
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of stretchable triboelectric material with strain-compensating ability using gradient interpenetrating polymer networks†
Do-Wan
Kim‡
a,
Hyeonwoo
Mun‡
b,
Yeonghun
Kang
b,
Weon-Guk
Kim
a,
Dahye
Ahn
b,
Seong-Yun
Yun
a,
Jeong-A
Han
a,
Do Hoon
Lee
a,
Taegoon
Lee
a,
Kihoon
Jeong
b,
Jihan
Kim
b,
Sung Gap
Im
 *b and
Yang-Kyu
Choi
*a
*b and
Yang-Kyu
Choi
*a
aSchool of Electrical Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: ykchoi@ee.kaist.ac.kr
bDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: sgim@kaist.ac.kr
First published on 25th February 2025
Abstract
Unlike conventional rigid triboelectric nanogenerators (TENGs), elastic TENGs are considered attractive for energy harvesting and sensing applications under mechanically harsh conditions. However, the practicality of elastic TENGs has been limited by the lack of elastic materials that simultaneously exhibit the desired mechanical and triboelectric properties. This paper introduces a complementary material synthesis strategy that uses a gradient interpenetrating polymer network (g-IPN) to address this issue. A sub-micron thick g-IPN was formed on a host elastomer (Ecoflex-CNT) that has high contact conformity using a highly chargeable guest polymer (pVP) with a low work function, through initiated chemical vapor deposition (iCVD). This complementary material synthesis effectively leveraged only the strengths of each component and resulted in a synergistic enhancement in output performance, with a short-circuit charge density (QSC) and an open-circuit voltage (VOC) up to 445 μC m−2 and 1335 V, respectively. These values were achieved without affecting the bulk mechanical properties of the host elastomer, such as high stretchability and a low bulk elastic modulus. Moreover, the depth-directional gradient profile of the g-IPN effectively prevented degradation in output performance under a severely stretched state (up to 100% strain), through a so-called strain-compensating ability. The effectiveness of the g-IPN in three-dimensional (3D)-structured elastic TENGs was successfully demonstrated by applying the g-IPN to a sponge-structured 3D elastic TENG (3D-IPN-TENG), which benefited from the exceptional deposition conformity of the iCVD process. The fabricated 3D-IPN-TENG showed stable operation with a short-circuit volume charge density (QSC,vol) of up to 267.2 mC m−3, which is a record-high value among 3D-structured TENGs that utilize contact electrification (CE) between solids. This work not only overcomes the limitations of existing material strategies for elastic TENGs, but also suggests a new universal material design principle for synthesizing high-performance triboelectric materials.
Broader contextTriboelectric nanogenerators (TENGs) are considered especially attractive among emerging renewable energy technologies because of their high design flexibility, wide material options, and low cost. Recently, elastic TENGs, which are readily deformable under mechanical stimuli, have been developed and show advantages over conventional rigid TENGs, extending their applicability. However, there is a shortage of elastic materials that simultaneously exhibit the desired mechanical properties as well as high and mechanically stable output performance, limiting the practicality of elastic TENGs. Moreover, most existing material strategies for elastic TENGs cannot be used in complex-structured elastic TENGs, especially three-dimensional (3D)-structured TENGs designed for high volumetric efficiency. To address these limitations, we propose a complementary material synthesis strategy that utilizes a sub-micron thick gradient interpenetrating polymer network (g-IPN) between a host elastomer (Ecoflex-CNT) and a highly chargeable guest polymer (pVP). This material, Ecoflex-CNT with g-IPN, not only showed superior mechanical properties, but also showed synergistically enhanced output performance that was maintained even under severe strain. Furthermore, forming the g-IPN using the initiated chemical vapor deposition (iCVD) process was fully compatible with 3D-structured TENGs. This work suggests a new concept for synthesizing high-performance triboelectric materials and provides insights into the factors that influence contact electrification (CE). |
Introduction
Contact electrification (CE) is a ubiquitous phenomenon that involves the separation of charges between two objects after mechanical contact.1,2 Because of its high energy density, versatile applications of CE have been demonstrated, ranging from potential to commercial areas such as laser printing,3 electrospraying,4 and X-ray generation.5 Triboelectric nanogenerators (TENGs),6 which also utilize CE, have been developed to harvest mechanical energy7–10 and to supply energy for various electronic devices.11–13 As a result, TENGs have powered a wide range of applications, including wearable electronics14,15 and implantable electronics.16,17Elastic TENGs that are readily deformable under mechanical stimuli have been developed to harvest mechanical energy from a wide range of circumstances.18–21 Due to their high design flexibility, these elastic TENGs can be easily engineered to efficiently convert mechanical energy from various types of sources.22–24 Recently, there has been increased interest in elastic TENGs with three-dimensional (3D) structures to avoid the limitations of elastic TENGs with two-dimensional (2D) planar structures, which suffer from low spatial efficiency and a poor ability to harvest energy from complex and multimodal mechanical motions.25–29 For example, elastic TENGs with a 3D sponge structure can harvest mechanical energy from multiple directions at various amplitudes, allowing them to scavenge more energy with improved spatial efficiency. This cannot be achieved with elastic TENGs with 2D structures.30,31
Despite the advantages of elastic TENGs, extending their applicability will require further boosting their output power. It is well known that the output power of TENGs mainly depends on their short-circuit charge density (QSC).32 Therefore, it is important to maximize QSC by appropriately designing the triboelectric materials. Compared to the triboelectric materials used in conventional rigid TENGs, the triboelectric materials utilized in elastic TENGs are often exposed to severe strain during operation.18,21,23 Accordingly, elastic triboelectric materials must not only possess a high QSC but also exhibit high stretchability beyond flexibility, have a low elastic modulus, maintain a consistent output regardless of strain, and harness high durability of output under stretched conditions. Developing elastic triboelectric materials that meet these requirements is highly challenging. However, most previous studies have focused solely on achieving high QSC in the unstretched state, by employing strategies such as mixing highly charging fillers with elastomers,33–37 introducing deep traps,38,39 or applying pre-strain.40–42 Consequently, they have been unable to simultaneously meet all of the aforementioned requirements, which limits their compatibility with various structures and working modes of elastic TENGs. To overcome this limitation, a different material engineering strategy, and a fabrication process that is compatible with various TENG structures, are required.
In this study, we introduce a new, facile strategy for synthesizing a high-performance elastic triboelectric material using a gradient interpenetrating polymer network (g-IPN), in order to synthesize an elastic triboelectric material that simultaneously satisfies the abovementioned requirements. The g-IPN is an IPN between a host polymer and a guest polymer that has a depth-directional gradient concentration profile, where the guest polymer refers to the polymer that penetrates and polymerizes after the polymerization of the host polymer.43 Here, we show that forming a thin g-IPN layer on a host elastomer using a highly chargeable guest polymer is an effective strategy to synthesize materials with both superior mechanical and triboelectric properties. The g-IPN synthesized in this work consists of a stretchable and conductive Ecoflex-carbon nanotube (CNT) composite (Ecoflex-CNT) as the host elastomer, and a tribo-positive polyvinylpyrrolidone (pVP) as the highly chargeable guest polymer.
For the one-step synthesis of this tribo-positive g-IPN, an initiated chemical vapor deposition (iCVD) process was employed.44 According to the depth profile analysis using time-of-flight secondary ion mass spectrometry (ToF-SIMS), the concentration of the pVP in the g-IPN gradually varied from high near the surface to low near the bulk, within a sub-micron thickness. Therefore, the synthesized g-IPN had minimal influence on the bulk mechanical properties of the Ecoflex-CNT samples, such as stretchability and bulk elastic modulus. Afterward, 2D-structured TENGs consisting of the Ecoflex-CNT with g-IPN and counter-contacting materials (hereafter abbreviated as 2D-IPN-TENG), were prepared to evaluate the triboelectric properties of the g-IPN. When Kapton was used as a counter-contacting material, the 2D-IPN-TENG showed a QSC of up to 445 μC m−2, an open-circuit voltage (VOC) of 1335 V, and a 12.6-fold reduction in charge accumulation cycle (τ90) compared to a 2D-structured TENG with pure Ecoflex-CNT, where τ90 represents the contact cycles taken to accumulate 90% of saturated QSC.45 This superior output performance was attributed to the synergistic effect of the high contact conformity of the Ecoflex-CNT and the low work function of the pVP.
Moreover, the Ecoflex-CNT with g-IPN retained its initial QSC even under 100% strain. This remarkable mechanical stability in output performance was ascribed to the gradient structure of the g-IPN. Note that the IPN layer with a high concentration of pVP near the surface is stiff, while the IPN layer with a low concentration of pVP near the bulk is intrinsically stretchable. When the Ecoflex-CNT with g-IPN was stretched, the stiff surface IPN layer cracked along the direction of stretch, and wrinkled along the direction perpendicular to the stretch. This exposed the intrinsically stretchable IPN layer buried beneath the stiff surface IPN layer to the counter-contacting material of the 2D-IPN-TENG. Consequently, this increased the contact area for CE, thereby resisting the degradation of output performance with increasing strain.
To demonstrate the effectiveness of the g-IPN in the 3D-structured elastic TENGs, we then applied the g-IPN to a sponge-structured 3D elastic TENG, comprised of multiple wires and a 3D-structured Ecoflex-CNT sponge. Leveraging the exceptional conformity of the iCVD process, the g-IPN was uniformly formed throughout the Ecoflex-CNT sponge, without any damage from heat or solvent. The resulting sponge-structured 3D elastic TENG with g-IPN (hereafter abbreviated as 3D-IPN-TENG) produced a short-circuit volume charge density (QSC,vol) of 267.2 mC m−3, which is the highest value among 3D-structured TENGs that utilize CE between solids to date. Moreover, it generated a high peak power density of 496.81 W m−3 and demonstrated durability over 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compression–relaxation cycles. Consequently, small electronic devices, such as a stopwatch and a thermohygrometer, could be simultaneously and continuously powered by a compact 3D-IPN-TENG with an effective volume of less than 2 cm3.
000 compression–relaxation cycles. Consequently, small electronic devices, such as a stopwatch and a thermohygrometer, could be simultaneously and continuously powered by a compact 3D-IPN-TENG with an effective volume of less than 2 cm3.
Results and discussion
Design strategy of g-IPN for high-performance elastic TENG
When two materials are brought into contact, contact conformity is defined, which indicates how closely the two materials are in contact with each other.46 On the molecular scale, CE between two molecules occurs only when they approach the repulsive regime.2,47 On the macroscale, this implies that the contact conformity between contacting surfaces is closely related to the magnitude of CE.46 On the other hand, the magnitude of CE is also influenced by the work function of the contacting materials.48,49 According to a previous study, when the contact conformity is improved by using liquid metal as one of the contacting materials, thereby avoiding the effects of incomplete contact, it was revealed that a material with a higher work function tends to acquire a higher negative charge.50Therefore, the magnitude of CE in triboelectric materials can be influenced by both their contact conformity and work function. Although elastomers have relatively high contact conformity due to their low elastic modulus, they typically have a moderate work function and thus are usually located at neutral positions in the triboelectric series.51 This counteraction on triboelectricity imposes a constraint on maximizing the magnitude of CE (Fig. 1a). In contrast, some polymers, capable of inducing high charging hence referred to as a highly chargeable polymer (HCP), exhibit an unusually high or low work function but typically possess an undesirable stiffness.48 Although they are positioned at both ends of the triboelectric series due to their extreme work functions, i.e., exhibiting two opposing work functions of either very low (tending to be highly tribo-positive) or very high (tending to be highly tribo-negative), the low contact conformity caused by their high elastic modulus hinders them from becoming extremely charged (Fig. 1b).
