
Catalyst–electrolyte interface engineering propels progress in acidic CO2 electroreduction
Yunling
Jiang
,
Linsen
Huang
,
Chaojie
Chen
,
Yao
Zheng
 * and
Shi-Zhang
Qiao
*
* and
Shi-Zhang
Qiao
*
School of Chemical Engineering, The University of Adelaide, Adelaide, SA 5005, Australia. E-mail: yao.zheng01@adelaide.edu.au; s.qiao@adelaide.edu.au
First published on 28th January 2025
Abstract
The electrocatalytic carbon dioxide reduction reaction (CO2RR) is a viable strategy that supports carbon neutrality via transforming the dominant greenhouse gas CO2 into high-value-added chemicals. The CO2RR in alkaline and neutral media has thrived in recent years owing to their high CO2 solubility and favourable CO2 activation ability. However, critical challenges have emerged, such as carbonate formation and subsequent CO2 crossover to the anodic sides, which decreases the carbon efficiency and stability of the system. Alternatively, acidic media provide an advantageous environment to prevent CO2 crossover into the anolyte but suffers from strong HER competition, which is significantly more active under acidic conditions, largely reducing the CO2 conversion efficiency. Research on acidic CO2RRs began with some basic studies, including testing various catalysts and electrolytes and designing diverse substrate structures. With advancements in characterization technologies, it has been found that the acidic CO2RR is not only influenced by variations in the composition of the catalyst, substrate or electrolyte, but also by internal changes at the catalyst–electrolyte interface. Thus, catalyst–electrolyte interface engineering, involving electrolyte engineering, catalyst modification, and interface optimization, provides many feasible solutions for acidic CO2RRs to weaken the competing HER. Importantly, it extends acidic CO2RR investigation to the exploration of electronic structures, interfacial adsorption of cations and anions, and surface hydrophobicity of catalysts in the presence of an electric field. However, there are limited articles reviewing acidic CO2RRs from this perspective, and thus, this review aims to discuss the challenges, history, evaluation, and breakthroughs in acidic CO2RRs regarding catalyst–electrolyte interface engineering, thereby providing insights for the future development of acidic CO2RRs.
 Yunling Jiang | Yunling Jiang received her BS degree and MS degree from Central South University. She is now a PhD candidate in the School of Chemical Engineering, The University of Adelaide. Her research focuses on the exploration of strategies for high-performance electrochemical CO2 reduction. |
 Linsen Huang | Linsen Huang is a PhD candidate in the School of Chemical Engineering at The University of Adelaide. His research is centred around the design and optimization of electrocatalysts, with a particular focus on understanding the underlying mechanisms involved in alkyne electrocatalysis. |
 Chaojie Chen | Chaojie Chen is currently a PhD student in the School of Chemical Engineering, The University of Adelaide. His research focuses on the design of electrocatalysts and mechanisms of electrocatalysis. |
 Yao Zheng | Yao Zheng received his PhD in 2014 from the University of Queensland (Australia). He is currently a Professor at The University of Adelaide. His current research focuses on fundamental studies of some key electrocatalysis processes by combining experiments and theoretical computations and the development of advanced electrocatalysts for energy conversion processes. |
 Shi-Zhang Qiao | Shi-Zhang Qiao received his PhD in Chemical Engineering from the Hong Kong University of Science and Technology in 2000. He is currently a Chair Professor, elected Fellow of the Australian Academy of Science, ARC Industry Laureate Fellow, and ARC Australian Laureate Fellow at the School of Chemical Engineering, The University of Adelaide (Australia). His research expertise is in nanomaterials for new energy technologies (electrocatalysis, photocatalysis and batteries). |
Broader contextRelentless utilization of fossil fuels has caused the accumulation of greenhouse gases, driving global warming and subsequent natural disasters. Carbon dioxide (CO2), as a major greenhouse gas, demands innovative strategies for efficient conversion to achieve carbon neutrality. Powered by renewable electricity, the electrocatalytic CO2 reduction reaction (CO2RR) provides promising technology to transform CO2 into high-value-added chemicals, thereby promoting a sustainable carbon cycle. In the advancement of CO2RRs, alkaline and neutral media took precedence owing to their high CO2 solubility, effective CO2 activation ability and excellent efficiency. However, alkaline and neutral CO2RR processes suffer from issues such as carbonate formation and carbon crossover, compromising system stability and conversion efficiency. Thus, the acidic CO2RR has emerged as a promising alternative, although it still faces challenges such as competing hydrogen evolution and limited CO2RR intermediate adsorption. Developments in catalyst–electrolyte interface engineering are crucial to overcoming the obstacles in acidic CO2RRs. Focusing on catalyst–electrolyte interface engineering, this review highlights the development challenges and trajectory of the acidic CO2RR, outlines its evaluation metrics, and summarizes the ongoing efforts in the development of the acidic CO2RR. Furthermore, a concluding overview of the current limitations in the acidic CO2RR and an outlook on the future development of this research field are proposed. |
1. Introduction
With the rapid development of global industrialization, the emission of global greenhouse gases has reached a historic high level.1 As a key greenhouse gas, carbon dioxide (CO2) emission has surged to as high as 540 billion tons annually, resulting in an unprecedented acceleration in global warming.2 On the path to achieving carbon neutrality and zero emissions, the electrochemical carbon dioxide reduction reaction (CO2RR) powered by renewable electricity offers a feasible technology for converting CO2 into high-value-added chemicals, thereby advancing carbon capture and utilization.3–5 Taking advantage of the high solubility of CO2 and superior ability for CO2 activation, alkaline and neutral CO2RRs, which can significantly suppress hydrogen evolution reaction (HER), dominate the development of CO2RRs.6,7 As device technology advances, including the development of flow cells coupled with gas diffusion electrodes (GDE) and membrane electrode assemblies (MEAs), CO2RR systems with excellent performance in alkaline and neutral environments proliferate.8–10 Notably, long-term stability of 2400 h (10 days) has been achieved by homogeneously alloyed Bi0.1Sn crystals for CO2-to-formate conversion under alkaline conditions.11 Meanwhile, as a significant breakthrough, pure-water-fed CO2RRs demonstrated over 1000 h of operation on a surface-step-rich Cu catalyst with a 50% faradaic efficiency (FE) for ethylene at a total current of 10 A using a scaled-up electrolyser stack.12 Advancements in the selectivity for single products have continuously emerged in alkaline and neutral media, including CO, formic acid, CH4, ethanol and ethylene, with FE exceeding 90% for single-carbon products, such as CO and formate, and FE of over 70% for double-carbon products, such as ethanol and ethylene.13–17 Particularly, several studies achieved promising progress in CO2RR toward triple-carbon products. The production of propane with a high FE of 91% was achieved on a 1-ethyl-3-methylimidazolium-functionalized Mo3P nanocatalyst.18 A co-electrodeposited CuAg alloy catalyst could produce 2-propanol with an FE of 56.7% at 59.3 mA cm−2.19 Nevertheless, a fatal weakness of alkaline and neutral CO2RR has exposed, i.e., the inevitable generation of carbonate due to the reaction between OH− and CO2, which can cross over into the anolyte, resulting in CO2 loss and reduced carbon conversion efficiency.20,21 Also, the generated carbonate increases the resistance of the operation system and blocks the gas diffusion channels, leading to additional reaction energy consumption and unnecessary cost of electrolyte reuse.21,22 Consequently, the industrialization of alkaline/neutral CO2RR encounters significant obstacles.In contrast to alkaline and neutral CO2RR, CO2RR in an acidic environment can prevent carbonate generation and CO2 crossover issues, enhancing the carbon efficiency and electrode stability. Thus, electrocatalytic CO2RR in acid media has gained increasing popularity recently.23–25 In fact, the initial attempt to perform acidic CO2RR was reported in 2001. Yano et al. pioneered an electrocatalytic reaction system featuring a gas–liquid–solid three-phase interface.26 Also, they examined the influence of different pH values and K+ ion concentrations in the electrolyte, as well as the halide composition in the electrodes on the CO2RR product selectivity.26–28 Thereafter, the development of acidic CO2RR was dormant for approximately ten years. However, with the identification of numerous practical challenges regarding alkaline and neutral CO2RR, acidic CO2RR has regained attention. Nevertheless, acidic CO2RR has encountered substantial hurdles, including intense competition with the HER and limited adsorption of the CO2RR intermediates, resulting in unsatisfactory product selectivity.29 Specifically, in acidic media, high-concentration H+ ions in the electrolyte compete with CO2RR-promoted intermediates to adsorb on the catalyst surface, aggravating the hydrogen evolution reaction (HER). Generally, the aggravated HER competition leads to a reduction in the FE for the CO2RR products, while increasing the selectivity for H2. Additionally, owing to the unstable adsorption of the CO2RR-promoted intermediates (*CO, *CHO, etc.) in acidic media, it becomes more difficult for the acidic CO2RR to support long-chain reaction pathways and produce multi-carbon (C2+) products compared to alkaline and neutral conditions. Therefore, it is crucial to develop strategies that can significantly enhance the acidic CO2RR, while effectively suppressing hydrogen evolution.
Unlike the initial development stage focusing on the basic exploration of simple component replacement in electrocatalysts and electrolytes for acidic CO2RR, advancements in various interface characterization techniques have further broadened the understanding of acidic CO2RR by highlighting the significant impacts of catalyst–electrolyte interfaces.30–32 Specifically, it is acknowledged that the alkali metal cations in alkaline and neutral CO2RR are closely related to the intermediate stabilization and reaction efficiency.33,34 However, unlike in alkaline and neutral electrolytes, in which alkali metal ions are inherently present, acidic electrolytes contain no alkali metal ions. Therefore, motivated by the role of alkali metal cations in alkaline and neutral CO2RR, the effects of varying concentrations and types of alkali metal cations on the catalyst–electrolyte interface were investigated in acidic CO2RR by introducing alkali metal salts into acidic electrolytes.35–37 Studies on catalyst–electrolyte engineering demonstrate that introducing high concentrations of alkali metal salts into acid electrolytes can induce cation aggregation on the catalyst surface under an electric field.25,37,38 The interfacial cations, especially K+ and Cs+, can effectively enhance the adsorption of *OH and impede proton transfer. Thus, a localized alkaline environment, namely a spatially confined and transient high-pH area on the catalyst surface, can be constructed, suppressing the competing HER.24,36,39 It should be noted that in contrast to the bulk pH governed by the electrolyte composition and concentration, the local alkalinity is subject to the reaction kinetics and local ion flux.40 Subsequent research integrated the cation effect with catalyst modification in electronic structures or interface hydrophobicity engineering to synergistically enhance the stability of the alkaline local environment and the adsorption of the relevant intermediates on the catalyst–electrolyte interface for acidic CO2RR.23,41–44 However, it is worth noting that due to a high local alkalinity, such high concentration of alkali metal cations in the electrolyte, can lead to the generation of carbonate and crystallization on the electrode, hindering gas transport and threatening the long-term stability of the whole reaction system.38,45,46 Thus, to thoroughly prevent salt crystallization caused by carbonate generation, some researchers have focused on developing acidic CO2RR systems using alkali metal ion-free electrolytes (pure acid) by directly immobilizing cation groups on the surface of the catalyst.45,47,48 Nevertheless, the stability of long-chain cation groups and the high energy barrier for CO2RR in pure acid electrolytes restrict the operation current density to below 300 mA cm−2. In short, catalyst–electrolyte interface engineering has advanced the development of acidic CO2RR; however, challenges are still emerging continuously.
Given that comprehensive reviews summarizing and categorizing these developments are limited, instead of focusing on the type of catalyst or electrolyte, this review systematically examines the developmental trajectory of acidic CO2RR by discussing the complex interactions between the catalyst and electrolyte, namely, the engineering of the catalyst–electrolyte interface. By discussing the reaction system and mechanism challenges for acidic CO2RR, the importance of modulating the catalyst–electrolyte interface is underscored. Moreover, the developmental history of acidic CO2RR highlights the significance of the investigation of the catalyst–electrolyte interfaces. Besides, beyond the common parameters for CO2RR, some evaluation metrics associated with interfacial properties specific to acidic CO2RR are also summarized. Extensively, the propelling effects on acidic CO2RR via catalyst–electrolyte interface engineering are comprehensively reviewed based on three key aspects including electrolyte engineering, catalyst modification and interface optimization. Finally, we present a conclusive overview of existing contradictions and constraints in acidic CO2RR, together with an outlook on the future research directions and perspectives in this field.
2. Challenges in acidic CO2RR
Despite the significant advancements and continuous emerging results in CO2 electroreduction under alkaline and neutral conditions, inevitable issues such as the formation of carbonate and subsequent carbon crossover continue to pose significant challenges. Thus, these drawbacks have driven the revitalization of acidic CO2RR. However, chronic problems and challenges of acidic CO2RR, such as competing HER, weak adsorption of CO2RR intermediates, and sluggish CO2RR kinetics, still exist and require the application of cutting-edge technologies for analysis. In this chapter, we comprehensively discuss these challenges with regards to the reaction systems and mechanisms.2.1 Reaction systems
From a kinetic perspective, it is preferable for the CO2RR process to take place in alkaline and near-neutral environments, owing to the effective activation of CO2, reduced HER competition and enhanced CO2RR product selectivity.21,35,49,50 However, although the primary species in the alkaline system at the start of electrolysis are just the KOH electrolyte and CO2 reactant, it is inevitable for these two species to react and form carbonate (KHCO3 or K2CO3) during prolonged electrolysis (OH− + CO2 → HCO3− and 2OH− + CO2 → CO32− + H2O, Fig. 1a).21,51 The generation of carbonate will lead to carbon crossover to the anode side and salt participation on the electrode, significantly reducing the carbon efficiency and blocking the gas transport channels.52 Also, when KHCO3 is utilized as the neutral electrolyte for CO2RR, OH− can be generated during CO2 reduction (CO2 + H2O + 2e− → CO + 2OH−) and H2O decomposition (H2O + 2e− → H2 + OH−).53 Therefore, carbonate formation and carbon crossover are also unavoidable (Fig. 1b). Additionally, the increase in cell voltage under neutral conditions, resulting from the high overpotential of the oxygen evolution reaction (OER), leads to additional energy consumption.54 Thus, based on the discussion above, carbonate generation is a common issue in both alkaline and neutral CO2RR, resulting in carbon crossover during continuous electrolysis. These issues can result in a significantly lower CO2 single-pass conversion efficiency (SPCE), threaten the device operation stability, and even cause higher energy consumption for large-scale applications. Consequently, addressing the carbonate formation issue is essential to achieve an economically viable CO2 electroreduction. When operating CO2RR in the acidic mode (Fig. 1c), OH− anions can still be generated during the reaction operation and react with CO2, leading to the formation of unwanted carbonate salts near the cathode. Ingeniously, the acidic environment will consume these carbonate salts and convert them back into CO2, preventing the carbon loss issue.55,56 Thus, in theory, the CO2 SPCE of strongly acidic CO2RR is 100%. Due to the high conductivity of strong acid and the efficient acidic OER kinetics, the increased cell voltage issue under neutral conditions can also be alleviated, benefiting the energy efficiency (EE).6 However, there are always two sides to consider. It is obvious that acidic CO2RR is challenging due to the intense HER competition, resulting from the kinetically favourable H+ transport in strong acids.57 Thus, HER tends to dominate on most catalysts in pure acid, significantly obstructing CO2RR.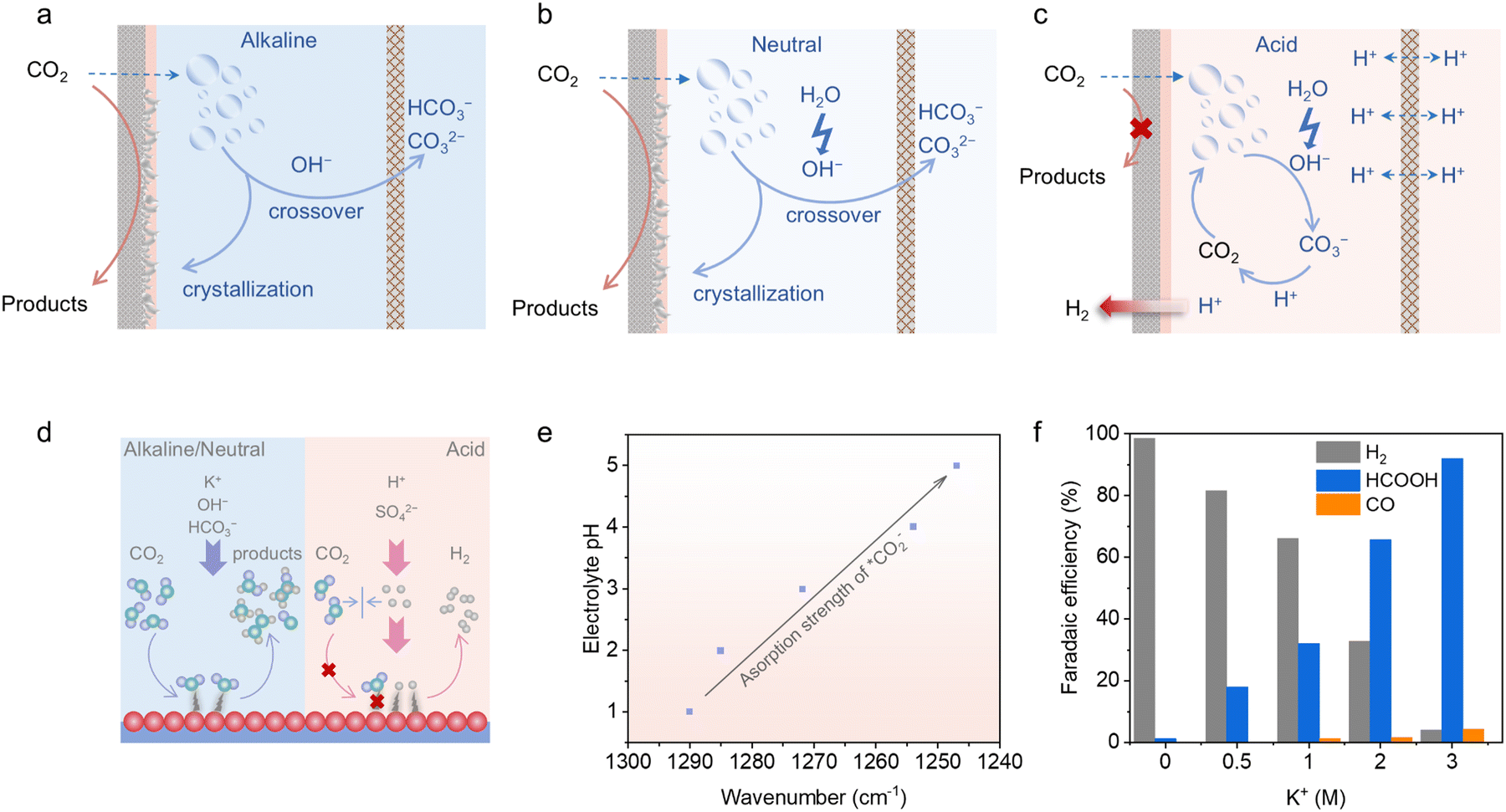 | ||
| Fig. 1 Schematic of the internal carbonate generation and crossover phenomena in (a) alkaline and (b) neutral CO2RRs. (c) Schematic of the internal carbonate-free operation in an acidic CO2RR. (d) Differences in intermediate adsorption in alkaline, neutral and acidic media. (e) Variations in CO2 binding strength with changes in electrolyte pH.58 (f) FEs of CO2RR products on a Bi nanosheet catalyst in a 0.05 M H2SO4 catholyte with different KCl concentrations, showing the effect of K+ on suppressing the HER.59 Reproduced with permission from ref. 59. Copyright 2022, the American Chemical Society. | ||
2.2 Reaction mechanisms
As mentioned above, alkaline and neutral environments are more advantageous for CO2 activation, resulting in high selectivity for the CO2RR products. In contrast, in acidic CO2RR, the bulk system will fill with H+ ions, which can be adsorbed on the cathode as *H during the reduction process, further leading to the dominance of H2 among the released products (Fig. 1d).45 Then, it is of high difficulty for the CO2 molecules to contact with the active sites and become adsorbed *CO2 species in pure acid. Besides, CO2RR-related intermediates tend to bind weakly and unstably in acidic media, making them prone to easy desorption, which further diminishes the reaction efficiency. As shown in Fig. 1e, the reaction intermediates of acidic CO2RR on Sn single-atom catalysts were manifested and compared under different pH values and reduction potentials through in situ attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy.58 It can be observed that the surface coverage of *CO2 was promoted with an increase in the pH value, and simultaneously the *CO2 adsorption strength on the catalyst was enhanced following the improvement in alkalinity. Therefore, the priority for advancing acidic CO2RR is to establish an interfacial alkaline microenvironment that strengthens *CO2 adsorption, while weakening *H adsorption even within acidic bulk electrolyte systems.In terms of the reaction system and mechanism challenges for acidic CO2RR, introducing high-concentration alkali metal cations in acidic electrolytes stands as an effective solution to suppress the competing HER and reduce *H adsorption. Research revealed that alkali cations from the electrolyte can adsorb onto the catalyst–electrolyte surface under the influence of an electric field during acidic CO2RR processes.37,49 This cation effect can facilitate the adsorption and accumulation of *OH, while suppressing the H+ diffusion, which subsequently elevates the local alkalinity. As a result, the HER competition can be suppressed, and the adsorption of CO2RR-related intermediates can be promoted, favouring the CO2RR process in acidic media (Fig. 1f).23,45,60,61 Further, an increasing number of studies combined catalyst structural optimization with the introduction of high-concentration alkali metal salts into acidic electrolytes to effectively regulate the catalyst–electrolyte interface and achieve high-performance acidic CO2RR.23,42,62 However, careful concentration modulation of the added alkali-metal cations is necessary. It was reported that an alkali-metal cation concentration of at least 2 M is typically sufficient to enhance CO2RR product formation, especially for the production of multi-carbon products.25 Nevertheless, the increase in local alkalinity driven by the addition of high-concentration alkali metal salts can result in generation and crystallization of carbonate on the catalyst–electrolyte interface. Therefore, even if the carbon crossover is well addressed in the acidic mode, the gas diffusion channel blocking issue still exists.38,45 Given this situation, it is required to explore strategies to decrease the concentration of alkali metal salts, while maintaining a comparable acidic CO2RR performance. Some studies introduced additional cation-enriched layers on the catalyst–electrolyte surface to increase the interfacial cation concentration in the bulk electrolyte with a low cation concentration, constructing a balanced trade-off between the cation concentration and acidic CO2RR performance.63 Furthermore, several researchers focused on alkali metal cation-free acidic CO2RR by immobilizing long-chain cation groups on the cathode surface.45,47,48 However, maintaining the stability of the cation-augment layers over long-term and high-current reaction processes is difficult. Additionally, it is extremely challenging to achieve high CO2RR product selectivity in pure acid. As a result, reports in this field are limited and this research direction remains in its early stages.
In short, although the research field of acidic CO2RR is developing rapidly as the understanding of the catalyst–electrolyte interface expands, challenges continue to emerge. Currently, a foolproof solution for achieving high-performance and long-term acidic CO2RR is still lacking.
3. Development history of acidic CO2RR
As feasible technology to achieve carbon neutrality, CO2RR has undergone decades of development with continuous innovations in terms of catalysts, electrolytes, and electrolysers. These advancements have enabled breakthroughs in the challenging acidic CO2RR. The acidic CO2RR has been in development for more than twenty years, undergoing different stages including preliminary attempts, subsequent stagnation, and recent resurgence (Fig. 2a). This chapter will retrospect the development history of acidic CO2RR chronologically, alongside the evolution of the electrolysers and key perspectives. | ||
| Fig. 2 (a) Development progress of the acidic CO2RR.26,64 Reproduced with permission from ref. 26. Copyright 2002, Elsevier. Schematic configuration of CO2RR electrolyser development: (b) H-cell, (c) flow cell, and (d) MEA. | ||
3.1 Preliminary attempts (2002–2004)
During the early research stage of acidic CO2RR, the main challenge was the limited CO2 concentration near the electrode due to the low solubility of CO2 in acid solution and the two-phase (liquid–solid) interface electrolysis system. In the early 21st century, to increase the CO2 concentration at the cathode interface, Yano et al. developed an electrolysis cell featuring a three-phase interface (gas–liquid–solid), in which CO2 gas can be pressed and contacted with the working cathode through a glass filter.65 This led to substantial advances in the acidic CO2RR. Capitalizing on the momentum, Yano and colleagues conducted further in-depth explorations, including the promoting effect of three-phase interfaces on both Cu and Ag-based catalysts, as well as the time and pH dependence of acidic CO2RR.26,65 Additionally, they studied CO2 conversion in electrolytes containing different types and concentrations of cations and anions.26 Furthermore, they delved into the effects of introducing Cu2+ ions in the electrolyte and doping different halogens into Cu on the acidic CO2RR.27,28 As pioneers in the field of acidic CO2RR, they proved that three-phase interface reaction system is more effective than the two-phase system in acidic CO2RR. In addition, they found that potassium cations (K+) and higher concentrations of cations are conducive to producing more multi-carbon products. Also, they demonstrated that feeding Cu2+ into electrolyte and doping halogens, especially Br, can promote ethylene production on Cu catalysts. Although their research merely focused on the direct relationship between the acidic CO2RR production and the catalyst/electrolyte composition, the findings laid the foundation for the subsequent advancement in the acidic CO2RR field.3.2 Subsequent stagnation (2005–2020)
After the initial attempt to perform acidic CO2RR, this research area remained dormant for nearly a decade. Following this decade-long hiatus, some intermittent studies on acidic CO2RR began to emerge in 2015, although most of them utilized CO2 reduction as an auxiliary process.Specifically, in the catalyst design aspect, Shen et al. reported the use of a cobalt protoporphyrin molecular catalyst supported on graphite for acidic CO2RR at pH values of 1–3 in a one-compartment electrochemical cell in 2015.66 The results from online electrochemical mass spectrometry demonstrated that the CO2RR products, CO and methane, originated from the cobalt protoporphyrin active sites. Also, Ni-doped covalent triazine frameworks were synthesized and utilized to compare CO production on GDE and PE (plate electrode) at the pH values of 2.0–2.5, showing the superiority of GDE.67 Additionally, although an alkaline environment is conducive to suppressing the competing HER, some products, such as formic acid, are dissociated in alkaline media, further complicating the separation process. Therefore, acidic CO2RR is also advantageous for the downstream separation process. Then, researchers attempted to utilize high pressure to boost the conversion of CO2 to formic acid in acid. Ramdin et al. demonstrated that the FE of formic acid could reach 80% with the assistance of 50 bar pressure at 30 mA cm−2 in acidic media.64 Furthermore, there has been research on novel CO2RR system designs and the mechanisms among the competing water-reduction HER, proton-reduction HER and CO2RR, which were explored under different pH conditions.68
During this period, the development of acidic CO2RR was accompanied by attempts to design different cells for both performance and mechanism research. Although the number of reports is limited, these studies illustrated the possibility of acidic CO2RR operation by designing catalysts and modulating the competitive relationship between CO2RR and HER.
3.3 Recent resurgence (2020–now)
In recent years, as the detrimental impact of carbon crossover and carbonate formation issues on the practical viability of CO2RR under alkaline and neutral conditions became increasingly apparent, the necessity of developing acidic CO2RR has been significantly recognized. Meanwhile, given the significant evolution of CO2RR reactors and characterization technologies, revisiting acidic CO2RR at this stage can potentially lead to further advancements.Firstly, regarding the transformation of CO2RR electrolysers (Fig. 2b–d), they evolved from two-phase interface reactors to three-phase interface reactors, including conventional H-cells, flow cells, and membrane electrode assemblies (MEA).69 The traditional two-phase interface reactors include single-chamber reactors and membrane-containing dual-chamber reactors (H-cells). When these two-phase interface reactors are utilized as electrolysers for CO2RR, the solubility of CO2 in the electrolyte becomes a decisive factor in determining the product selectivity.9 However, the solubility and diffusion coefficient of CO2 in acidic electrolyte are limited, resulting in low mass transfer efficiency and slow reaction rates.69,70 In addition, the distances between the anode and cathode are always large in H-cells, leading to increased internal resistance.9,69 As a further refinement, flow cells are designed with a porous hydrophobic gas diffusion electrode (GDE), cathode and anode chambers, and an ion exchange membrane. CO2 can directly contact the GDE layer, allowing the CO2RR process to take place at the gas–liquid–solid three-phase interface, largely addressing the issues of low mass transfer efficiency and slow reaction rates.71 Also, the distance between the anode and cathode is shortened, reducing the resistance of the entire system.6,9 Although flow-cell electrolysers are capable of high-current CO2RR operation, their stability resulting from the unstable GDE poses a risk of the electrolyte “flooding” issue.6 The flooding issue originates from the loss of hydrophobicity in the GDE under high-current and long-term operation, causing penetration of the bulk electrolyte into the gas diffusion channels and failure of the CO2RR operation. Thus, to alleviate the flooding issue, it is critical to design GDE with exceptional hydrophobicity, porosity and mechanical strength. Operating with no electrolyte passing through the cathode chamber, MEA can partially alleviate the gas diffusion layer blockage and electrolyte flooding issues. Nevertheless, it is likely that the anolyte will diffuse to the cathode side, causing electrolyte flooding. Typically, MEA operating in a two-electrode system retains the high mass transfer efficiency characteristic of the flow cell, enabling a high current density operation. Humid CO2 passes through GDE and the gas–solid reaction occurs directly. Moreover, compared to flow cells, the shorter anode–cathode distance in MEA further reduces the system impedance, enhancing the reaction rate and energy efficiency. Meanwhile, experimental characterization technologies and theoretical simulation calculation methods for electrocatalytic reactions gradually flourished during this period. Specifically, in situ ATR-FTIR and Raman spectroscopy are powerful tools to provide strong evidence for the adsorption of the reaction intermediates and guide the inference of reaction pathway mechanism.31,72,73 With the continuous development and widespread adoption of in situ electrolysers, laboratory-level in situ characterization has ultimately achieved near-complete simulation of the CO2RR reaction environment, broadening the understanding of the reaction interfaces. Furthermore, an increasing number of researchers advocated the combination of experimental work and theoretical calculations; therefore, theoretical calculations have also been adopted for simulating the interfacial adsorption behaviours and reaction pathways.73,74
Based on the improvement in CO2RR reactors and characterization technologies, the resurgence of acidic CO2RR is bound to drive further breakthroughs. In 2021, the first breakthrough came from Huang et al.24 They focused on the interfacial characteristics between the catalyst and the electrolyte, modelling the local pH values at their interface during acidic CO2RR under different reduction current densities. Subsequently, they identified the key factor, namely, the cation effect, and leveraged the CO2 electrostatic stabilization by high-concentration K+ together with a cation-augmenting layer to induce a local alkaline environment and mitigate the HER competition issue in strongly acidic CO2RR. Based on this strategy, high multi-carbon product efficiency (FEC2+, >50%) and SPCE (77% at 3.0 sccm) could be achieved in flow cell operation at high current density.24 Since then, an increasing number of researchers have shifted their focus to acidic CO2RR, leading to a surge in catalyst–electrolyte interface engineering research. Specifically, researchers have begun designing various catalyst electric structures for acidic CO2RR assisted by high-concentration cation electrolytes to promote the interfacial cation effect and alkalinity.23,42 Additionally, efforts have also been made to develop methods for acidic CO2RR at lower cation concentrations to mitigate the salt crystallization effects.61,63,75,76 Further, there are also sporadic reports on acidic CO2RR in cation-free electrolytes to completely eradicate salt crystallization.45,47,48 The descriptions of these advancements on acidic CO2RR, particularly in catalyst–electrolyte interface engineering, will be categorized and discussed in detail in Section 5.
4. Evaluation metrics for acidic CO2RR
As emerging technologies advance, an in-depth and precise understanding of their evaluation metrics becomes increasingly important. Numerous direct and indirect evaluation parameters have been reported to assess the electrochemical activity, conversion efficiency, and stability within CO2RR reaction systems. Different from the evaluation for neutral and alkaline CO2RR, acidic CO2RR requires special consideration of the HER competition and local pH-value variation at the reaction interface. Besides, the CO2 conversion efficiency is generally highlighted as a main advantage of acidic CO2RR compared to its neutral and alkaline counterparts. This chapter will explore the special parameters related to acidic CO2RR, including methods for detecting H+ diffusion at the catalyst–electrolyte interface, localized alkalinity and CO2 single-pass conversion efficiency.4.1 H+ diffusion
As a fundamental technique, voltammetry, including linear sweep voltammetry (LSV) and cyclic voltammetry (CV), is a common characterization for most electrocatalytic reactions to investigate the catalyst activity, overpotential, the redox transformation process of the catalyst phase, and electrochemically active surface area (ECSA).55,77 Specially, in this section, LSV, and CV tests will be discussed for assessing the H+ diffusion and analysing the competition between HER and CO2RR in acidic media.By conducting LSV measurements using a rotating disk electrode (RDE) in different acidic electrolytes or with varying catalysts, typical LSV curves exhibiting an obvious plateau region, followed by a current density increasing region, can be observed.37,38 The plateau region represents the H+ reduction, while the subsequent slope-increasing part corresponds to H2O reduction.38 Through RDE-LSV experiments, the diffusion situation of H+ during acidic CO2RR can be revealed. Also, the lowest limiting diffusion current density of H+ reduction (jlimit) can be calculated using the Levich equation, as follows:38
| jlimit = −0.62nFADH+2/3ω1/2ν−1/6cH+ | (4.1) |
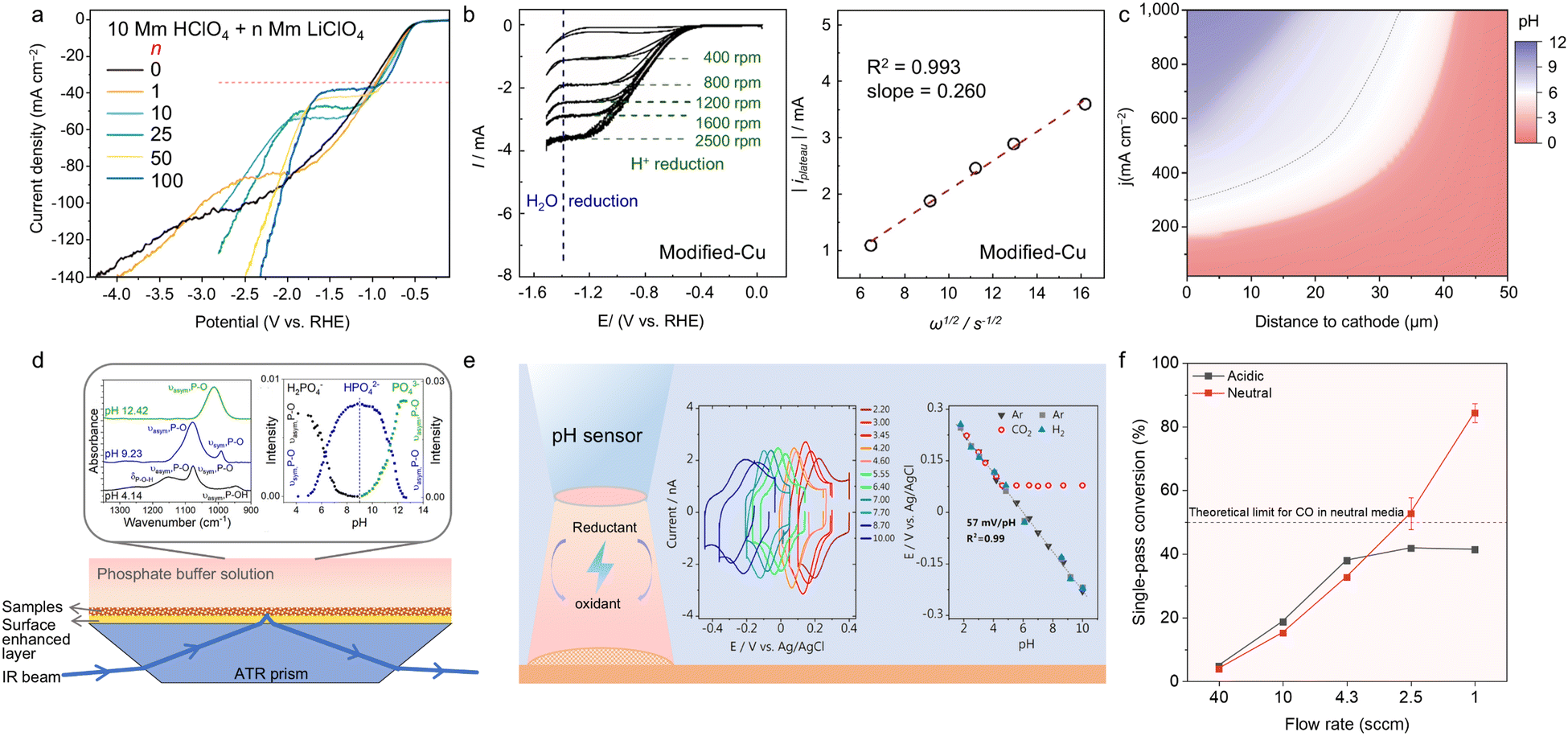 | ||
| Fig. 3 (a) LSV curves of Au RDE in Ar-saturated acidic electrolytes with different concentrations of Li+. The red dashed line represents the limiting diffusion current density of H+ reduction using the Levich equation.38 Reproduced with permission from ref. 38. Copyright 2023, the American Chemical Society. (b) CV curves of modified-Cu RDE in N2-saturated 0.1 M KClO4/HClO4 solutions at different rotation rates and linear fitting of iplateauvs. ω1/2 for modified-Cu RDE based on Levich equation.63 Reproduced with permission from ref. 63. Copyright 2023, Wiley-VCH. (c) pH modelling results at different current densities and cathode distances in 1 M H3PO4 and 3 M KCl.24 Reproduced with permission from ref. 24. Copyright 2021, the American Association for the Advancement of Science. (d) Schematic of working mechanism of ATR-SEIRA, SEIRA spectra of phosphate standard solutions, and the corresponding calibration curves.79 (e) Schematic of working mechanism of SECM, voltammetry curves of pH sensor in different pH-based Li2SO4 standard solutions, and corresponding calibration curves.80 (f) SPCE comparison of Ag/PTFE under acidic and neutral conditions.81 Reproduced with permission from ref. 81 Copyright 2022, the American Chemical Society. | ||
As an extended application, the transformed form of the Levich equation can be utilized to compare the DH+ values based on different catalysts in the same electrolyte formula and highlight the positive influence of catalyst engineering for acidic CO2RR on suppressing the competing HER. Specifically, the CV curves of different electrocatalysts in a certain electrolyte can be obtained by RDE experiments at different rotation rates (Fig. 3b). Further, the ratio of the H+ diffusion coefficients (DH+) of the experimental group to the control group can be calculated using the following equation:63
 | (4.2) |
Furthermore, it is worth noting that the voltage loss due to the electrolyte between the working and reference electrode should be considered and corrected using iR compensation in the analysis of the CV and LSV results.82 Simultaneously, the scan rate also significantly impacts CV and LSV measurements, and thus maintaining a consistent scan rate is crucial for effectively controlling the variables in comparisons.83
4.2 Interfacial pH
To date, the importance of interfacial properties is increasingly emphasized in CO2RR, and thus methods for the characterization of interfaces have continuously diversified through the dedicated efforts by researchers. As mentioned before, interfacial alkalinity is a key parameter to predict the potential CO2RR performance in acidic media, which can affect the reaction selectivity, efficiency and product distributions. Monitoring the local pH value allows researchers to deeply understand the reaction environment at the catalyst–electrolyte interfaces. However, unlike the bulk pH level, directly measuring the interfacial pH value is challenging, making it necessary to rely on other methods for assistance. To date, various techniques have been developed to monitor the interfacial pH indirectly, including theoretical calculation modelling, in situ spectroscopy (FTIR and Raman), fluorescent probes and electrochemical measurements.As a crucial breakthrough of acidic CO2RR, Huang et al. pioneered the investigation of the interfacial pH levels on the catalyst interfaces, examining how the pH level changes with variations in the operating current density and distances to the cathode through theoretical modelling.24 Specifically, they employed the COMSOL Multiphysics software to account for all the species interactions within the phosphate buffer electrolytes (CO2, HCO3−, CO32−, H3PO4, H2PO4−, HPO42−, PO43−, OH−, H+ and H2O). Also, the model incorporated all homogeneous and heterogeneous reactions throughout the system. The concentrations and pH levels of the bulk electrolytes were identified by experiments and applied to the model. Additionally, the thickness of the diffusion layer, porosity and catalyst length were set as fixed parameters. After setting the boundary conditions of the model, the local pH values were calculated at different current densities, revealing the pH variation trend on the catalyst–electrolyte interface. The results indicated that in the bulk electrolyte with a pH of 1, the interfacial pH (distance to cathode was 0 μm) is almost similar to that in the bulk solution, and the interfacial pH level increases to neutrality and even alkalinity as the current density increases (Fig. 3c). This effect originates from the local rate of the H+ consumption exceeding the H+ mass transfer rate from the bulk. However, the pH level can reduce to acid within the cathodic distance of 50 μm, avoiding CO2 loss and carbon crossover to the anolyte.
Further, in situ spectroscopy (ATR-FTIR and Raman) has also been applied to investigate the surface pH changes during the acidic CO2RR process.79 Specifically, to enhance the signal intensity and sensitivity, surface-enhanced techniques for both ATR-FTIR and Raman spectroscopy, named attenuated total reflection surface-enhanced infrared adsorption spectroscopy (ATR-SEIRAS) and surface-enhanced Raman spectroscopy (SERS), are implemented by coating a polycrystalline Ag/Au underlayer on the cathode substrate (Fig. 3d).10,79,84 During the pH detection process, a phosphate buffer solution serves as an indicator of the pH variation. Firstly, it is necessary to record the spectra of the phosphate species (H2PO4−, HPO42−, and PO43−) within different pH-value phosphate standard solutions, creating a calibration spectrum. After peak deconvolution, the peak intensities attributed to H2PO4−, HPO42−, and PO43− can be identified and plotted as a function of pH value to generate a calibration curve (Fig. 3d, inset). In the case of experimental samples, it is necessary to detect the ATR-SEIRAS or SERS spectra and calculate the intensity of the phosphate-related peaks. Subsequently, the interfacial pH is estimated by matching the peak intensities to the calibration curve. It should be noted that in neutral and alkaline systems, carbonate species can also act as a local pH indicator; however, carbonates are unstable in acid media, and thus phosphate species are more suitable for monitoring the interfacial pH in the acidic CO2RR.
Fluorescence confocal laser scanning microscopy (CLSM) measurement has been extended to monitor the interfacial pH during acidic CO2RR.79 After selecting an appropriate fluorescent probe that is sensitive to the required pH range, the probe is dissolved in standard solutions with varying pH values. Subsequently, the probe is excited by lasers at two different wavelengths. By collecting the fluorescence emission from both excitations, the resulting emission ratios serve as pH indicators, which can be plotted against pH value to create a calibration curve. Similarly, to characterize the interfacial pH in acidic CO2RR, fluorescent probes are deposited on the CO2RR cathode, and adsorption spectra are acquired during the acidic CO2RR process. Finally, the emission ratio is calculated and compared with the calibration curve to determine the interfacial pH level.
Also, electrochemical signals can also be applied as a response to interfacial pH level. Under mass transport control, the local pH level changes during CO2RR can be measured using the rotating ring-disk electrode (RRDE) technique, with an Au disk and Pt ring electrode.85 In this method, the CO2RR product, CO molecule, serves as the probe, which can be generated at the Au disk during the CO2RR process, and then oxidized into CO2 on the Pt ring electrode by CO oxidation. The oxidation of CO at the Pt ring electrode causes a pH-sensitive peak shift, which can be used to track the local pH changes. To measure the local pH values, the CV curves are first recorded in standard solutions with known pH values. A calibration curve can be created based on the correlation between the known solution pH and CO oxidation peak potential. By matching the measured CO oxidation peak potential in experimental acidic CO2RR systems, the interfacial pH can be estimated. Scanning electrochemical microscopy (SECM) based on electrochemical mechanisms can drive the microelectrode to scan the sample interface and detect current variations induced by the oxidation and reduction of substances within the micro-zone. With advancements in SCEM technology, it has been utilized as a high-resolution tool for local pH measurements in CO2RR, using specialized pH-sensitive probes (Fig. 3e).80,86 Typically, the probes consist of redox species with distinct oxidation and reduction peaks observable in CV tests, where the mid-peak potentials shift in response to the pH changes. Thus, a calibration curve can be constructed by correlating the pH values and their corresponding mid-peak potentials, and the interfacial pH of the experimental systems can be determined.
In summary, advances in characterization techniques have driven a deeper understanding of electrocatalytic reactions. As the importance of creating surface alkaline microenvironments continues to be emphasized in acidic CO2RR, more interfacial pH measurement methods have been developed, certainly driving further progress in catalyst–electrolyte engineering.
4.3 CO2 single-pass conversion efficiency
When evaluating the efficiency of CO2RR, three key dimensions are involved, i.e., electron transfer, electricity consumption, and carbon conversion, which correspond to the faradaic efficiency (FE), energy efficiency (EE), and CO2 single-pass conversion efficiency (SPCE), respectively. As the most common metrics of product selectivity and electricity cost for CO2RR, FE and EE are used to evaluate CO2RR across all pH levels, respectively. SPCE, defined as the ratio of the carbon content in the CO2RR products to the total input CO2, serves as a measure of the carbon utilization efficiency.25 Due to the generation of carbonate and the subsequent carbon crossover phenomenon, the input CO2 gas in neutral and alkaline media is mostly transferred to the anode side, and theoretically the SPCE is typically lower in alkaline and neutral CO2RR, which is less than 50% for C1 products and below 25% for C2+ products.41,81 Thus, in most reported works on alkaline and neutral CO2RR, SPCE is rarely discussed. In contrast, carbon crossover is significantly reduced in acidic media, and the SPCE values are more widely discussed in acidic CO2RR studies, with values up to 90% achieved (Fig. 3f).81The SPCE can be calculated using the following equation:
 | (4.3) |
5. Catalyst–electrolyte interface engineering for promoting acidic CO2 electroreduction
Recently, an increasing number of strategies have been proposed to promote CO2RR-related intermediate adsorption and enhance the performance of acidic CO2RR. In general, these strategies revolve around catalyst–electrolyte engineering and can be categorized into three main approaches, catalyst modification, electrolyte engineering and interface optimization (Fig. 4). In this chapter, we summarize these modification strategies and highlight the recent advancements in this field.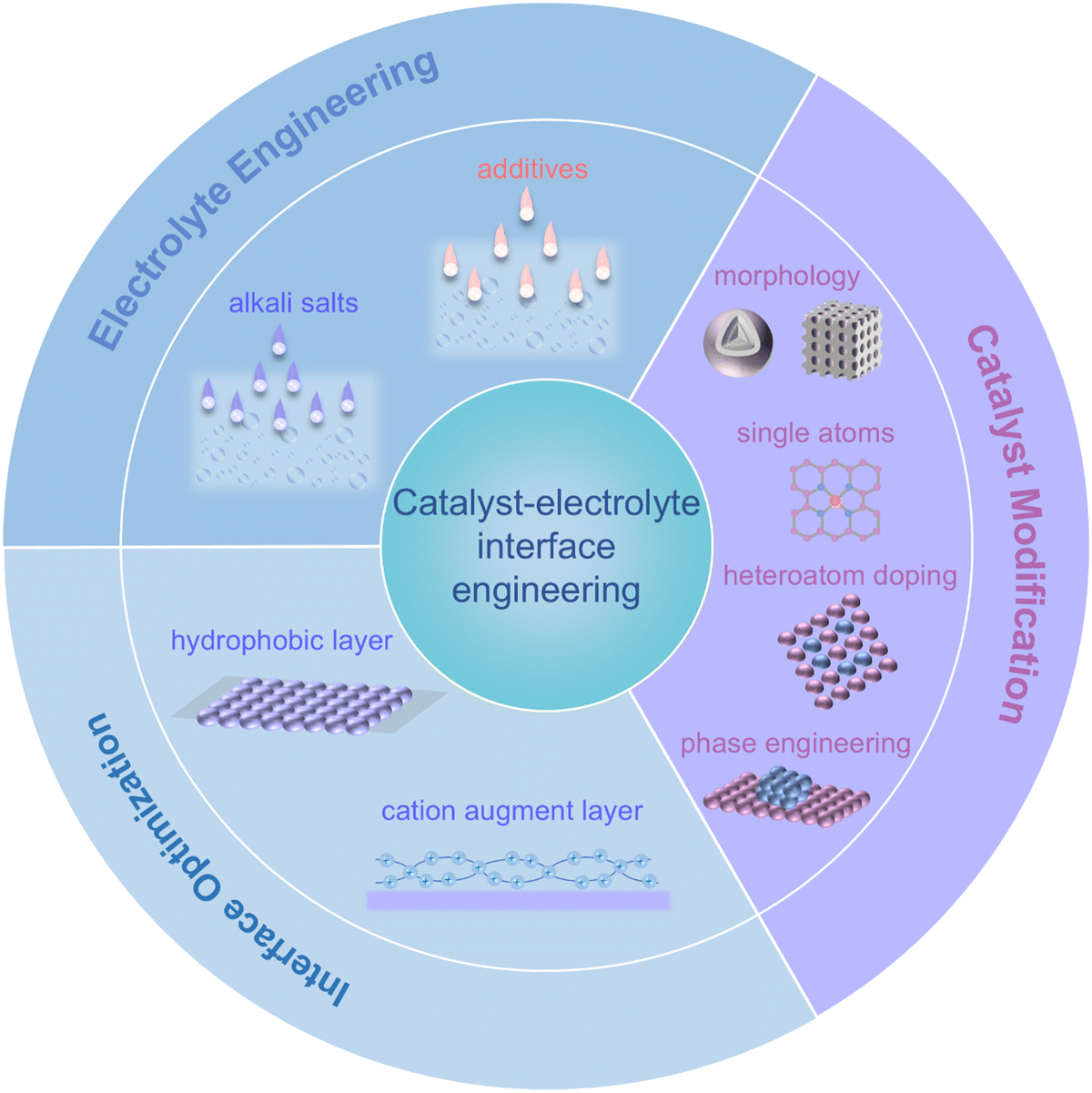 | ||
| Fig. 4 Schematic of catalyst–electrolyte interface engineering strategies for promoting acidic CO2RRs. | ||
5.1 Electrolyte engineering
In the CO2 electroreduction reaction, the electrolyte directly interacts with the electrocatalyst, significantly affecting the overall reaction environment, the distribution of active sites and the reaction efficiency. Thus, a thorough investigation of the electrolyte properties, including its pH level, composition and concentration, is meaningful for understanding and optimizing the reaction process. Building on the comprehensive understanding of the influence of electrolyte pH level, composition and cation concentration on acidic CO2RR processes, some studies have been dedicated to integrating additives into acidic electrolytes, reconstructing the electrolyte structure and driving a CO2RR-promoted interface in acid media. The aforementioned methods are all involved in electrolyte engineering, which involves deliberate modification of the electrolyte properties to effectively optimize the reaction microenvironment, promote the adsorption of CO2RR-related intermediates, and ultimately enhance the CO2RR performance in acidic media. Different from pure electrolyte engineering, electrolyte engineering at the interface specifically influences the cathodic region within the electrical double layer during the acidic CO2RR process, impacting the ion transport, local pH and intermediate adsorption.87 In this section, electrolyte engineering at the interface will be discussed according to the cation effect, hydrogen bond (H-bond) reconstruction and maintaining the high valence of catalysts. The reported acidic CO2RR performance driven by electrolyte engineering is summarized in Table 1.| Category | Catalyst | Electrolyte | Main products | FE | SPCE | Stability (h) | Ref. |
|---|---|---|---|---|---|---|---|
| Cation effect | Cu catalyst | 1 M H3PO4 + 3 M KCl (pH < 1) | CH4 | 28% (400 mA cm−2) | — | — | 24 |
| Bi nanosheet | 0.05 M H2SO4 + 3 M KCl (pH < 1) | HCOOH | 92.2% (257 mA cm−2) | 27.4% (3 sccm) | 8 | 59 | |
| Ni–N–C catalyst | 0.5 M K2SO4 + H2SO4 (pH = 0.5) | CO | 95% (500 mA cm−2) | — | 8 | 76 | |
| Ag/PTFE | 0.01 M H2SO4 + 0.01 M Cs2SO4 (pH ≤ 3) | CO | 80% (60 mA cm−2) | 90% (1 sccm) | 50 | 81 | |
| 10 cm2 Au GDE | 1 M Cs2SO4 (pH = 2–4) | CO | 80–90% (200 mA cm−2) | — | — | 88 | |
| Cu–PTFE | Na2SO4 + H2SO4 (pH = 2) | CH4 | 48% (220 mA cm−2) | — | — | 89 | |
| Cu–PTFE | Na2SO4 + K2SO4 + H2SO4 (pH = 2; Na+/K+ ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
C2+ | 46% (220 mA cm−2) | — | — | 89 | |
| Cu–PTFE | K2SO4 + H2SO4 (pH = 2) | C2+ | 70% (220 mA cm−2) | — | — | 89 | |
| Reconstruction of H-bonds | Ag catalyst | 0.1 M H2SO4 + 0.4 M K2SO4 + 0.1 M SPS (pH ≈ 1.42) | CO | 97.8% (250 mA cm−2) | 66.3% (2 sccm) | 20 | 90 |
| Ag powder | 0.1 M H2SO4 + 0.2 M K2SO4 + 2 wt% PSS (pH ≈ 1.42) | CO | 93.9% (250 mA cm−2) | 72.2% (2 sccm) | 12 | 91 | |
| Maintaining the high valence of catalysts | Cu particles | 0.3 M KI + 0.05 M H2SO4 + 20 mM I2 (pH = 1.2) | C2+ | 70% (600 mA cm−2) | 54% (2.5 sccm) | 8 | 75 |
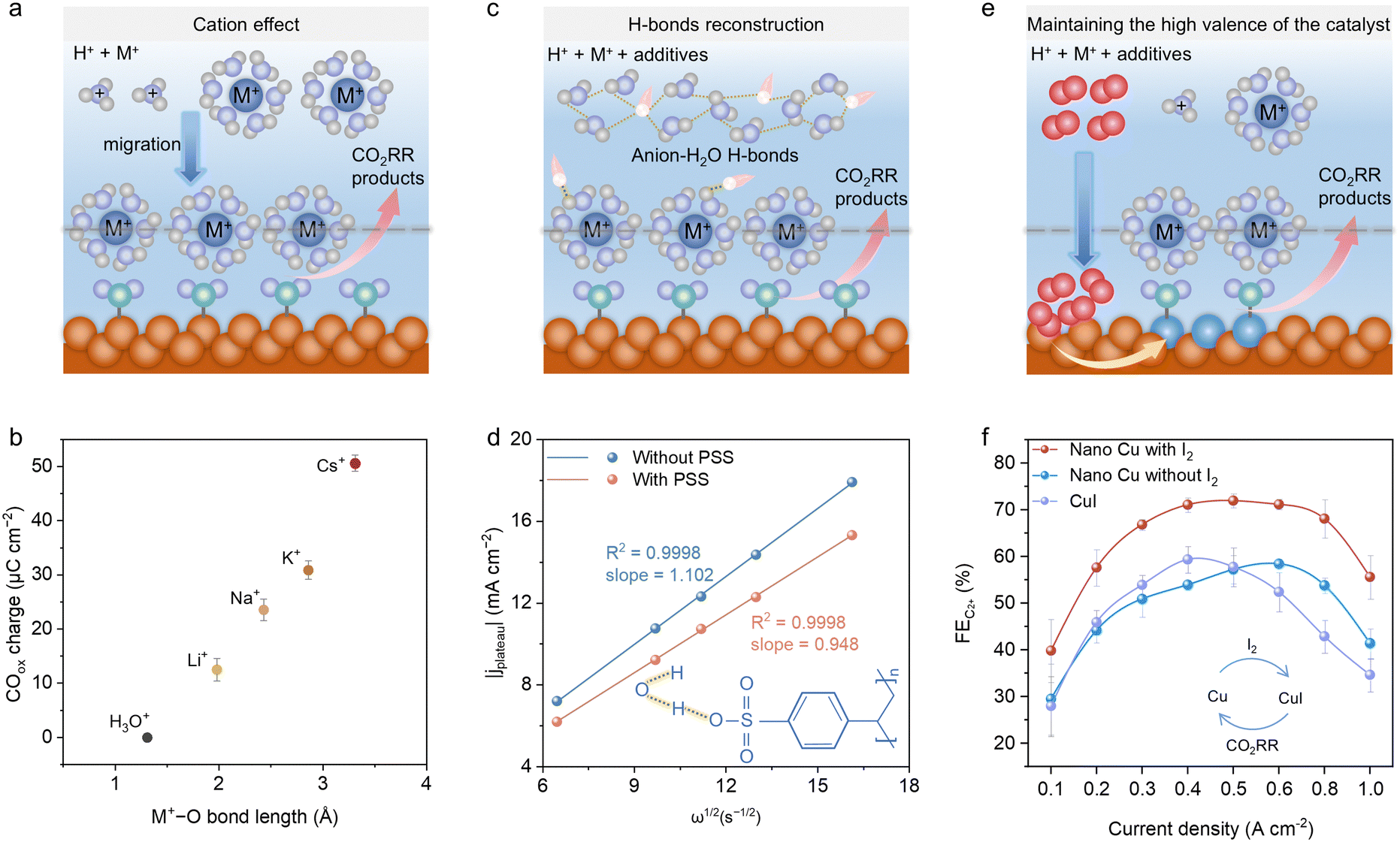 | ||
| Fig. 5 (a) Illustration of the cation effect on acidic CO2RRs with alkali metal salts. (b) Correlation between cation–water radial distribution (M+–O bond length) and acidic CO2RR production.36 Reproduced with permission from ref. 36. Copyright 2021, Springer Nature. (c) Illustration of H-bond reconstruction in acidic CO2RR electrolytes by additives. (d) Comparison of H+ diffusion coefficients of electrolytes with/without PSS, which can break the H-bond network of water molecules.91 Reproduced with permission from ref. 91. Copyright 2024, Wiley-VCH. (e) Illustration of the high-valent catalyst maintaining effect by special oxidant additives. (f) Comparison of FEC2+ of acidic CO2RR reaction systems among nano-Cu with I2, nano-Cu without I2, and CuI without I2, showing the oxidation effect of I2 on the acidic CO2RR performance.75 Reproduced with permission from ref. 75. Copyright 2024, the American Chemical Society. | ||
Moreover, it has been recognized that under alkaline conditions, the electrolyte composition, namely alkali cation (Li+, Na+, K+ and Cs+), influences the CO2RR performances, following the order of Cs+ > K+ > Na+ > Li+.92,93 Therefore, it is worthwhile to analyse and compare the trend in the variation of CO2RR product selectivity in different alkali-cation electrolytes under acidic condition as well. Gu et al. investigated acidic CO2RR using electrolytes composed of 0.1 M HOTf + 0.4 M MOTf (M = Li, Na, K, and Cs) with both SnO2/C and Cu/C catalysts, exhibiting that all the alkali cations enhanced CO2RR by suppressing HER, with the selectivity following the order of Li+ < Na+ < K+ < Cs+ on SnO2/C and Li+ < Na+ < K+ ≈ Cs+ on Cu/C.37 Also, the mechanistic study revealed that hydrated alkali cations (M+–H2O) and H3O+ competed for adsorption at the outer Helmholtz plane (OHP) at the cathode side. Promptly, a chemically inert layer of alkali cations formed at the OHP, shielding the cathodic electric field over an extended potential range, which restricted the movement of H3O+ ions. Without alkali cations, the migration of H3O+ under the influence of an electric field accelerates their replenishment at the outer Helmholtz plane, thereby initiating HER. Besides, Pan et al. utilized an acid-fed MEA with Ag catalysts and observed an increase in CO FE from 8% to 77% at 60 mA cm−2, following the cation sequence of H+ < Li+ < Na+ ≈ K+ ≤ Cs+ in a mild acid electrolyte.81 A CO FE of 80% and SPCE of 90% could be achieved at 60 mA cm−2 in 0.01 M H2SO4 + 0.01 M Cs2SO4 electrolyte with a stability of 50 h. The feasibility of the cation promotion effect was further evaluated using practical electrode geometries with a 10 cm2 gold electrode. The results suggest that the FE for CO can reach 80% at current densities ranging from 50 to 200 mA cm−2 in 1 M CsSO4 electrolyte at pH values of 2, 3, and 4. In contrast, when using 1 M Li2SO4, almost no CO product was produced at all the applied current densities.88 As an interesting attempt, Xu et al. modified an acidic electrolyte by adjusting the concentration ratio of K+ and Na+, discovering that this ratio can influence the product distribution on the Cu-based catalyst.89 Specifically, in an electrolyte containing only Na+, CH4 was the dominant product, achieving an FE of 48%, together with an FE of 39% for H2. With the gradual addition of K+, the FE for CH4 and H2 progressively decreased, while the selectivity for C2+ products increased. Ultimately, when the electrolyte was adjusted to only contain K+ cations, the FE for the C2+ products reached 70%. These findings indicate that the type of alkali metal cation in the acidic electrolyte can directly influence the CO2RR product distribution, warranting a deeper investigation into the underlying mechanisms. Monteiro et al. applied CV measurements of CO2 reduction and CO oxidation on Au catalysts in different cation composition electrolytes to quantify the CO production during acidic CO2RR.36 The results suggested that the CO production was correlated with the size of the solvated cations following the order of Li+ < Na+ < K+ < Cs+ (Fig. 5b). Based on the experimental results, ab initio molecular dynamics (AIMD) simulations were carried out using an alkali metal cation-inserted Au model (Au–H2O–M+). The calculation result indicated that a larger cation ionic radius corresponded to a softer solvation shell and the average cation coordination number was determined to be 2.8, 3.2, 3.5 and 5.8 for Li+, Na+, K+ and Cs+, respectively. Additionally, Li+ exhibited a limited CO2 coordination number due to its hard solvation shell; meanwhile, other alkali metal cations can continuously bind to the oxygen atoms in CO2 and the coordination number followed the order of Li+ < Na+ < K+ < Cs+. Also, it was concluded that the CO2RR activity trend depends on the cation surface concentrations at the OHP and the differing ability of cations to coordinate with the *CO2− intermediate, ranging from minimal bonding for Li+ to double bonds for Cs+. This cation effect topic was further expanded to multivalent cations (Li+, Cs+, Be2+, Mg2+, Ca2+, Ba2+, Al3+, Nd3+, and Ce3+) in acidic electrolyte at pH 3.94 It was found that cations with soft hydration shells (Cs+, Ba2+, and Nd3+) exhibited minimal cation–cation repulsion, allowing them to effectively accumulate at OHP and coordinate with *CO2−. However, trivalent cations can enhance both CO2RR and water dissociation. Thus, Nd3+ can only activate CO2RR at the potential range below H2O reduction, while Cs+ and Ba2+ can promote CO2RR at higher overpotentials. Furthermore, the Levich equation was applied to investigate the cation effect on suppressing the mass transport of H+ reduction during acidic CO2RR.38 Following LSV tests of Au RDE in HClO4 acidic electrolyte with different LiClO4 concentrations, the current density for H+ reduction in the plateau region decreased with an increase in the cation concentration, and gradually approached the limiting diffusion current density for H+ reduction. To more intuitively observe the impact of different concentrations and types of cations in the electrolyte on the acidic CO2RR process, ATR-SEIRAS was applied to reveal the differences in the intermediate adsorption.95 Firstly, the in situ ATR-SEIRAS tests were conducted on a Cu film in both 0.05 M H2SO4 and 0.05 M H2SO4/1 M Na2SO4. The results showed that adsorption peaks appeared at 2056, 1466 and 1416 cm−1 during different potential reduction processes, which can be assigned to atop-bound CO, asymmetry stretching of adsorbed CO2 and bidentate COO−, respectively. Subsequently, the cation effect was quantified by comparing the peak intensity of adsorbed CO2 at 1466 cm−1 in acidic electrolytes containing differing cations. Combining AIMD simulations and the spectroscopic features of water, the findings indicated that the CO2 adsorption in the Li-based electrolyte was more efficient than that with other cations. Nevertheless, the hard hydrated shell of Li+ impeded hydrogen atoms from approaching the oxygen atoms in the adsorbed CO2, decelerating the further hydrogenation of CO2. On the contrary, larger cations possessed much softer hydration shells, facilitating the interaction of hydrogen atoms and CO2 and leading to more effective CO2RR.95
Briefly, the internal mechanism of the cation promotion effect may originate from three viewpoints, as follows: (1) adjustment of the electronic field near the OHP; (2) activating and stabilizing the CO2RR intermediate by coordination and (3) regulating the local pH level. Besides the cation effect, it is worth exploring the anion impact on acidic CO2RR processes. Research shows that most anions have an insignificant influence on the CO2RR, referring to the negligible production distribution differences by substituting SO42− with I−, Cl− and Br−.24,59 However, if special anion chemisorption occurs on the catalyst surface, the electronic structure of catalysts can be optimized, favouring the CO2RR kinetics.96,97 Also, anions with buffering ability can influence the local pH on the cathode surface, modifying the product distribution.98
5.2 Catalyst modification
Following the resolution and investigation of electrolyte issues, designing electrocatalysts to further promote the performance of acidic CO2RR has become a prominent focus. Research has revealed that an ideal electrocatalyst for CO2RR should possess moderate adsorption capability for the CO2RR intermediates, while maintaining a lower *H adsorption ability to favour CO2RR over HER.58 Thus, rationally modulating the electronic and geometric structures of catalysts is conducive to the mass and electronic transport during acidic CO2RR. Similar to alkaline systems, catalyst modification strategies also involve morphology optimization, atomic-scale regulation, and material phase engineering in acidic CO2RR. A summary of the detailed acidic CO2RR performances driven by catalyst modification is presented in Table 2, and the corresponding elaboration is discussed below.| Category | Catalyst | Electrolyte | Main products | FE | SPCE | Stability | Ref. |
|---|---|---|---|---|---|---|---|
| Morphology optimization | Electro-reduced porous Cu nanosheets | 0.05 M H2SO4 + 3 M KCl (pH < 1) | C2+ | 83% (670 mA cm−2) | 54.4% (2 sccm) | 30 h | 23 |
| Porous carbon-coated Fe nanoparticles | 0.2 M H2SO4 + 0.1 M KCl (pH = 3.4) | CO | 90% (−0.6 VRHE) | — | — | 100 | |
| Vertically grown Bi nanosheets | 0.05 M H2SO4 + 0.5 M K2SO4 (pH = 2) | HCOOH | 96.3% (489 mA cm−2) | 79% (1 sccm) | 45 h | 101 | |
| Hollow-structured Ag@C | 0.5 M K2SO4 (pH = 1.1) | CO | 95% (300 mA cm−2) | 46.2% (2 sccm) | 9 h | 102 | |
| N-doped carbon nanocages | 0.25 M Na2SO4 + 1 mM H2SO4 (pH = 2.5) | CO | 84.3% (−0.6 VAgCl/Ag) | — | 15 h | 103 | |
| High-curvature Cu nanoneedle catalyst | 3 M KCl + HCl (pH = 1) | C2+ | 90.69% (1400 mA cm−2) | 25.49% (7 sccm) | 8 h | 61 | |
| Cu hollow fibre penetration electrode | H2SO4 + 3 M KCl (pH = 0.71) | C2+ | 73.4% (3000 mA cm−2) | 51.8% (2 sccm) | 100 h | 104 | |
| Ag penetration electrodes | 0.05 M H2SO4 + 3 M KCl (pH = 1) | CO | 95% (4300 mA cm−2) | 85% (5 sccm) | 200 h | 105 | |
| 3D porous electrode with Bi/C nanoparticles | 1 M Na2SO4 + H2SO4 (pH = 2.7) | HCOOH | 89.2% (−0.6 Vfullcell) |
— | 24 h | 106 | |
| Atomic-scale regulation | Ni–N–C catalyst | H2SO4 + 0.5 M K2SO4 (pH = 0.5) | CO | 95% (500 mA cm−2) | — | 8 h | 76 |
| Ni SAs | 0.5 M H3PO4 + 0.5 M KH2PO4 + 1.5 M KCl (pH = 2) | CO | 99.9% (300 mA cm−2) | 45.9% (2 sccm) | 12 h | 107 | |
| NU-1000-Sn with ditin sites | 0.005 M H2SO4 + 3 M KCl (pH = 1.67) | HCOOH | 100% (260 mA cm−2) | 95% | 15 h | 55 | |
| Cu-based cluster compound (Inz-Cu3) | H2SO4 + 0.5 M K2SO4 (pH = 2) | C2+ | 42.2% (320 mA cm−2) | — | — | 108 | |
| Pd–Cu catalyst | H2SO4 + 0.5 M K2SO4 (pH = 2) | C2+ | 89% (500 mA cm−2) | 68% (2 sccm) | 4.5 h | 42 | |
| La-doped Cu hollow sphere | 0.05 M H2SO4 + 3 M KCl (pH = 1) | C2+ | 86.2% (900 mA cm−2) | 74.5% (3 sccm) | 40 h | 109 | |
| Phase engineering | Cu6Sn5 catalysts | 0.05 M H2SO4 + 3 M KCl (pH = 1) | HCOOH | 91% (1200 mA cm−2) | 77.4% (3 sccm) | 130 h | 110 |
| High-roughness Ag–Cu alloy catalyst | 0.05 M H2SO4 + 3 M KCl (pH = 1) | C2+ oxygenates | 56.5% (−1.9 VRHE) | 49.3% (1.5 sccm, C2+ oxygenates) | 8 h | 111 | |
| Zn-incorporated Cu alloy catalysts | H2SO4 + 3 M KCl (pH = 4) | C2+ | 70% (400 mA cm−2) | 31% (12 sccm, C2+) | 150 h | 112 | |
| n-Butylamine modified Cu | 0.005 M H2SO4 + 0.5 M K2SO4 (pH = 2) | C2+ | 81.8% (410 mA cm−2) | 60% (1 sccm, C2+) | 80 h | 113 | |
| Ni SAs on Cu tandem catalyst (Cu PTFE/Ni–N4) | 0.05 M H2SO4 + 0.5 M K2SO4 (pH = 2) | C2+ | 82.4% (400 mA cm−2) | — | 24 h | 114 | |
| π-SnS | H2SO4 + 0.5 M K2SO4 (pH = 3) | HCOOH | 92.15% (200 mA cm−2) | 36.43% (5 sccm) | 14 h | 62 | |
| Specialized types of catalysts | Cobalt protoporphyrin immobilized on a pyrolytic graphite electrode | 0.1 M perchlorate solution saturated with CO2 (pH = 3) | CO | 60% (−0.6 VRHE) | — | — | 66 |
| CoPc@HC/Cu–PFSA | 0.5 M H3PO4 + 0.5 M KH2PO4 + 2.5 M KCl | C2+ | 82% (800 mA cm−2) | 90% (2 sccm) | 16 h | 115 | |
| Nickel phthalocyanine electrocatalysts on carbon nanotubes | H2SO4 + 0.5 M K2SO4 (pH = 2) | CO | 98% (400 mA cm−2) | — | 12 h | 116 | |
| EDTA/CuPc/CNP | 0.005 M H2SO4 (pH = 2) | CH4 | 71% (100 mA cm−2) | 78% (0.1 sccm) | 300 min | 117 | |
| CoTAAPc@CNT | 0.05 M H2SO4 + 3 M KCl (pH = 1) | CO | 93.3% (609.7 mA cm−2) | 84% (1 sccm) | 8 h | 118 | |
| CoPc@CNT catalyst | H2SO4 (pH = 1) | CO | 60% (30 mA cm−2) | — | 13 h | 60 | |
| Imidazole-linked COF (PcNi-im) | 0.01 M H2SO4 + 3 M KCl (pH = 1) | CO | 100% (320 mA cm−2) | — | 10 h | 119 | |
| CoPc-CTF | 0.5 M H3PO4 + 0.5 M KH2PO4 + 1.5 M KCl (pH = 2) | CO | 94.3% (−1.5 VRHE) | — | 10 h | 120 | |
| NiPc–NiTAA | 0.5 M K2SO4 + H2SO4 (pH = 2) | CO | 95.1% (−1.5 VRHE) | — | 6 h | 121 | |
| Cs3Bi2Br9/C | 3 mM HBr + 0.5 M CsBr (pH = 2.5) | HCOOH | >80% (−0.75 to −1.25 VRHE) | 47.2% (2 sccm) | 20 h | 122 | |
 | ||
| Fig. 6 (a) Illustration of morphology optimization for trapping K+ and OH−. (b) and (c) Needle structure of CuNNs shown using TEM and comparison of surface-adsorbed K+ concentration between CuNNs and the Cu film.61 Reproduced with permission from ref. 61. Copyright 2023, Wiley-VCH. (d) Illustration of atomic-scale regulation on catalysts for promoting acidic CO2RRs. (e) and (f) EXAFS fitting curve, confirming the existence of the ditin sites and corresponding performance.55 Reproduced with permission from ref. 55. Copyright 2023, the American Chemical Society. (g) Illustration of phase engineering on catalysts to synergistically promote acidic CO2RRs. (h) and (i) SEM image of Cu PTFE/Ni–N4 composite and in situ Raman results of Cu PTFE/Ni–N4 in an acidic CO2RR.114 (j) Illustration of precise modulation of specialized catalysts to promote CO2RR intermediate adsorption. (k) and (l) In situ extended X-ray absorption fine structure spectra of EDTA/CuPc/CNP before and during the CO2RR, showing the multidentate chelation constraining effect for preventing Cu agglomeration and the existence of Cu–N/O single sites, and corresponding performance.117 | ||
Thus, this bridging ditin structure displayed exceptional activity, delivering almost 100% FE for formic acid at 260 mA cm−2 in an acidic electrolyte (pH = 1.67) with a high SPCE of 95% (Fig. 6e and f). By further increasing the number of atoms, clusters were formed. Wang et al. designed a stable, cyclically symmetrical Cu-based cluster compound (Inz-Cu3) by linking the indazole ligand with Cu salts. The asymmetric Cu active sites in Inz-Cu3 helped stabilize the C2H5OH-related intermediate *CHOHCH3, leading to a C2+ selectivity of 42.2% in an acidic electrolyte.108 Furthermore, heteroatom doping can trigger the redistribution of charge and adjustment of intermediate adsorption energy. To balance the *H and *CO coverage on the cathode surface, Xie et al. explored the incorporation of various metals with different *CO affinity into Cu catalysts to reduce the *H binding.42 After screening based on DFT calculations of the Gibbs free energies for the formation of CHO* (C1 pathway) and OCCOH* (C2 pathway) on the 111 facets, Pd–Cu emerged as the most promising candidate for C2+ production in acidic CO2RR. Using the optimally designed Pd–Cu catalyst, a crossover-free system was realized, yielding a C2+ FE of 89% at 500 mA cm−2 and a C2+ SPCE of 60%. Combining the microenvironment regulation through morphology optimization with intrinsic activity enhancement via heteroatom doping, an La-doped Cu channel-filled hollow sphere was synthesized using the solvothermal method, followed by electrochemical reduction.109 The Fourier-transformed extended X-ray absorption fine structure spectrum (FT-EXAFS) results revealed that La on the Cu surface existed as La–Ox. The resulting La–O–Cu sites facilitated the adsorption of *CO and the further C–C coupling process, and simultaneously the channel-filled structure drove the accumulation of K+ at the cathode, constructing an alkaline local environment, which suppressed HER. Consequently, a C2+ product FE of 86.2% and a C2+ SPCE of 52.8% were achieved at the current density of 900 mA cm−2.
5.3 Interface optimization
Interface optimization focuses on directly tailoring the interaction zone between the catalyst surface and the electrolyte. It can effectively restrict the transport of H+ from the bulk electrolyte to the cathode, which in turn promotes the accumulation of alkali cations at the catalyst–electrolyte interface, thereby enhancing the acidic CO2RR process. The published studies employed three primary approaches to shift the acidic reaction interfaces in favour of CO2RR, enhancing the hydrophobicity/*OH adsorption, increasing the interfacial cations, and immobilizing the linked cation groups for low-cation or cation-free acidic CO2RR. The reported works on acidic CO2RR driven by optimizing interfaces are concluded in Table 3 and elaborated in detail as follows.| Category | Catalyst | Electrolyte | Main products | FE | SPCE | Stability | Ref. |
|---|---|---|---|---|---|---|---|
| Enhancing hydrophobicity/*OH adsorption | Ni–N–C/PTFE | 1 M Cs2SO4 + H2SO4 (pH = 2) | CO | 100% (250 mA cm−2) | 75.7% (20 sccm) | 36 h | 127 |
| Cu/SiO2 | 0.05 M H2SO4 + 3 M KCl (pH = 1) | C2+ | 76.9% (900 mA cm−2) | 34.8% (5 sccm) | 12.5 h | 128 | |
| Cu/1-octadecanethiol GDE | 1 M KCl + 1 M HCl (pH = 1) | C2+ | 87% (1600 mA cm−2) | 42% (3.5 sccm) | 10 h | 44 | |
| Electrodeposited Cu catalyst | 0.05 M H2SO4 + 2.5 M KCl (pH = 1) | C2+ | 90% (250 mA cm−2) | 70% (0.5 sccm) | 10 h | 39 | |
| SiC-Nafion/SnBi/PTFE | 0.05 M H2SO4 + 3 M KCl (pH = 1) | HCOOH | 92% (100–800 mA cm−2) | 65% (3 sccm) | 125 h | 20 | |
| SiC-Nafion/Cu/PTFE | 0.05 M H2SO4 + 3 M KCl (pH = 1) | C2+ | 65–70% (100–800 mA cm−2) | — | 35 h | 20 | |
| SiC-Nafion/Ag/PTFE | 0.05 M H2SO4 + 3 M KCl (pH = 1) | CO | 92% (100 mA cm−2) | — | 52 h | 20 | |
| Electrostatic carbon/Cu/PTFE | 0.5 M K2SO4 + H2SO4 (pH = 2) | C2H4 | 64.5% (300 mA cm−2) | — | — | 129 | |
| Increasing interfacial cations | Cu/PFSA | 1 M H3PO4 + 3 M KCl (pH < 1) | C2+ | 50% (1200 mA cm−2) | 77% (3 sccm) | 12 h | 24 |
| COF:PFSA-modified PTFE–Cu electrode | 1 M H3PO4 + 3 M KCl (pH < 1) | C2+ | 75% (200 mA cm−2) | 75% (0.5 sccm) | 30 h | 25 | |
| CoPc@HC/Cu–PFSA | 0.5 M H3PO4 + 0.5 M KH2PO4 + 2.5 M KCl | C2+ | 82% (800 mA cm−2) | 90% (2 sccm) | 16 h | 115 | |
| Surface iodine and PGDY co-modified Bi catalyst | 0.5 M K2SO4 (pH = 3.5) | HCOOH | 90% (110 mA cm−2) | — | 240 h | 130 | |
| Phenyldiazonium-modified Cu | 1 M H3PO4/1 M KCl (pH = 1) | C2+ | 65% (100 mA cm−2) | — | 6 h | 43 | |
| 18-Crown-6/Cu | 0.5 M K2SO4 + H2SO4 (pH = 2) | CH4 | 51.2% (600 mA cm−2) | 50.4% (2 sccm) | 16000 s |
131 | |
| Immobilizing cation group for low-cation or cation-free acidic CO2RR | N-Tolyl pyridinium modified-Cu | 1.0 M H3PO4 + 0.1 M KH2PO4 (pH = 1) | C2+ | 55% (50 mA cm−2) | — | 5 h | 63 |
| QAPPT and PTFE-modified Ag catalyst | 0.1 M H2SO4 + 0.1 M K2SO4 (pH ≈ 1) | CO | 95.6% (100 mA cm−2) | — | 400 min | 132 | |
| CuNP-EMIM | 0.1 M H2SO4 (pH = 0.7) | All CO2RR products | 11% (100 mA cm−2) | — | — | 133 | |
| Cationic-group-functionalized Cu | 0.2 M H2SO4 (pH = 0.4) | C2+ | 80% (100 mA cm−2) | 90% (0.2 sccm) | 150 h | 45 | |
| PDDA/GO-modified Ag catalyst | 0.01 M H2SO4 (pH ≈ 2) | CO | 85% (100 mA cm−2) | — | 50 h | 47 | |
| c-PDDA-modified Ag catalyst | 0.1 M H2SO4 (pH = 1) | CO | 95% (100 mA cm−2) | — | — | 48 | |
| c-PDDA-modified indium catalyst | 0.1 M H2SO4 (pH = 1) | HCOOH | 76% (200 mA cm−2) | — | 35 h | 48 | |
 | ||
| Fig. 7 (a) Illustration of the effect of enhancing hydrophobicity/*OH adsorption on an acidic CO2RR. (b) Product distribution of Cu/SiO2 at different current densities in an acidic CO2RR (inset: the contact angle image of Cu/SiO2, showing its hydrophobicity).128 Reproduced with permission from ref. 128. Copyright 2024, the American Chemical Society. (c) Illustration of promoted cation accumulation on the catalyst interface. (d) CH4 FE comparison of Cu catalysts with different ratios of 18-crown-6 (inset: the structure of 18-crown-6, showing its ability to coordinate with K+).131 Reproduced with permission from ref. 131. Copyright 2023, Wiley-VCH. (e) Illustration of the effect of immobilizing linked cation groups for a low-cation or cation-free acidic CO2RR. (f) Product distribution on bare Cu and CG-low-Cu in acidic electrolytes with/without K+, showing the influence of CG on acidic CO2RR performance.45 Reproduced with permission from ref. 45. Copyright 2023, Springer Nature. | ||
6. Conclusions and outlook
To date, CO2 electroreduction under alkaline and neutral conditions has rapidly developed; however, the associated problems have also become evident. For example, the formation of carbonates can cause severe CO2 crossover into the anolyte in alkaline and neutral media, significantly reducing the carbon utilization efficiency, and thus driving the development of CO2RR under acidic conditions. Fortunately, leveraging the advancements in alkaline and neutral CO2RR, related components such as electrodes, electrolysers, and membranes have been thoroughly researched and optimized. Therefore, it is a great opportunity to advance the development of acidic CO2RR. In recent years, studies on acidic CO2RR have mainly focused on the regulation of the catalyst–electrolyte interface, promoting CO2 activation and CO2RR-related intermediate adsorption in acidic media. The developing strategies can be classified into three aspects including electrolyte engineering for affecting the interfacial catalyst environment and electrolyte structures, modification of the electronic and geometric structures of catalysts to favour the adsorption of the CO2RR-related intermediates, and interface optimization for hindering proton diffusion and inducing cation augmentation. Additionally, there are also some problems and suggestions regarding this research field as follows.(1) Development of electrolysers and related accessories. Despite the continuous development of CO2RR electrolysers ranging from H-cells and flow cells to MEA, the operability of these devices is still limited, and experimental errors remain significant, which affects the scientific validity of the experiments. Also, the electrolyte flooding phenomenon still easily occurs and disrupts the entire operation of the reaction. Thus, it is still urgent to further develop next-generation CO2RR electrolysers and GDE to enhance the scientific validity and accuracy of experimental results.
(2) Low-cation/cation-free electrolytes for acidic CO2RR. Alkali cations play a vital role in the performance of acidic CO2RR, however, a high concentration of cations causes carbonate crystallization and blocks the gas transport channels. Given these challenges, only a limited number of studies have successfully achieved high CO2RR product selectivity in pure acid, primarily by modifying the interface through the immobilization of large cation groups. However, the operation current density is limited to below 300 mA cm−2 to date. To advance CO2RR in pure acid, it is essential to develop new strategies focused on catalyst structure design and microenvironment modification that enable operation at a higher current density.
(3) Seawater electrolysis. Recently, electrolysis in seawater has emerged as practical technology for conserving freshwater resources and decreasing the technique cost. Various electrocatalytic processes, such as HER, OER, and the oxygen reduction reaction, have made significant progress in acidic/alkaline seawater electrolysis.134 Reports indicate that chloride anions and alkali cations, which are abundant in seawater, positively influence CO2RR.24,97,135 Also, the salt-rich and complex ionic composition of seawater provides a unique electrolyte environment for acidic CO2RR, simplifying the electrolyte preparation process. Also, the acidic electrolyte can react with CO32+/OH−-based participation species to reduce the blockage of the active sites by sediment deposition. Therefore, seawater-based acidic CO2RR is anticipated to become an important research area in the future development of CO2RR research.
(4) Characterization technologies for dynamic observation of the interfacial cation accumulation during acidic CO2RR. Cation accumulation has been verified to effectively promote the performance of CO2RR in acidic media. However, most existing techniques, such as zero-potential measurement, nuclear magnetic resonance spectroscopy, and inductively coupled plasma optical emission spectroscopy, are limited to ex situ observations. Real-time monitoring of the cation accumulation phenomenon remains a significant challenge. Fluorescent probes and differential interference contrast microscopy, using ion-selective probes and local light refraction as indicators, offer promising options for monitoring real-time cation distributions. However, these techniques require the integration of specialized micro-electrochemical flow cells capable of accommodating the probes and optical lenses, posing significant technical challenges. Thus, developing real-time observation technologies for investigating interfacial composition variations during acidic CO2RR is crucial for deeper mechanistic understanding and further optimization of this reaction.
(5) C–N coupling and cascade reaction. Some studies focus on the coupling reaction of CO2RR and NO2−/NO3−/N2/NH3 reduction to produce compounds such as urea and amide under neutral and alkaline conditions.136,137 Thus, it is worthwhile to explore the C–N coupling reaction and the possibility for product diversity in acidic media. Additionally, cascading CO2RR with other reactions is conducive to producing long-chain carbon products, such as chlorohydrin, propyl alcohol, ethylene oxide, and ethyl acetate. However, this exploration is still undeveloped in acidic CO2RR. Consequently, developing cascade electrocatalytic systems based on acidic CO2RR to produce long-chain carbon products presents a meaningful avenue for further research.
Author contributions
Y. J., Y. Z., and S.-Z. Q. proposed the topic of the review. Y. J., L. H., and C. C. collaboratively designed and corrected the figures. Y. J. drafted the manuscript. Y. Z., and S.-Z. Q. corrected and reviewed the manuscript prior to submission. All authors contributed to discussions of the review.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support provided by the Australian Research Council through Discovery Project and Linkage Project Programs (FT200100062, DP230102027, DP240102575, CE230100032, LP210301397). Y. J. is supported by the Chinese CSC Scholarship Program.References
- K. O. Yoro and M. O. Daramola, Advances in Carbon Capture, 2020, 3–28, DOI:10.1016/B978-0-12-819657-1.00001-3.
- P. Singh, D. Yadav and S. Pandian, Global Climate Change, 2021, 79–108, DOI:10.1016/B978-0-12-822928-6.00009-5.
- M. Jiang, H. Wang, M. Zhu, X. Luo, Y. He, M. Wang, C. Wu, L. Zhang, X. Li, X. Liao, Z. Jiang and Z. Jin, Chem. Soc. Rev., 2024, 53, 5149–5189 RSC.
- Z. Wang, Y. Zhou, P. Qiu, C. Xia, W. Fang, J. Jin, L. Huang, P. Deng, Y. Su, R. Crespo-Otero, X. Tian, B. You, W. Guo, D. Di Tommaso, Y. Pang, S. Ding and B. Y. Xia, Adv. Mater., 2023, 35, 2303052 CrossRef CAS PubMed.
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A. C. Roger, R. Amal, H. He and S. E. Park, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- W. Lai, Y. Qiao, J. Zhang, Z. Lin and H. Huang, Energy Environ. Sci., 2022, 15, 3603–3629 RSC.
- P. P. Yang and M. R. Gao, Chem. Soc. Rev., 2023, 52, 4343–4380 RSC.
- X. Tan, C. Yu, Y. Ren, S. Cui, W. Li and J. Qiu, Energy Environ. Sci., 2021, 14, 765–780 RSC.
- Z. Yao, X. He and R. Lin, Electrochem. Energy Rev., 2024, 7, 8 CrossRef CAS.
- M. Zheng, P. Wang, X. Zhi, K. Yang, Y. Jiao, J. Duan, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2022, 144, 14936–14944 CrossRef CAS PubMed.
- L. Li, A. Ozden, S. Guo, A. D. A. F. P. Garci, C. Wang, M. Zhang, J. Zhang, H. Jiang, W. Wang, H. Dong, D. Sinton, E. H. Sargent and M. Zhong, Nat. Commun., 2021, 12, 5223 CrossRef CAS PubMed.
- X. She, L. Zhai, Y. Wang, P. Xiong, M. M.-J. Li, T.-S. Wu, M. C. Wong, X. Guo, Z. Xu, H. Li, H. Xu, Y. Zhu, S. C. E. Tsang and S. P. Lau, Nat. Energy, 2024, 9, 154–162 CrossRef.
- F. Huang, X. Chen, H. Sun, Q. Zeng, J. Ma, D. Wei, J. Zhu, Z. Chen, T. Liang, X. Yin, X. Liu, J. Xu and H. He, Angew. Chem., Int. Ed., 2024, 64, e202415642 CrossRef PubMed.
- Y. J. Ko, C. Lim, J. Jin, M. G. Kim, J. Y. Lee, T. Y. Seong, K. Y. Lee, B. K. Min, J. Y. Choi, T. Noh, G. W. Hwang, W. H. Lee and H. S. Oh, Nat. Commun., 2024, 15, 3356 CrossRef CAS PubMed.
- W. Li, C. Yu, X. Tan, Y. Ren, Y. Zhang, S. Cui, Y. Yang and J. Qiu, ACS Catal., 2024, 14, 8050–8061 CrossRef CAS.
- B. Shao, D. Huang, R. K. Huang, X. L. He, Y. Luo, Y. L. Xiang, L. B. Jiang, M. Dong, S. Li, Z. Zhang and J. Huang, Angew. Chem., Int. Ed., 2024, 63, e202409270 CrossRef CAS PubMed.
- H. Wu, L. Huang, J. Timoshenko, K. Qi, W. Wang, J. Liu, Y. Zhang, S. Yang, E. Petit, V. Flaud, J. Li, C. Salameh, P. Miele, L. Lajaunie, B. Roldán Cuenya, D. Rao and D. Voiry, Nat. Energy, 2024, 9, 422–433 CrossRef CAS.
- M. Esmaeilirad, Z. Jiang, A. M. Harzandi, A. Kondori, M. Tamadoni Saray, C. U. Segre, R. Shahbazian-Yassar, A. M. Rappe and M. Asadi, Nat. Energy, 2023, 8, 891–900 CrossRef CAS.
- K. Qi, Y. Zhang, N. Onofrio, E. Petit, X. Cui, J. Ma, J. Fan, H. Wu, W. Wang, J. Li, J. Liu, Y. Zhang, Y. Wang, G. Jia, J. Wu, L. Lajaunie, C. Salameh and D. Voiry, Nat. Catal., 2023, 6, 319–331 CrossRef CAS.
- L. Li, Z. Liu, X. Yu and M. Zhong, Angew. Chem., Int. Ed., 2023, 62, e202300226 CrossRef CAS PubMed.
- W. Wu, L. Xu, Q. Lu, J. Sun, Z. Xu, C. Song, J. C. Yu and Y. Wang, Adv. Mater., 2024, 37, 2312894 CrossRef PubMed.
- T. Zhang, J. Zhou, T. Luo, J. Q. Lu, Z. Li, X. Weng and F. Yang, Chem. – Eur. J., 2023, 29, e202301455 CrossRef CAS PubMed.
- Z. Ma, Z. Yang, W. Lai, Q. Wang, Y. Qiao, H. Tao, C. Lian, M. Liu, C. Ma, A. Pan and H. Huang, Nat. Commun., 2022, 13, 7596 CrossRef CAS PubMed.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. Garcia de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C. T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- Y. Zhao, L. Hao, A. Ozden, S. Liu, R. K. Miao, P. Ou, T. Alkayyali, S. Zhang, J. Ning, Y. Liang, Y. Xu, M. Fan, Y. Chen, J. E. Huang, K. Xie, J. Zhang, C. P. O’Brien, F. Li, E. H. Sargent and D. Sinton, Nat. Synth., 2023, 2, 403–412 CAS.
- H. Yano, F. Shirai, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2002, 519, 93–100 CrossRef CAS.
- K. Ogura, H. Yano and F. Shirai, J. Electrochem. Soc., 2003, 150, D163–D168 CrossRef CAS.
- H. Yano, T. Tanaka, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2004, 565, 287–293 CrossRef CAS.
- T. Lee, Y. Lee, J. Eo and D. H. Nam, Nanoscale, 2024, 16, 2235–2249 RSC.
- X. Cao, D. Tan, B. Wulan, K. S. Hui, K. N. Hui and J. Zhang, Small Methods, 2021, 5, 2100700 CrossRef CAS PubMed.
- J. Li and J. Gong, Energy Environ. Sci., 2020, 13, 3748–3779 RSC.
- J. Wang, H. Y. Tan, M. Y. Qi, J. Y. Li, Z. R. Tang, N. T. Suen, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2023, 52, 5013–5050 RSC.
- S. Banerjee, Z.-Q. Zhang, A. S. Hall and V. S. Thoi, ACS Catal., 2020, 10, 9907–9914 CrossRef CAS.
- H. Khani, A. R. Puente Santiago and T. He, Angew. Chem., Int. Ed., 2023, 62, e202306103 CrossRef CAS PubMed.
- F. P. Garcia de Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef CAS PubMed.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 Search PubMed.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS.
- H.-G. Qin, F.-Z. Li, Y.-F. Du, L.-F. Yang, H. Wang, Y.-Y. Bai, M. Lin and J. Gu, ACS Catal., 2023, 13, 916–926 CrossRef CAS.
- Y. Cao, Z. Chen, P. Li, A. Ozden, P. Ou, W. Ni, J. Abed, E. Shirzadi, J. Zhang, D. Sinton, J. Ge and E. H. Sargent, Nat. Commun., 2023, 14, 2387 CrossRef CAS PubMed.
- X. Zhu, J. Huang and M. Eikerling, Acc. Chem. Res., 2024, 57, 2080–2092 CrossRef CAS PubMed.
- C. P. O’Brien, R. K. Miao, S. Liu, Y. Xu, G. Lee, A. Robb, J. E. Huang, K. Xie, K. Bertens, C. M. Gabardo, J. P. Edwards, C.-T. Dinh, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 2952–2959 CrossRef.
- Y. Xie, P. Ou, X. Wang, Z. Xu, Y. C. Li, Z. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. Wang, T. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Nat. Catal., 2022, 5, 564–570 CrossRef CAS.
- N. B. Watkins, Y. Wu, W. Nie, J. C. Peters and T. Agapie, ACS Energy Lett., 2023, 8, 189–195 CrossRef CAS.
- M. Sun, J. Cheng and M. Yamauchi, Nat. Commun., 2024, 15, 491 CrossRef CAS PubMed.
- M. Fan, J. E. Huang, R. K. Miao, Y. Mao, P. Ou, F. Li, X.-Y. Li, Y. Cao, Z. Zhang, J. Zhang, Y. Yan, A. Ozden, W. Ni, Y. Wang, Y. Zhao, Z. Chen, B. Khatir, C. P. O’Brien, Y. Xu, Y. C. Xiao, G. I. N. Waterhouse, K. Golovin, Z. Wang, E. H. Sargent and D. Sinton, Nat. Catal., 2023, 6, 763–772 CrossRef CAS.
- S. Garg, Q. Xu, A. B. Moss, M. Mirolo, W. Deng, I. Chorkendorff, J. Drnec and B. Seger, Energy Environ. Sci., 2023, 16, 1631–1643 RSC.
- J. Fan, B. Pan, J. Wu, C. Shao, Z. Wen, Y. Yan, Y. Wang and Y. Li, Angew. Chem., Int. Ed., 2024, 63, e202317828 CrossRef CAS PubMed.
- H. G. Qin, Y. F. Du, Y. Y. Bai, F. Z. Li, X. Yue, H. Wang, J. Z. Peng and J. Gu, Nat. Commun., 2023, 14, 5640 CrossRef CAS PubMed.
- M. Zeng, W. Fang, Y. Cen, X. Zhang, Y. Hu and B. Y. Xia, Angew. Chem., Int. Ed., 2024, 63, e202404574 CrossRef CAS PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- C. Chen, Y. Li and P. Yang, Joule, 2021, 5, 737–742 CrossRef.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- X. Zou and J. Gu, Chin. J. Catal., 2023, 52, 14–31 CrossRef CAS.
- N. Ziv, A. N. Mondal, T. Weissbach, S. Holdcroft and D. R. Dekel, J. Membr. Sci., 2019, 586, 140–150 CrossRef CAS.
- H. Xue, Z. H. Zhao, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2023, 145, 16978–16982 CrossRef CAS PubMed.
- N. Ling, J. Zhang, M. Wang, Z. Wang, Z. Mi, S. Bin Dolmanan, M. Zhang, B. Wang, W. Ru Leow, J. Zhang and Y. Lum, Angew. Chem., Int. Ed., 2023, 62, e202308782 CrossRef CAS PubMed.
- J. Li and N. Kornienko, Chem Catal., 2022, 2, 29–38 CrossRef CAS.
- B. Sun, Z. Li, D. Xiao, H. Liu, K. Song, Z. Wang, Y. Liu, Z. Zheng, P. Wang, Y. Dai, B. Huang, A. Thomas and H. Cheng, Angew. Chem., Int. Ed., 2024, 63, e202318874 CrossRef CAS PubMed.
- Y. Qiao, W. Lai, K. Huang, T. Yu, Q. Wang, L. Gao, Z. Yang, Z. Ma, T. Sun, M. Liu, C. Lian and H. Huang, ACS Catal., 2022, 12, 2357–2364 CrossRef CAS.
- S. Feng, X. Wang, D. Cheng, Y. Luo, M. Shen, J. Wang, W. Zhao, S. Fang, H. Zheng, L. Ji, X. Zhang, W. Xu, Y. Liang, P. Sautet and J. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202317942 CrossRef CAS PubMed.
- X. Zi, Y. Zhou, L. Zhu, Q. Chen, Y. Tan, X. Wang, M. Sayed, E. Pensa, R. A. Geioushy, K. Liu, J. Fu, E. Cortes and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309351 CrossRef CAS PubMed.
- H. Shen, H. Jin, H. Li, H. Wang, J. Duan, Y. Jiao and S. Z. Qiao, Nat. Commun., 2023, 14, 2843 CrossRef CAS PubMed.
- W. Nie, G. P. Heim, N. B. Watkins, T. Agapie and J. C. Peters, Angew. Chem., Int. Ed., 2023, 62, e202216102 CrossRef CAS PubMed.
- M. Ramdin, A. R. T. Morrison, M. de Groen, R. van Haperen, R. de Kler, E. Irtem, A. T. Laitinen, L. J. P. van den Broeke, T. Breugelmans, J. P. M. Trusler, W. D. Jong and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2019, 58, 22718–22740 CrossRef CAS.
- H. Yano, F. Shirai, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2002, 533, 113–118 CrossRef CAS.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed.
- Y. Wu, K. Kamiya, T. Hashimoto, R. Sugimoto, T. Harada, K. Fujii and S. Nakanishi, Electrochemistry, 2020, 88, 359–364 CrossRef CAS.
- H. Ooka, M. C. Figueiredo and M. T. M. Koper, Langmuir, 2017, 33, 9307–9313 CrossRef CAS PubMed.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- L. Yuan, S. Zeng, X. Zhang, X. Ji and S. Zhang, Mater. Rep.: Energy, 2023, 3, 100177 CAS.
- D. Xu, K. Li, B. Jia, W. Sun, W. Zhang, X. Liu and T. Ma, Carbon Energy, 2022, 5, e230 CrossRef.
- S. Zhu, T. Li, W.-B. Cai and M. Shao, ACS Energy Lett., 2019, 4, 682–689 CrossRef CAS.
- A. Vasileff, X. Zhi, C. Xu, L. Ge, Y. Jiao, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 9411–9417 CrossRef CAS.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- Y. Jiang, H. Li, C. Chen, Y. Zheng and S.-Z. Qiao, ACS Catal., 2024, 14, 8310–8316 CrossRef CAS.
- H. Li, H. Li, P. Wei, Y. Wang, Y. Zang, D. Gao, G. Wang and X. Bao, Energy Environ. Sci., 2023, 16, 1502–1510 RSC.
- Y. Fu, Q. Xie, L. Wan, Q. Huang and J. Luo, Mater. Today Energy, 2022, 29, 101105 CrossRef CAS.
- P. Moreno-Garcia, N. Kovacs, V. Grozovski, M. J. Galvez-Vazquez, S. Vesztergom and P. Broekmann, Anal. Chem., 2020, 92, 4301–4308 CrossRef CAS PubMed.
- M. H. Hicks, W. Nie, A. E. Boehme, H. A. Atwater, T. Agapie and J. C. Peters, J. Am. Chem. Soc., 2024, 146, 25282–25289 CrossRef CAS PubMed.
- M. C. O. Monteiro, A. Mirabal, L. Jacobse, K. Doblhoff-Dier, S. C. Barton and M. T. M. Koper, JACS Au, 2021, 1, 1915–1924 CrossRef CAS PubMed.
- B. Pan, J. Fan, J. Zhang, Y. Luo, C. Shen, C. Wang, Y. Wang and Y. Li, ACS Energy Lett., 2022, 7, 4224–4231 CrossRef CAS.
- W. Zheng, ACS Energy Lett., 2023, 8, 1952–1958 CrossRef CAS.
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu, S. Chu and Y. Cui, Nat. Catal., 2018, 1, 592–600 CrossRef CAS.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2020, 4, 20–27 CrossRef.
- F. Zhang and A. C. Co, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 CrossRef CAS PubMed.
- S. Dieckhofer, D. Ohl, J. R. C. Junqueira, T. Quast, T. Turek and W. Schuhmann, Chem. – Eur. J., 2021, 27, 5906–5912 CrossRef PubMed.
- T. Luo, K. Liu, J. Fu, S. Chen, H. Li, H. Pan and M. Liu, Adv. Energy Sustainability Res., 2022, 4, 2200148 CrossRef.
- M. C. O. Monteiro, M. F. Philips, K. J. P. Schouten and M. T. M. Koper, Nat. Commun., 2021, 12, 4943 CrossRef CAS PubMed.
- Z. Xu, M. Sun, Z. Zhang, Y. Xie, H. Hou, X. Ji, T. Liu, B. Huang and Y. Wang, ChemCatChem, 2022, 14, e202200052 CrossRef CAS.
- W. Ge, L. Dong, C. Wang, Y. Zhu, Z. Liu, H. Jiang and C. Li, ACS Catal., 2024, 14, 10529–10537 CrossRef CAS.
- A. Wang, W. Ge, W. Sun, X. Sheng, L. Dong, W. Zhang, H. Jiang and C. Li, Angew. Chem., Int. Ed., 2024, 64, e202412754 CrossRef PubMed.
- Q. Zhang, X. Shao, J. Yi, Y. Liu and J. Zhang, Chin. J. Chem. Eng., 2020, 28, 2549–2554 CrossRef CAS.
- C. M. Gunathunge, V. J. Ovalle and M. M. Waegele, Phys. Chem. Chem. Phys., 2017, 19, 30166–30172 RSC.
- M. C. O. Monteiro, F. Dattila, N. Lopez and M. T. M. Koper, J. Am. Chem. Soc., 2022, 144, 1589–1602 CrossRef CAS PubMed.
- Z.-M. Zhang, T. Wang, Y.-C. Cai, X.-Y. Li, J.-Y. Ye, Y. Zhou, N. Tian, Z.-Y. Zhou and S.-G. Sun, Nat. Catal., 2024, 7, 807–817 CrossRef CAS.
- J. J. Masana, B. Peng, Z. Shuai, M. Qiu and Y. Yu, J. Mater. Chem. A, 2022, 10, 1086–1104 RSC.
- T. Kim and G. T. R. Palmore, Nat. Commun., 2020, 11, 3622 CrossRef CAS PubMed.
- J. Resasco, Y. Lum, E. Clark, J. Z. Zeledon and A. T. Bell, ChemElectroChem, 2018, 5, 1064–1072 CrossRef CAS.
- X. Zhou, J. Shan, L. Chen, B. Y. Xia, T. Ling, J. Duan, Y. Jiao, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2022, 144, 2079–2084 CrossRef CAS PubMed.
- Q. Fan, G. Bao, X. Chen, Y. Meng, S. Zhang and X. Ma, ACS Catal., 2022, 12, 7517–7523 CrossRef CAS.
- L. P. Chi, Z. Z. Niu, Y. C. Zhang, X. L. Zhang, J. Liao, Z. Z. Wu, P. C. Yu, M. H. Fan, K. B. Tang and M. R. Gao, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2312876120 CrossRef CAS PubMed.
- X. Li, P. Zhang, L. Zhang, G. Zhang, H. Gao, Z. Pang, J. Yu, C. Pei, T. Wang and J. Gong, Chem. Sci., 2023, 14, 5602–5607 RSC.
- Z. Liu, T. Yan, H. Shi, H. Pan, Y. Cheng and P. Kang, ACS Appl. Mater. Interfaces, 2022, 14, 7900–7908 CrossRef CAS PubMed.
- C. Zhu, G. Wu, A. Chen, G. Feng, X. Dong, G. Li, S. Li, Y. Song, W. Wei and W. Chen, Energy Environ. Sci., 2024, 17, 510–517 RSC.
- S. Li, X. Dong, G. Wu, Y. Song, J. Mao, A. Chen, C. Zhu, G. Li, Y. Wei, X. Liu, J. Wang, W. Chen and W. Wei, Nat. Commun., 2024, 15, 6101 CrossRef CAS PubMed.
- T. Yan, H. Pan, Z. Liu and P. Kang, Small, 2023, 19, 2207650 CrossRef CAS PubMed.
- Q. Wu, J. Liang, L. L. Han, Y. B. Huang and R. Cao, Chem. Commun., 2023, 59, 5102–5105 RSC.
- R. Wang, L.-Z. Dong, J.-W. Shi, M. Zhang, S.-L. Li, Y.-Q. Lan and J. Liu, ACS Catal., 2024, 14, 741–750 CrossRef CAS.
- J. Feng, L. Wu, X. Song, L. Zhang, S. Jia, X. Ma, X. Tan, X. Kang, Q. Zhu, X. Sun and B. Han, Nat. Commun., 2024, 15, 4821 CrossRef CAS PubMed.
- X. Yu, Y. Xu, L. Li, M. Zhang, W. Qin, F. Che and M. Zhong, Nat. Commun., 2024, 15, 1711 CrossRef CAS PubMed.
- Y. Yang, J. Zhang, Z. Tan, J. Yang, S. Wang, M. Li and Z. Su, Angew. Chem., Int. Ed., 2024, 63, e202408873 CrossRef CAS PubMed.
- J. Zhang, C. Guo, S. Fang, X. Zhao, L. Li, H. Jiang, Z. Liu, Z. Fan, W. Xu, J. Xiao and M. Zhong, Nat. Commun., 2023, 14, 1298 CrossRef CAS PubMed.
- B. Han, Y. Wu, C. Chen, S. Liu, Q. Qian, Q. Zhu, R. Feng, L. Jing, X. Kang and X. Sun, Angew. Chem., Int. Ed., 2024, 63, e202410659 CrossRef PubMed.
- M. Wang, B. Wang, J. Zhang, S. Xi, N. Ling, Z. Mi, Q. Yang, M. Zhang, W. R. Leow, J. Zhang and Y. Lum, Nat. Commun., 2024, 15, 1218 CrossRef CAS PubMed.
- Y. Chen, X. Y. Li, Z. Chen, A. Ozden, J. E. Huang, P. Ou, J. Dong, J. Zhang, C. Tian, B. H. Lee, X. Wang, S. Liu, Q. Qu, S. Wang, Y. Xu, R. K. Miao, Y. Zhao, Y. Liu, C. Qiu, J. Abed, H. Liu, H. Shin, D. Wang, Y. Li, D. Sinton and E. H. Sargent, Nat. Nanotechnol., 2023, 19, 311–318 CrossRef PubMed.
- Z. Jiang, Z. Zhang, H. Li, Y. Tang, Y. Yuan, J. Zao, H. Zheng and Y. Liang, Adv. Energy Mater., 2023, 13, 2203603 CrossRef CAS.
- M. Fan, R. K. Miao, P. Ou, Y. Xu, Z. Y. Lin, T. J. Lee, S. F. Hung, K. Xie, J. E. Huang, W. Ni, J. Li, Y. Zhao, A. Ozden, C. P. O'Brien, Y. Chen, Y. C. Xiao, S. Liu, J. Wicks, X. Wang, J. Abed, E. Shirzadi, E. H. Sargent and D. Sinton, Nat. Commun., 2023, 14, 3314 CrossRef CAS PubMed.
- Q. Zhang, C. B. Musgrave, Y. Song, J. Su, L. Huang, L. Cheng, G. Li, Y. Liu, Y. Xin, Q. Hu, G. Ye, H. Shen, X. Wang, B. Z. Tang, W. A. Goddard and R. Ye, Nat. Synth., 2024, 3, 1231–1242 CrossRef CAS.
- M. D. Zhang, J. R. Huang, W. Shi, P. Q. Liao and X. M. Chen, Angew. Chem., Int. Ed., 2023, 62, e202308195 CrossRef CAS PubMed.
- Q. Wu, D. H. Si, J. Liang, Y. B. Huang and R. Cao, Appl. Catal., B, 2023, 333, 122803 CrossRef CAS.
- C. P. Wan, H. Guo, D. H. Si, S. Y. Gao, R. Cao and Y. B. Huang, JACS Au, 2024, 4, 2514–2522 CrossRef CAS PubMed.
- Y. Wang, C. Wang, Y. Wei, F. Wei, L. Kong, J. Feng, J. Q. Lu, X. Zhou and F. Yang, Chem. – Eur. J., 2022, 28, e202201832 CrossRef CAS PubMed.
- S. Li, G. Wu, J. Mao, A. Chen, X. Liu, J. Zeng, Y. Wei, J. Wang, H. Zhu, J. Xia, X. Wang, G. Li, Y. Song, X. Dong, W. Wei and W. Chen, Angew. Chem., Int. Ed., 2024, 63, e202407612 CrossRef CAS PubMed.
- J.-N. Zhang, N. K. Niazi, J. Qiao, L. Li, I. M. U. Hasan, R. He, L. Peng, N. Xu and F. Farwa, Nano Res. Energy, 2022, 1, e9120015 Search PubMed.
- Y. Jiang, J. Shan, P. Wang, L. Huang, Y. Zheng and S.-Z. Qiao, ACS Catal., 2023, 13, 3101–3108 CrossRef CAS.
- G. Neri, I. M. Aldous, J. J. Walsh, L. J. Hardwick and A. J. Cowan, Chem. Sci., 2016, 7, 1521–1526 RSC.
- X. Sheng, W. Ge, H. Jiang and C. Li, Adv. Mater., 2022, 34, 2201295 CrossRef CAS PubMed.
- M. Wang, Z. Wang, Z. Huang, M. Fang, Y. Zhu and L. Jiang, ACS Nano, 2024, 18, 15303–15311 CrossRef CAS PubMed.
- Z. Wang, Y. Li, X. Zhao, S. Chen, Q. Nian, X. Luo, J. Fan, D. Ruan, B. Q. Xiong and X. Ren, J. Am. Chem. Soc., 2023, 145, 6339–6348 CrossRef CAS PubMed.
- M. Zhang, J. Wang, X. Rong, X.-L. Lu and T.-B. Lu, Nano Res., 2024, 17, 2381–2387 CrossRef CAS.
- K. Xu, J. Li, F. Liu, X. Chen, T. Zhao and F. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202311968 CrossRef CAS PubMed.
- X. Zhao, H. Xie, B. Deng, L. Wang, Y. Li and F. Dong, Chem. Commun., 2024, 60, 542–545 RSC.
- E. Vichou, A. Perazio, Y. Adjez, M. Gomez-Mingot, M. W. Schreiber, C. M. Sánchez-Sánchez and M. Fontecave, Chem. Mater., 2023, 35, 7060–7068 CrossRef CAS.
- H. Jin, J. Xu, H. Liu, H. Shen, H. Yu, M. Jaroniec, Y. Zheng and S. Z. Qiao, Sci. Adv., 2023, 9, eadi7755 CrossRef CAS PubMed.
- C. Peng, H. Shen, M. Zheng, M. Jaroniec, Y. Zheng and S.-Z. Qiao, ACS Catal., 2024, 15, 468–476 CrossRef.
- X. Fan, C. Liu, X. He, Z. Li, L. Yue, W. Zhao, J. Li, Y. Wang, T. Li, Y. Luo, D. Zheng, S. Sun, Q. Liu, L. Li, W. Chu, F. Gong, B. Tang, Y. Yao and X. Sun, Adv. Mater., 2024, 36, 2401221 CrossRef CAS PubMed.
- Z. Zhang, D. Li, Y. Tu, J. Deng, H. Bi, Y. Yao, Y. Wang, T. Li, Y. Luo, S. Sun, D. Zheng, S. A. C. Carabineiro, Z. Chen, J. Zhu and X. Sun, SusMat, 2024, 4, e193 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |