
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConducting polymer transforms hydrophobic porous membranes into robust gas diffusion layers in electrochemical applications†
Hwiyoon
Noh
 ,
Hyunki
Yeo
,
Bryan W.
Boudouris
* and
Brian M.
Tackett
*
,
Hyunki
Yeo
,
Bryan W.
Boudouris
* and
Brian M.
Tackett
*
Charles D. Davidson School of Chemical Engineering, Purdue University, West Lafayette, IN 47907, USA. E-mail: boudouris@purdue.edu; bmtacket@purdue.edu; Fax: +1-765-494-0805; Tel: +1-765-496-6056 Tel: +1-765-496-7235
First published on 11th December 2024
Abstract
The increasing demand for sustainable chemical production due to strict regulations for carbon emission aligns with growing availability of solar and wind energy, making electrochemical manufacturing a viable route toward decarbonized chemical syntheses. Electrodes with gas diffusion layers (GDLs) critically enhance reaction efficiency for continuous-flow electrochemical reactors with liquid electrolytes fed with gaseous reactants, but they currently suffer from challenges like electrolyte flooding and poor long-term stability. Porous polytetrafluoroethylene (PTFE) membrane-based GDLs overcome some of these issues, but they require additional functionality to enable conductivity. Herein, we demonstrate a novel GDL structure, introducing a porous conductive polymer, poly(3,4-ethylenedioxythiophene) (PEDOT), onto a porous PTFE membrane. Compared to a carbon-based GDL, the PEDOT-coated PTFE GDL exhibited similar electrochemical performance with enhanced stability under industrially relevant conditions for the CO2 reduction reaction. PEDOT-coated PTFE GDL demonstrates remarkable resistance to electrolyte flooding, making it a promising candidate for various gas-fed electrocatalytic reactions.
Broader contextThe push for sustainable chemical production is accelerating in response to increasingly stringent carbon-emission regulations. Simultaneously, global expansion of renewable energy makes decarbonized electrochemical synthesis a viable route to achieve this goal. A significant portion of possible industrial electrochemical transformations involves gaseous reactants, necessitating the use of electrodes with gas diffusion layers (GDLs) to achieve industrially relevant reaction rates. To date, the commonly used GDLs in electrochemical cells that employ liquid electrolytes and gaseous reactants either fail after several hours of operation or they require thick metal layers to conduct current, which is impractical for many electrocatalytic reactions. These hurdles impede progress toward sustainable chemical manufacturing. Here, we developed a new GDL configuration that adds a conductive polymer on top of an inherently robust polytetrafluoroethylene layer to enable efficient catalysis and exceptional long-term stability, overcoming the major issues with previous GDL designs. This new paradigm – a self-conductive all-polymer GDL – is versatile and tunable, providing a technological backbone to enable decarbonization of a wide swath of the chemical industry that currently contributes roughly 4% of global CO2 emissions. This innovation sits at the intersection of electrocatalysis and optoelectronics fields, highlighting the importance of interdisciplinary work in pursuit of a sustainable future. |
Introduction
The demand for sustainable chemical production is rapidly increasing as carbon-emission regulations tighten. At the same time, the rapid global influx of solar and wind energy facilitates decarbonized chemical synthesis routes via electrification. In particular, electrochemical manufacturing is a promising way to produce valuable chemical commodities, powered by renewable energy.1–4 A substantial fraction of these envisioned decarbonized electrochemical pathways involves gaseous reactants, which require electrodes that incorporate gas diffusion layers (GDLs) in continuous flowing electrolyzers to mitigate low solubility and the slow diffusion of reactants.5–8 The most prominent example of this technology in recent work is for the electrochemical CO2 reduction reaction (CO2RR), where GDLs enable conversion of CO2 into high-value products at much greater current densities, compared to standard electrodes in batch cells. This is achieved by porous channels in the GDL transporting reactants from a gas stream to continuously replenish CO2 in the aqueous phase near the electrode–electrolyte interface. Critically, the GDL also acts as a conductive current collector to facilitate electron transfer through the external circuit, making porous carbon paper the traditional GDL material of choice. The major challenge for this technology, however, is to prevent the liquid electrolyte from filling the gas channels and increasing mass transport resistance of gaseous reactant to the electrocatalyst surface – this is known as flooding in the GDL. This often causes undesired side reactions, and it decreases energy efficiency.6,7,9 In traditional carbon-based GDLs, flooding tends to occur at high reaction rates after several hours of operation, posing a critical barrier to desired performance for industrial chemicals manufacturing.Recently, porous polytetrafluoroethylene (PTFE) membranes were used to replace carbon-based GDLs, taking advantage of the chemical stability and inherent hydrophobicity of PTFE. Due to the non-conductive nature of PTFE, magnetron sputtering of metal layers was required in previous work to impart conductivity, and this strategy showed enhanced catalytic performance and improved stability in CO2RR when the metal layer was catalytically suitable for the desired reaction.10–20 Despite these advances, the typical preparation method of sputtering metal with a thickness of 300–500 nm results in complications and drawbacks for general use in electrochemical devices. For example, these thin metal films coated on porous PTFE still suffered from low electrical conductivity, requiring additional processing steps to improve performance, like adding additional current collecting layers composed of carbon, thick copper, polymer-coated grids, or aluminum.19–22 These methods require several additional steps to make current collecting layers, and the metal or carbon layers can catalyze reactions, possibly resulting in unwanted side products. The most significant drawback of PTFE GDLs that impedes progress for CO2RR and other decarbonized electrochemical reactions, is its severe limitation on electrocatalyst composition and morphology. Rational design of electrocatalysts, relying on structure–function relationships, has resulted in improved performances that employ various metal and alloy particles with a range of sizes, compositions, morphologies, and preferentially exposed facets.23 But exploiting these concepts in the current configuration of porous PTFE GDL is essentially impossible when conductivity is established by a sputtered contiguous metal film. Thus, enabling the best catalyst performance in combination with the robust and hydrophobic porous PTFE GDL requires a non-metal conductive layer, which will not contribute to the reaction, is compatible with any electrocatalyst motif, and maintains desired gas diffusion properties. To achieve this, we designed a new GDL structure composed of a porous conductive polymer layer assembled on a microporous PTFE layer in the first demonstration of a self-conductive PTFE-based all-polymer GDL.
