chains bonded by intrinsic H-bonds. At a PA concentration of 10 wt% within BE, the electrolyte transitions to a semisolid GPE at room temperature (referred to as PAE hereafter), characterized by pronounced mechanical properties (elevated viscoelasticity and modulus), rapid ion transport kinetics (ionic conductivity: 1.23 × 10−3 S cm−1 at 20 °C; Li+ transference number: 0.71), and a high oxidation voltage threshold exceeding 5.1 V. Importantly, a highly nitrided SEI is formed on the Li-metal anode, bestowing a completely dendrite-free Li anode morphology and high reversibility. Consequently, the Li‖NCM811 cell with a practical N/P (negative capacity/positive capacity) ratio of 3.5 demonstrates an ultra-long cycling life exceeding 800 cycles with over 70% capacity retention and a high energy density near 400 W h kg−1. These novel chemistries of Nylon in high-energy LMBs are vividly depicted in Fig. 1c and will be detailed in the subsequent content.
Results and discussion
Mechanical properties
Macromolecular polymers are typically characterized by high chemical and thermodynamic stability, rendering them resistant to aprotic and even protic solvents. It is widely recognized that only specific aggressive chemical agents, such as anhydrous formic acid and sulfuric acid, are capable of disrupting or weakening the intermolecular H-bonds between PA chains, thereby dissolving PA.26,27 Interestingly, while PA shows resistance to typical carbonate solvents (Fig. S4, ESI†), it exhibits high solubility in LiPF6-based carbonate electrolytes. As demonstrated in Fig. 2a, PA can be dissolved in BE with concentrations of up to 10 wt% at 60 °C. Notably, when the temperature decreases to 20 °C, a 10 wt% PA solution undergoes a phase transition into a semisolid and transparent state (namely PAE, Fig. 2b). In contrast, solutions at lower concentrations maintain a liquid state, whereas PA precipitates at higher concentrations such as 15 wt%. The dissolution mechanism will be discussed later in detail, and the semisolid PAE (10 wt% PA in BE) will serve as the primary focus in subsequent studies.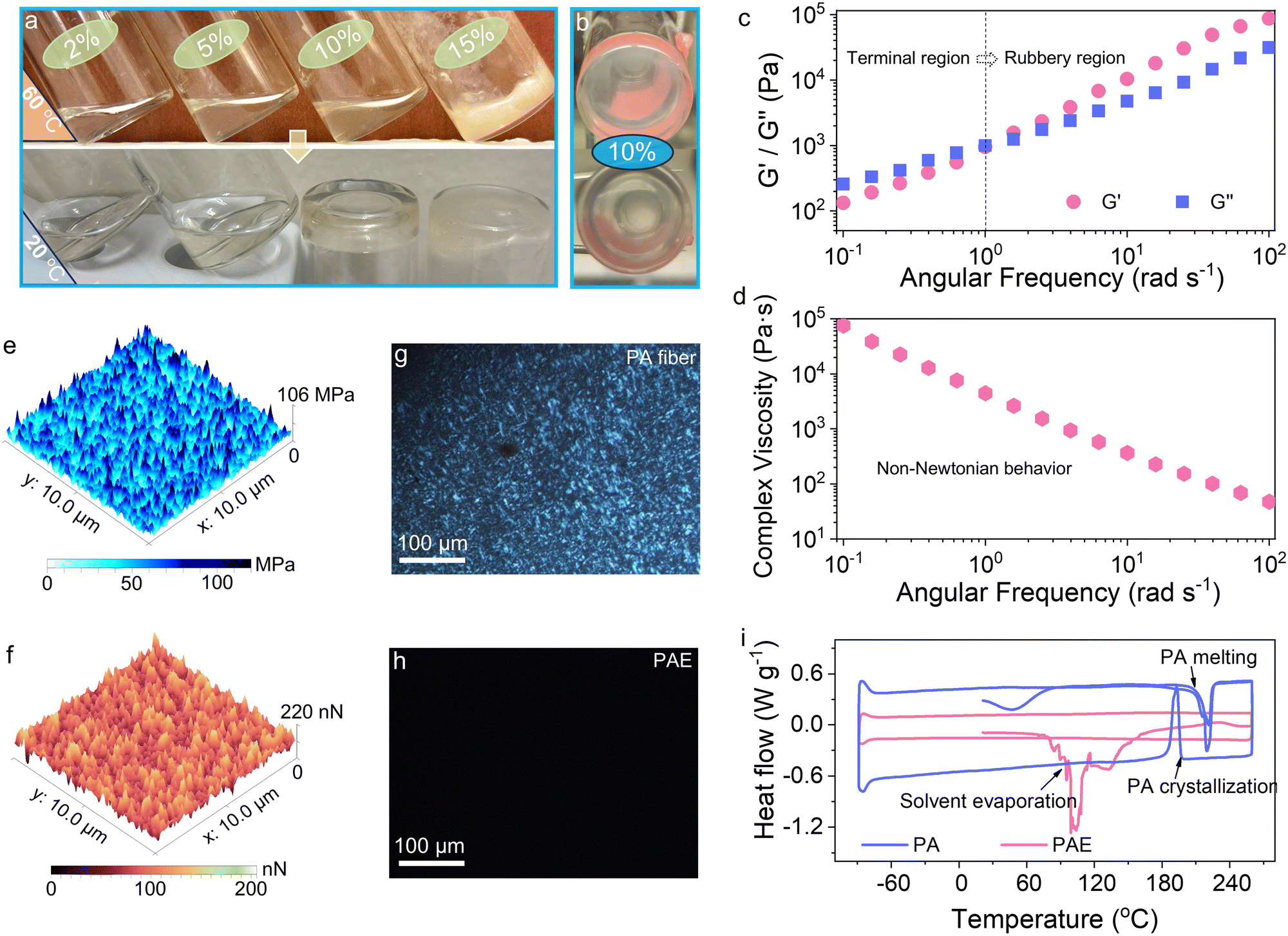 | ||
| Fig. 2 Nylon electrolyte preparation and mechanical properties. (a) Photographic images illustrating the dissolution of PA in BE at different weight concentrations, observed at 60 °C and 20 °C. (b) Photographic images depicting the dissolution of PA in BE at a weight concentration of 10%. (c) Rheological study of PAE, with G′ representing the storage modulus and G′′ representing the loss modulus. (d) Plot of complex viscosities of PAE against angular frequency. Measurement of (e) Young's modulus and (f) adhesion force of PAE obtained via atomic force microscopy (AFM). Polarized light microscopy images of (g) PA 6 fiber and (h) PAE. (i) DSC results for PA and PAE over the temperature range of −90 to 260 °C. | ||
Solid-state batteries commonly employ a layered sandwich architecture, in which the mechanical rigidity at the interface coupling electrode and electrolyte often results in relative displacement and severe contact failure. To effectively mitigate the interfacial challenges, the electrolyte should ideally demonstrate viscoelastic properties, providing it with the flexibility to accommodate volume changes in metallic anodes at high areal capacities and to respond synchronously with the electrode under mechanical stresses (Fig. S5, ESI†). A rheological analysis was conducted to assess the elastic and viscous dimensions of PAE, characterized by storage modulus (G′) and loss modulus (G′′), respectively.28 The evolution of these moduli with frequency sweeping is depicted in Fig. 2c. At lower frequencies (<1 rad s−1), G′′ surpasses G′, indicating viscosity prevails in a terminal region that predominantly responds to weak strains. However, as the frequency exceeds the threshold of 1 rad s−1, a notable escalation in G′ occurs, marking a transition to a rubbery phase where elasticity predominates. This demise from a viscous to an elastic dominance illustrates that PAE will stiffen when subjected to significant forces, which is an essential feature for resisting external deformation in solid batteries. Moreover, PAE displays a complex viscosity that aligns linearly with frequency on a logarithmic scale, highlighting its non-Newtonian behavior and inherent solid-state nature (Fig. 2d).29 Atomic force microscopy (AFM) was applied to further gauge the stiffness and adhesion characteristics of PAE. Encouragingly, even with merely 10 wt% PA in PAE, an average Young's modulus of 84 MPa was measured (Fig. 2e), along with a high average adhesion force of 132 nN (Fig. 2f). These mechanical properties of viscoelasticity and resilience are anticipated to enhance solid-state batteries by providing compatible interfaces and mitigating structural dislocations.
To scrutinize the origin of these favorable mechanical qualities, polarized light microscopy was employed, which can distinguish isotropic amorphous from crystalline regions in polymers through polarized light, visible as dark and bright areas, respectively.30 Our observations reveal large bright spherulites indicative of a semicrystalline structure of pure PA fibers (Fig. 2g), whereas PAE shows a uniform amorphous structure devoid of any crystalline regions (Fig. 2h). The amorphous structure of PAE is further verified using differential scanning calorimetry (DSC). Fig. 2i exhibits the DSC thermograms ranging from −90 to 260 °C. For pristine PA, a pronounced exothermic peak at approximately 198 °C indicates rapid crystallization during cooling, characteristic of highly crystalline polymers. However, the crystallization peak completely disappears in PAE, substantiating the amorphous structure. These results validate that the ordered semicrystalline structure in pristine PA, arising from organized H-bond arrangements, is totally disrupted by the BE solution. These reconstructed amorphous motifs in PAE contribute to the ductile behavior with a high elastic and viscous modulus.
Nylon dissolution mechanism
Having shown the mechanical robustness of PAE, we turned our attention to disclosing the PA dissolution mechanism and mapping the elusive microstructure hidden within. In carbonate electrolytes, EC molecules excel in their ability to dissociate Li salt due to their high dielectric constant, constituting the primary solvation shell around Li+.31 Raman spectroscopy was deployed to differentiate between coordinated and free EC molecules, with Raman shifts located at 903.5 and 893.2 cm−1, respectively (Fig. 3a).32 An increase in the PA moiety within the electrolyte is correlated with a decrease in the peak intensity for coordinated EC, suggesting a withdrawal of EC molecules from the Li+ solvation shell, accompanied by the substitution of polymer fragments. This subtle transformation is further reinforced by analyzing the NMR chemical shift evolution in 7Li. As apparent from Fig. 3b, a downfield shift in 7Li resonance emerges with an increase in PA, indicative of the deshielding effect caused by the displacement of EC molecules with PA chains around Li+. This alteration could be attributed to the replacement of EC molecules by PA chains, influenced by fewer coordination sites between PA and Li+ due to the steric effects of the macromolecular chains. | ||
| Fig. 3 Analysis of the nylon dissolution mechanism. (a) Raman spectra in the ranges of 860–930 cm−1 (the peak intensity was normalized by the peak located at 893.2 cm−1). (b) 7Li and (c) 19F NMR spectra of various samples. 2D 19F-1H HOESY spectra showing the interaction of F with H-nuclei in the electrolytes for (d) PAE 5% (BE with 5 wt% PA dissolved) and (e) PAE (BE with 10 wt% PA dissolved). The insets show the details of the cross signals between the H-nuclei of N–H groups and the F nuclei of PF6− anions. The external reference for the NMR spectra is 0.02 M LiPF6 in a deuterated-DMSO (dimethyl sulfoxide-d6) solution. | ||
With the understanding of Li+ solvation evolution, we extended our inquiry to the chemical environment of PF6− anions, unraveling yet another layer of delicate molecular interactions in PAE. In dilute BE, anions typically do not engage in pronounced interactions with the solvent, remaining predominantly unbound.33,34 This facilitates their mobility and results in a low Li+ transference number. As illustrated in Fig. 3c, a substantial downshift in the 19F signal is displayed with an increase in PA content, indicative of a deshielding effect. This is attributed to the interactions between free PF6− anions and electron-deficient segments within PAE. Notably, the electron-deficient N–H groups in PA, which have a partially positively charged proton, can serve as H-bond donors. Concurrently, the high electronegativity of the fluorine atoms in PF6− renders them effective H-bond acceptors. It is thus reasonable that PF6− anions form H-bonds with the N–H groups of PA. This hypothesis is corroborated by 2D heteronuclear Overhauser effect spectroscopy (HOESY), which provides direct evidence of close internuclear distances and is invaluable for identifying spatial inter- and intramolecular contacts.35 We note that while HOESY can discern both inter- and intramolecular atomic interactions, correlation spectroscopy (COSY) is limited to identifying through-bond couplings within the same type of nuclei, and nuclear Overhauser effect spectroscopy (NOESY) is restricted to detecting interactions involving identical nuclei.36 The 2D 19F-1H HOESY maps for PAE concentrations of 5 wt% and 10 wt% are depicted in Fig. 3d and e, respectively. The cross signals in these maps correspond to the heteronuclear Overhauser effect (HOE) between H- and F-nuclei in PAE caused by magnetization transfer. It is important to note that the 1D line graph should derive from 2D signal projections, rather than from direct transfer of 1D NMR spectra from separate experiments. The presence of a peak in a 1D projection typically correlates with a coupling signal in 2D HOE. The resonance at ∼6.57 ppm in the 1H spectrum is attributed to the proton in N–H groups of PA (for detailed assignment of H-nuclei, please refer to Fig. S6, ESI†), while the resonance at −124 ppm in the 19F spectrum corresponds to FEC.37,38 Clear HOE cross peaks between the F-nuclei of PF6− and the H-nuclei of the N–H groups in PA, along with sharp signals in the 1D 1H projection, are evident, confirming the formation of crosslinks via H-bonding between PF6− and PA. However, the absence of any cross signals suggests no interaction between the H-nuclei of the N–H group in PA and the F-nuclei of FEC solvents. When the proximity between the F-nuclei of PF6− and the H-nuclei of the N–H group in PA approaches ∼5 Å, magnetization transfer through space via cross-relaxation occurs amongst other H-nuclei in PA and F-nuclei of PF6− anions due to the dense H-nuclei environment in PA, explaining the observed additional cross signals.