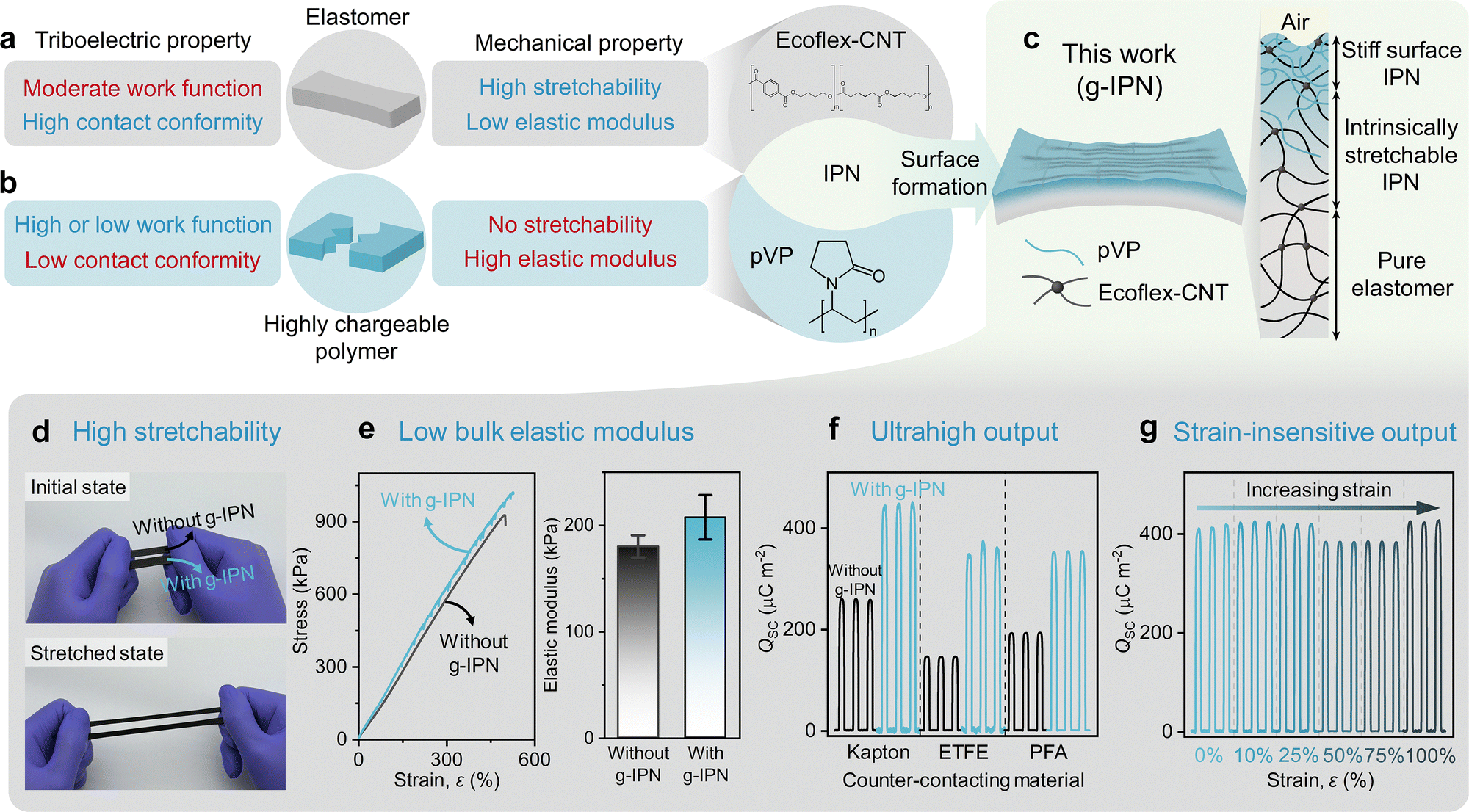 | ||
| Fig. 1 Concept of g-IPN for high-performance elastic TENGs. (a) Various counteracting factors which affect the triboelectric and mechanical properties of elastomers. (b) Various counteracting factors which influence the triboelectric and mechanical properties of HCPs. (c) Schematic of the Ecoflex-CNT with g-IPN and its cross-section schematic showing a gradiently changing concentration of pVP along the depth direction. (d) Photographs of pure Ecoflex-CNT and Ecoflex-CNT with g-IPN, demonstrating their high stretchability. (e) Comparison of the stress–strain curve (left) and bulk elastic modulus (right) between Ecoflex-CNT with and without g-IPN. The values of elongation at break for the Ecoflex-CNT with and without g-IPN were 530% and 501%, respectively. The bulk elastic modulus was extracted from stress–strain curves measured from three randomly selected samples. (f) Comparison of measured QSC between Ecoflex-CNT with and without g-IPN. (g) Measured QSC while applying various strains on Ecoflex-CNT with g-IPN, ranging from 0% to 100%. In (g), the counter-contacting material was Kapton. For (d)–(g), the tIPN,equiv of Ecoflex-CNT with g-IPN was 75 nm. | ||
To address the conflicting tendencies of work function and contact conformity in triboelectric materials, a material design strategy that can synergistically combine an extreme work function and a high contact conformity is necessary. It is worth noting that synthetic polymers are usually tribo-negative, while fewer options are available for tribo-positive polymers, such as Nylon 66, poly(vinyl alcohol) (PVA), etc.48 Moreover, most tribo-positive polymers possess high elastic modulus, limiting their contact conformity, and thus, limiting their magnitude of CE. Since tribo-positive materials are as important as tribo-negative materials to maximize the output performance of TENGs, a tribo-positive material with a higher magnitude of CE is still necessary.48 In response to this, here, we demonstrate a complementary material synthesis strategy by synthesizing a tribo-positive g-IPN between a host elastomer (Ecoflex-CNT) and a highly chargeable guest polymer (pVP) (Fig. 1c). This effectively combined the high contact conformity of the Ecoflex-CNT and the low work function of the pVP, resulting in a high magnitude of CE. Hereafter, unless otherwise specified, g-IPN denotes the gradient-IPN, which is composed of pVP and Ecoflex-CNT as a guest polymer and a host polymer, respectively.
At the same time, bulk mechanical properties themselves are also important to ensure compatibility with various TENG structures and practical applications. In this regard, elastomers are evidently superior to HCPs, as they can undergo elastic deformation within a wide range of strain. Therefore, a g-IPN with a sub-micron thick gradient profile, gradually transitioning from a stiff surface IPN (IPN with a high concentration of pVP) to a soft bulk elastomer, was used for the proposed 2D-IPN-TENG and 3D-IPN-TENG (Fig. 1c). This made it possible to leverage the outstanding triboelectric properties (high magnitude of CE) of the g-IPN and the excellent bulk mechanical properties (high stretchability and low bulk elastic modulus) of the Ecoflex-CNT. Note that conductive Ecoflex-CNT has been widely used for 2D- and 3D-structured elastic TENGs due to its superior bulk mechanical properties.26,28
Based on the proposed strategy, the Ecoflex-CNT with g-IPN exhibited superior triboelectric properties while maintaining the mechanical properties of the pure Ecoflex-CNT. As shown in Fig. 1d and e, the bulk elastic modulus and elongation at break for the Ecoflex-CNT with and without g-IPN were similar. Additionally, the 2D-IPN-TENG showed greatly enhanced output performance compared to the same 2D-structured TENG without the g-IPN, i.e., that with the pure Ecoflex-CNT (Fig. 1f). In particular, the 2D-IPN-TENG with Kapton as a counter-contacting material showed a QSC of up to 445 μC m−2. This output performance was maintained even with increasing strain on the Ecoflex-CNT with g-IPN, and exhibited almost no degradation up to 100% strain (Fig. 1g). These characteristics confirm that the Ecoflex-CNT with g-IPN is a promising material candidate for elastic TENGs. Detailed explanations of the ultrahigh output performance and the strain-insensitive output performance of the 2D-IPN-TENG are described later.
Strategy for selecting the guest polymer
The selection of a guest polymer is crucial for enhancing electron transfer from the g-IPN to the counter-contacting materials, while inhibiting it from the counter-contacting materials to the g-IPN. Thus, to achieve a highly tribo-positive g-IPN with a low work function, a guest polymer with relatively high levels of the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) is desirable.52,53 According to density functional theory (DFT) calculations, pVP (Fig. 2a) can satisfy those requirements. As illustrated in the energy band diagram (Fig. 2b), the relatively higher HOMO and LUMO levels of pVP compared to Nylon 66 are advantageous for electron donation. This means that pVP can be even more tribo-positive than Nylon 66, which is a commonly used reference material placed near the positive end of the triboelectric series.54,55 These results were consistent regardless of the chain lengths of both pVP and Nylon 66 (Fig. S1, ESI†). | ||
| Fig. 2 Formation of g-IPN on Ecoflex-CNT and comparison with drop-cast samples. (a) Molecular structure of pVP used in the DFT calculations. Grey, red, blue, and white represent carbon, oxygen, nitrogen, and hydrogen atoms, respectively. (b) Energy band diagram of the pVP monomer and Nylon 66 monomer obtained from the DFT calculations. (c) Measured ISC showing the relative position of pVP in the triboelectric series (left) and a simplified cross-sectional schematic of the measurement for identifying the relative position in the triboelectric series (right). The thickness of the pure pVP film used in this experiment was 3 μm. (d) Synthesis procedure of g-IPN by iCVD. (e) ToF-SIMS depth profile of pure Ecoflex-CNT (tIPN,equiv = 0 nm). (f) ToF-SIMS depth profile of Ecoflex-CNT with g-IPN (tIPN,equiv = 75 nm). (g) OM image of case I prior to applying strain, along with its schematic illustration (left). OM image of case I released after applying 100% strain, along with its schematic illustration (right). (h) OM image of case II (tIPN,equiv = 75 nm) prior to applying strain, along with its schematic illustration (left). OM image of case II (tIPN,equiv = 75 nm) released after applying 100% strain, along with its schematic illustration (right). Scale bars in (g) and (h) are 50 μm. (i) Measured QSC for pure Ecoflex-CNT, case I with commercial pVP (Mw = 10000 g mol−1 and 29000 g mol−1), case I with pVP synthesized by iCVD (Mw ∼ 18600 g mol−1), and case II with tIPN,equiv = 75 nm. | ||
To experimentally confirm the strong electron-donating tendency of pVP, its relative position in the triboelectric series was investigated.56 As shown in Fig. 2c, seven materials ranging from the positive side to the negative side of the triboelectric series were selected as counter-contacting materials. To identify the relative position of pVP in the triboelectric series, all were brought into contact with and separated from a pure pVP film deposited on an Au substrate. The resulting waveforms of the short-circuit current (ISC) show that the pVP was consistently positively charged, irrespective of the counter-contacting material, including Nylon 66. Pure Ecoflex-CNT was placed between polyethylene terephthalate (PET) and Kapton in the triboelectric series, due to its moderate work function (Fig. S2 and S3, ESI†).