We selected poly(3,4-ethylenedioxythiophene) (PEDOT) as the conductive layer, due to its high electrical conductivity, mechanical durability, and successful implementation for diverse applications.24,25 Moreover, PEDOT is low-cost and has wide availability.26 Recent work also employs PEDOT for electrocatalytic reactions in multiple ways, for example, as an anchoring structure or coordination modifier, but the usage of PEDOT as a GDL component on PTFE has not been reported.27,28 PEDOT can also be fabricated in any form factor, making it a logical choice to add electrical conductivity, while maintaining the original properties of the PTFE GDL.
In this work, we first synthesized PEDOT doped with PF6− (PEDOT:PF6) on commercial PTFE membranes to form a thin, porous, and electrically conductive layer via electropolymerization, resulting in a PEDOT-coated PTFE (PEDOT–PTFE) GDL. The self-conductive GDL was then evaluated in a continuous flowing electrolyzer, with CO2RR as a probe reaction. The CO2RR performance of nanoparticle electrocatalysts assembled in this configuration was evaluated in acidic, neutral, and alkaline electrolytes. Importantly, PEDOT–PTFE GDLs showed comparable results with those of the commercial carbon-based GDL, Sigracet 22bb, during short-term testing. Lastly, CO2RR stability tests were executed at industrially relevant current densities (i.e., −100 mA cm−2 and −200 mA cm−2), in which PEDOT–PTFE GDLs exhibited remarkable resistance to electrolyte flooding compared to the carbon-based GDL in all electrolytes tested. These results highlight that the PEDOT layer imparts sufficient electrical conductivity to enable reaction on electrocatalyst nanoparticles, while maintaining the best properties of robust porous PTFE. Thus, PEDOT–PTFE can be widely used for any gas-fed electrocatalytic reactions requiring the application of a GDL, while providing better stability than carbon-based GDLs.
Results and discussion
Synthesis and microscopic characterization of PEDOT–PTFE
The electrochemically synthesized PEDOT on PTFE membrane (PEDOT–PTFE) was fabricated through a three-step process (details in Experimental section) depicted in Fig. S1 (ESI†). Briefly, 150 nm of Au was deposited onto the PTFE membrane via thermal evaporation. Then, EDOT was electrochemically oxidized on the Au-coated PTFE membrane to initiate polymerization,29 using the setup shown in Fig. S2 (ESI†), and forming a thin and porous PEDOT layer. Molecular structures of monomer, EDOT, and polymer, PEDOT, are shown in Fig. S3 (ESI†) for a summary of this process. The Au layer was selectively dissolved by aqua regia while keeping the PEDOT layer attached to the top of the PTFE membrane. A schematic illustration of the structure of PEDOT–PTFE is shown in Fig. 1(a), indicating two different layers, PEDOT (dark blue) and PTFE (white). The top-view scanning electron microscopy (SEM) image of the pristine PTFE membrane exhibited a comparatively thick and interconnected fibrous structure of PTFE, making pores for gas diffusion (Fig. S4, ESI†). After the electropolymerization, the morphology of the GDL surface was significantly altered, as shown in Fig. 1(b). The image revealed thin PEDOT chains interconnected to each other to form a contiguous polymeric network. Critically, this morphology forms void spaces that should maintain gas permeability while enabling electrical conductivity (1590 ± 140 S cm−1), shown in Fig. S5 (ESI†), through the PEDOT network. Notably, the change in electrical conductivity induced by aqua regia treatment was marginal (Fig. S5, ESI†). Such structure is likely characteristic of the electropolymerized PEDOT with PF6− dopant, as prior work reported a similar appearance for PEDOT:PF6.26,30,31 Thus, electropolymerization created a conductive porous PEDOT layer on top of inherently hydrophobic PTFE, which should function well as a GDL. | ||
| Fig. 1 Microscopic characterization of PEDOT–PTFE. (a) Illustration of PEDOT–PTFE. SEM images of PEDOT–PTFE from (b) top-view and (c) cross-section using FIB. The black scale bars in (b) and (c) indicate 2 μm. (d) HAADF-STEM image of cross-section of PEDOT–PTFE and (e) overlapped EDX mapping image of C and S. | ||
The PEDOT structure and its interface with PTFE were probed with cross-sectional electron microscopy. The SEM images of the GDL cross-section obtained by focused ion beam (FIB) are shown in Fig. 1(c). The two distinct PEDOT and PTFE layers were easily distinguished. The top layer shows the interconnected PEDOT network with void spaces distributed evenly throughout the cross-section thickness. The bottom layer exhibits image distortion and contrast irregularity, general characteristics of non-conductive material, consistent with the non-conductive nature of PTFE. The thickness of the PEDOT layer is around 1 μm based on the cross-section SEM image. To further probe the structure of the PEDOT fibers, a cross-sectional high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of the PEDOT layer was obtained and shown in Fig. 1(d). The PEDOT fibers were ∼20 nm thick with randomly distributed voids. The overlapped energy dispersive X-ray (EDX) mapping image of carbon and sulfur in Fig. 1(e) indicated the fiber composition is consistent with that of sulfur-containing PEDOT. The presence of phosphorus in the fibers (Fig. S6, ESI†) was a result of PF6−, which is doped into the PEDOT backbone, facilitating conductivity. It is also notable that some Au species were observed in HAADF-STEM EDX (Fig. S6, ESI†) even after the aqua regia treatment. To better quantify the extent of Au remaining, SEM/EDX analysis covering a comparatively wide area (Fig. S7, ESI†), revealed the atomic percent of Au decreased from 12.6% before aqua regia treatment to 0.3% after the treatment, confirming that the majority of Au was removed. Thus, even if Au is distributed in small quantities throughout the PEDOT fiber, this GDL preparation method avoids undesirable properties of sputtered metal current collectors exposed to electrolyte.
Spectroscopic characterization of PEDOT–PTFE
The results of Raman, Fourier transform infrared (FTIR), and X-ray photoemission spectroscopy (XPS) are shown in Fig. 2 to elucidate structural features of PEDOT and confirm its formation on the PTFE membrane. The Raman spectrum (Fig. 2(a)) shows three bands consistent with a PTFE signature, likely due to laser penetration through the thin and porous PEDOT layer. The spectrum mainly depicts some bands originating from oxyethylene and thiophene rings and characteristic vibrational bands of PEDOT from 1250 to 1560 cm−1, which is associated with the stretching between Cα and Cβ. Moreover, the FTIR spectrum (Fig. 2(b)) exhibits multiple peaks related to the vibrations within those rings and carbon–carbon bonds. Both Raman and FTIR spectra suggest the successful formation of the thin PEDOT layer effectively covering the PTFE membrane. | ||
| Fig. 2 Spectroscopic characterization of PEDOT–PTFE. (a) Raman spectra of PTFE (black) and PEDOT–PTFE (blue). (b) FTIR spectrum of PEDOT–PTFE. The inset represents the spectrum of pristine PTFE. (c) Survey scan XPS spectrum of PEDOT–PTFE. | ||
The XPS survey spectrum in Fig. 2(c) provides the elemental composition of PEDOT at the surface, indicating the existence of six components (C, F, O, S, Cl, and Au), in good agreement with the SEM/EDX result (Fig. S7, ESI†). In addition, N species were additionally observed, likely due to the overoxidation of PEDOT during the electropolymerization,32 but this is not expected to significantly alter the physical properties of PEDOT. To define the chemical states of each component, high-resolution XPS spectra were further obtained in Fig. S8 and S9 (ESI†) and quantification of each component with peak assignation is shown in Table S1 (ESI†). It is noteworthy that both Au and Cl were not detected in high-resolution XPS spectra before the aqua regia treatment in Fig. S9 (ESI†). This indicates that the aqua regia treatment is responsible for Cl introduction, and migration and introduction of Au into the PEDOT structure. Considering the observation of both the oxidized Au and Cl ions, it is likely that ionic species (e.g., AuCl4−) act as an additional dopant in the PEDOT structure. This is consistent with the presence of Au in STEM/EDX results, and it may explain why not all of the Au was removed with aqua regia. The combination of the spectroscopic results indicates that PEDOT maintained its expected chemical and structural composition after aqua regia treatment, although minor structural changes with respect to addition of dopants and overoxidation were detected.
Comparison of CO2RR performance between PEDOT–PTFE and carbon-based GDL
To demonstrate the utility of PEDOT–PTFE as an effective gas diffusion layer, its performance was compared to that of commercial carbon-based Sigracet 22bb in a continuous-flow electrochemical cell (Fig. S10, ESI†). CO2RR on Ag was selected as a representative test case, because Ag selectively produces CO as the only carbon product from the gaseous CO2 reactant. Thus, tracking the faradaic efficiency in a flow cell with Ag electrocatalysts indicates the mass transfer performance – more CO means CO2 mass transport is facilitated through the GDL, while more H2 indicates gas channels may be flooded, resulting in reaction of the aqueous electrolyte. This simple system enables direct evaluation of GDL mass transport properties during a catalytic reaction.The CO2RR performance of the PEDOT–PTFE with Ag nanoparticle (AgNP) mass loading of 1.0 mg cm−2 in different electrolytes is shown in Fig. 3, to examine its performance compared to a carbon-based GDL. Three electrolytes were selected; 0.8 M KOH, 0.8 M KHCO3, and 0.01 M H2SO4 with 0.4 M K2SO4, representing alkaline, neutral, and acidic electrolytes, respectively, to show the versatility of the PEDOT–PTFE GDL across a range of common aqueous electrolysis conditions. Sigracet was also tested under identical conditions and compared as a representative of a widely used carbon-based GDL.33,34 During constant current experiments, PEDOT–PTFE showed comparable results to Sigracet in terms of potential required to achieve a specific current and product selectivity for all electrolytes (Fig. 3). This indicates that PEDOT–PTFE imparts sufficient conductivity and gas permeability to perform efficient CO2RR without the need for a contiguous sputtered metal layer. Additionally, conducting the same experiment on bare PEDOT–PTFE without catalyst present (Fig. S11, ESI†) showed negligible electrocatalytic reaction, indicating the contribution from PEDOT–PTFE and remaining Au species can be excluded from observed results. The faradaic efficiency (FE) of H2 from Ag NP on PEDOT–PTFE was less than 1.5% in acidic and alkaline electrolytes up to currents of −150 mA cm−2, whereas H2 FE exceeded 1.7% in the case of Sigracet in the same regime (Fig. 3). At the high current of −200 mA cm−2, PEDOT–PTFE exhibited slightly increased H2 FE (ca. 1.8%), which was still on-par with Sigracet at that current. The H2 FE was higher on PEDOT–PTFE in the neutral electrolyte (between 1.9 and 4.2%), but it was still comparable to Sigracet. Other than H2, CO was the only gas product detected in all current densities, and the CO faradaic efficiencies at each current density were comparable for both GDLs in all electrolytes.
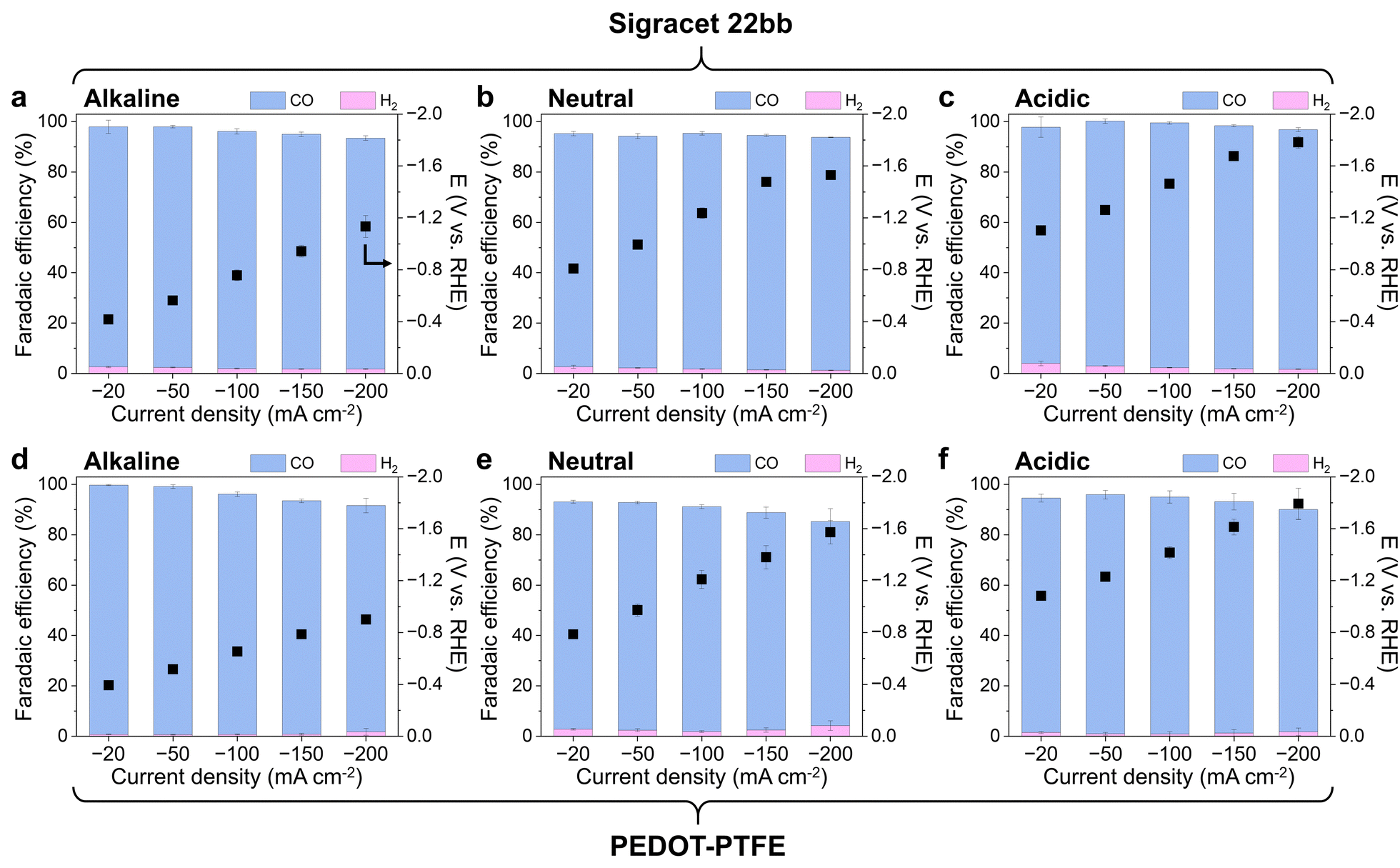 | ||
| Fig. 3 CO2RR performance in various electrolytes. CO2RR activity at constant current densities of −20, −50, −100, −150, and −200 mA cm−2 of AgNP 1 mg cm−2 on (a)–(c) Sigracet 22bb and (d)–(f) PEDOT–PTFE in terms of potential (right y-axis) and faradaic efficiencies (left y-axis) for CO (blue) and H2 (pink). For alkaline, neutral, and acidic electrolytes, 0.8 M KOH, 0.8 M KHCO3, and 0.01 M H2SO4 with 0.4 M K2SO4 were used, respectively. The error bars indicate standard deviations from three independent electrodes. For short-term CO2RR measurement, each current step was maintained for 20 min. | ||
The sum of faradaic efficiencies reveals the effectiveness of product mass transfer through the GDL. Assuming no liquid products are formed, FE should sum to 100% for each measurement. A lower summation means some gas products were not efficiently transported through the GDL and were instead trapped in the catholyte stream and not detected by GC.35 In all cases, the FE summation decreases at higher current densities, but this effect is more pronounced on PEDOT–PTFE. This result is expected, because a higher flux of gas products will experience greater mass transfer resistance through GDL pores. While the PEDOT–PTFE configuration performs exceptionally well at low current densities, the pore size and structure of the PEDOT layer have not yet been optimized, leading to greater diffusion barriers at high current density. Specifically, the structure of PEDOT–PTFE consists of two microporous layers in series – PEDOT and PTFE – to achieve both conductivity and hydrophobicity. This adds resistance to gas diffusion, but this effect could be mitigated by controlling the PEDOT pore size and structure. Such tuning may be achieved by modifying electropolymerization conditions or dopant identity,26,36 which is a target of future work. Even without such optimization, the PEDOT–PTFE captures roughly 90% of gas products at high current density, proving its efficacy as a GDL.
Stability comparison between PEDOT–PTFE and carbon-based GDL
The main benefit of a PTFE-based GDL is enhanced stability against electrolyte flooding, so 20 h CO2RR stability tests were carried out to evaluate the durability of PEDOT–PTFE compared to Sigracet 22bb in alkaline, neutral, and acidic electrolytes. The results in Fig. 4 show CO FE and CO2 mass flow rate at constant current over time. Current densities of −100 and −200 mA cm−2 were chosen for stability tests, because they represent industrially relevant operating conditions.37,38 These conditions also create an environment conducive to electrolyte flooding phenomena observed for carbon-based GDL failure. Electrolyte flooding becomes severe at high current due to the decrease in breakthrough pressure, attributed to electrolyte carbonation and salt formation, leading to decrease of hydrophobicity of GDLs.34,39,40 In these tests, the CO FE provides an evaluation of the extent of gas channel blockage (either by flooding or some other mechanism), where it is assumed that decreased CO FE in favor of H2 results from an increase in the diffusion path length of reactant CO2 from the gas–liquid interface to the liquid–electrocatalyst interface. The flowrate of CO2 through the gas channel, measured by the mass flow controller (Fig. 4 and Fig. S12–S16, ESI†), was also used as a GDL flooding indicator. Observation of uncontrolled flow rate oscillations suggested severe flooding of the entire GDL thickness, resulting in liquid impeding CO2 flow on the backside of the GDL. Each condition was triplicated with independent GDLs (Fig. S12 and S13, ESI†), confirming these trends in CO FE and electrolyte flooding are repeatable during stability tests. | ||
| Fig. 4 Stability tests in various electrolytes. CO2RR stability test of AgNP 1 mg cm−2 on PEDOT–PTFE (blue) and Sigracet 22bb (black) at constant current densities of (a), and (b) −100 mA cm−2 and (c), and (d) −200 mA cm−2 in alkaline (0.8 M KOH), neutral (0.8 M KHCO3), and acidic (0.01 M H2SO4 with 0.4 M K2SO4) electrolytes. (a), and (c) CO faradaic efficiency obtained from GC and (b), and (d) CO2 mass flow rate recorded by MFC along with the operating time. | ||
In alkaline environment (0.8 M KOH), CO FE decreases continuously over time for both GDLs, but this is substantially mitigated by PEDOT–PTFE. The CO FE decreases below 60% on Sigracet in less than 16 hours of operation at both −100 and −200 mA cm−2, at which point the experiment was stopped. On the other hand, the PEDOT–PTFE enabled high CO FE for the duration of the 20-hour test, maintaining 93.9% at −100 mA cm−2 and 90.0% at −200 mA cm−2 at the end of testing. This clearly shows the improved resistance to flooding for the PEDOT–PTFE configuration in the alkaline electrolyte. It should be noted, however, that severe flooding affecting the gas flow rate was not observed for either GDL (Fig. 4 and Fig. S14, ESI†) in these conditions.
The stability tests in neutral electrolyte (0.8 M KHCO3) showed different trends compared to the alkaline case, but still demonstrated enhanced durability of PEDOT–PTFE over Sigracet. Notably, Sigracet maintained very high CO FE for both currents (near 100%) in the first four hours, while PEDOT–PTFE decreased moderately from 90% to 80% in the same time. Sigracet held this performance until 16 hours at −100 mA cm−2, and then decreased continuously to the end of the 20 h test. This decrease was coincident with CO2 flowrate oscillations (Fig. 4 and Fig. S15a1, ESI†), indicative of severe flooding. The same flowrate oscillations for Sigracet were observed around 7 h at −200 mA cm−2, which was followed by a precipitous drop in CO FE that is considered GDL failure (Fig. 4 and Fig. S15b1, ESI†). PEDOT–PTFE avoided severe flooding under these conditions, showing stable CO FE for the duration of the 20-hour test at both currents (Fig. 4 and Fig. S15c1, d1, ESI†). The performance enhancement of PEDOT–PTFE is especially evident for −200 mA cm−2, where the Sigracet flooded and failed at an early stage.
Stability tests in acidic electrolyte are of particular interest, because CO2RR in acidic conditions has much higher maximum theoretical energy efficiency than neutral or alkaline electrolytes. This is due to homogeneous consumption of CO2 by hydroxide ions that occurs to a lesser extent in acids.10,41Fig. 4 shows that performance with both GDLs exhibited lower CO FE over the stability tests in acid compared to the other electrolytes, but again, PEDOT–PTFE outperformed the carbon-based GDL. Both GDLs showed similar performance under −100 mA cm−2, decreasing from 90% to 80% CO FE in the first 14 h. Then, CO FE decreases rapidly for Sigracet, while PEDOT–PTFE continues a smooth but shallower decrease to the end of the 20 h test. The point at which Sigracet performance rapidly decreases is accompanied by some moderate CO2 flowrate oscillations, which may indicate severe flooding (Fig. 4 and Fig. S16a1, ESI†). These flowrate oscillations were not observed for PEDOT–PTFE. The same phenomenon was seen on Sigracet at −200 mA cm−2, but it occurred after only 5 hours, and caused rapid GDL failure (Fig. 4 and Fig. S16b1, ESI†). This is in stark contrast to PEDOT–PTFE, which showed no signs of severe flooding for the 20-hour duration at the same current density (Fig. 4 and Fig. S16d1, ESI†). It is notable that the acidic condition did cause more significant and continuous reduction in CO FE on PEDOT–PTFE, compared to other electrolytes, and this may be related to low solubility of K2SO4 (12 g/100 mL H2O) compared to the solubility of KHCO3 (22.4 g/100 mL H2O) and KOH (55 g/100 mL H2O). High current densities could create high local ion concentrations near the electrode, resulting in salt precipitation that could impede CO2 transport to the electrocatalyst. This problem should be addressable by judicious choice of supporting electrolyte as well as controlling PEDOT pore structures. Despite this, it remains clear that PEDOT–PTFE is a more robust GDL option in the acidic electrolyte than Sigracet.
All cases in Fig. 4 exhibited CO FE decrease during operation, which can be attributed to three different phenomena: mild flooding, severe flooding, and pore blockage via solid precipitate (which can be exacerbated as a result of flooding). Considering precipitate formation should occur similarly on both GDLs in non-flooded conditions, this points to flooding as a key differentiator of GDL stability. Severe flooding clearly afflicted Sigracet's stability in neutral and acidic electrolytes, but the enhanced performance of PEDOT–PTFE in alkaline electrolyte was not attributed to this same effect. Instead, mild flooding that does not fully penetrate the GDL thickness, could also reduce CO FE by increasing CO2 mass transfer resistance. To analyze this effect, cross-section SEM/EDX was conducted after stability tests to detect traces of electrolyte or precipitates. The majority of K species was observed within remaining catalyst layer in PEDOT–PTFE, while K species were prominently distributed throughout the microporous layer of Sigracet (Fig. S17–S19, ESI†). Even though none of signals related to severe flooding were observed in some cases of Sigracet, the results showed the presence of electrolyte penetration to GDL, considered mild flooding. This shows that PEDOT–PTFE has higher resistance to any kind of flooding, whereas Sigracet allows greater extent of flooding, leading to decreased GDL performance. This type of mild flooding can also cause compounding GDL degradation on carbon-based GDLs, which can also catalyze unwanted HER. Such reaction can decrease pressure needed for electrolyte breakthrough and can change wetting characteristics of the carbon, causing faster electrolyte flooding.39,42 PTFE, on the other hand, is highly hydrophobic and non-conductive, which prohibiting its participation in electrochemical reaction. This explains why PEDOT–PTFE did not show electrolyte flooding characteristics for all electrolytes regardless of current density whereas Sigracet experienced both mild and severe electrolyte flooding. This makes PEDOT–PTFE a preferable GDL candidate for extended operation.
As an example of its applicability in this regard, PEDOT–PTFE was further tested at −100 mA cm−2 in the neutral electrolyte for much longer time (80 h), shown in Fig. S20 (ESI†). PEDOT–PTFE did not show any characteristics related to continuous electrolyte flooding for more than 3 days. The CO faradaic efficiency did decrease to 64.3% at 81 h, with a corresponding increase in H2 faradaic efficiency. This behavior suggests the decrease is due to catalyst detachment and decreased gas diffusion ability attributed to salt precipitation during the extended operation, which were both observed in post-mortem SEM/EDX characterization of the GDL (Fig. S21, ESI†). Despite the decrease in CO faradaic efficiency, the origin of the decrease is clearly distinct from the severe flooding that caused GDL failure for Sigracet. Thus, PEDOT–PTFE shows versatile application as a GDL across a range of operating conditions and electrolyte pH, making it a promising candidate to replace carbon-based GDLs. Its hydrophobic nature imparts enhanced stability against electrolyte flooding and the conductive polymer enables current collection without catalytic contributions from a sputtered metal layer.
Conclusion
In this work, we synthesized PEDOT:PF6 as a conductive layer on a PTFE membrane through electropolymerization to create a self-conductive, hydrophobic, and robust GDL. The PEDOT layer was composed of interconnected polymer chains, roughly 20 nm thick, that created a porous structure with 1 μm thickness, suitable for efficient gas transport. With the addition of a catalyst layer, PEDOT–PTFE showed comparable results with carbon-based GDL for CO2RR in terms of faradaic efficiency and measured potential during short-term constant-current evaluation. In stability tests, PEDOT–PTFE exhibited superior stability of catalytic performance without electrolyte flooding across a wide range of electrolyte pH at industrially relevant currents. This is the first demonstration of a conductive, PTFE-based, all-polymer GDL, and it overcomes the most substantial challenges associated with previous iterations of carbon-based and PTFE-based GDLs. Further investigation for variation in PEDOT properties, for example, dopant modification and control of thickness and porosity, may lead to enhanced mass transport at high current with improved stability, enabling broad application of this configuration. The versatility of this design makes it suitable to improve the electrocatalytic performance of any device that involves gas-phase reactants and products, which is a critical step toward enabling decarbonized, electrified processes that are powered by the increasing availability of wind and solar energy.Experimental section
Electropolymerization of PEDOT on PTFE membrane
The electropolymerization of PEDOT:PF6 was inspired by previous work using the monomer, 3,4-ethylenedioxythiophene (EDOT, 97%, Sigma Aldrich), and the dopant, 1-butyl-3-methylimidazolium hexafluorophosphate (BMIMPF6, 98+%, Thermo Scientific).26 The schematic illustration in Fig. S1 (ESI†) shows the procedure of electropolymerization of PEDOT:PF6 and corresponding photograph of the membrane at each stage. In detail, a porous PTFE membrane with polypropylene backer (QL822, Sterlitech) was first placed in a thermal evaporator installed in an N2 atmosphere glovebox. Then, an Au layer with a thickness of 150 nm was deposited on the PTFE side at a pressure of ca. 10−6 bar, with the deposition rate of 1.5 Å s−1. The Au-coated PTFE membrane was cut into small pieces (2.0 × 3.3 cm2) and the edge of each side was painted with silver conductive paint (SPI supplies) to minimize potential drop through the membrane for the electropolymerization step. A Cu tape was put on the top side of the membrane for the current collector. Then, Kapton tape was used to mask the Ag-covered areas to prevent undesired electropolymerization of PEDOT:PF6 on the areas and limit the exposed area to 1.2 × 2.5 cm2.The three-electrode configuration was applied for the electropolymerization of PEDOT:PF6 (Fig. S2, ESI†); the Au-coated PTFE membrane was the working electrode, carbon paper was the counter electrode, and Ag/AgCl in saturated KCl was the reference electrodes. All electrodes were immersed in 100 mL of acetonitrile (HPLC grade, Fisher Scientific) containing 0.01 M EDOT and 0.01 M BMIMPF6, and the beaker was tightly covered by parafilm to prevent evaporation of acetonitrile. For the electropolymerization of PEDOT:PF6 layer on the Au-coated PTFE membrane, a VSP potentiostat (BioLogic) was utilized to apply constant potential at 2.1 V vs. Ag/AgCl until the total charge reached 200 mC. The chemical equation of PEDOT formation is suggested in Fig. S3 (ESI†). The resulting electrode was then thoroughly washed with acetonitrile and dried in a hood to remove residual solvent. After drying, all sides were trimmed to remove the Ag-covered areas. Subsequently, the electrodes were immersed in aqua regia for 10 min to dissolve the Au layer, and repeated two more times. The final products were washed with copious amounts of water, dried in an oven at 60 °C for 30 min, and placed in a fume hood, at least overnight. For convenience, the PEDOT:PF6-coated PTFE membrane is denoted as PEDOT–PTFE in this work.
Electrochemical measurement of CO2RR performance
To test the CO2RR activity of Ag nanoparticles (Ag NPs) loaded on PEDOT–PTFE and Sigracet 22bb (Fuel Cell Store) containing 5 wt% PTFE in its microporous layer, the drop-casting method was used to introduce electrocatalyst to the GDLs, and the performance was measured in the lab-made custom flow cell, controlled by a SP-200 potentiostat (BioLogic). The structural details of whole cell assembly, cell dimensions, scheme of catalyst-loaded PEDOT–PTFE, and working electrode assembly are provided in Fig. S10 (ESI†). Specifically, the catalyst ink, containing 3 mg of Ag NPs (20–40 nm, Fisher Scientific), 350 μL of deionized (DI) water (18.2 MΩ), 150 μL of 2-propanol (HPLC grade, Fisher Scientific), and 30 μL of 5 wt% Nafion dispersion (D520CS, Ion Power), was first sonicated at room temperature (RT) for 30 min. Then, a small portion of the ink was cast on the exposed GDL area (1.2 × 2.5 cm2), masked by Kapton tape for both PEDOT–PTFE and Sigracet 22bb. GDLs were carefully moved to the oven at 70 °C for drying and repeated until 1.0 mg cm−2 of catalyst loading was achieved.The GDL was then placed in the flow cell with a separator, counter, and reference electrodes. For all electrocatalytic measurements, Hg/Hg2SO4 in saturated K2SO4 was used as a reference, and the potential vs. reversible hydrogen electrode (RHE) was calculated by the following equation:
| E(V vs. RHE) = E(V vs. Hg/Hg2SO4) + 0.62 V + 0.0591 V × pH |
Chronopotentiometry (CP) at different current densities (−20, −50, −100, −150, and −200 mA cm−2) was used for measuring the CO2RR performance of Ag NPs on GDLs. Each constant current step was held for 20 min. The electrolyte was circulated at a constant rate of 5 mL min−1 by a peristaltic pump for both catholyte and anolyte and the circulated volume for each channel was 15 mL. CO2 (99.99%, Indiana Oxygen) controlled by a mass flow controller (Alicat) was continuously fed to the gas chamber with the mass flow rate at 20 standard cm3 min−1 (sccm) during CO2RR measurements. The in-line gas chromatograph (GC, Agilent 7890A) was equipped with a thermal conductivity detector (TCD) and flame ionization detector (FID) with Jetanizer (Activated Research Company) for the detection of gas products. Faradaic efficiencies for gas products were calculated using the following equations:
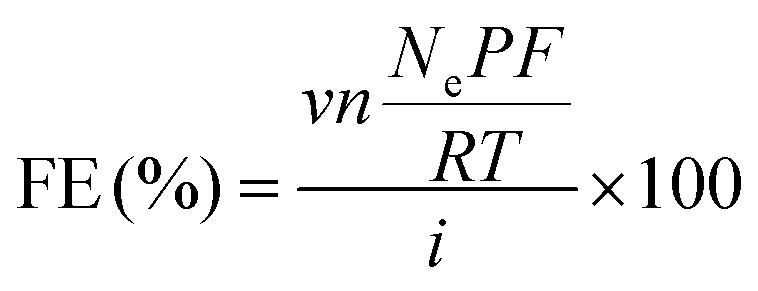
CP at two different current densities (−100 and −200 mA cm−2) was carried out for evaluation of the stability of GDLs in different electrolytes. The general experimental setup was similar to the procedure mentioned above except for the counter electrode and the circulated volume of electrolytes. For stability tests, Pt foil (0.05 mm thick, 99.99%, Thermo Scientific) was used instead of IrO2 on Sigracet 22bb, and the volume of electrolyte circulating the catholyte and anolyte chambers was increased to 30–50 mL.
Characterization
Top-view scanning electron microscopy (SEM) images and energy dispersive X-ray spectroscopy (EDX) spectra were obtained from a field emission scanning electron microscope (Hitachi S-4800) with EDX detector (Oxford X-MaxN 80 SDD). For the cross-section SEM images, an FEI Quanta 3D FEG dual-beam electron microscope with Ga ion source was utilized for focused ion beam (FIB) milling. A Talos F200X (Thermo Scientific) was used for obtaining high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and EDX mapping results. Electrical conductivity was calculated by measuring the resistance value using a Keithley 2400 source meter in the range of −0.2 V ≤ V ≤ +0.2 V. Raman spectra were collected from the DXR Raman microscope (Thermo Scientific) with a 633 nm laser. Fourier transform infrared (FTIR) spectra were recorded by using a Thermo Nicolet 6700 FT-IR spectrometer equipped with Diamond attenuated total reflectance (ATR). For X-ray photoelectron spectroscopy (XPS), a Kratos AXIS Ultra DLD X-ray photoelectron spectrometer equipped with a monochromatic Al Kα radiation X-ray source and a built-in standard charge neutralizer was used.Data availability
The data supporting this article have been included in this article or as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
The XPS data was obtained at the Surface Analysis Facility in the Birck Nanotechnology Center at Purdue University. This work was supported in part by the Research Instrumentation Center in the Department of Chemistry at Purdue University. This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science program, under award number DE-SC0023257.References
- P. De Luna, et al., What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364, eaav3506 CrossRef CAS.
- C. Liu, F. Chen, B.-H. Zhao, Y. Wu and B. Zhang, Electrochemical hydrogenation and oxidation of organic species involving water, Nat. Rev. Chem., 2024, 8, 277–293 CrossRef CAS PubMed.
- B. M. Tackett, E. Gomez and J. G. Chen, Net reduction of CO2via its thermocatalytic and electrocatalytic transformation reactions in standard and hybrid processes, Nat. Catal., 2019, 2, 381–386 CrossRef CAS.
- S. M. Jordaan and C. Wang, Electrocatalytic conversion of carbon dioxide for the Paris goals, Nat. Catal., 2021, 4, 915–920 CrossRef.
- N. T. Nguyen and C.-T. Dinh, Gas diffusion electrode design for electrochemical carbon dioxide reduction, Chem. Soc. Rev., 2020, 49, 7488–7504 RSC.
- H. Rabiee, et al., Gas diffusion electrodes (GDEs) for electrochemical reduction of carbon dioxide, carbon monoxide, and dinitrogen to value-added products: a review, Energy Environ. Sci., 2021, 14, 1959–2008 RSC.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Gas diffusion electrodes and membranes for CO2 reduction electrolysers, Nat. Rev. Mater., 2022, 7, 55–64 CrossRef CAS.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, Gas-Diffusion Electrodes for Carbon Dioxide Reduction: A New Paradigm, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- A. Reyes, et al., Managing Hydration at the Cathode Enables Efficient CO2 Electrolysis at Commercially Relevant Current Densities, ACS Energy Lett., 2020, 5, 1612–1618 CrossRef CAS.
- J. E. Huang, et al., CO2 electrolysis to multicarbon products in strong acid, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- Y. Xie, et al., High carbon utilization in CO2 reduction to multi-carbon products in acidic media, Nat. Catal., 2022, 5, 564–570 CrossRef CAS.
- C. Kim, et al., Tailored catalyst microenvironments for CO2 electroreduction to multicarbon products on copper using bilayer ionomer coatings, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- Y. Cao, et al., Surface hydroxide promotes CO2 electrolysis to ethylene in acidic conditions, Nat. Commun., 2023, 14, 2387 CrossRef CAS.
- F. Huq, et al., Influence of the PTFE Membrane Thickness on the CO2 Electroreduction Performance of Sputtered Cu-PTFE Gas Diffusion Electrodes, ChemElectroChem, 2022, 9, e202101279 CrossRef CAS.
- C.-T. Dinh, F. P. García de Arquer, D. Sinton and E. H. Sargent, High Rate, Selective, and Stable Electroreduction of CO2 to CO in Basic and Neutral Media, ACS Energy Lett., 2018, 3, 2835–2840 CrossRef CAS.
- F. P. García de Arquer, et al., CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2, Science, 2020, 367, 661–666 CrossRef.
- F. Li, et al., Molecular tuning of CO2-to-ethylene conversion, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- J. Zhang, et al., Accelerating electrochemical CO2 reduction to multi-carbon products via asymmetric intermediate binding at confined nanointerfaces, Nat. Commun., 2023, 14, 1298 CrossRef CAS.
- C.-T. Dinh, et al., CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface, Science, 2018, 360, 783–787 CrossRef CAS.
- H.-P. Iglesias van Montfort, et al., Non-invasive current collectors for improved current-density distribution during CO2 electrolysis on super-hydrophobic electrodes, Nat Commun., 2023, 14, 6579 CrossRef CAS.
- M. Filippi, et al., Scale-Up of PTFE-Based Gas Diffusion Electrodes Using an Electrolyte-Integrated Polymer-Coated Current Collector Approach, ACS Energy Lett., 2024, 9, 1361–1368 CrossRef CAS PubMed.
- S. Yamaguchi, H. Ebe, T. Minegishi and M. Sugiyama, Introduction of a Conductive Layer into Flood-Resistant Gas Diffusion Electrodes with Polymer Substrate for an Efficient Electrochemical CO2 Reduction with Copper Oxide, ACS Appl. Mater. Interfaces, 2024, 16, 17371–17376 CrossRef CAS.
- S. Nitopi, et al., Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- N. Ahmad Shahrim, Z. Ahmad, A. W. Azman, Y. F. Buys and N. Sarifuddin, Mechanisms for doped PEDOT:PSS electrical conductivity improvement, Mater. Adv., 2021, 2, 7118–7138 RSC.
- Y. Seki, M. Takahashi and M. Takashiri, Effects of different electrolytes and film thicknesses on structural and thermoelectric properties of electropolymerized poly(3,4-ethylenedioxythiophene) films, RSC Adv., 2019, 9, 15957–15965 RSC.
- M. Culebras, C. M. Gómez and A. Cantarero, Enhanced thermoelectric performance of PEDOT with different counter-ions optimized by chemical reduction, J. Mater. Chem. A, 2014, 2, 10109–10115 RSC.
- H. Liu, et al., PEDOT decorated CoNi2S4 nanosheets electrode as bifunctional electrocatalyst for enhanced electrocatalysis, Chem. Eng. J., 2022, 428, 131183 CrossRef CAS.
- X. Chen, et al., Highly Stable Layered Coordination Polymer Electrocatalyst toward Efficient CO2-to-CH4 Conversion, Adv. Mater., 2024, 36, 2310273 CrossRef CAS PubMed.
- A. Benoudjit, M. M. Bader and W. W. A. Wan Salim, Study of electropolymerized PEDOT:PSS transducers for application as electrochemical sensors in aqueous media, Sens. Bio-Sens. Res., 2018, 17, 18–24 CrossRef.
- A. Roy, A. Ponnam, V. Varade, G. V. Honnavar and R. Menon, Evidence of Lampert Triangle and Jonscher's Double Power Law in Doped Poly(3,4-ethylenedioxythiophene) Devices, J. Phys. Chem. C, 2023, 127, 5502–5512 CrossRef CAS.
- A. Roy, S. Mandal and R. Menon, Lampert triangle formation and relaxation behavior in doped poly(3,4-ethylenedioxythiophene) devices, J. Appl. Phys., 2021, 129, 195501 CrossRef CAS.
- R. Holze, Overoxidation of Intrinsically Conducting Polymers, Polymers, 2022, 14, 1584 CrossRef CAS PubMed.
- H. Wu, et al., Selective and energy-efficient electrosynthesis of ethylene from CO2 by tuning the valence of Cu catalysts through aryl diazonium functionalization, Nat. Energy, 2024, 9, 422–433 CrossRef CAS.
- Y. Wu, et al., Effects of microporous layer on electrolyte flooding in gas diffusion electrodes and selectivity of CO2 electrolysis to CO, J. Power Sources, 2022, 522, 230998 CrossRef CAS.
- Z.-Z. Niu, L.-P. Chi, R. Liu, Z. Chen and M.-R. Gao, Rigorous assessment of CO2 electroreduction products in a flow cell, Energy Environ. Sci., 2021, 14, 4169–4176 RSC.
- J. F. Serrano-Claumarchirant, et al., Textile-based Thermoelectric Generator Produced Via Electrochemical Polymerization, Adv. Mater. Interfaces, 2023, 10, 2202105 CrossRef CAS.
- M. Fan, et al., Cationic-group-functionalized electrocatalysts enable stable acidic CO2 electrolysis, Nat. Catal., 2023, 6, 763–772 CrossRef CAS.
- T. Burdyny and A. W. Smith, CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, Investigating Electrode Flooding in a Flowing Electrolyte, Gas-Fed Carbon Dioxide Electrolyzer, ChemSusChem, 2020, 13, 400–411 CrossRef CAS.
- L. M. Baumgartner, C. I. Koopman, A. Forner-Cuenca and D. A. Vermaas, When Flooding Is Not Catastrophic—Woven Gas Diffusion Electrodes Enable Stable CO2 Electrolysis, ACS Appl. Energy Mater., 2022, 5, 15125–15135 CrossRef CAS.
- J. Gu, et al., Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium, Nat. Catal., 2022, 5, 268–276 CrossRef CAS.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, Role of the Carbon-Based Gas Diffusion Layer on Flooding in a Gas Diffusion Electrode Cell for Electrochemical CO2 Reduction, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04163a |
| This journal is © The Royal Society of Chemistry 2025 |