To offer a theoretical juxtaposition to the above experimental results, molecular dynamics (MD) simulations were applied. A representative Li+ solvation arrangement in BE is displayed in Fig. S7b (ESI†), extracted from the equilibrium trajectory of the simulation box for BE (Fig. S7a, ESI†). As anticipated, the solvation shell of Li+ is predominantly formed by EC and DEC molecules, while PF6− solvation appears more dispersed due to modest ion–dipole interactions and dispersion forces with the solvents (Fig. S7c, ESI†). Transitioning from BE to PAE, the simulation reveals notable alterations in the local chemical milieu. A snapshot from the MD simulation of PAE shows a uniform isosurface distribution of PA (Fig. 4b), indicative of a homogeneous structural fabric within the electrolyte. The solvation environment surrounding Li+ in PAE undergoes a significant shift from that in BE; here, CO groups of PA predominantly make up the first coordination sphere (Fig. 4a). Simultaneously, PF6− is closely solvated by N–H groups from PA, with H-atoms of N–H oriented towards the F-atoms of PF6− supported by the H-bond (Fig. 4c).
 | ||
| Fig. 4 Molecular dynamics (MD) simulations. (a) Typical Li+ chemical environment in PAE. (b) Equilibrium MD simulation snapshots of PAE. In the MD box, PA chains were highlighted with a blue isosurface. (c) Typical PF6− chemical environment in PAE. (d) Radial distribution function (RDF, g[r], solid lines) and coordination number (dashed lines) for Li+ in BE. (e) RDF (solid lines) and coordination number (dashed lines) for Li+ in PAE. (f) RDF (solid lines) and coordination number (dashed lines) for PF6− in PAE. (g) Schematic diagrams for the dissolution of PA. | ||
Further insights were gleaned from radial distribution functions (RDFs), which quantify the likelihood of locating a species at a specified radial distance from a reference point. The results reveal that in BE, a single Li+ typically interacts with about four O-atoms from the CO groups of EC and DEC, occasionally interacting with fewer than one F-atom from PF6− anions, at an average distance of approximately 2 Å (Fig. 4d). In contrast, in PAE, two O-atoms from the CO groups of PA dominate the first solvation shell at a closer average distance of 1.94 Å, relegating ester coordination to a secondary role (Fig. 4e). The most compelling findings pertain to the PF6− anions in PAE (Fig. 4f). The introduction of PA results in a pronounced RDF peak at 1.96 Å, signaling strong bonds between the N–H groups of PA and PF6− anions, while interactions with H-atoms of carbonate molecules are markedly less frequent and more distant. This compellingly corroborates the formation of H-bonds between N–H groups of PA and PF6− anions. These theoretical analyses agree well with the experimental observations.
On the basis of the combined experimental and computational results, it becomes possible to figure out the PA dissolution mechanism and delineate PAE configuration (Fig. 4g). In its pure form, macromolecular PA presents a semicrystalline structure, where PA chains are methodically aligned through H-bonds between CO and N–H groups, rendering it insoluble in common solvents. However, once PA is introduced into BE solutions, a significant transformation occurs: Li+ aggressively interacts with CO groups on its chains, while simultaneously, PF6− anions form H-bonds with the N–H groups. These interactions disrupt the original H-bonds within PA, effectively “unzipping” its structured assembly. Consequently, PA becomes soluble in BE solutions, leading to the formation of semi-solid electrolytes within the PAE matrix. These interactions unveil a new chemistry for dissolving PA polymers in mild Li+ solutions. From the perspective of electrolytes, a portion of small molecules in the Li+ solvation shell is replaced by PA macromolecular chains, substantially enhancing the oxidation resistance of the resulting PAE due to the higher thermodynamic stability of macromolecules compared to small molecules. The remaining small molecules serve a ‘lubricant’ role, facilitating Li+ conduction along the PA chains. Simultaneously, the anions are immobilized by amido groups, which likely leads to a high Li+ transference number and rapid Li+ transport kinetics. Moreover, the potential for homogeneous Li+ flux through PA networks, combined with the semi-solid properties, may inhibit Li dendrite formation. These promising electrochemical effects will be disclosed in subsequent sections.
Dendrite-free Li metal anode and the highly nitrided interphases
Upon delineating the structure of PAE, we embarked on an extensive investigation into its ramifications for SEI development and the resultant behavior of the Li-metal anode. Morphological assessment of Li deposits on Cu foil was undertaken via scanning electron microscopy (SEM). In BE, Li deposits on Cu foil manifest a porous and dendritic architecture, as depicted in Fig. 5a and b. In marked contrast, under the PAE regimen, Li deposits exhibit a uniformly flat and compact morphology, featuring a completely dendrite-free surface (Fig. 5c), with the deposited Li particle resembling closely packed, smooth pebbles (Fig. 5d). These morphological distinctions are primarily attributable to the uniformity of interfacial Li+ flux, which is inherently influenced by the microstructural properties of the SEI and its homogeneity across the 2D surface.39,40 | ||
| Fig. 5 A completely dendrite-free Li metal anode and the highly nitrided interphases. Typical SEM images of Li deposits on Cu substrates with a deposition capacity of 4 mA h cm−2, using different electrolytes: (a) BE (wide view), (b) BE (detailed view), (c) PAE (wide view), and (d) PAE (detailed view). Cryo-TEM images of Li deposits in different electrolytes: (e) overall view featuring a single Li strip deposited from BE, (f) high-magnification of the SEI formed in BE on Li, (g) high-magnification image of the SEI formed in PAE on Li. The assignment of the crystal lattice is detailed in Fig. S8 and S9 (ESI†). 2D N 1s contour HR-XPS data of the SEI on the Li metal formed in: (h) BE and (i) PAE. 2D Li 1s contour HR-XPS data of the SEI on the Li metal formed in: (j) BE and (k) PAE. Etching times range from 0 to 7 min. | ||
To draw correlations between the electrolyte architecture and the ensuing interphasial chemistry, cryogenic transmission electron microscopy (cryo-TEM) was employed. This method exploits the stability of Li metal against electron-beam irradiation damage at liquid nitrogen temperatures, enabling depth analysis of interphase constructs. The evident darker shell layer, differentiated from the bulk area, is recognized as the SEI layer emerging due to electrolyte decomposition (Fig. 5e). Quantitative assessments indicate that the BE-engendered SEI exhibits a thickness of approximately 23 nm (Fig. S8, ESI†) and is primarily composed of amorphous materials interspersed with sporadic inorganic islands, building a mosaic-like nanostructure (Fig. 5f). These inorganic components, as determined by analyzing fast Fourier-transform (FFT) patterns from high-resolution imaging, primarily correspond to the crystal indices of Li2CO3, LiF, and Li2O (the crystal lattice assignment is detailed in Fig. S8 and Tables S1–S4, ESI†). In contrast, the PAE-induced SEI exhibits a more streamlined profile with a thickness of about 21 nm, affording a more conformal coating over the Li metal deposits (Fig. S9, ESI†). Distinctively, the SEI produced in PAE is distinctly darker under TEM, showcasing a more pronounced tone compared to its BE analogue (Fig. S8 and S9, ESI†). This intensity in darkness, inherent to the TEM imaging process, points to the possible presence of denser elements, likely signaling N-containing inorganic derivatives over C/H-containing organic compounds. FFT results validate this corollary, which images distinct Li3N, Li2CO3, and Li2O crystal signatures (Fig. S9, ESI†). Of particular note, Li3N appears in a monolithic form, permeating the entire SEI (Fig. 5g). Prior research accentuates the advantageous properties of Li3N for Li metal anodes, including thermodynamic stability, elevated Li+ conductivity, and effective dendrite suppression.41 This enrichment with Li3N is pivotal for the observed dendrite-free morphology of Li deposits in PAE. Synthesizing these visual insights, it becomes evident that PAE confers upon the Li-metal anode an interphase of remarkable robustness, benefiting from predominant Li3N-rich constituents.
Further validation of the interphasial chemistry conferred by PAE was achieved through nanoscale depth-profiling high resolution X-ray photoelectron spectroscopy (HR-XPS) combined with time-of-flight secondary ion mass spectrometry (ToF-SIMS). Displayed in Fig. 5h–k are N 1s and Li 1s 2D contour XPS spectra with Ar+ etching of the retrieved Li-metal form Li‖NCM811 cells with various electrolytes (will be discussed later). Specifically, no N 1s signals are detected in the interphase generated in BE, aligning with the absence of nitrogen elements in the electrolyte (Fig. 5h). In contrast, abundant N 1s signals are observed in the interphase produced in PAE (Fig. 5i), where peaks at 399.5 eV, 398.3 eV, and 397.2 eV are attributed to Li–N–C, Li3N, and LixNy, respectively.42 These reduction products pervade the interphase, providing pervasive benefits for reversible Li+/Li transformation, whether in guiding Li+ electrodeposition or offering thermodynamic protection. Li 1s HR-XPS analysis confirms these findings. For the interphase formed in BE, LiF signals predominate (Fig. 5j). In comparison, alongside LiF, Li2CO3, and Li2O, dominant signals from Li–N–C, Li3N, and LixNy originating from PAE are extensively detected (Fig. 5k).43 Additionally, complex carbonaceous organic species were identified in the C 1s spectra for both electrolytes (Fig. S10, ESI†), originating from inevitable solvent decomposition. The SEI chemistry is further elucidated by employing ToF-SIMS on the Li metal anode recovered from cycled cells, with Li2N− selected as a representative nitride component (Fig. S11, ESI†). Its pervasive distribution in the interphase is vividly depicted in three-dimensional reconstruction images, illustrating how PA orchestrates the formation of a novel interphasial chemistry.44
Practical LMBs
The oxidative robustness of electrolytes is crucial for their deployment in high-voltage scenarios. As delineated in Fig. S12 (ESI†), the absence of decomposition currents at voltages ascending to 5.1 V, versus a mere 4.3 V for BE, underscores the superior oxidation stability of PAE. This improvement stems from replacing oxidizable small ester molecules with robust PA macromolecules in the Li+ solvation shell. PAE also exhibits notable reduction stability compared to both BE and ether electrolytes (Fig. S13, ESI†). Moreover, the ionic conductivity of PAE at room temperature registers at 1.23 × 10−3 S cm−1 at 20 °C (Fig. S14, ESI†), slightly lower than that of BE (2.73 × 10−3 S cm−1) (Fig. S15, ESI†). Nonetheless, the markedly lower activation energy for ion migration within PAE (1.217 vs. 16.66 kJ mol−1 in BE) highlights a reduced barrier of ion mobility. Furthermore, the temperature-dependent ionic conductivity of PAE fits the Vogel–Tammann–Fulcher (VTF) empirical equation, indicating that Li+ conduction in PAE is akin to polymer motion-assisted transport, where the polymer matrix can facilitate uniform Li+ flow.45,46 Concurrently, the Li+ transference number in PAE escalates to 0.71 from 0.51 in BE (Fig. S16, ESI†), primarily due to the effective immobilization of anions via H-bonding along PA chains. The ionic conductivity of the solvent-free pure PAE membrane was also tested, achieving 2.16 × 10−6 S cm−1 at room temperature (Fig. S17, ESI†).Motivated by the fast Li+ kinetics in PAE and the beneficial nitrided SEI rich in Li–N compounds on Li-metal, we endeavor to harness PAE to construct robust LMBs for practical viability. Preliminary assessments employing the modified Aurbach's method reveal a high average Coulombic efficiency (CE) of 98.71% for Li‖Cu cells utilizing PAE, while cells with BE manifest a lower CE of 96.61% (Fig. S18, ESI†). These outcomes imply that cells integrated with PAE should exhibit extended durability, particularly on the Li-metal side. Symmetrical LMBs were assembled and tested under constant current conditions. Illustrated in Fig. 6a, the symmetrical LMB utilizing PAE sustains stable cycling for over 2000 hours at a current density of 0.5 mA cm−2, whereas the counterpart with BE exhibits escalating polarization voltages after merely 550 hours. Additionally, PAE proves robust against elevated current densities, supporting up to 3 mA cm−2 (Fig. S19, ESI†). The limiting current was also assessed in symmetrical Li cells (Fig. S20, ESI†), where the cell utilizing PAE exhibits a significantly higher limiting current density than its BE counterpart (17.1 vs. 6.6 mA cm−2). This pronounced disparity underscores the capacity of the SEI formed in PAE to accommodate substantial ion fluxes, thereby mitigating dendrite formation at the Li anode. In contrast, cells employing BE are more susceptible to reaching safety thresholds, indicative of potential dendrite formation. This assertion is further substantiated by the higher exchange current density (3.5 vs. 2.0 mA cm−2) (Fig. S21, ESI†) and comparably lower activation energy (27.3 vs. 38.5 kJ mol−1) for Li+ transport across the nitrided SEI (Fig. S22, ESI†), ensuring a homogeneous Li+-flux crucial for uniform, nondendritic Li+ deposition.
 | ||
| Fig. 6 Practical electrochemical performances. (a) Cycling performance of symmetric LMBs with various electrolytes at a current density of 0.5 mA cm−2 (0.5 mA h cm−2). The inset provides a detailed view from the 1000th to the 1010th cycle. (b) Cycling performance of 50 μm Li‖NCM811 (4.06 mA h cm−2) cells with various electrolytes cycled at 0.2C charge and 0.3C discharge (1C = 200 mA g−1). (c) Charge–discharge voltage profiles of the 50 μm Li‖NCM811 (4.06 mA h cm−2) cell using BE. (d) Charge–discharge voltage profiles of the 50 μm Li‖NCM811 (4.06 mA h cm−2) cell using PAE. (e) Cycling performance of the Li‖NCM811 (692 mA h) pouch cell with an energy density of 397 W h kg−1 (inset: the digital photo of the pouch cell; 0.2C charge/0.3C discharge). (f) Performance comparison of the cell with PAE against published studies involving SPEs (more data are detailed in Table S5, ESI†). | ||
Full LMBs configured with 50 μm Li‖NCM811 (4.06 mA h cm−2, single crystal) and a practical negative-to-positive (N/P) capacity ratio of 3.5 were assembled. Operated under a protocol of 0.2C charge and 0.3C discharge with a high cutoff voltage of 4.4 V, the cells incorporating PAE demonstrate markedly enhanced rechargeability, resulting in a capacity retention of over 70% after 800 cycles, in contrast to 17% for the control electrolyte (Fig. 6b). The progressive increase in voltage polarization noted in cells employing BE indicates the deteriorating interphases at both cathode and anode interfaces (Fig. 6c), coupled with potential phase transitions within NCM811 (which will be elaborated upon later). In contrast, cells using PAE exhibit minimal polarization widening over extensive cycling, underscoring the stability of the constructed interphases on electrodes and superior energy efficiency (Fig. 6d). Furthermore, when subjected to an escalated current rate of 5C, the cell with BE displays negligible capacity, while a PAE-based cell maintains a decent discharge capacity of approximately 65 mA h g−1 (Fig. S23, ESI†). More importantly, a pouch-type Li‖NCM811 cell with a capacity of ∼700 mA h and a high energy density of 397 W h kg−1 (calculated using data from the 10th cycle, see details in Methods) also presents excellent cycling stability, achieving 87% capacity retention after 60 cycles at a current density of 0.2C/0.3C (Fig. 6e and Fig. S24, ESI†). To the best of our knowledge, our PAE here serves as the best (gel/semi-solid) polymer electrolytes for LMBs, considering factors such as cycle life, areal capacity, and operational voltage (Fig. 6f and Table S5, ESI†).47–64
Intrigued by the exemplary electrochemical performance and practicality of LMBs pairing NCM811, a thorough postmortem analysis was undertaken to assess the structure integrity of the NCM811 cathode side. High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images uncover significant transformations in the NCM811 particles retrieved from diverse electrolytes. Particles from BE exhibit pronounced microcracks (Fig. 7a), signaling extensive mechanical degradation under electrochemical stress, which ultimately results in particle pulverization. These fractured particles, characterized by an expanded specific surface area vulnerable to electrolyte interaction, are predisposed to acidic erosion and accelerated transition metal (TM) dissolution.65 Additionally, the CEI formed in BE is notably thick (Fig. 7b), approximately 9 nm, indicating severe electrolyte oxidation and decomposition necessitating interface stabilization. Atomic-resolution HAADF-STEM imaging provides direct evidence of atomic disarray and phase transitions within NCM811. As illustrated in Fig. 7c, a proliferation of disordered rock-salt phases emerges in the near-surface area, characterized by a cubic arrangement of atoms with deviations in their original positioning, indicative of TM-atom dissolution into interstitial layers. This displacement of TM atoms obstructs Li+ passage, exacerbating voltage polarization and undermining reversible Li+ storage.
 | ||
| Fig. 7 Cathode integrity and sustainability. (a) Low-magnification HAADF-STEM image of the cycled NCM811 particles in BE. (b) TEM image of the CEI after cycling in BE. (c) Atomic-resolution HAADF-STEM image of the cycled NCM811 in BE. (d) Low-magnification HAADF-STEM image of the cycled NCM811 particles in PAE. (e) TEM image of the CEI after cycling in PAE. (f) Atomic-resolution HAADF-STEM image of the cycled NCM811 in PAE. (g) Cycling performance of the 50 μm Li‖NCM811 (4.06 mA h cm−2) cell with a cut-off voltage of 4.4 V, charged at 0.2C and discharged at 0.3C before cathode recycling. (h) Recycling process of the NCM811 cathode. (i) Cycling performance of a 50 μm Li‖NCM811 (4.06 mA h cm−2) cell with recycled NCM811, charged at 0.2C and discharged at 0.3C; cut-off voltage: 4.4 V. (j) Regeneration process of the PA polymer. | ||
In stark contrast, particles extracted from PAE retain a pristine morphology devoid of discernible cracks (Fig. 7d). This structural resilience is primarily ascribed to the anti-oxidative properties of PAE and the durability of the CEI it forms. The CEI formed in PAE is considerably thinner than that in BE, measured at approximately 4 nm (Fig. 7e). A thinner CEI denotes rapid and effective stabilization of the interface during the electrochemical cycle, reflecting superior interphase quality and integrity. Further insights into the CEI composition were gleaned from HR-XPS analyses (Fig. S25, ESI†). Mirroring observations at the anode side, abundant Li3N, Li–N–C, and N-containing organic species were identified from N 1s and Li 1s spectra. This analogous nitride-rich composition in both the CEI and SEI facilitates kinetic matching between the Li-metal anode and the NCM811 cathode. Benefiting from the efficacious electrolyte and a compatible, robust CEI, the layered structure of NCM811 after cycling in PAE is meticulously preserved, with only a thin Li/TM mixture layer observed on the near-surface zone (Fig. 7f).
The extensive utilization of metal elements such as Ni, Co, and Li in batteries poses a long-term risk of depletion due to their inherent scarcity, emphasizing the critical need for renewability in sustainable, large-scale deployment.66 However, recycling processes for solid-state batteries are inherently complex and costly, owing to the intricate admixture of electrolyte and cathode particles.67 As previously established, PA macromolecules exhibit high solubility in BE, thereby potentially enabling feasible cathode and electrolyte separation for recycling. As a proof of concept, we demonstrated the recyclability of the batteries endowed by our electrolytes. A Li‖NCM811 cell with PAE was assembled and subjected to cycling at 0.2C/0.3C for 50 cycles, achieving a capacity retention of 90.8% of its initial capacity (Fig. 7g). After cycling, the cell was disassembled, and the NCM811 cathode was immersed in BE at 60 °C to dissolve the PA macromolecular adhering to the cathode (Fig. 7h). This treatment yielded a purified NCM811 free from PA polymers. We then reconstituted this cleaned NCM811 into a recycled Li‖NCM811 cell, subjecting it once more to reversible cycling using the same current protocol. The rejuvenated cell manifests an initial capacity of ∼98% relative to its pre-recycling capacity and maintains an 84% capacity retention after 200 additional cycles (Fig. 7i). These findings underscore the feasibility of direct cathode recycling facilitated by the simple dissolution of PAE in liquid BE. Furthermore, PA within PAE showcases additional recyclability merits. Upon the introduction of water into PAE, the PA polymer precipitates immediately (Fig. 7j), a reaction attributed to the reformation of H-bonds between the N–H and CO groups of PA chains, mediated by water molecules. Given its cost-effectiveness and low toxicity relative to conventional, highly fluorinated electrolytes, our PAE here offers a promising alternative for sustainable electrolyte solutions.
Conclusions
To fulfill the demanding specifications of high-energy LMBs, PEs prove essential, notably for their flexibility, versatility, and inherent safety features. Traditional polyether PEs like PEO face constraints due to the oxidation susceptibility of ether groups at voltages exceeding 4 V. By contrast, PEs incorporating carbonyl groups with high-voltage resilience, warrant consideration on par with ester and ether electrolytes prevalent in liquid formulations. However, to date, no PEs featuring carbonyl groups have demonstrated effective ion incorporation and satisfactory ion transport. In this study, we propose repurposing well-established PA as the matrix for PEs in chaotropic Li+ solutions. The interaction of Li+ with carbonyl groups and anions with amido groups disrupts the original H-bonds among these groups, allowing PA to dissolve in mild Li+ carbonate solutions. This alteration enhances Li+ kinetics and increases the Li+ transference number in the resultant semisolid electrolytes. Additionally, the amido groups facilitate the formation of a nitrided SEI on the Li-metal, yielding a completely dendrite-free anode with an ultra-long cycling life exceeding 2000 hours. With a high voltage tolerance surpassing 5.1 V, the assembled high-voltage Li‖NCM811 battery achieves an energy density close to 400 W h kg−1 and sustains over 800 cycles. Notably, our battery design avoids heavily fluorinated species, significantly reducing costs and enhancing environmental friendliness. Both the cathode and the polyamide matrix within the electrolytes are recyclable, enhancing considerable sustainability. This work offers new avenues for high-voltage PEs and innovative chemistry for regenerating PA.Author contributions
Z. Z. and H. N. A. conceptualized the study and designed the experiments. G. M. executed the cryo-TEM and HRTEM experiments and analyzed the results. M. N. H. performed the XPS tests. X. G. provided guidance on the NMR tests. Z. Z., D. G., Y. L., Z. S., Y. W., J. K. E., and W. Z. carried out the electrochemical experiments and their subsequent analyses. Z. Z. conducted all additional characterization studies. Z. Z. and G. M. prepared the manuscript. H. N. A. supervised the project. All authors contributed to interpreting the results and revising the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this publication was supported by King Abdullah University of Science and Technology (KAUST)-Center of Excellence for Renewable Energy and Storage Technologies (CREST) under award number 5937.References
- Y. S. Meng, V. Srinivasan and K. Xu, Science, 2022, 378, eabq3750 CrossRef PubMed.
- L. Schweiger, K. Hogrefe, B. Gadermaier, J. L. Rupp and H. M. R. Wilkening, J. Am. Chem. Soc., 2022, 144, 9597–9609 CrossRef PubMed.
- L.-Z. Fan, H. He and C.-W. Nan, Nat. Rev. Mater., 2021, 6, 1003–1019 CrossRef.
- R. Lundberg, F. Bailey and R. Callard, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 1563–1577 CrossRef.
- Z. Song, F. Chen, M. Martinez-Ibañez, W. Feng, M. Forsyth, Z. Zhou, M. Armand and H. Zhang, Nat. Commun., 2023, 14, 4884 CrossRef PubMed.
- W. Gorecki, M. Jeannin, E. Belorizky, C. Roux and M. Armand, J. Phys.: Condens. Matter, 1995, 7, 6823 CrossRef.
- J. Wan, J. Xie, X. Kong, Z. Liu, K. Liu, F. Shi, A. Pei, H. Chen, W. Chen and J. Chen, Nat. Nanotechnol., 2019, 14, 705–711 CrossRef PubMed.
- H. Pitawala, M. Dissanayake and V. Seneviratne, Solid State Ionics, 2007, 178, 885–888 CrossRef.
- Q. Zhou, J. Ma, S. Dong, X. Li and G. Cui, Adv. Mater., 2019, 31, 1902029 CrossRef PubMed.
- V. Vijayakumar, B. Anothumakkool, S. Kurungot, M. Winter and J. R. Nair, Energy Environ. Sci., 2021, 14, 2708–2788 RSC.
- Z. Zhao, J. Wang, Z. Lv, Q. Wang, Y. Zhang, G. Lu, J. Zhao and G. Cui, Chem. Eng. J., 2021, 417, 128096 CrossRef.
- X. Wang, C. Zhang, M. Sawczyk, J. Sun, Q. Yuan, F. Chen, T. C. Mendes, P. C. Howlett, C. Fu and Y. Wang, Nat. Mater., 2022, 21, 1057–1065 CrossRef PubMed.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J.-P. Bonnet, T. N. Phan, D. Bertin, D. Gigmes and D. Devaux, Nat. Mater., 2013, 12, 452–457 CrossRef.
- L. Yue, J. Ma, J. Zhang, J. Zhao, S. Dong, Z. Liu, G. Cui and L. Chen, Energy Storage Mater., 2016, 5, 139–164 CrossRef.
- X. Xie, Z. Wang, S. He, K. Chen, Q. Huang, P. Zhang, S. M. Hao, J. Wang and W. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202218229 CrossRef PubMed.
- J. Mindemark, R. Mogensen, M. J. Smith, M. M. Silva and D. Brandell, Electrochem. Commun., 2017, 77, 58–61 CrossRef.
- P. N. Stockmann, D. Van Opdenbosch, A. Poethig, D. L. Pastoetter, M. Hoehenberger, S. Lessig, J. Raab, M. Woelbing, C. Falcke and M. Winnacker, Nat. Commun., 2020, 11, 509 CrossRef PubMed.
- P. Morgan, Macromolecules, 1977, 10, 1381–1390 CrossRef.
- L. P. Manker, M. A. Hedou, C. Broggi, M. J. Jones, K. Kortsen, K. Puvanenthiran, Y. Kupper, H. Frauenrath, F. Marechal and V. Michaud, Nat. Sustainability, 2024, 7, 640–651 CrossRef.
- X. Luo, X. Lu, G. Zhou, X. Zhao, Y. Ouyang, X. Zhu, Y.-E. Miao and T. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 42198–42206 CrossRef.
- J. Chen, Q. Liu, B. Wang, F. Li, H. Jiang, K. Liu, Y. Wang, M. Li, Z. Lu and D. Wang, J. Electrochem. Soc., 2017, 164, A1526 CrossRef.
- C. F. Francis, I. L. Kyratzis and A. S. Best, Adv. Mater., 2020, 32, 1904205 CrossRef.
- S. De Vrieze, P. Westbroek, T. Van Camp and K. De Clerck, J. Appl. Polym. Sci., 2010, 115, 837–842 CrossRef.
- M. Chen, C. Ma, Z. Ding, L. Zhou, L. Chen, P. Gao and W. Wei, ACS Energy Lett., 2021, 6, 1280–1289 CrossRef.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 25, 1–30 CrossRef.
- P. Saunders, J. Polym. Sci., 1962, 57, 131–139 CrossRef.
- K. Behler, M. Havel and Y. Gogotsi, Polymer, 2007, 48, 6617–6621 CrossRef.
- O. Chaudhuri, S. H. Parekh and D. A. Fletcher, Nature, 2007, 445, 295–298 CrossRef.
- F. Irgens, Rheology and non-newtonian fluids, Springer, 2014 Search PubMed.
- L. Sawyer, D. T. Grubb and G. F. Meyers, Polymer microscopy, Springer Science & Business Media, 2008 Search PubMed.
- K. Xu, Electrolytes, Interfaces and Interphases: Fundamentals and Applications in Batteries, Royal Society of Chemistry, 2023 Search PubMed.
- Y.-H. Tseng, Y.-H. Lin, R. Subramani, Y.-H. Su, Y.-L. Lee, J.-S. Jan, C.-C. Chiu, S.-S. Hou and H. Teng, J. Power Sources, 2020, 480, 228802 CrossRef.
- Z. Zhao and H. N. Alshareef, Adv. Mater., 2024, 36, 2309223 CrossRef.
- Z. Zhao, B. Nian, Y. Lei, Y. Wang, L. Shi, J. Yin, O. F. Mohammed and H. N. Alshareef, Adv. Energy Mater., 2023, 13, 2300063 CrossRef.
- G. Hilmersson, P. I. Arvidsson, Ö. Davidsson and M. Håkansson, J. Am. Chem. Soc., 1998, 120, 8143–8149 CrossRef.
- C. Yu and G. C. Levy, J. Am. Chem. Soc., 1984, 106, 6533–6537 CrossRef.
- C.-C. Su, M. He, R. Amine, T. Rojas, L. Cheng, A. T. Ngo and K. Amine, Energy Environ. Sci., 2019, 12, 1249–1254 RSC.
- M. García, G. van Vliet, M. G. ten Cate, F. Chavez, B. Norder, B. Kooi, W. E. van Zyl, H. Verweij and D. H. Blank, Polym. Adv. Technol., 2004, 15, 164–172 CrossRef.
- B. Jin, T. Lai and A. Manthiram, ACS Energy Lett., 2023, 8, 3767–3774 CrossRef.
- Z. Zhao, B. Nian, Y. Lei, L. Zhao, M. N. Hedhili, D. Guo, Z. Shi, W. Zhao, J. K. El-Demellawi and Y. Wang, Adv. Mater., 2024, 2402626 CrossRef PubMed.
- M. S. Kim, Z. Zhang, J. Wang, S. T. Oyakhire, S. C. Kim, Z. Yu, Y. Chen, D. T. Boyle, Y. Ye and Z. Huang, ACS Nano, 2023, 17, 3168–3180 CrossRef PubMed.
- J. Yang, M. Li, Z. Sun, X. Lian, Y. Wang, Y. Niu, C. Jiang, Y. Luo, Y. Liu and Z. Tian, Energy Environ. Sci., 2023, 16, 3837–3846 RSC.
- Q. Wang, Z. Yao, C. Zhao, T. Verhallen, D. P. Tabor, M. Liu, F. Ooms, F. Kang, A. Aspuru-Guzik and Y.-S. Hu, Nat. Commun., 2020, 11, 4188 CrossRef.
- B. Jin, Z. Cui and A. Manthiram, Angew. Chem., 2023, 135, e202301241 CrossRef.
- K. M. Diederichsen, H. G. Buss and B. D. McCloskey, Macromolecules, 2017, 50, 3831–3840 CrossRef.
- N. A. Stolwijk, M. Wiencierz, C. Heddier and J. Kösters, J. Phys. Chem. B, 2012, 116, 3065–3074 CrossRef PubMed.
- L. Porcarelli, A. S. Shaplov, F. Bella, J. R. Nair, D. Mecerreyes and C. Gerbaldi, ACS Energy Lett., 2016, 1, 678–682 CrossRef.
- Q. Wang, X. Xu, B. Hong, M. Bai, J. Li, Z. Zhang and Y. Lai, Chem. Eng. J., 2022, 428, 131331 CrossRef.
- J. Xiang, Y. Zhang, B. Zhang, L. Yuan, X. Liu, Z. Cheng, Y. Yang, X. Zhang, Z. Li and Y. Shen, Energy Environ. Sci., 2021, 14, 3510–3521 RSC.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef.
- C. Z. Zhao, Q. Zhao, X. Liu, J. Zheng, S. Stalin, Q. Zhang and L. A. Archer, Adv. Mater., 2020, 32, 1905629 CrossRef.
- F.-Q. Liu, W.-P. Wang, Y.-X. Yin, S.-F. Zhang, J.-L. Shi, L. Wang, X.-D. Zhang, Y. Zheng, J.-J. Zhou and L. Li, Sci. Adv., 2018, 4, eaat5383 CrossRef PubMed.
- S.-J. Tan, J. Yue, Y.-F. Tian, Q. Ma, J. Wan, Y. Xiao, J. Zhang, Y.-X. Yin, R. Wen and S. Xin, Energy Storage Mater., 2021, 39, 186–193 CrossRef.
- J. Chai, Z. Liu, J. Ma, J. Wang, X. Liu, H. Liu, J. Zhang, G. Cui and L. Chen, Adv. Sci., 2017, 4, 1600377 CrossRef PubMed.
- S. Li, Y.-M. Chen, W. Liang, Y. Shao, K. Liu, Z. Nikolov and Y. Zhu, Joule, 2018, 2, 1838–1856 CrossRef.
- R. Lin, Y. He, C. Wang, P. Zou, E. Hu, X.-Q. Yang, K. Xu and H. L. Xin, Nat. Nanotechnol., 2022, 17, 768–776 CrossRef PubMed.
- J. Han, M. J. Lee, K. Lee, Y. J. Lee, S. H. Kwon, J. H. Min, E. Lee, W. Lee, S. W. Lee and B. J. Kim, Adv. Mater., 2023, 35, 2205194 CrossRef PubMed.
- M. J. Lee, J. Han, K. Lee, Y. J. Lee, B. G. Kim, K.-N. Jung, B. J. Kim and S. W. Lee, Nature, 2022, 601, 217–222 CrossRef PubMed.
- J. Yu, X. Lin, J. Liu, J. T. Yu, M. J. Robson, G. Zhou, H. M. Law, H. Wang, B. Z. Tang and F. Ciucci, Adv. Energy Mater., 2022, 12, 2102932 CrossRef.
- F. He, W. Tang, X. Zhang, L. Deng and J. Luo, Adv. Mater., 2021, 33, 2105329 CrossRef.
- H. X. Yang, Z. K. Liu, Y. Wang, N. W. Li and L. Yu, Adv. Funct. Mater., 2023, 33, 2209837 CrossRef CAS.
- T. Deng, L. Cao, X. He, A.-M. Li, D. Li, J. Xu, S. Liu, P. Bai, T. Jin and L. Ma, Chem, 2021, 7, 3052–3068 CAS.
- Y. Zheng, X. Li, W. R. Fullerton, Q. Qian, M. Shang, J. Niu and C. Y. Li, ACS Appl. Energy Mater., 2021, 4, 5639–5648 CrossRef CAS.
- P. Zhai, Z. Yang, Y. Wei, X. Guo and Y. Gong, Adv. Energy Mater., 2022, 12, 2200967 CrossRef CAS.
- Z. Cui, P. Zuo, Z. Guo, C. Wang and A. Manthiram, Adv. Mater., 2024, 2402420 CrossRef CAS.
- Z. Zhao, Y. Lei, L. Shi, Z. Tian, M. N. Hedhili, Y. Khan and H. N. Alshareef, Angew. Chem., Int. Ed., 2022, 61, e202212941 CrossRef CAS.
- L. Azhari, S. Bong, X. Ma and Y. Wang, Matter, 2020, 3, 1845–1861 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee05739b |
| This journal is © The Royal Society of Chemistry 2025 |