Formation of g-IPN on Ecoflex-CNT and comparison with drop-cast samples
The g-IPN was formed using a facile one-step gas-phase polymerization with the iCVD process on pure Ecoflex-CNT. As depicted in Fig. 2d, the gas-phase N-vinyl-2-pyrrolidone (NVP) monomer and tert-butyl peroxide (TBPO) initiator were initially released into the vacuum chamber, where the pure Ecoflex-CNT samples were placed. The injected vapor-phase monomer and initiator molecules could penetrate and diffuse through the free volumes within the elastomeric Ecoflex-CNT. Simultaneously, a hot filament remotely placed over the Ecoflex-CNT samples activated the TBPO and initiated the polymerization of NVP. Consequently, the iCVD process created a depth-directional concentration gradient of pVP on the Ecoflex-CNT substrate, with a higher concentration of pVP near the surface and a lower concentration near the bulk. This in situ process created the g-IPN in a damage-free manner.We confirmed the gradient profile of the g-IPN along the depth direction using ToF-SIMS, as shown in Fig. 2e and f.57 While the intensity of the characteristic fragment ion of Ecoflex-CNT (i.e., Si+) remained constant throughout the entire depth profile, that of pVP (i.e., C6H10NO+) decreased exponentially from the surface (Fig. 2f). This confirms that the g-IPN was formed near the surface, as intended. In addition, the gradient profile of the g-IPN could be tuned systematically by controlling the iCVD process parameters, such as deposition time (Fig. S4, ESI†). However, an excessively long deposition time resulted in an unintended additional top layer of pure pVP over the g-IPN (Fig. S5, ESI†). Hereafter, we denote this excessively deposited sample as an over-deposited sample. Since the thickness of the g-IPN cannot be precisely defined due to its gradient profile, we define its equivalent thickness (tIPN,equiv) as the thickness of the pure pVP film deposited on the surface of a Si wafer placed in the same batch when forming g-IPN on the Ecoflex-CNT.
The Ecoflex-CNT with g-IPN (case II) has several advantages in terms of mechanical and triboelectric properties, compared to the Ecoflex-CNT with the pure pVP film deposited by a conventional solution-based drop-casting process (case I). For comparison between them, pure pVP synthesized by iCVD was dissolved in 2-propanol, and the solution was dropped onto pure Ecoflex-CNT samples to prepare drop-cast samples. In this case, there was practically no intermixing between the drop-cast pVP and the underlying pure Ecoflex-CNT, as confirmed by ToF-SIMS analyses (Note S1 and Fig. S6, ESI†). Samples for case II were prepared by forming g-IPN on Ecoflex-CNT, with tIPN,equiv = 75 nm.
Firstly, the mechanical characteristics of the two cases were comparatively analyzed. As shown in the optical microscopy (OM) image in case I (Fig. 2g, left), a double-layer structure was created. However, the drop-cast pVP layer in case I was easily delaminated and fractured after being released from 100% uniaxial strain, exhibiting characteristics of strain localization (Fig. 2g, right).58,59 This indicates poor interfacial toughness in case I.59,60 In contrast to case I, preexisting wrinkles were found in case II (Fig. 2h, left), indicating the formation of a relatively stiff surface layer tightly bound to the elastomeric substrate.61 Interestingly, after being released from 100% uniaxial strain, no delamination occurred, and only small traces of microcracks appeared, exhibiting characteristics of effective strain delocalization (Fig. 2h, right).58,59 This suggests that the physical anchoring of the stiff surface IPN layer into the Ecoflex-CNT substrate, through a molecularly intertwined structure, resulted in a tough interface.59,60 Additionally, the difference in interfacial toughness between case I and case II was further confirmed by a tape peeling experiment (Note S1 and Fig. S6, ESI†).60
Secondly, the triboelectric characteristics were comparatively analyzed for the two cases. QSC was measured in both cases while using Kapton as a counter-contacting material. Two commercial pVP samples with different weight-average molecular weights (Mw) and one pVP sample synthesized by iCVD were used to prepare case I. To make conditions as similar as possible for a fair comparison, commercial pVP samples with Mw of 10000 g mol−1 and 29000 g mol−1 were chosen, since the Mw of the pVP synthesized by iCVD was approximately 18600 g mol−1 (Fig. S7, ESI†). As shown in Fig. 2i, there was no significant difference in QSC between the pure Ecoflex-CNT and case I. However, the QSC in case II was notably enhanced. The major reason for the difference between case I and case II was attributed to the degradation of the output performance in case I, due to the severe mechanical wear resulting from its inferior interfacial toughness.
Case II also has significant advantages in terms of bulk mechanical and triboelectric properties compared to the uniform IPN (case III), in which the IPN was uniformly formed throughout the entire Ecoflex-CNT sample (Note S2 and Fig. S8, ESI†). The bulk elastic modulus and the degree of plasticity in case III were significantly higher compared to those of the pure Ecoflex-CNT (Fig. S8c–f, ESI†). In contrast, the bulk mechanical properties in case II were almost identical to those of the pure Ecoflex-CNT, due to the sub-micron thickness of the g-IPN. Moreover, the QSC in case III, measured using Kapton as a counter-contacting material, was even lower than that of the pure Ecoflex-CNT (Note S2 and Fig. S8g, ESI†). Therefore, among the three cases (cases I, II, and III), only case II exhibited both superior mechanical and triboelectric properties due to its depth-directional gradient profile.
The output performance characterization of 2D-IPN-TENG
A 2D-structured TENG with a contact-separation mode was fabricated to comprehensively evaluate its output performance. This 2D-structured TENG consisted of two parts: a bottom triboelectric layer and a top counter-contacting layer. For the bottom layers, Ecoflex-CNT with g-IPN with various tIPN,equiv were prepared. For the control groups, both extremes: pure Ecoflex-CNT, and pure pVP deposited on Au instead of Ecoflex-CNT were also prepared as bottom layers to evaluate the triboelectric properties of the pure materials which comprise g-IPN. For the counter-contacting layers, Kapton, ethylene tetrafluoroethylene (ETFE), and perfluoroalkoxy (PFA) films attached to the copper induction electrode were used (Fig. 3a). The measured QSC and VOC are plotted in Fig. 3b. For the Ecoflex-CNT with g-IPN, these values were significantly larger than those of the control groups, irrespective of the counter-contacting materials. Similarly, Ecoflex-CNT with g-IPN synthesized using guest polymers other than pVP also demonstrated the highest output performance compared to their respective control groups (Note S3 and Fig. S9 and S10, ESI†). These results indicate that a synergistic performance enhancement was achieved by forming the g-IPN on the Ecoflex-CNT. The underlying mechanism of this synergistic performance enhancement is described later. | ||
| Fig. 3 Output performance characterization of 2D-IPN-TENG. (a) Schematic of a 2D-structured TENG with contact-separation mode to evaluate output performance. (b) Measured QSC and VOC for Ecoflex-CNT with g-IPN (tIPN,equiv = 75 nm) and its control groups, using various counter-contacting materials. (c) and (d) Measured QSC and VOC for various tIPN,equiv. (e) Measured charging ratio (ηQ) for extreme tIPN,equiv (tIPN,equiv = 0 nm and 1000 nm) and for the optimized tIPN,equiv of 75 nm. (f) Extracted charge accumulation cycle (τ90) for various tIPN,equiv. For (c)–(f), Kapton was used as the counter-contacting material. (g) Measured QSC of Ecoflex-CNT with g-IPN (tIPN,equiv = 75 nm) with various counter-contacting Kapton film thicknesses, superimposed on the theoretical limit derived from Paschen's law.62 (h) Comparison of peak power density between a 2D-structured TENG with pure Ecoflex-CNT and a 2D-IPN-TENG (tIPN,equiv = 75 nm). (i) Benchmarking comparison graph of peak power density among previously reported stretchable triboelectric materials and Ecoflex-CNT with g-IPN (tIPN,equiv = 75 nm). (j) Comparison of mechanical and triboelectric properties between commonly used tribo-positive reference materials (Nylon 66 and Al) and Ecoflex-CNT with g-IPN (tIPN,equiv = 75 nm). | ||
To determine the optimal range of the tIPN,equiv when the Ecoflex-CNT with g-IPN contacts Kapton, the QSC and VOC were characterized for various tIPN,equiv, which were achieved by controlling the deposition time in the iCVD process. As shown in Fig. 3c and d, the optimal range of tIPN,equiv was in between 25 nm and 100 nm. In particular, QSC and VOC reached up to 445 μC m−2 and 1335 V at tIPN,equiv of 75 nm, respectively. Within this optimal range, dynamic performance was also notably improved. By defining the charging ratio (ηQ) as the present QSC divided by the saturated QSC, ηQ was plotted versus the number of contacts, to show how quickly a triboelectric layer is charged (Fig. 3e). As shown in Fig. 3e, ηQ rapidly increased to over 0.9 within only about 44 contacts when tIPN,equiv was 75 nm. However, ηQ slowly increased and reached 0.9 after a few hundred contacts when tIPN,equiv was either too low (near 0 nm) or too high (near 1000 nm). Accordingly, τ90 at tIPN,equiv = 75 nm was reduced by approximately an order of magnitude compared to that at tIPN,equiv = 0 nm and 1000 nm (Fig. 3f). An optimal thickness range from approximately 25 nm to 100 nm was also observed when using different counter-contacting materials (ETFE and PFA), demonstrating the versatility of g-IPN (Fig. S11 and S12, ESI†). On the other hand, it is also important to investigate how QSC can be varied by the thickness of the counter-contacting material (Kapton). As shown in Fig. 3g, the measured QSC from a 2D-IPN-TENG with tIPN,equiv = 75 nm was as high as the limit of air breakdown, irrespective of the Kapton thickness, which ranged from 25 μm to 90 μm.63
The power generation capability of the 2D-IPN-TENG with tIPN,equiv = 75 nm was evaluated by measuring the peak power density with various load resistances (Rload). When using Kapton as a counter-contacting material, the peak power density reached 22.06 W m−2 at an Rload of 12 MΩ. This maximum peak power density was approximately 2.3-fold higher than that of the 2D-structured TENG with pure Ecoflex-CNT (Fig. 3h). Moreover, this maximum peak power density value of the 2D-IPN-TENG was substantially greater than those in previous works on stretchable triboelectric materials (Fig. 3i).33–42,64,65 In summary, both the triboelectric and mechanical properties of the Ecoflex-CNT with g-IPN (tIPN,equiv = 75 nm) were compared with those of the conventional reference tribo-positive materials (Nylon 66 and Al) as shown in Fig. 3j. For the measurement of triboelectric properties, Kapton was used as a counter-contacting material in all cases. Compared to the reference materials, the Ecoflex-CNT with g-IPN not only exhibited better bulk mechanical properties, including high stretchability and low bulk elastic modulus, but also superior triboelectric properties in terms of QSC, VOC, and τ90. A systematic comparison of triboelectric and mechanical properties with those from previous works on stretchable TENGs is also shown in Table S1 (ESI†).
Origin of the synergistic performance enhancement in 2D-IPN-TENG
As shown in Fig. 3c and d, bell-shaped profiles of the QSC and VOC were observed as tIPN,equiv varied; they were maximized between both extremes of pure Ecoflex-CNT and pure pVP. It is inferred that this synergistic enhancement of output performance is due to the formation of the g-IPN between the Ecoflex-CNT and pVP. As mentioned earlier, the magnitude of CE in g-IPN, which determines the output performance of the 2D-IPN-TENG, is expected to be dominated by the factors closely related to the charge transfer process in CE, including contact conformity and work function. Other factors, such as surface roughness, maximum contact capacitance (Cmax),62 and series resistance (Rs),23,64 can also contribute to the output performance. Although the surface roughness slightly increased with the increase in tIPN,equiv, its magnitude was not sufficient to significantly impact the output performance (Fig. S13, ESI†).66,67 Moreover, as shown in Fig. S14 and S15 (ESI†), Cmax and Rs remained nearly constant across the entire range of tIPN,equiv. Therefore, the primary factors that influenced the output performance of the 2D-IPN-TENG were the contact conformity and the work function (Fig. 4a).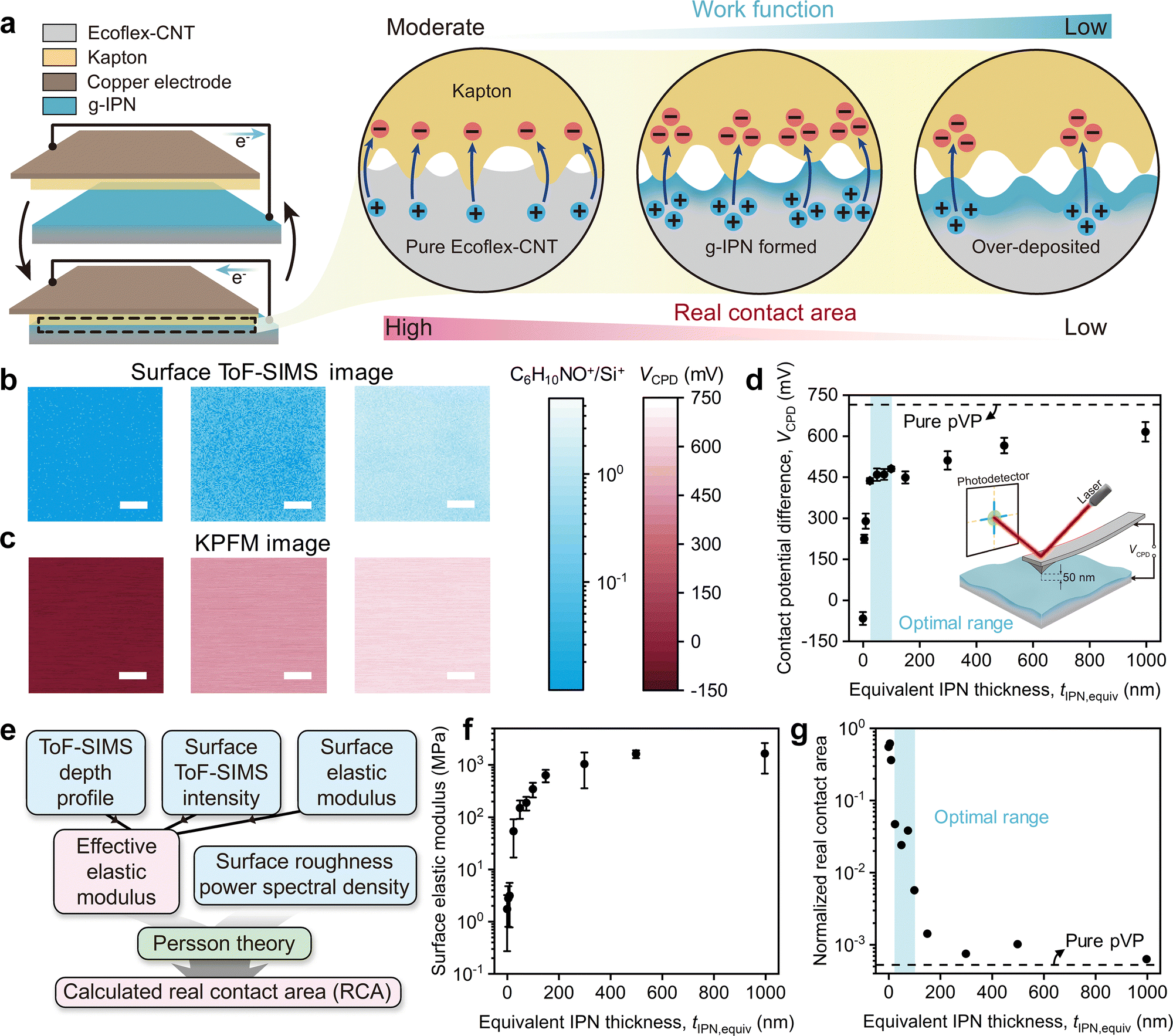 | ||
| Fig. 4 Origin of synergistic performance enhancement in 2D-IPN-TENG. (a) Cross-sectional schematics of 2D-IPN-TENG for the separated (top) and contacted (bottom) states, with the close-up views of the contact interfaces. While the counter-contacting material is Kapton, the bottom triboelectric layers are divided into three groups: pure Ecoflex-CNT (tIPN,equiv = 0 nm), Ecoflex-CNT with g-IPN (tIPN,equiv = 75 nm), and over-deposited Ecoflex-CNT (tIPN,equiv = 1000 nm). (b) and (c) Surface ToF-SIMS intensity and KPFM images for the three groups. The images in the left, middle, and right columns are for pure Ecoflex-CNT (tIPN,equiv = 0 nm), Ecoflex-CNT with g-IPN (tIPN,equiv = 75 nm), and over-deposited Ecoflex-CNT (tIPN,equiv = 1000 nm), respectively. Scale bars for the surface ToF-SIMS intensity images and KPFM images are 20 μm and 1 μm, respectively. (d) Measured VCPD of Ecoflex-CNT with g-IPN for various tIPN,equiv. A dashed line denotes the VCPD of the pure pVP. The measurement setup is illustrated in the inset. (e) Process for calculating RCA using the Persson theory. Experimentally measured parameters are colored blue, while the calculated parameters are colored pink. (f) Measured surface elastic modulus of Ecoflex-CNT with g-IPN for various tIPN,equiv. (g) Calculated RCA between Ecoflex-CNT with g-IPN and Kapton for various tIPN,equiv. It is normalized by the nominal contact area (A0). A normalized RCA of 1 indicates perfect contact conformity. A dashed line represents the normalized RCA between the pure pVP and Kapton. | ||
While Ecoflex-CNT is a soft polymer with a moderate work function, pVP is a stiff polymer with a low work function. Therefore, both the contact conformity and work function of the g-IPN may vary with tIPN,equiv. As shown in the surface ToF-SIMS images (Fig. 4b), the concentration of pVP at the surface of the g-IPN increased with increasing tIPN,equiv. Accordingly, the contact potential difference (VCPD) measured by Kelvin probe force microscopy (KPFM) shifted to a more positive value (Fig. 4c). Referring to the relationship between VCPD and the work function of the g-IPN (eqn (1)),68 the work function of the g-IPN decreased as tIPN,equiv increased:
| Φg-IPN = Φprobe − eVCPD, | (1) |
To quantify contact conformity, the real contact area (RCA) between the g-IPN and Kapton was calculated using the Persson theory,70–73 where a larger RCA indicates better contact conformity. The RCA between the g-IPN and Kapton may notably change between the two extremes: the RCA between pure Ecoflex-CNT and Kapton with a high level of contact conformity, and the RCA between pure pVP and Kapton with a low level of contact conformity. For this reason, the Persson theory, known for its accuracy across a wide range of RCA values, was employed.70,74
The procedure for calculating RCA with experimental input parameters, such as the effective elastic modulus (Eeff)70 and the surface roughness power spectral density (C(q)),75,76 where q is a wavevector, is depicted in Fig. 4e.70 Considering that the g-IPN has a gradient profile, Eeff should be obtained from the depth profile of the elastic modulus. However, since the depth profile of elastic modulus cannot be measured directly, it was indirectly obtained based on the composition of three relations: (1) the calibrated ToF-SIMS depth profile (Fig. S16, ESI†), (2) the relation between surface ToF-SIMS intensity and tIPN,equiv (Fig. S17a, ESI†), and (3) the relation between the surface elastic modulus and tIPN,equiv (Fig. 4f). Here, the surface elastic modulus refers to the elastic modulus measured at the surface of the sample using force–distance (F–d) spectroscopy,77 and is clearly distinguished from the bulk elastic modulus shown in Fig. 1e. Together with the C(q) obtained from multi-scale atomic force microscopy (AFM) topography measurements (Fig. S19b, ESI†), the RCA was calculated using eqn (2)–(4), where AR and A0 represent the RCA and nominal contact area, respectively.
 | (2) |
 | (3) |
 | (4) |
Generally, charge generation by CE and spontaneous charge dissipation cause QSC to reach dynamic equilibrium.63,78 Since CE occurs only at the contact asperities which approach the repulsive regime, the magnitude of charge generation at each contact cycle should be proportional to the RCA.46 Therefore, as the RCA sharply decreases with increasing tIPN,equiv, the output performance of the 2D-IPN-TENG should accordingly decrease as tIPN,equiv increases, if all other factors, such as ΔΦ, remain the same.
The existence of an optimal range of tIPN,equiv from 25 nm to 100 nm (Fig. 3c and d), where the output performance of 2D-IPN-TENG was maximized, can be explained by the synergistic effect of the work function and RCA, as illustrated in the center circle of Fig. 4a. As tIPN,equiv increased, the work function of the g-IPN quickly lowered until it reached the optimal range of tIPN,equiv, then slowly approached the work function of the pure pVP (Fig. 4d). On the other hand, the RCA between the g-IPN and Kapton began to drop at the optimal range of tIPN,equiv and slowly approached the RCA between the pure pVP and Kapton (Fig. 4g). Therefore, within the optimal range of tIPN,equiv from 25 nm to 100 nm, the output performance was maximized due to the saturated low work function and partially maintained high RCA.
