
Photomechanically accelerated degradation of perovskite solar cells†
Haonan
Wang
a,
Qing
Li
a,
Yan
Zhu
a,
Xinyuan
Sui
 a,
Xiulian
Fan
c,
Miaoyu
Lin
a,
Yifeng
Shi
d,
Yichu
Zheng
e,
Haiyang
Yuan
a,
Yu
Zhou
c,
Haibao
Jin
*b,
Hua Gui
Yang
a,
Yu
Hou
*a and
Shuang
Yang
*a
a,
Xiulian
Fan
c,
Miaoyu
Lin
a,
Yifeng
Shi
d,
Yichu
Zheng
e,
Haiyang
Yuan
a,
Yu
Zhou
c,
Haibao
Jin
*b,
Hua Gui
Yang
a,
Yu
Hou
*a and
Shuang
Yang
*a
aKey Laboratory for Ultrafine Materials of Ministry of Education, Shanghai Engineering Research Center of Hierarchical Nanomaterials, School of Materials Science and Engineering, East China University of Science and Technology, 200237, Shanghai, China. E-mail: yhou@ecust.edu.cn; syang@ecust.edu.cn
bShanghai Key Laboratory of Advanced Polymeric Materials, Frontiers Science Center for Materiobiology and Dynamic Chemistry, School of Materials Science and Engineering, East China University of Science and Technology, 200237, Shanghai, China. E-mail: haibaojin@ecust.edu.cn
cHunan Key Laboratory of Nanophotonics and Devices, School of Physics, Central South University, Changsha, Hunan 410083, P. R. China
dKey Laboratory of Materials for High-Power Laser, Shanghai Institute of Optics and Fine Mechanics, Chinese Academy of Sciences, 201800, Shanghai, China
eSchool of Mechatronic Engineering and Automation, Shanghai University, Shanghai, 200444, China
First published on 7th January 2025
Abstract
Understanding the origin of intrinsic instability of metal halide perovskites is indispensable for their advancement in opto-electronic applications. Here, we report a photomechanically accelerated degradation mechanism of perovskite thin films, in which the lattice expansion driven by light illumination has been found to govern the degradation kinetics. The dynamic lattice evolution under illumination causes crowding of the perovskite grains, leading to large local strains near the grain boundaries (GBs), which thereby facilitates defect formation and iodine component loss in the region. We show that the physical separation of each perovskite grain using trans-polyisoprene (TPI) could circumvent photomechanical damage at the GBs, achieving a T97 of 1000 h under continuous one-sun illumination at 55 °C in solar cell devices. Our results emphasize the nontrivial role of dynamic lattice deformation in the decomposition of perovskite thin films and open up new possibilities to further improve the intrinsic stability of solar cells.
Broader contextThe certificated power conversion efficiency (PCE) of perovskite solar cells (PSCs) has surpassed 26%, yet their widespread application is still constrained by long-term reliability, particularly under light and thermal conditions. Softness is a fundamental characteristic of perovskite materials, which undergo remarkable dynamic structural evolution under environmental stress, such as light irradiation, temperature, and electrical fields. This can manifest in many unique dynamic properties, including giant photostriction, electrostriction, and large thermal expansivity. Here, we present the first study on the impact of the photomechanical effect on the stability of PSCs. We demonstrated a photomechanically accelerated degradation mechanism of perovskite thin films, where lattice expansion driven by light illumination governs the degradation kinetics. Experimental and theoretical studies revealed that perovskite grains accumulated strain at grain boundaries under illumination, which favored the formation of iodine-related defects and subsequent degradation. We further used soft polymers, such as TPI, to separate perovskite grains, stabilizing PSCs with 97.17% retained PCE after exposure to AM 1.5G irradiation and 55 °C illumination for 1000 h. We anticipate that the discovery of photomechanically accelerated degradation will open new avenues for designing long-lifetime PSCs for practical applications. |
Introduction
Metal halide perovskites are regarded as the leading semiconductors for next-generation solid-state opto-electronics, having demonstrated unprecedented progress with over 26% certified efficiency in photovoltaic devices.1–6 However, the widespread implementation of this technology is still challenged by its unsatisfactory long-term stability during operation under light illumination and thermal conditions.7–13 Although progress has been made in recent years in stabilizing alternative components, such as charge transport layers and electrodes in photovoltaic devices, the perovskite layer itself still suffers from unsatisfactory intrinsic stability.14–17 Before devising measures to enhance the photostability of perovskite solar cells (PSCs), it is crucial to understand the degradation mechanisms under operating conditions. Previous studies have identified several degradation pathways in perovskites, such as ion migration, redox reactions, phase segregation, and trapped charge-driven degradation, which severely compromise device performance.18–22 Moreover, softness and ionicity are fundamental features of the perovskite lattice that facilitate its dynamic structural evolution under environmental stress, such as light irradiation, temperature, and electrical field, contributing to its unique dynamic properties, including giant photostriction, electrostriction, photoflexoelectric effect and large thermal expansivity.23–25 When operated under illumination, the lattice of perovskite polycrystalline films undergoes remarkable photostriction, for instance, grazing incidence wide-angle X-ray scattering (GIWAXS) studies have demonstrated lattice expansion ranging from 6.290 Å to 6.330 Å in a ternary cation perovskite film under a one-sun source.26 Such phenomenon is generally accompanied by inconspicuous morphological changes and mechanical stress that should, in principle, be closely coupled with the subsequent degradation process. However, it is not yet clear how lattice dynamics affect the intrinsic stability of perovskite films.Here, we investigate the impact of photo-induced lattice deformation on the degradation behavior of polycrystalline perovskite films. We find that the lattice expansion accumulates large strain at the contacted GBs and significantly facilitates their degradation, while the non-contacted GB region, without local strain, retains the perovskite structure for a longer duration under light exposure. We ascribe these observations to photomechanically accelerated degradation—an important phenomenon that, to our knowledge, has previously been ignored with respect to the instability of PSCs. We further demonstrate that the spatial isolation of these perovskite grains using soft polymers can migrate the photomechanical damage, thus establishing an effective pathway towards advancing the operational durability of perovskite devices.
Results and discussion
Photomechanical phenomena in perovskites
Lattice expansion under light illumination and high temperatures has been observed in many kinds of metal halide perovskite materials; it is closely associated with the inherent ion migration, defect formation, and redox reactions, and further degrades perovskites (Fig. 1A).25,26In situ X-ray diffraction (XRD) measurements were performed using a white LED that simulated 1-sun illumination (100 mW cm−2) to evaluate the structural evolution of perovskite thin films. A ternary cation perovskite thin film with a nominal chemical composition of Cs0.05FA0.81MA0.14PbI2.85Br0.15 (CsFAMA) was adopted because of the negligible residual strain at the interface, as observed from the comparison of its powder XRD and thin film XRD data (Fig. S1, ESI†). The duration of the XRD experiment was 125 min, and the film temperature was increased from about 25 °C to 40 °C. Upon illumination, there was a gradual shift in the diffraction vector q towards lower values for all diffraction peaks, corresponding to an isotropic increase in interplanar spacings; for instance, the (002) and (012) planes expanded from 3.160 Å to 3.168 Å and 2.822 Å to 2.829 Å with an increase factor of ∼0.25% (Fig. 1B), respectively. The lattice dynamics were found to be isotropic along the in-plane and out-of-plane orientations, according to the GIWAXS results (Fig. S2, ESI†). The in-plane and out-of-plane lattice parameters were similar, indicating negligible residual stress within the film. To check whether the lattice evolution is recoverable, we measured the XRD patterns of perovskite films under alternating light and dark cycles. Specifically, a single cycle consisted of 125 min of illumination followed by 30 min of darkness. As illustrated in Fig. S3 (ESI†), the photo-induced deformation of the perovskite films was rather elastic; the lattice parameters reverted back to the values of the initial state during these cycling tests. We also performed an XRD test on perovskite films pre-illuminated for 300 h. We found that the recovery of the perovskite lattice of the aged film was slower in the dark, accompanied by a slight increase in the full width at half maximum, suggesting that the degradation of this perovskite would affect the dynamic structural evolution (Fig. S4, ESI†). | ||
| Fig. 1 Photomechanical expansion effect in perovskites. (A) Schematic illustrations of the photomechanical expansion effect in a perovskite film upon illumination. (B) In situ XRD patterns of the ternary cation perovskite film under AM 1.5G illumination for up to 125 min. The insets show the enlarged patterns of the (002) and (012) crystal planes extracted from the XRD spectra. (C) AFM topographic images (left) and height profiles of selected regions (right) of the perovskite film before and after light soaking. (D) DMT modulus maps (left) and extracted modulus distributions (right) of the perovskite film before and after light soaking. | ||
Given that the thermal expansion coefficient of Cs0.05FA0.81MA0.14PbI2.85Br0.15 perovskite is ∼5 × 10−5 K−1, the temperature-induced expansion was only 0.06% (Fig. S5, ESI†).27,28 Disregarding the temperature effect, the photostrictive efficiency of the ternary perovskite was estimated to be approximately 9.5 × 10−13 m3 W−1, which is significantly higher than that of conventional single-crystalline silicon (−3.7 × 10−20 m3 W−1).29 This discrepancy is a crucial factor that contributes to the poor photostability of the perovskite active layer since the photomechanical behavior would result in lattice expansion, grain crowding and thus stress accumulation near the GBs. To examine the light-induced morphological evolution of perovskite films, we conducted AFM measurements of the same scanned area before and after illumination for 60 min. The extracted height profiles in Fig. 1C reveal geometrical narrowing of the GBs together with a slight increase in the height of the perovskite films after illumination, which agrees with our hypothesis about the large structural deformation of perovskite grains. Further inspection by nanomechanical imaging revealed an overall augmentation in the Derjaguin–Muller–Toporov (DMT) modulus after light soaking for 60 min (Fig. 1D). However, the improvement in modulus was particularly concentrated at the GBs. By extracting the data from nanomechanical mapping, it was found that an additional peak emerged at around 11 GPa under light soaking likely from the component of GBs. In principle, the DMT modulus is interpreted as the resistance of a material to elastic deformation under mechanical load. In polycrystalline perovskite films, the spatially asynchronous evolution of the surface modulus reflects the non-uniform distribution of strain or phase with disparate stiffness under light illumination. Nevertheless, the possibility of phase segregation in the as-prepared perovskite was excluded based on photoluminescence (PL) measurements: there was no notable spectral splitting or widening after light irradiation for 120 min (Fig. S7, ESI†).30 Considering the potential emergence of a non-emissive second phase, we conducted XRD tests of the films before and after illumination, which also did not show any evidence of phase segregation (Fig. S8, ESI†).
The effect of photomechanical behavior on GBs
The decomposition of the perovskite polycrystalline films is initiated at the GBs in most circumstances, making it challenging to directly quantify the contribution of photomechanical expansion to the decomposition process.31,32 To distinguish these effects, two types of perovskite films were prepared: the first one had a normal polycrystalline morphology with full coverage, while the second type was incompletely covered polycrystalline films. Details of sample preparation are provided in the Experimental Section. The scanning electron microscopy (SEM) images in Fig. S9 and S10 (ESI†) show the formation and accumulation of flake-shaped decomposition products between the GBs of both perovskite films during the irradiation time of 100 h. These impurities were verified to be PbI2 based on XRD analysis (Fig. S11, ESI†). Notably, most PbI2 impurities nucleated only at the contacted GBs but were absent at the uncontacted GBs (Fig. 2B). Meanwhile, we employed high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) to observe the perovskite sample after 500 h of light exposure, which offered more accurate and clear information than SEM. As shown in Fig. 2A and Fig. S12 (ESI†), lamellar PbI2 was predominantly observed at the contacted GBs, consistent with the conclusion drawn from the SEM analysis. We also conducted PL and time-resolved photoluminescence (TRPL) measurements on both types of perovskite films, and the results are shown in Fig. S13 and S14 (ESI†). Although the PL intensity and carrier lifetime of the initial state of the semi-covered perovskite film were slightly lower compared to those of the pristine film, it exhibited a slow decline in both parameters during the entire light soaking process. As the major difference between the two films is the connectivity of the perovskite grains, this set of experiments led to the conclusion that the interaction between grains, whether elastic or chemical, significantly accelerates the degradation kinetics. Furthermore, we fabricated perovskite films on polydimethylsiloxane (PDMS); during the illumination process, the temperature of the perovskite film and substrate increased by approximately 15 °C. Considering the relatively high thermal expansion coefficient of the PDMS substrate (3 × 10−4 K−1), this led to a lateral expansion of about 0.3%, which alleviated the grain compression experienced by the perovskite film during light soaking.33 The absence of PbI2 in this film after light soaking for 500 h again validated the critical impact of grain crowding and micro-stress on perovskite decomposition (Fig. S15, ESI†).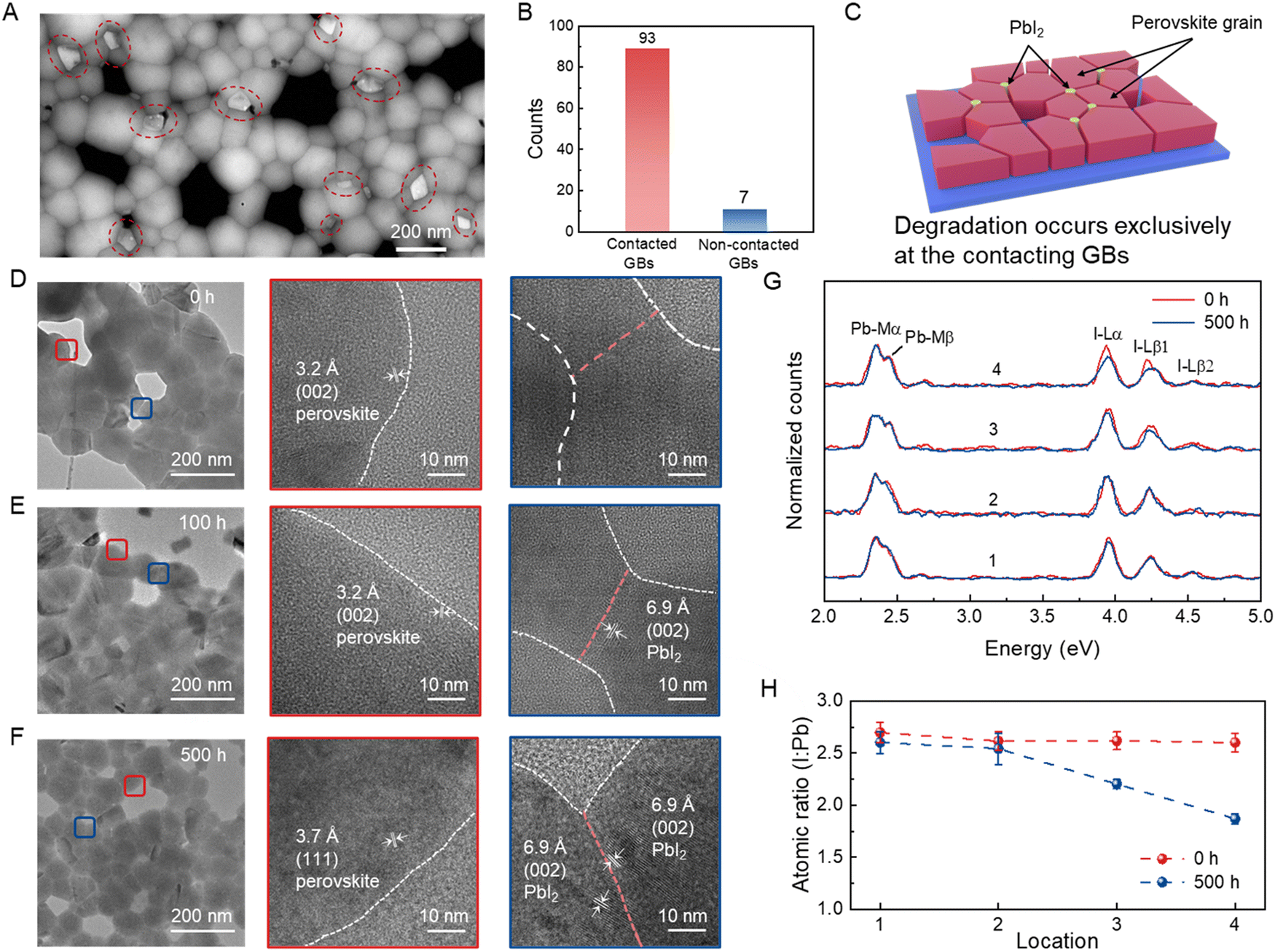 | ||
Fig. 2 Quantification of damage to the perovskite induced by photomechanical behavior. (A) HAADF-STEM images of the semi-covered perovskite film after light soaking for 500 h. The regions highlighted in red circles indicate PbI2. (B) Random statistical distribution of the positions of 100 PbI2 in the semi-covered perovskite films after light soaking for 500 h. (C) Schematic of the degraded perovskite film showing the presence of PbI2 at the contacted GBs. TEM images and the corresponding enlarged views of the perovskite films under AM 1.5G illumination for (D) 0 h, (E) 100 h, and (F) 500 h. The regions highlighted in red and blue boxes indicate the non-contacted and contacted GBs, respectively. (G) EDS profiles of the four different regions before and after light illumination. Bulk grains, non-contacted GBs, contacted GBs without PbI2, and contacted GBs with PbI2 are represented by 1, 2, 3, and 4, respectively. (H) Atomic I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb ratios in the four regions determined by EDS before and after light illumination. :Pb ratios in the four regions determined by EDS before and after light illumination. | ||
Transmission electron microscopy (TEM) and energy-dispersive X-ray spectroscopy (EDS) were used to monitor the microscopic structural and component evolution at different positions of the perovskite films upon light soaking. We prepared semi-covered perovskite specimens via a similar spinning procedure on poly(bis(4-phenyl)(2,4,6-trimethylphenyl)amine) (PTAA)-coated carbon support films. Before illumination, all measured spots, including contacted GBs, non-contacted GBs, and bulk grains, showed typical lattice fringes of the hybrid perovskite without observable degradation (Fig. 2D). After 100 h of light soaking, a small number of regions with a lattice spacing of 6.9 Å, which corresponds to the characteristic (002) crystal plane of lead iodide, appeared specifically at the contacted GBs (Fig. 2E). At 500 h of light soaking duration, the degraded regions at the contacted GBs became more pronounced and undesirably propagated into the bulk grains (Fig. 2F). In stark contrast, almost no lead iodide existed within the non-contacted GBs when more than 20 individual areas were inspected, consistent with the SEM observations (Fig. S17 and S18, ESI†). The elemental ratios of I to Pb in the representative non-contacted GBs, as determined by EDS, were similar at 2.63:1 and 2.54:1, respectively, before and after light soaking. Similar trends in the stoichiometric ratio were also observed in the intragranular region. However, the I-to-Pb ratio at the contacted GBs reduced from 2.61:1 to 1.89:1 with the PbI2 phase and 2.62:1 to 2.2:1 without the PbI2 phase after light soaking, highlighting the iodide-deficient nature of these contacted GB areas (Fig. 2G and H). Although perovskite degradation has been related to multiple possible mechanisms, such as redox reaction and ionic migration; our results unequivocally substantiate that the photomechanical effect progressively promotes these processes at the GBs in polycrystalline perovskites, thus increasing the iodine loss ratio at the GBs by around 8 times under standard light illumination conditions (Fig. S19, ESI†).
Photomechanical acceleration of iodine defect formation
We conducted finite element analysis to simulate the stress and strain distributions in the semi-covered polycrystalline films, which included the geometric structures of voids and GBs. (Fig. 3A and B). Isotropic grain expansion fractions of 0%, 0.1%, 0.2%, and 0.4% were employed in the model to represent films with varied photostriction ratios. The detailed procedure is described in the Experimental Section. Initially, the film with intact GBs was stress- and strain-free, whereas the expanded film displayed evident stress and strain gradient in the grain. In grains enclosed by GBs, the stress increased exponentially from the grain center outward and reached its maximum near the GB, while the corresponding tensile strain decreased from the grain center outward gradually and was converted into compressive strain at the GB, indicating the crowding effect from GBs. In contrast, for those with voids, the stress value in the region remained even lower than that at the grain center due to unrestricted expansion. Importantly, the overall stress level of the perovskite GBs was determined by the photo-induced expansion ratios and grain connectivity; for expansion ratios of 0.1%, 0.2%, and 0.4%, stress in the grain center was ∼3, 7, and 15 MPa, respectively and increased to ∼35, 70, and 120 MPa at the GBs, but were ∼1, 3, and 5 MPa near the voids (Fig. 3D and Fig. S20, ESI†). | ||
| Fig. 3 Effect of strain accumulation on the formation energy of iodine defects. (A) Strain and (B) stress distributions of the semi-covered polycrystalline perovskite films under varied expansion ratios. Crowding of the perovskite grains results in compression of the contacted GBs with large accumulated stress. (C) Atomic model of the FAPbI3 perovskite used for probing the impact of uniaxial strain on defect formation. Strain is applied along the perpendicular direction (blue arrow) to the ab-plane. (D) Statistical mean stress at different regions as a function of the lattice expansion ratio. (E) Evolution of the formation energy of V+I defects in (001) strained perovskites compared with the unstrained counterparts at 300 K. | ||
Considering that strain accumulation often exacerbates defect formation and ion migration kinetics in perovskites, density functional theory (DFT) simulation was conducted to further investigate the evolution of defect formation energetics upon compression. Iodine vacancy (V+I) was selected for the analysis due to its broad prevalence and very low migration energy (∼0.1 eV) in iodine-based perovskites.34–36 We constructed structural models of cubic FAPbI3 with and without iodine vacancy defects under various uniaxial compressive strains along the [100] axis (Fig. S21, ESI†). Under a compressive strain of 0.2%, the defect formation energy gradually decreased by 16.07 meV compared with the unstrained state, resulting in a 1.3-fold increase in V+I defect density. Further compressing the perovskite with up to 0.6% strain monotonously declined the defect formation energy by 35.58 meV, along with a 1.8-fold increase in defect density (Fig. 3E and Fig. S22, ESI†). To investigate the reasons behind the reduced formation energy of the defects due to high compressive strain, we calculated the structural formation energies of various FAPbI3 configurations (Table S1, ESI†). The results demonstrate that, with an increase in strain, the structural formation energy significantly rises. Generally, an increase in structural formation energy indicates a tendency towards an unstable state, which is more conducive to the generation of defects.37 These calculations thus elucidate that the photomechanical strain accumulates near the GBs and favours the generation of iodine-related defects that degrade the performance of solar cells.
The effect of photomechanical strain on the optoelectronic properties of perovskite films
To identify how photomechanical strain impacts the optoelectronic properties of perovskite films, the surface potential and current of the perovskite films before and after light soaking of 500 h were visualized using Kelvin probe force microscopy (KPFM) and conductive atomic force microscopy (c-AFM) techniques. As shown in Fig. 4A, the pristine perovskite film exhibited a uniform distribution of the Contact potential difference (CPD), indicating consistent work functions (WF) at both intragranular and intergranular positions. However, after light soaking, spatial heterogeneity in surface potential gradually occurred with significant CPD reduction at the iodine-deficient GBs. At this stage, most perovskite phases were not d, as confirmed by XRD measurements, that is, only a few varied CPD signals were derived from the decomposed PbI2 phase. Such evolution in the uniformity of physical properties was also observed by PL intensity mapping, in which the GBs of the perovskite film tuned to dark upon light soaking (Fig. S23, ESI†).38 Based on microscopic characterization and the simulation results, we suspect that the predominant factor that governs the WF of the perovskite film should be the formation of iodine-related defects near the GBs. Furthermore, the current maps in Fig. 4B reveal that the mean local photocurrent approximately decreases by half after light soaking, and the changes are particularly pronounced at the GBs along with the existence of many near-zero photocurrent regions, suggestive of the nucleation of iodine defects and PbI2 phases.39,40 | ||
| Fig. 4 Variations in the optoelectronic properties of the perovskite films upon light soaking. (A) KPFM images (left) and extracted CPD distribution (right) of the perovskite film before and after light soaking. (B) c-AFM images (left) and extracted current distribution (right) of the perovskite film before and after light soaking. The applied voltage was 5 V in the test. (C) Schematic showing the spatial heterogenization behavior in terms of the work function and photocurrent of the perovskite polycrystalline film upon light soaking. | ||
The underlying mechanism of perovskite film evolution under light soaking is summarized in Fig. 4C. During the initial stages of light soaking, localized stress accumulation induced by the photomechanical expansion effect reduces the formation energy of iodine vacancy defects in the perovskite film, thereby accelerating the escape of iodine components from this region and leading to the establishment of initial degradation sites within the film (Stage II). As the duration of light soaking increases, the further depletion of iodine components results in the formation of PbI2 (Stage III). Along with decomposition, a heterogenization process involving the electrical potential, photocurrent and PL emission happens and is responsible for performance loss in solar cell devices. Particularly, the changes in the physical properties of GBs observed in our study, including charge transport and recombination processes, are jointly detrimental to device operation.
The universality of the photomechanically accelerated degradation mechanism
The photostriction phenomenon occurs in a variety of perovskite materials, and therefore, the photomechanically accelerated degradation mechanism can be extended to all kinds of perovskite polycrystalline thin films. We tested the photostriction of three more perovskite films, i.e., Cs0.1FA0.9PbI3 (CsFA), FAPbI3, and MAPbI3 by in situ XRD measurements under standard one-sun illumination. As shown in Fig. S24 to S26 (ESI†), all diffraction vectors q shifted to higher values, corresponding to an increase in the lattice constant. For instance, for the typical (100) lattice plane (or (110) for tetragonal MAPbI3), the interplanar lattice spacing expanded from 6.343 Å to 6.359 Å in Cs0.05FA0.81MA0.14PbI2.85Br0.15, 6.329 Å to 6.348 Å in Cs0.1FA0.9PbI3, 6.326 Å to 6.346 Å in FAPbI3, and 6.310 Å to 6.338 Å in MAPbI3, respectively yielding photostriction ratios of 0.252%, 0.3%, 0.316% and 0.444% (Fig. 5A and B). The driving mechanisms of photostriction in various materials can be of different microscopic origins. In polar materials, photoexcited carriers screen the polarization and change the internal electric field, which is commonly interpreted as a consequence of the converse piezoelectric effect.41,42 In contrast, above-bandgap illumination on non-polar materials creates an excess of electron–hole pairs in the conduction band, leading to the deformation of the sample directly or variations in atomic bonds.43 The employed halide perovskites, such as MAPbI3, undergo carrier transition from the hybridized Pb 6s–I 5p orbital to the Pb 6p orbital, which leads to electron density reduction on the iodine sites and weakening of the hydrogen bonds with ammonium molecules.44 As a result, the Pb–I–Pb bonds are straightened with increased interatomic spacing. When MA is replaced with larger FA or multiple cations, the perovskite lattice evolves from a tetragonal to a cubic structure, in which octahedral tilting becomes less pronounced. Zhou et al. also observed a much lower photostriction ratio in the cubic MAPbI3 single crystal at 70–90 °C than the tetragonal crystal at 30–50 °C, in good agreement with the above discussions.45 Furthermore, the tolerance factors of CsMAFA, CsFA, and FA perovskites were calculated to be 0.967, 0.970, and 0.987, respectively. Given that the optimal values range from 0.8–1.0, the calculated results indicate that the ternary cation perovskite has a close to ideal cubic structure, in which the reduced octahedral tilting might contribute to a low photostriction ratio.46 | ||
| Fig. 5 Consistency of the photostriction ratio and PCE decay ratio. (A) Variations in the lattice parameter of the Cs0.05FA0.81MA0.14PbI2.85Br0.15, Cs0.1FA0.9PbI3, FAPbI3, and MAPbI3 perovskite films under illumination. (B) The photostriction ratio of the four types of perovskite films. (C) Light-soaking stability of the PSCs under open-circuit conditions at 45 °C. The efficiency of the PSCs is normalized to the initial values. (D) Relationship between the photostriction ratio and PCE decay ratio. | ||
We hypothesize that such a discrepancy in the photomechanical photostriction ratio between the perovskite thin films might engender very different degradation rates under operation. Solar cells were fabricated with power conversion efficiencies (PCEs) >21% and light-soaked under one-sun illumination under open-circuit conditions (Fig. S27, ESI†). As expected, the ternary cation perovskite with the smallest photostriction ratio was the most stable, with only a 4.61% loss in PCE, among all tested cells after 500 h of solar exposure, whereas the MA cells with the highest photostriction ratio exhibited the largest PCE loss of 7.82% (Fig. 5C). Despite the severe intrinsic volatility of the MA cations, the device degradation statistics of the non-MA and less-MA cells complied with our assumption that perovskites with larger photostriction ratios experience faster PCE loss (Fig. 5D).
Notably, the photo-induced expansion of the perovskite lattice may improve efficiency via local strain relaxation in solar cell devices, but this phenomenon appears within the initial 50 h of light soaking. In our case, the stability tests spanned over a few hundred hours, exceeding this timescale. In addition, we found that the PbI2 products appeared at the GBs of these perovskite films, providing evidence for the same degradation mechanism (Fig. S28, ESI†). Further examination of transient photovoltage (TPV) decay pinpointed defect formation with reduced carrier lifetime in these devices after light soaking (Fig. S29, ESI†).47
Physical separation overcomes photomechanical phenomena in perovskites
The attention was then shifted to the release of photomechanical strain at the GBs to stabilize the perovskite layers. We hypothesized that isolating the granular grains using soft polymeric materials will avoid direct GB contact and release the photomechanical strain into the soft macromolecular phase. Trans-polyisoprene (TPI), a major component of natural rubber with a small Young's modulus of 17.7 MPa, was incorporated as an additive into the perovskite precursor solution (Fig. S30, ESI†).48 Due to its immiscibility with the crystalline perovskite structure, TPI was excluded and localized at the GBs after perovskite formation (Fig. 6A). In the overall-view TEM images, we found that most perovskite grains were physically isolated by the TPI phase with an average spacing of ∼3 nm between each grain (Fig. 6B). The corresponding EDS elemental distributions in Fig. 6C showcase that Pb and I were primarily located in the perovskite grains and did not appear at the GBs, indicating that TPI was outstanding in grain segregation. As the introduction of TPI at the GBs may impact the light absorption properties of the film, we conducted UV-vis absorption measurements of the modified films, and the results indicated that there was no significant decrease in the light absorption capacity (Fig. S31, ESI†). The cross-sectional SEM images indicated that the thickness of the modified film had slightly increased, which should leverage the light-harvesting capacity of the TPI-modified film (Fig. S32, ESI†). Besides, the employment of an appropriate amount of TPI rendered a champion PCE of 25.10% with negligible hysteresis (Fig. 6D and Fig. S33, ESI†). | ||
| Fig. 6 Improvement in device stability caused by the TPI strategy. (A) Schematic representation of the perovskite films with each grain isolated by TPI. (B) TEM image of perovskite grains surrounded by TPI. The crystal lattice spacing of 3.2 Å is derived from the (002) planes of the perovskite, and the SAED pattern of this region is shown in the inset. (C) Enlarged STEM image and the corresponding EDS elemental mapping of Pb and I. (D) J–V characteristics of the best-performing solar cell device with TPI. (E) Light-soaking stability of PSCs under open-circuit conditions at 45 °C. (F) MPP tracking of an encapsulated PSC with TPI under constant 1-sun illumination at 55 °C in air. | ||
We then investigated long-term device stability under one-sun irradiation at open-circuit conditions at 45 °C. As illustrated in Fig. 6E, the efficiency loss in the solar cell with TPI was only 1.45% after 500 h of light exposure, whereas the solar cell without TPI suffered a PCE loss of 4.60%. More importantly, solar cell with TPI retained 97.17% of the initial PCE after operation at the maximum power point (MPP) under AM 1.5G irradiation and 55 °C for 1000 h (Fig. 6F) and 93.15% of the initial PCE after operation at 85 °C for 300 h (Fig. S35, ESI†), again confirming the effectiveness of the GB strain isolation strategy in stabilizing perovskite devices against photomechanical damage. The SEM and XRD analyses shown in Fig. S36 and S37 (ESI†) further reveal no decomposition species in the perovskite film with TPI after prolonged light soaking for 300 h. Additionally, thermal admittance spectroscopy (TAS) measurements indicated that the trap-state density of the TPI devices remained almost unchanged after light soaking (Fig. S38, ESI†). These tests provide direct evidence of the stabilizing effect of the TPI additive in solar cell devices. Moreover, the TPI grain isolation strategy demonstrated excellent universality in PSCs with varying perovskite compositions (Fig. S39 and S40, ESI†).
Conclusion
In summary, we discover that perovskite degradation at GBs under illumination is predominantly dependent on whether they are in contact with the neighboring grains. Our experimental and theoretical studies together reveal a crucial degradation photo-induced mechanism that has not been explored previously, i.e., photomechanically accelerated degradation. We find that photo-induced lattice expansion leads to grain crowding and strain accumulation at the GBs, which favors iodine-related defect formation in the perovskite. A soft TPI layer has been used to separate the perovskite grains, which stabilizes PSCs with 97.17% PCE retention after irradiation at AM 1.5G and 55 °C for 1000 h. In fact, many reported ultrastable PSCs, such as those based on 2D/3D heterostructures and surface ligand engineering with long alkyl chains, also feature physical separation of the perovskite grains. Our results implicate that these interfaces not only protect/passivate perovskite grains but also mitigate the dynamic strain for improved device longevity. We anticipate that the discovery of photomechanically accelerated degradation will open up new avenues for designing long-lifetime PSCs for practical applications.Author contributions
H. Jin, Y. Hou and S. Yang directed the research. H. Wang performed the experiments and data analyses. Q. Li, Y. Zhu and M. Lin provided assistance with the SEM characterization. X. Sui, H. Yuan and Y. Zheng performed the calculations. H. Jin assisted with the AFM tests. X. Fan and Y. Zhou assisted with the KPFM and c-AFM tests. Y. Shi participated in the PL mapping characterization studies. All the authors participated in writing and editing the manuscript and contributed their efforts to the discussion.Data availability
The data for the plots presented in this paper and other findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (22379044, 52203330, 12304109, 52373114), National Ten Thousand Talent Program for Young Top-notch Talent, the Science and Technology Commission of Shanghai Municipality (21DZ1207101, 23520710700), the Key Program of the National Natural Science Foundation of China (22239001), Shanghai Pilot Program for Basic Research (22TQ1400100-5), “Dawn” Program of Shanghai Education Commission (22SG28), Shanghai Municipal Natural Science Foundation (22ZR1418000), Shanghai Sailing Program (22YF1410000, 22YF1413100), the Fundamental Research Funds for the Central Universities (JKD01241607, JKVD1241041), Shanghai Engineering Research Center of Hierarchical Nanomaterials (18DZ2252400), Shanghai Frontiers Science Center of Optogenetic Techniques for Cell Metabolism (Shanghai Municipal Education Commission).References
- Z. Huang, Y. Bai, X. Huang, J. Li, Y. Wu, Y. Chen, K. Li, X. Niu, N. Li, G. Liu, Y. Zhang, H. Zai, Q. Chen, T. Lei, L. Wang and H. Zhou, Nature, 2023, 623, 531–537 CrossRef CAS PubMed.
- Z. Liang, Y. Zhang, H. Xu, W. Chen, B. Liu, J. Zhang, H. Zhang, Z. Wang, D.-H. Kang, J. Zeng, X. Gao, Q. Wang, H. Hu, H. Zhou, X. Cai, X. Tian, P. Reiss, B. Xu, T. Kirchartz, Z. Xiao, S. Dai, N.-G. Park, J. Ye and X. Pan, Nature, 2023, 624, 557–563 CrossRef CAS.
- H. Chen, C. Liu, J. Xu, A. Maxwell, W. Zhou, Y. Yang, Q. Zhou, A. S. R. Bati, H. Wan, Z. Wang, L. Zeng, J. Wang, P. Serles, Y. Liu, S. Teale, Y. Liu, M. I. Saidaminov, M. Li, N. Rolston, S. Hoogland, T. Filleter, M. G. Kanatzidis, B. Chen, Z. Ning and E. H. Sargent, Science, 2024, 384, 189–193 CrossRef CAS PubMed.
- S. Liu, J. Li, W. Xiao, R. Chen, Z. Sun, Y. Zhang, X. Lei, S. Hu, M. Kober-Czerny, J. Wang, F. Ren, Q. Zhou, H. Raza, Y. Gao, Y. Ji, S. Li, H. Li, L. Qiu, W. Huang, Y. Zhao, B. Xu, Z. Liu, H. J. Snaith, N.-G. Park and W. Chen, Nature, 2024, 632, 536–542 CrossRef PubMed.
- M. J. Paik, Y. Y. Kim, J. Kim, J. Park and S. I. Seok, Joule, 2024, 8, 2073–2086 CrossRef CAS.
- Y. Shen, T. Zhang, G. Xu, J. A. Steele, X. Chen, W. Chen, G. Zheng, J. Li, B. Guo, H. Yang, Y. Wu, X. Lin, T. Alshahrani, W. Yin, J. Zhu, F. Wang, A. Amassian, X. Gao, X. Zhang, F. Gao, Y. Li and Y. Li, Nature, 2024, 635, 882–889 CrossRef CAS.
- H. Zhu, S. Teale, M. N. Lintangpradipto, S. Mahesh, B. Chen, M. D. McGehee, E. H. Sargent and O. M. Bakr, Nat. Rev. Mater., 2023, 8, 569–586 CrossRef.
- Y. Zhao, T. Heumueller, J. Zhang, J. Luo, O. Kasian, S. Langner, C. Kupfer, B. Liu, Y. Zhong, J. Elia, A. Osvet, J. Wu, C. Liu, Z. Wan, C. Jia, N. Li, J. Hauch and C. J. Brabec, Nat. Energy, 2022, 7, 144–152 CrossRef CAS.
- M. Wang, Z. Shi, C. Fei, Z. J. D. Deng, G. Yang, S. P. Dunfield, D. P. Fenning and J. Huang, Nat. Energy, 2023, 8, 1229–1239 CrossRef CAS.
- L. V. Torres Merino, C. E. Petoukhoff, O. Matiash, A. S. Subbiah, C. V. Franco, P. Dally, B. Vishal, S. Kosar, D. Rosas Villalva, V. Hnapovskyi, E. Ugur, S. Shah, F. Peña Camargo, O. Karalis, H. Hempel, I. Levine, R. R. Pradhan, S. Kralj, N. Kalasariya, M. Babics, B. K. Yildirim, A. A. Said, E. Aydin, H. Bristow, S. Mannar, W. Raja, A. R. Pininti, A. Prasetio, A. Razzaq, H. Al Nasser, T. G. Allen, F. H. Isikgor, D. Baran, T. D. Anthopoulos, M. M. Masis, U. Schwingenschlögl, T. Unold, M. Stolterfoht, F. Laquai and S. De Wolf, Joule, 2024, 8, 2585–2606 CrossRef CAS.
- H. Liu, Y. Gao, F. Xu, X. Zhang, A. Ullah, L. Xu, S. Zhang, J. Wang, S. De Wolf and H.-L. Wang, Adv. Funct. Mater., 2024, 34, 2315843 CrossRef CAS.
- X. Leng, Y. Zheng, J. He, B. Shen, H. Wang, Q. Li, X. Liu, M. Lin, Y. Shi, Z. Wei, Y. Peng, H. G. Yang, Q. Niu, S. Yang and Y. Hou, Energy Environ. Sci., 2024, 17, 4295–4303 RSC.
- X. Ren, J. Wang, Y. Lin, Y. Wang, H. Xie, H. Huang, B. Yang, Y. Yan, Y. Gao, J. He, J. Huang and Y. Yuan, Nat. Mater., 2024, 23, 810–817 CrossRef CAS.
- C. Fei, A. Kuvayskaya, X. Shi, M. Wang, Z. Shi, H. Jiao, T. J. Silverman, M. Owen-Bellini, Y. Dong, Y. Xian, R. Scheidt, X. Wang, G. Yang, H. Gu, N. Li, C. J. Dolan, Z. J. D. Deng, D. N. Cakan, D. P. Fenning, Y. Yan, M. C. Beard, L. T. Schelhas, A. Sellinger and J. Huang, Science, 2024, 384, 1126–1134 CrossRef CAS.
- S. Yu, Z. Xiong, H. Zhou, Q. Zhang, Z. Wang, F. Ma, Z. Qu, Y. Zhao, X. Chu, X. Zhang and J. You, Science, 2023, 382, 1399–1404 CrossRef CAS PubMed.
- Z. Li, X. Sun, X. Zheng, B. Li, D. Gao, S. Zhang, X. Wu, S. Li, J. Gong, J. M. Luther, Z. a Li and Z. Zhu, Science, 2023, 382, 284–289 CrossRef CAS PubMed.
- X. Lin, H. Su, S. He, Y. Song, Y. Wang, Z. Qin, Y. Wu, X. Yang, Q. Han, J. Fang, Y. Zhang, H. Segawa, M. Grätzel and L. Han, Nat. Energy, 2022, 7, 520–527 CrossRef CAS.
- T. Kim, S. Park, V. Iyer, B. Shaheen, U. Choudhry, Q. Jiang, G. Eichman, R. Gnabasik, K. Kelley, B. Lawrie, K. Zhu and B. Liao, Nat. Commun., 2023, 14, 1846 CrossRef CAS.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef.
- D. J. Slotcavage, H. I. Karunadasa and M. D. McGehee, ACS Energy Lett., 2016, 1, 1199–1205 CrossRef CAS.
- N. Ahn, K. Kwak, M. S. Jang, H. Yoon, B. Y. Lee, J.-K. Lee, P. V. Pikhitsa, J. Byun and M. Choi, Nat. Commun., 2016, 7, 13422 CrossRef CAS.
- W. Li, M. Hao, A. Baktash, L. Wang and J. Etheridge, Nat. Commun., 2023, 14, 8523 CrossRef CAS PubMed.
- T.-C. Wei, H.-P. Wang, T.-Y. Li, C.-H. Lin, Y.-H. Hsieh, Y.-H. Chu and J.-H. He, Adv. Mater., 2017, 29, 1701789 CrossRef PubMed.
- B. Chen, T. Li, Q. Dong, E. Mosconi, J. Song, Z. Chen, Y. Deng, Y. Liu, S. Ducharme, A. Gruverman, F. D. Angelis and J. Huang, Nat. Mater., 2018, 17, 1020–1026 CrossRef CAS.
- D.-J. Xue, Y. Hou, S.-C. Liu, M. Wei, B. Chen, Z. Huang, Z. Li, B. Sun, A. H. Proppe, Y. Dong, M. I. Saidaminov, S. O. Kelley, J.-S. Hu and E. H. Sargent, Nat. Commun., 2020, 11, 1514 CrossRef CAS.
- H. Tsai, R. Asadpour, J.-C. Blancon, C. C. Stoumpos, O. Durand, J. W. Strzalka, B. Chen, R. Verduzco, P. M. Ajayan, S. Tretiak, J. Even, M. A. Alam, M. G. Kanatzidis, W. Nie and A. D. Mohite, Science, 2018, 360, 67–70 CrossRef CAS PubMed.
- J. Zhao, Y. Deng, H. Wei, X. Zheng, Z. Yu, Y. Shao, J. E. Shield and J. Huang, Sci. Adv., 2017, 3, eaao5616 CrossRef PubMed.
- N. Rolston, K. A. Bush, A. D. Printz, A. Gold-Parker, Y. Ding, M. F. Toney, M. D. McGehee and R. H. Dauskardt, Adv. Energy Mater., 2018, 8, 1802139 CrossRef.
- B. Kundys, Appl. Phys. Rev., 2015, 2, 011301 Search PubMed.
- X. Liu, H. Lian, Z. Zhou, C. Zou, J. Xie, F. Zhang, H. Yuan, S. Yang, Y. Hou and H. G. Yang, Adv. Energy Mater., 2022, 12, 2103933 CrossRef CAS.
- A.-F. Castro-Méndez, J. Hidalgo and J.-P. Correa-Baena, Adv. Energy Mater., 2019, 9, 1901489 CrossRef.
- D.-Y. Son, J.-W. Lee, Y. J. Choi, I.-H. Jang, S. Lee, P. J. Yoo, H. Shin, N. Ahn, M. Choi, D. Kim and N.-G. Park, Nat. Energy, 2016, 1, 16081 CrossRef CAS.
- J. Bang, W. S. Lee, B. Park, H. Joh, H. K. Woo, S. Jeon, J. Ahn, C. Jeong, T.-I. Kim and S. J. Oh, Adv. Funct. Mater., 2019, 29, 1903047 CrossRef.
- S. G. Motti, D. Meggiolaro, S. Martani, R. Sorrentino, A. J. Barker, F. De Angelis and A. Petrozza, Adv. Mater., 2019, 31, 1901183 CrossRef CAS PubMed.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 RSC.
- W.-Q. Wu, P. N. Rudd, Z. Ni, C. H. Van Brackle, H. Wei, Q. Wang, B. R. Ecker, Y. Gao and J. Huang, J. Am. Chem. Soc., 2020, 142, 3989–3996 CrossRef CAS PubMed.
- A. R. Nirjhar, S. J. Tan-Ema, M. A. Sahriar, M. N. Ahsan Dipon, M. R. Hasan Abed, D. B. Gainza, A. Koneru, S. S. Nishat, K. M. Shorowordi and S. Ahmed, Int. J. Hydrogen Energy, 2023, 48, 37273–37285 CrossRef CAS.
- C. Wang, Y. Jiang, H. Xu, N. Zheng, G. Bai, Y. Zha, H. Qi, Z. Bian, X. Zhan and Z. Liu, eScience, 2023, 3, 100113 CrossRef.
- W. Nie, J.-C. Blancon, A. J. Neukirch, K. Appavoo, H. Tsai, M. Chhowalla, M. A. Alam, M. Y. Sfeir, C. Katan, J. Even, S. Tretiak, J. J. Crochet, G. Gupta and A. D. Mohite, Nat. Commun., 2016, 7, 11574 CrossRef CAS PubMed.
- T. Wang, T. Fang, X. Li, L. Xu and J. Song, J. Phys. Chem. C, 2021, 125, 5475–5484 CrossRef CAS.
- C. Paillard, B. Xu, B. Dkhil, G. Geneste and L. Bellaiche, Phys. Rev. Lett., 2016, 116, 247401 CrossRef PubMed.
- C. Paillard, S. Prosandeev and L. Bellaiche, Phys. Rev. B, 2017, 96, 045205 CrossRef.
- B. Peng, D. Bennett, I. Bravić and B. Monserrat, Phys. Rev. Mater., 2022, 6, L082401 CrossRef CAS.
- C. Chen and Z. Yi, Adv. Funct. Mater., 2021, 31, 2010706 CrossRef.
- Y. Zhou, L. You, S. Wang, Z. Ku, H. Fan, D. Schmidt, A. Rusydi, L. Chang, L. Wang, P. Ren, L. Chen, G. Yuan, L. Chen and J. Wang, Nat. Commun., 2016, 7, 11193 CrossRef CAS.
- M. Saliba, T. Matsui, K. Domanski, J.-Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J.-P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- B. Chen, J. Song, X. Dai, Y. Liu, P. N. Rudd, X. Hong and J. Huang, Adv. Mater., 2019, 31, 1902413 CrossRef PubMed.
- J.-S. Song, B.-C. Huang and D.-S. Yu, J. Appl. Polym. Sci., 2001, 82, 81–89 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04878d |
| This journal is © The Royal Society of Chemistry 2025 |