Output performance of the 2D-IPN-TENG (tIPN,equiv = 75 nm) with its bottom layer under strain
The materials used in elastic TENGs are often exposed to severe strain. Therefore, it is important to characterize the surface deformation behavior and output performance of the elastic triboelectric materials under stretching (high strain, usually far greater than 10%) rather than under bending (low strain, generally less than 2%). As illustrated in Fig. 5a, when pure Ecoflex-CNT was uniaxially stretched, it uniformly elongated along the direction of stretch (a–a′) and uniformly compressed along the direction perpendicular to the stretch (b–b′) due to the Poisson effect.79 In contrast, when the Ecoflex-CNT with g-IPN (tIPN,equiv = 75 nm) was uniaxially stretched, cracks formed along the direction of stretch (a–a′), and wrinkles appeared along the direction perpendicular to the stretch (b–b′) due to the non-uniform depth profile of the elastic modulus (Fig. 5b). In this case, both cracks and wrinkles were identified from the OM images. Before stretching the Ecoflex-CNT with g-IPN, preexisting directionless wrinkles were observed (Fig. 5c). However, after stretching the same sample with 100% strain, the preexisting wrinkles disappeared. Instead, the surface cracked along the direction of stretch, and new wrinkles formed along the direction perpendicular to the stretch (Fig. 5d). | ||
| Fig. 5 Output performance of 2D-IPN-TENG (tIPN,equiv = 75 nm) with its bottom layer under strain. (a) Schematic illustrations of pure Ecoflex-CNT, with and without strain. (b) Schematic illustrations of Ecoflex-CNT with g-IPN, with and without strain. For (a) and (b), a–a′ and b–b′ indicate the direction of stretch and the direction perpendicular to the stretch, respectively. (c) OM image of Ecoflex-CNT with g-IPN, without strain. Scale bar is 50 μm. (d) OM image of stretched (ε = 100%) Ecoflex-CNT with g-IPN on the left, AFM topography image of a single microcrack on the top-right, and its surface elastic modulus mapping on the bottom-right. Scale bars on the left and right are 50 μm and 0.5 μm, respectively. (e) Distribution of surface elastic modulus in stretched (ε = 100%) pure Ecoflex-CNT. The average value of the surface elastic modulus was 3.66 MPa. (f) Distribution of the surface elastic modulus inside the microcrack of stretched (ε = 100%) Ecoflex-CNT with g-IPN. The average value of the surface elastic modulus inside the microcrack was 26.4 MPa. (g) Normalized surface ToF-SIMS intensity measured at the surface of Ecoflex-CNT with g-IPN under various strains. A dashed line represents the crack-onset strain of the stiff surface IPN. (h) Measured QSC of pure Ecoflex-CNT and Ecoflex-CNT with g-IPN under various strains. (i) Measured VOC of pure Ecoflex-CNT and Ecoflex-CNT with g-IPN under various strains. (j) Measured QSC and VOC of Ecoflex-CNT with g-IPN for various stretching cycles. One cycle involves stretching (ε = 100%) and releasing. (k) Durability data extracted from the stretched (ε = 100%) Ecoflex-CNT with g-IPN for various numbers of contacts. In (h)–(k), Kapton was used as the counter-contacting material. For (b)–(d) and (f)–(k), the tIPN,equiv of Ecoflex-CNT with g-IPN was 75 nm. | ||
In order to characterize the surface newly exposed by the formation of microcracks, AFM topography imaging was used to identify a single microcrack in a close-up view, and surface elastic modulus mapping was performed at the same site (Fig. 5d). The significant difference in surface elastic modulus between the inside and outside of the microcrack indicates that the stiff surface IPN did not exist inside the microcrack. On the other hand, the average surface elastic modulus inside the microcrack was higher than that of pure Ecoflex-CNT under 100% strain, but was similar to that of the g-IPN with tIPN,equiv = 25 nm under 100% strain (Fig. 5e, f and Fig. S20, ESI†). This indicates that an intrinsically stretchable IPN with a low pVP concentration was newly exposed to the ambient by the cracking. Note that g-IPN with tIPN,equiv of 25 nm or less is intrinsically stretchable, and had a spatially uniform surface elastic modulus in the stretched state, even though pure pVP itself is stiff and brittle (Fig. S20–S22, ESI†).
The uniaxial stretching led to an increase in the surface area of the Ecoflex-CNT with g-IPN. The surface ToF-SIMS intensity measurement showed that the characteristic fragment ion intensity of the pVP initially decreased until the crack-onset strain, due to the flattening of preexisting wrinkles. Subsequently, it continued to increase with further strain, implying that the surface area increased with further strain (Fig. 5g). This was attributed to the cracking and wrinkling of the stiff surface IPN and the additional surface area supplied by the exposure of the buried intrinsically stretchable IPN. The increase in surface area with increasing strain can also be quantitatively explained by a simple analytical model (Note S5 and Fig. S23, ESI†).
In Fig. 5h and i, the output performance was compared between the 2D-structured TENG with pure Ecoflex-CNT and 2D-IPN-TENG with tIPN,equiv = 75 nm for various strains applied to the bottom layers. For the 2D-structured TENG with pure Ecoflex-CNT, output performance decreased monotonically as the strain on the pure Ecoflex-CNT increased. In contrast, for the 2D-IPN-TENG, the output performance slightly degraded at a low strain of 10%; however, it interestingly returned to the initial values as the strain increased further. It is well known that a decrease in output performance is due to the increased Rs in stretched TENGs, which are based on elastomer-CNT composites.80,81 Although Rs of Ecoflex-CNT with g-IPN also increased with increasing strain (Fig. S24, ESI†), the surface area continuously increased with increasing strain. Consequently, both effects compensated for each other and resulted in strain-insensitive output performance. Hereafter, we refer to this spontaneous recovery of output performance under severe strain as the strain-compensating ability of the g-IPN.
The reliability of the output performance related to mechanical deformation was further evaluated. As shown in Fig. 5j, the output performance of the 2D-IPN-TENG was measured after iterative stretching and releasing of the bottom layer for up to 10000 cycles. The output performance degraded slightly after one cycle but nearly maintained its level up to 10000 cycles. This slight degradation in output performance arose from a small residual strain after release. As shown in Fig. S25 (ESI†), a residual strain below 10% was not sufficient to expose the buried intrinsically stretchable IPN to the ambient; the strain was too small to spontaneously recover the output performance. Meanwhile, the preexisting randomly oriented wrinkles were smoothed out, leading to a slight degradation in output performance due to the decreased surface area. In Fig. 5k, the QSC of the 2D-IPN-TENG was measured during 50000 contact–separation cycles while applying 100% strain to its bottom layer. As a result, no degradation occurred, demonstrating the high output durability of the Ecoflex-CNT with g-IPN, even under severely stretched conditions.
Demonstration of sponge-structured 3D-IPN-TENG with high performance
Ecoflex-CNT with g-IPN, which is stretchable and capable of high, strain-insensitive output performance, offers an appealing option for the development of 3D-structured elastic TENGs with high, reliable output performance and mechanical elasticity. In this work, to demonstrate the effectiveness of the g-IPN in 3D-structured elastic TENGs, the g-IPN was applied to a sponge-structured 3D elastic TENG.30,31 As shown in Fig. 6a, the sponge-structured 3D elastic TENG consists of a densely packed structure, designed to maximize volumetric efficiency. Specifically, it is composed of a conductive elastic sponge made of Ecoflex-CNT and multiple copper wires coated with Kapton, which are inserted into the well-ordered, lengthy pores of the Ecoflex-CNT sponge. The copper core of the wire and the Ecoflex-CNT sponge serve as induction electrodes, while the Kapton coating on the wire and the Ecoflex-CNT sponge serve as triboelectric layers. The two electrodes are connected through an external load, enabling power generation under mechanical stimuli (Fig. 6b). | ||
| Fig. 6 Sponge-structured 3D-IPN-TENG and its output characteristics. (a) Schematics of a 3D-TENG (left) and its close-up view showing a single wire inserted inside a pore (right). (b) Working principle of a 3D-TENG. (c) Photograph of a pure Ecoflex-CNT sponge (left) and an Ecoflex-CNT sponge with g-IPN (right). (d) Cross-sectional scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) images of a single wire consisting of a copper core with a Kapton coating around the copper core. Scale bar is 50 μm. (e) Photograph of the fabricated 3D-IPN-TENG (left). Scale bar is 1 cm. The device could be stretched (top-right) or twisted (bottom-right) without noticeable degradation. (f) Comparison of QSC,vol between the 3D-TENG and 3D-IPN-TENG. (g) Comparison of VOC between the 3D-TENG and 3D-IPN-TENG. (h) Durability characteristic of the 3D-IPN-TENG. (i) Comparison of the QSC,vol between previously reported 3D-structured TENGs and the 3D-IPN-TENG. All of them utilized CE between solids. (j) Comparison of peak power density between the 3D-TENG and 3D-IPN-TENG. (k) Configuration of the 3D-IPN-TENG with a PMC for supplying power to electronic devices. (l) Measured voltage of an output capacitor (Vcap) while simultaneously powering a stopwatch and a thermohygrometer. Both the stopwatch and the thermohygrometer operated continuously during the working period, shaded in red. (m) Photographs of a stopwatch and a thermohygrometer in the off-state (top) and in the on-state (bottom). | ||
To boost the output performance of the sponge-structured 3D elastic TENG, it is crucial to increase the magnitude of CE between the Ecoflex-CNT and Kapton. Therefore, g-IPN with tIPN,equiv = 75 nm was uniformly formed throughout the pores of a pure Ecoflex-CNT sponge, using the iCVD process. Note that the iCVD process stands out as one of the most promising candidates for forming g-IPN throughout all the surfaces of complex 3D structures, owing to its superior deposition conformity.82 Hereafter, for convenience, in the sponge-structured 3D elastic TENG, 3D-IPN-TENG refers to the case where g-IPN is formed throughout the entire Ecoflex-CNT sponge, and 3D-TENG refers to the case where g-IPN is not formed. The detailed fabrication processes for the 3D-TENG and 3D-IPN-TENG are described in the Experimental section and Fig. S26 (ESI†).
Since the Ecoflex-CNT sponge not only serves as an induction electrode and triboelectric layer but also as a structural support, maintaining its dimensions and mechanical properties after the formation of the g-IPN is important. Because the formation process of the g-IPN using iCVD does not involve any heat or solvent, there were no differences in either the overall dimensions or pore sizes of the Ecoflex-CNT sponge before and after the formation of the g-IPN (Fig. 6c and Fig. S27, ESI†). Additionally, it did not affect the bulk mechanical properties of the Ecoflex-CNT sponge (Fig. S28, ESI†). Finally, a complete 3D-IPN-TENG was fabricated by inserting multiple copper wires coated with Kapton (Fig. 6d) into the pores of the Ecoflex-CNT sponge with g-IPN (Fig. 6e). The resulting device was highly elastic, able to be stretched and twisted without any noticeable degradation (Fig. 6e).
The output performance of a 3D-IPN-TENG with an optimized pore diameter of 600 μm was evaluated. The theoretical and experimental optimization of the pore diameter is described in Note S6 and Fig. S29 (ESI†). For a 3D-structured TENG, QSC,vol, which represents the short-circuit volume charge density, is more convenient for making a fair comparison with the charge densities reported in other works. As shown in Fig. 6f and g, the maximum QSC,vol and VOC for 3D-IPN-TENG were 267.2 mC m−3 and 455 V, respectively. These QSC,vol and VOC in 3D-IPN-TENG were about 6.1-fold and 3.1-fold larger, respectively, compared to those of the 3D-TENG.
As shown in Fig. 3c and d, the QSC and VOC of the 2D-IPN-TENG were about 1.5-fold and 1.5-fold larger, respectively, than those of the 2D-structured TENG with pure Ecoflex-CNT. One of the main reasons for the larger difference in output performance between the 3D-structured TENGs compared to that between the 2D-structured TENGs is the local strain that occurs during compression of the 3D-structured TENGs. A numerical simulation based on the finite element method (FEM) showed that high local strain occurs during compression of the Ecoflex-CNT sponge, indicating that CE may occur when the pure Ecoflex-CNT or Ecoflex-CNT with g-IPN are under the stretched state (Fig. S30, ESI†). Therefore, since the difference in output performance between the pure Ecoflex-CNT and Ecoflex-CNT with g-IPN increases with increasing strain due to the strain-compensating ability (Fig. 5h and i), the difference in output performance between the 3D-structured TENGs should be larger than that between the 2D-structured TENGs.
Moreover, owing to the high output durability of the Ecoflex-CNT with g-IPN under the stretched state (Fig. 5k), the QSC,vol of 3D-IPN-TENG scarcely fluctuated during iterative compression–relaxation up to 50000 cycles (Fig. 6h). To the best of our knowledge, the QSC,vol of 267.2 mC m−3 is the highest value compared to other 3D-structured TENGs that utilize CE between solids (Fig. 6i).27–31,83–87 We also evaluated the peak and average power density with various Rload for the 3D-IPN-TENG and 3D-TENG. As shown in Fig. 6j and Fig. S31 (ESI†), the maximum peak power density and maximum average power density of the 3D-IPN-TENG were 496.81 W m−3 and 48.9 W m−3 Hz−1, respectively, which were about 9.5-fold and 8.5-fold higher than those of the 3D-TENG. This high output performance of the 3D-IPN-TENG indicates its high volumetric efficiency due to the 3D sponge structure (Table S2, ESI†).
The feasibility of driving small electronic devices was demonstrated using a 3D-IPN-TENG as a portable power source with a small size (effective volume of 2 cm × 2.2 cm × 0.44 cm in length, width, and height, respectively). To efficiently store and utilize the energy generated from the 3D-IPN-TENG, a power management circuit (PMC) was used, which was composed of a full-wave rectifier and a buck converter88 (Fig. 6k and Fig. S32, ESI†). The energy generated from the 3D-IPN-TENG was finally stored in an output capacitor (Cout) with a capacitance of 3.3 mF. The fabricated 3D-IPN-TENG was able to simultaneously turn on both a commercial stopwatch and a thermohygrometer. It generated enough power to drive them continuously without the aid of any additional power supply, as shown in Fig. 6l, m and Movie S1 (ESI†).
Conclusions
In summary, we report a complementary material synthesis strategy for the development of elastic triboelectric materials with high and reliable output performance. In this work, a synergistic enhancement of triboelectricity was achieved by forming a sub-micron thick g-IPN on a stretchable host elastomer (Ecoflex-CNT) that has high contact conformity using a highly chargeable guest polymer (pVP) with a low work function. This was accomplished without affecting the superior bulk mechanical properties of the host elastomer. For the 2D-structured TENG with g-IPN (2D-IPN-TENG), the QSC and VOC of up to 445 μC m−2 and 1335 V, respectively, were achieved when Kapton was used as a counter-contacting material.In addition, the depth-directional gradient structure of g-IPN, with a high concentration of pVP near the surface and a low concentration of pVP near the bulk, effectively prevented the degradation of output performance under increasing strain, which was impossible without the g-IPN. This spontaneous recovery of output performance under severe strain, referred to as the strain-compensating ability, was attributed to the increasing surface area of the g-IPN with increasing strain, as confirmed by ToF-SIMS characterization and analytical modeling.
We demonstrated the effectiveness of the g-IPN in 3D-structured elastic TENGs by fabricating a sponge-structured 3D elastic TENG with g-IPN (3D-IPN-TENG). Owing to the excellent deposition conformity of the iCVD process, the g-IPN could be conformally formed throughout the Ecoflex-CNT sponge. The 3D-IPN-TENG generated a high QSC,vol of 267.2 mC m−3, which is the highest value among previously reported 3D-structured TENGs that utilize CE between solids. A compact 3D-IPN-TENG with an effective volume of less than 2 cm3 was able to simultaneously and continuously power a commercial stopwatch and a thermohygrometer without any additional power supply. We believe that the proposed complementary material synthesis strategy will pave the way for holistic engineering in the field of polymer synthesis and its application to elastic triboelectric materials.
Experimental section
Fabrication of Ecoflex-CNT samples for 2D-structured TENGs
To prepare the conductive and stretchable Ecoflex-CNT composite, both prepolymers (part A and part B) of Ecoflex 0050 (Smooth-On Inc., USA) were first mixed at a weight ratio of 1:1 to obtain uncured Ecoflex 0050. Then, 4 wt% of conductive multi-walled CNT (Carbon Nano-material Technology Inc., Korea) was immediately added to the uncured Ecoflex 0050 and stirred for 1 minute. It was then placed in a vacuum chamber for 1 minute to remove any trapped bubbles. The resulting uncured Ecoflex-CNT was poured into an acrylic mold (4 cm × 4 cm × 0.1 cm in length, width, and height, respectively) and was cured at room temperature for 24 hours. After curing, the planar Ecoflex-CNT sample was peeled from the mold and was used without any further treatment.
Formation of g-IPN
In this work, g-IPN was conformally formed on the Ecoflex-CNT samples by depositing pVP using the iCVD process (Fig. 2d). Before deposition, the monomer (NVP, 99%, Tokyo Chemical Industry) and the initiator (TBPO, 99%, Sigma-Aldrich) were embedded in separate canisters, which were connected to a custom-built iCVD reactor through mass flow controllers. The monomer (NVP) and the initiator (TBPO) were used as received without further purification. The Ecoflex-CNT samples were then placed on the substrate inside the vacuum chamber of the iCVD reactor. To induce the vaporized monomer and initiator with the designed flow rates in the vacuum chamber, the canisters containing NVP and TBPO were heated to 55 °C and 30 °C, respectively. During deposition, the temperatures of the hot filament and the substrate of the iCVD reactor were maintained at 150 °C and 30 °C, respectively, while the flow rates of NVP and TBPO were kept at 1.08 sccm and 0.88 sccm, respectively. The chamber pressure was maintained at 250 mTorr during deposition. The deposition rate of the pVP, based on the thickness of the pVP deposited on a Si wafer, was 3 nm min−1.Fabrication of drop-cast samples
To prepare drop-cast samples, firstly, the powders of (i) pVP synthesized by iCVD and (ii) commercial pVP with Mw of 10000 g mol−1 and 29000 g mol−1 (Sigma-Aldrich) were prepared. As mentioned earlier, commercial pVP powders with similar Mw were chosen to provide a fair comparison with the pVP synthesized by iCVD (Mw ∼ 18600 g mol−1). To prepare the powder of (i), pVP was deposited by iCVD on a slide glass with a thickness of 3 μm, and then the pVP was scraped from the slide glass using a commercial razor blade. Each pVP powder sample was dissolved individually in 2-propanol to prepare solutions for drop-casting. The prepared solutions were drop-cast onto pure Ecoflex-CNT substrates which were placed on a hot plate at 80 °C. Afterwards, they were left to dry at room temperature for 24 hours, and then placed inside a vacuum chamber for another 24 hours to remove any residual solvent from the drop-cast pVP.
Fabrication of pure pVP on Au
To evaluate the triboelectric properties of pure pVP, pure pVP deposited on Au was used. First, Ni and Au were sequentially thermally evaporated onto a diced Si wafer (2 cm × 2.4 cm in length and width, respectively), with thicknesses of 10 nm and 100 nm, respectively. Then, pVP was deposited on the Au/Ni-coated Si wafer using the iCVD process with a thickness of 3 μm.Fabrication of stretched bottom layers for 2D-structured TENGs
To evaluate the output performance of the 2D-structured TENGs with stretched bottom layers, planar samples of pure Ecoflex-CNT and Ecoflex-CNT with g-IPN were first mounted on a bench vise and stretched with various strains. Subsequently, acrylic supports (2 cm × 2 cm × 0.3 cm in length, width, and height, respectively) were adhered to the backside of the stretched samples using a gasket maker. After curing the gasket maker for 24 hours, the samples were released from the bench vise. The area of the samples where the acrylic support was placed on the backside remained stretched. These stretched samples with acrylic supports were then used as the stretched bottom layers for the 2D-structured TENGs, after cutting them into the desired dimensions (2 cm × 2.4 cm in length and width, respectively).Fabrication of top counter-contacting layers for 2D-structured TENGs
For the 2D-structured TENG, the top counter-contacting layer consisted of a counter-contacting material and an induction electrode. Kapton (Alphaflon, Korea), ETFE (Goodfellows, UK), and PFA (Goodfellows, UK) films, each with a thickness of 25 μm, and an Al plate with a thickness of 300 μm were cleaned twice by carefully submerging and swiping them in baths of acetone, 2-propanol, and ethanol to eliminate any organic contaminants. Afterwards, they were dried by blowing N2 gas before use. To prepare the induction electrode, double-sided conductive copper tape was seamlessly attached to the cleaned Al plate. The top counter-contacting layers for the 2D-structured TENGs were fabricated by attaching the cleaned films of the counter-contacting materials to the induction electrodes.Fabrication of Ecoflex-CNT sponges for 3D-structured TENGs
The overall process used to fabricate the Ecoflex-CNT sponges is briefly described in Fig. S26 (ESI†). Initially, a sacrificial PVA mold with the inverse structure of the desired Ecoflex-CNT sponge was fabricated using a 3D printer (S3, Ultimaker). The PVA mold was then completely submerged in uncured liquid-state Ecoflex-CNT. A cup containing the PVA mold and the uncured Ecoflex-CNT was placed in a vacuum chamber for 30 minutes to seamlessly inject the uncured Ecoflex-CNT into the PVA mold. After curing at room temperature for 24 hours, the cured Ecoflex-CNT which wrapped around the PVA mold, was cut out. To selectively dissolve the PVA mold, the PVA mold containing the cured Ecoflex-CNT sponge was submerged in a hot (∼50 °C) deionized water bath for at least 72 hours. The resulting Ecoflex-CNT sponge was then rinsed with deionized water and dried by blowing N2 gas. An Ecoflex-CNT sponge with g-IPN, which was used to fabricate the 3D-IPN-TENG, was prepared by forming g-IPN throughout the pure Ecoflex-CNT sponge, with tIPN,equiv = 75 nm, using the iCVD process.Fabrication of 3D-TENG and 3D-IPN-TENG
The complete 3D-TENG and 3D-IPN-TENG were fabricated by inserting multiple copper wires (with a copper core diameter of 250 μm) coated with Kapton (with a thickness of 25 μm), into the lengthy pores of the conductive elastic sponges. A pure Ecoflex-CNT sponge and an Ecoflex-CNT sponge with g-IPN were used as the conductive elastic sponges for the 3D-TENG and 3D-IPN-TENG, respectively. To measure the output performance of the 3D-TENG and 3D-IPN-TENG, they were bonded to acrylic frameworks (4 cm × 4 cm in length and width, respectively) using a gasket maker.Electrical measurements
A mechanical shaker (ET-140-2, Labworks Inc.) connected to a power amplifier (PA-141, Labworks Inc.) and a function generator (33120A, Hewlett Packard) were used to apply the desired mechanical pressure to the TENGs. Before each measurement, the pressure applied by the mechanical shaker was calibrated using a high-precision pressure sensor (208C02, PCB Piezotronics). The peak pressure of 150 kPa was applied to characterize the output performance of all the TENGs used in this work. The QSC and ISC were measured using a system electrometer (model 6514, Keithley). The VOC was measured using a high-power sourcemeter (model 2657, Keithley). The Cmax was measured using an LCR meter (E4980A, Agilent technologies) at an applied frequency of 1 kHz. The Rs was measured using a digital multimeter (model 289, Fluke).Material characterizations
The bulk mechanical properties of the planar Ecoflex-CNT samples were measured using a universal testing machine (Z005, Zwick Roell). The mechanical properties of the Ecoflex-CNT sponges were measured using the same equipment, but with jigs for the compression test. The tIPN,equiv was measured using an ellipsometer (M2000U with auto angle ESM-300 Base, J. A. Woollam). A stretching machine (Han-tech, Co., Ltd) was used to periodically stretch (100% strain) and release the Ecoflex-CNT samples with g-IPN. Gel permeation chromatography (GPC) measurements were performed on commercial equipment (1260 Infinity system, Agilent Technologies) to identify the Mw of the pVP synthesized by iCVD. The Mw was calculated relative to linear polystyrene (PS) standards (Agilent Technologies). A copper wire sample for the cross-sectional SEM/EDS images was prepared using an ion milling system (ArBlade 5000, Hitachi). Cross-sectional SEM/EDS images were taken using commercial field emission SEM equipment (JSM-IT800, JEOL).Analyses using atomic force microscopy (AFM)
The imaging of surface morphology, the imaging of VCPD, and the measurement/mapping of surface elastic modulus were carried out using commercial AFM equipment (NX-10, Park Systems). For imaging of surface morphology, non-contact mode AFM was used, equipped with a silicon probe (PR-T190, tip radius of <10 nm, spring constant of ∼48 N m−1, and resonance frequency of ∼190 kHz). For the extraction of C(q) from the measured surface morphology, a publicly available application75 was used. For the imaging of VCPD, amplitude modulation KPFM mode was used, equipped with a Pt-coated probe (PR-E75, tip radius of <30 nm, spring constant of ∼2.8 N m−1, and resonance frequency of ∼75 kHz). For the measurement and mapping of surface elastic modulus, the F–d spectroscopy mode was used, equipped with a silicon probe (PPP-FMR, tip radius of <7 nm, spring constant of ∼2.8 N m−1, and resonance frequency of ∼75 kHz). Before every AFM, KPFM, and F–d spectroscopy mode measurement, the spring constants of the probes were calibrated using the thermal tune method for PR-E75 and PPP-FMR, and the Sader method for PR-T190. For F–d spectroscopy mode, the force slope and cantilever sensitivity were also calibrated before every measurement.Analyses using time-of-flight secondary ion mass spectrometry (ToF-SIMS)
All measurements of surface ToF-SIMS intensities and ToF-SIMS depth profiles were conducted using a commercial time-of-flight secondary ion mass spectrometer (ToF-SIMS5 iontof, ION-TOF GmbH) equipped with a Bi3+ primary ion source. For all experiments, primary ion bombardment was performed with 30 keV Bi3+ ions. For the measurement of surface ToF-SIMS intensity, data were acquired from at least three different spots on the surface of a sample, at a scan area of 100 μm × 100 μm and a total acquisition time of 3 seconds. For the measurement of ToF-SIMS depth profile, a 5 keV Ar1450+ gas cluster ion beam (GCIB) was used as a sputtering ion source, and depth profiling was carried out in non-interlaced mode with a sputter time of 1 second and a sputtering area of 300 μm × 300 μm. The process of calibration between the sputter time and depth is described in Note S4 (ESI†).DFT calculations
All DFT calculations were performed using the Gaussian 16 package. To determine the global minimum structure, 100 distinct initial configurations for pVP and Nylon 66 were generated using the RDKit library. Each configuration underwent geometry optimization calculations using the B3LYP/6-311++G(d,p) functional and basis set. Vibrational frequency calculations were performed using the B3LYP/6-311++G(d,p) framework for the configuration with the lowest energy after geometric optimization. The highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and the energy gap between them were determined based on the optimized geometric structures. The procedure was repeated for dimers (pVP and Nylon 66) and tetramer (pVP) to obtain their HOMO and LUMO, and bandgap.FEM simulation
An FEM simulation was conducted to analyze the compression of the 3D-structured sponge using commercially available software (ANSYS Mechanical). A unit cell structure of the 3D-structured sponge, which was designed and imported from SpaceClaim, was used for the FEM simulation (Fig. S30a, ESI†). The material for the sponge used in the simulation was customized by fitting the 3-parameter Mooney-Rivlin model to the experimental stress–strain curve of Ecoflex-CNT with g-IPN. Two rigid plates were employed to apply boundary conditions: while one plate was fixed, the other was used to apply external pressure to the sponge only in the compression direction. The interfaces between the plates and the sponge were set to be bonded.Author contributions
D.-W. Kim and H. Mun contributed equally to this work. D.-W. Kim conceived the idea, conducted formal analysis, fabricated devices, performed electrical measurements, characterized electrical and material properties, curated data, conducted investigations, constructed figures, and wrote the original draft. H. Mun conducted an iCVD process, fabricated devices, curated data, conducted investigations, and contributed to reviewing, and editing. Y. Kang conducted DFT calculations, curated data, conducted investigations, and contributed to reviewing, and editing. W.-G. Kim curated data, and conducted investigations. D. Ahn conducted the iCVD process, and contributed to reviewing, and editing. S.-Y. Yun, J.-A. Han, D. H. Lee, T. Lee, K. Jeong, and J. Kim participated in data curation and analyses. S. G. Im and Y.-K. Choi supervised all experiments, reviewed and edited the draft, and interpreted the results. All authors contributed to data analyses and discussions.Data availability
All data that support the findings of this study are available in the main text or the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) under grants RS-2023-00260637, 2022M3F3A2A01072851, and RS-2023-00217888 and by the 2018 Open R&D project of the Korea Electric Power Corporation (KEPCO) (R18EO01). This work was also supported by Samsung Science and Technology Foundation under project number SRFC-IT2102-04.References
- D. J. Lacks and T. Shinbrot, Nat. Rev. Chem., 2019, 3, 465–476 CAS.
- Z. L. Wang and A. C. Wang, Mater. Today, 2019, 30, 34–51 CAS.
- E. S. Polsen, A. G. Stevens and A. J. Hart, ACS Appl. Mater. Interfaces, 2013, 5, 3656–3662 CAS.
- A. Jaworek and A. T. Sobczyk, J. Electrost., 2008, 66, 197–219 CAS.
- C. G. Camara, J. V. Escobar, J. R. Hird and S. J. Putterman, Nature, 2008, 455, 1089–1092 CAS.
- F.-R. Fan, Z.-Q. Tian and Z. L. Wang, Nano Energy, 2012, 1, 328–334 CAS.
- W. Xu, H. Zheng, Y. Liu, X. Zhou, C. Zhang, Y. Song, X. Deng, M. Leung, Z. Yang, R. X. Xu, Z. L. Wang, X. C. Zeng and Z. Wang, Nature, 2020, 578, 392–396 CAS.
- R. Hinchet, H.-J. Yoon, H. Ryu, M.-K. Kim, E.-K. Choi, D.-S. Kim and S.-W. Kim, Science, 2019, 365, 491–494 CAS.
- S.-W. Kim, J.-K. Kim, J. Y. Park, J. Mun, S. Jung, S. E. Yang, G. Lee, P. S. Lee, H.-C. Song, C. Yang, H. S. Park, J. S. Son and J. M. Baik, Adv. Energy Mater., 2023, 13, 2202987 CrossRef CAS.
- S. Jung, J. Oh, U. J. Yang, S. M. Lee, J. Lee, M. Jeong, Y. Cho, S. Kim, J. M. Baik and C. Yang, Nano Energy, 2020, 77, 105271 CrossRef CAS.
- X. Li, L. Xu, P. Lin, X. Yang, H. Wang, H. Qin and Z. L. Wang, Energy Environ. Sci., 2023, 16, 3040–3052 RSC.
- Y. Song, J. Min, Y. Yu, H. Wang, Y. Yang, H. Zhang and W. Gao, Sci. Adv., 2020, 6, eaay9842 CrossRef CAS PubMed.
- J. Oh, J.-K. Kim, J. Gao, S. Jung, W. Kim, G. Park, J. Park, J. M. Baik and C. Yang, ACS Nano, 2024, 18, 12146–12157 CrossRef CAS PubMed.
- L. Yin, K. N. Kim, J. Lv, F. Tehrani, M. Lin, Z. Lin, J.-M. Moon, J. Ma, J. Yu, S. Xu and J. Wang, Nat. Commun., 2021, 12, 1542 CrossRef CAS PubMed.
- J. Han, N. Xu, J. Yu, Y. Wang, Y. Xiong, Y. Wei, Z. L. Wang and Q. Sun, Energy Environ. Sci., 2022, 15, 5069–5081 RSC.
- H. Ouyang, Z. Liu, N. Li, B. Shi, Y. Zou, F. Xie, Y. Ma, Z. Li, H. Li, Q. Zheng, X. Qu, Y. Fan, Z. L. Wang, H. Zhang and Z. Li, Nat. Commun., 2019, 10, 1821 CrossRef PubMed.
- H. Ryu, H.-M. Park, M.-K. Kim, B. Kim, H. S. Myoung, T. Y. Kim, H.-J. Yoon, S. S. Kwak, J. Kim, T. H. Hwang, E.-K. Choi and S.-W. Kim, Nat. Commun., 2021, 12, 4374 CrossRef CAS PubMed.
- L. Chen, C. Chen, L. Jin, H. Guo, A. C. Wang, F. Ning, Q. Xu, Z. Du, F. Wang and Z. L. Wang, Energy Environ. Sci., 2021, 14, 955–964 RSC.
- Z. Zhou, K. Chen, X. Li, S. Zhang, Y. Wu, Y. Zhou, K. Meng, C. Sun, Q. He, W. Fan, E. Fan, Z. Lin, X. Tan, W. Deng, J. Yang and J. Chen, Nat. Electron., 2020, 3, 571–578 CrossRef.
- K. Parida, G. Thangavel, G. Cai, X. Zhou, S. Park, J. Xiong and P. S. Lee, Nat. Commun., 2019, 10, 2158 CrossRef PubMed.
- T. H. Wong, Y. Liu, J. Li, K. Yao, S. Liu, C. K. Yiu, X. Huang, M. Wu, W. Park, J. Zhou, S. K. Nejad, H. Li, D. Li, Z. Xie and X. Yu, Adv. Funct. Mater., 2022, 32, 2111269 CrossRef CAS.
- S. Li, W. Peng, J. Wang, L. Lin, Y. Zi, G. Zhang and Z. L. Wang, ACS Nano, 2016, 10, 7973–7981 CrossRef CAS PubMed.
- K. Yao, Y. Liu, D. Li, J. He, J. Li, R. H. W. Lam, Z. Xie, L. Wang and X. Yu, Nano Energy, 2020, 76, 105017 CrossRef CAS.
- B. Cheng, S. Niu, Q. Xu, J. Wen, S. Bai and Y. Qin, ACS Appl. Mater. Interfaces, 2021, 13, 59975–59982 CrossRef CAS PubMed.
- C. Dong, A. Leber, D. Yan, H. Banerjee, S. Laperrousaz, T. D. Gupta, S. Shadman, P. M. Reis and F. Sorin, Sci. Adv., 2022, 8, eabo0869 CrossRef PubMed.
- J. Wang, S. Li, F. Yi, Y. Zi, J. Lin, X. Wang, Y. Xu and Z. L. Wang, Nat. Commun., 2016, 7, 12744 CrossRef CAS PubMed.
- B. Chen, W. Tang, T. Jiang, L. Zhu, X. Chen, C. He, L. Xu, H. Guo, P. Lin, D. Li, J. Shao and Z. L. Wang, Nano Energy, 2018, 45, 380–389 CrossRef CAS.
- S. Li, J. Wang, W. Peng, L. Lin, Y. Zi, S. Wang, G. Zhang and Z. L. Wang, Adv. Energy Mater., 2017, 7, 1602832 CrossRef.
- S. Tang, W. Chang, G. Li, J. Sun, Y. Du, X. Hui, Q. Tang, Z. Hu, J. Li, J. Chen, W. He and H. Guo, Nano Res., 2023, 16, 6933–6939 CrossRef CAS.
- W.-G. Kim, D. Kim, S.-B. Jeon, S.-J. Park, I.-W. Tcho, I.-K. Jin, J.-K. Han and Y.-K. Choi, Adv. Energy Mater., 2018, 8, 1800654 Search PubMed.
- W.-G. Kim, J.-K. Kim, D.-W. Kim, I.-W. Tcho and Y.-K. Choi, Nano Energy, 2022, 96, 107090 CAS.
- S. Niu and Z. L. Wang, Nano Energy, 2015, 14, 161–192 CrossRef CAS.
- L. Yang, C. Liu, W. Yuan, C. Meng, A. Dutta, X. Chen, L. Guo, G. Niu and H. Cheng, Nano Energy, 2022, 103, 107807 CrossRef CAS.
- S. K. Ghosh, J. Kim, M. P. Kim, S. Na, J. Cho, J. J. Kim and H. Ko, ACS Nano, 2022, 16, 11415–11427 CrossRef CAS PubMed.
- J. Qian, J. He, S. Qian, J. Zhang, X. Niu, X. Fan, C. Wang, X. Hou, J. Mu, W. Geng and X. Chou, Adv. Funct. Mater., 2020, 30, 1907414 CrossRef CAS.
- S. M. S. Rana, M. A. Zahed, M. R. Islam, O. Faruk, H. S. Song, S. H. Jeong and J. Y. Park, Chem. Eng. J., 2023, 473, 144989 CrossRef.
- M. Salauddin, S. M. S. Rana, M. Sharifuzzaman, H. S. Song, M. S. Reza, S. H. Jeong and J. Y. Park, Adv. Energy Mater., 2023, 13, 2203812 CrossRef CAS.
- J. Zhao, Y. Xiao, W. Yang, S. Zhang, H. Wang, Q. Wang, Z. Sun, W. Li, M. Gao, Z. Wang, Y. Xu, H. Chen and J. Wang, Adv. Mater. Technol., 2023, 8, 2201769 CrossRef CAS.
- D. W. Kim, J. H. Lee, I. You, J. K. Kim and U. Jeong, Nano Energy, 2018, 50, 192–200 CrossRef CAS.
- H. Li, X. Fang, R. Li, B. Liu, H. Tang, X. Ding, Y. Xie, R. Zhou, G. Zhou and Y. Tang, Nano Energy, 2020, 78, 105288 CrossRef CAS.
- H. Chen, Y. Xu, J. Zhang, W. Wu and G. Song, Nano Energy, 2019, 58, 304–311 CrossRef CAS.
- W. Wu, X. Peng, Y. Xiao, J. Sun, L. Li, Y. Xu, S. Zhang, K. Dong and L. Wang, Mater. Today Chem., 2023, 27, 101286 CAS.
- Y. S. Lipatov and L. V. Karabanova, J. Mater. Sci., 1995, 30, 1095–1104 CAS.
- K. K. Gleason, Nat. Rev. Phys., 2020, 2, 347–364 CAS.
- Z. Li, L. Zhang, L. Guo, W. Hu, A. Yu and J. Zhai, Nano Res., 2023, 16, 11855–11861 CAS.
- G. Min, Y. Xu, P. Cochran, N. Gadegaard, D. M. Mulvihill and R. Dahiya, Nano Energy, 2021, 83, 105829 CAS.
- S. Li, Y. Zhou, Y. Zi, G. Zhang and Z. L. Wang, ACS Nano, 2016, 10, 2528–2535 CrossRef CAS PubMed.
- X. Tao, X. Chen and Z. L. Wang, Energy Environ. Sci., 2023, 16, 3654–3678 RSC.
- D. K. Davies, J. Phys. D: Appl. Phys., 1969, 2, 1533 CrossRef.
- H. Zou, L. Guo, H. Xue, Y. Zhang, X. Shen, X. Liu, P. Wang, X. He, G. Dai, P. Jiang, H. Zheng, B. Zhang, C. Xu and Z. L. Wang, Nat. Commun., 2020, 11, 2093 CrossRef CAS PubMed.
- H. Zou, Y. Zhang, L. Guo, P. Wang, X. He, G. Dai, H. Zheng, C. Chen, A. C. Wang, C. Xu and Z. L. Wang, Nat. Commun., 2019, 10, 1427 CrossRef PubMed.
- Y. Shang, Z. Wang, C. Yu, W. Xu, Z. Chen, B. Jiang and H. Zhang, Nano Energy, 2022, 103, 107847 CrossRef CAS.
- J. W. Lee, S. Jung, T. W. Lee, J. Jo, H. Y. Chae, K. Choi, J. J. Kim, J. H. Lee, C. Yang and J. M. Baik, Adv. Energy Mater., 2019, 9, 1901987 CrossRef.
- M. Li, H.-W. Lu, S.-W. Wang, R.-P. Li, J.-Y. Chen, W.-S. Chuang, F.-S. Yang, Y.-F. Lin, C.-Y. Chen and Y.-C. Lai, Nat. Commun., 2022, 13, 938 CrossRef CAS PubMed.
- J. Kim, D. Kang, H.-K. Lee, J.-H. Hwang, H. Y. Lee, S. Jeon, D. Kim, S. Kim and S.-W. Kim, Adv. Funct. Mater., 2023, 33, 2209648 CrossRef CAS.
- X. Zhang, L. Chen, Y. Jiang, W. Lim and S. Soh, Chem. Mater., 2019, 31, 1473–1478 CrossRef CAS.
- K. Jeong, Y. Lee, Y. Kim, H. Mun, K.-U. Kyung and S. G. Im, Chem. Eng. J., 2022, 429, 132250 CrossRef CAS.
- T. Li, Z. Y. Huang, Z. C. Xi, S. P. Lacour, S. Wagner and Z. Suo, Mech. Mater., 2005, 37, 261–273 CrossRef.
- T. Yang, X. Li, X. Jiang, S. Lin, J. Lao, J. Shi, Z. Zhen, Z. Li and H. Zhu, Mater. Horiz., 2016, 3, 248–255 RSC.
- J. Kang, J. Mun, Y. Zheng, M. Koizumi, N. Matsuhisa, H.-C. Wu, S. Chen, J. B.-H. Tok, G. H. Lee, L. Jin and Z. Bao, Nat. Nanotechnol., 2022, 17, 1265–1271 CrossRef CAS PubMed.
- M.-W. Moon, S. H. Lee, J.-Y. Sun, K. H. Oh, A. Vaziri and J. W. Hutchinson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1130–1133 CrossRef CAS PubMed.
- Y. Liu, W. Liu, Z. Wang, W. He, Q. Tang, Y. Xi, X. Wang, H. Guo and C. Hu, Nat. Commun., 2020, 11, 1599 CrossRef CAS PubMed.
- C. Zhang, L. Zhou, P. Cheng, X. Yin, D. Liu, X. Li, H. Guo, Z. L. Wang and J. Wang, Appl. Mater. Today, 2020, 18, 100496 CrossRef.
- W. Zhang, Q. Liu, S. Chao, R. Liu, X. Cui, Y. Sun, H. Ouyang and Z. Li, ACS Appl. Mater. Interfaces, 2021, 13, 42966–42976 CrossRef CAS PubMed.
- J. X. Liu, G. Liu, Z. H. Guo, W. Hu, C. Zhang and X. Pu, Chem. Eng. J., 2023, 462, 142167 CrossRef CAS.
- T. Prada, V. Harnchana, A. Lakhonchai, A. Chingsungnoen, P. Poolcharuansin, N. Chanlek, A. Klamchuen, P. Thongbai and V. Amornkitbamrung, Nano Res., 2022, 15, 272–279 CrossRef CAS.
- K. C. Pradel and N. Fukata, Nano Energy, 2021, 83, 105856 CrossRef CAS.
- S. S. Kwak, S. M. Kim, H. Ryu, J. Kim, U. Khan, H.-J. Yoon, Y. H. Jeong and S.-W. Kim, Energy Environ. Sci., 2019, 12, 3156–3163 RSC.
- Y. S. Zhou, S. Wang, Y. Yang, G. Zhu, S. Niu, Z.-H. Lin, Y. Liu and Z. L. Wang, Nano Lett., 2014, 14, 1567–1572 CrossRef CAS PubMed.
- B. N. J. Persson, Surf. Sci. Rep., 2006, 61, 201–227 CrossRef CAS.
- C. Yang and B. N. J. Persson, J. Phys.: Condens. Matter, 2008, 20, 215214 CrossRef.
- B. N. J. Persson, O. Albohr, U. Tartaglino, A. I. Volokitin and E. Tosatti, J. Phys.: Condens. Matter, 2005, 17, R1 CrossRef CAS PubMed.
- B. N. J. Persson, Phys. Rev. Lett., 2001, 87, 116101 CrossRef CAS PubMed.
- Y. Xu, G. Min, N. Gadegaard, R. Dahiya and D. M. Mulvihill, Nano Energy, 2020, 76, 105067 CrossRef CAS.
- M. C. Röttger, A. Sanner, L. A. Thimons, T. Junge, A. Gujrati, J. M. Monti, W. G. Nöhring, T. D. B. Jacobs and L. Pastewka, Surf. Topogr.: Metrol. Prop., 2022, 10, 035032 CrossRef.
- T. D. B. Jacobs, T. Junge and L. Pastewka, Surf. Topogr.: Metrol. Prop., 2017, 5, 013001 CrossRef.
- S. Kim, Y. Lee, M. Lee, S. An and S.-J. Cho, Nanomaterials, 2021, 11, 1593 CrossRef CAS PubMed.
- C. Wang, H. Guo, P. Wang, J. Li, Y. Sun and D. Zhang, Adv. Mater., 2023, 35, 2209895 CrossRef CAS PubMed.
- Y. Yang, J. Han, J. Huang, J. Sun, Z. L. Wang, S. Seo and Q. Sun, Adv. Funct. Mater., 2020, 30, 1909652 CrossRef CAS.
- Q. Guan, G. Lin, Y. Gong, J. Wang, W. Tan, D. Bao, Y. Liu, Z. You, X. Sun, Z. Wen and Y. Pan, J. Mater. Chem. A, 2019, 7, 13948–13955 RSC.
- H. Wang, Z. Rao, Y. Liu, L. Shan, T. Guo, H. Chen and R. Wang, Nano Energy, 2023, 107, 108170 CrossRef CAS.
- K. K. Gleason, Adv. Mater., 2024, 36, 2306665 CrossRef CAS PubMed.
- W. Zhang, W. He, S. Dai, F. Ma, P. Lin, J. Sun, L. Dong and C. Hu, Nano Energy, 2023, 111, 108432 CrossRef CAS.
- W. Zhong, L. Xu, H. Wang, D. Li and Z. L. Wang, Nano Energy, 2019, 66, 104108 CrossRef CAS.
- G. Li, J. Wang, S. Fu, C. Shan, H. Wu, S. An, Y. Du, W. He, Z. Wang, W. Liu, Y. Nie, S. Liu, P. Wang and C. Hu, Adv. Funct. Mater., 2023, 33, 2213893 CrossRef CAS.
- G. Li, S. Fu, C. Luo, P. Wang, Y. Du, Y. Tang, Z. Wang, W. He, W. Liu, H. Guo, J. Chen and C. Hu, Nano Energy, 2022, 96, 107068 CrossRef CAS.
- L. N. Y. Cao, E. Su, Z. Xu and Z. L. Wang, Mater. Today, 2023, 71, 9–21 CrossRef.
- W. Harmon, D. Bamgboje, H. Guo, T. Hu and Z. L. Wang, Nano Energy, 2020, 71, 104642 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee03110e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |