
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid construction of a tellurium artificial interface to form a highly reversible zinc anode†
Young-Hoon
Lee‡
ab,
Eunbin
Park‡
ab,
Yunseo
Jeoun
ab,
Sung-Ho
Huh
c,
Kwang-Soon
Ahn
*d,
Yung-Eun
Sung
 *ab and
Seung-Ho
Yu
*c
*ab and
Seung-Ho
Yu
*c
aSchool of Chemical and Biological Engineering, Seoul National University, 1 Gwanak-ro, Seoul 08826, Republic of Korea. E-mail: ysung@snu.ac.kr
bCenter for Nanoparticle Research, Institute for Basic Science (IBS), 1 Gwanak-ro, Seoul 08826, Republic of Korea. E-mail: ysung@snu.ac.kr
cDepartment of Chemical and Biological Engineering, Korea University, 145 Anam-ro, Seoul 02841, Republic of Korea. E-mail: seunghoyu@korea.ac.kr
dSchool of Chemical Engineering, Yeungnam University, 280 Daehak-ro, Gyeongsan 38541, Republic of Korea. E-mail: kstheory@ynu.ac.kr
First published on 14th January 2025
Abstract
Aqueous zinc-ion batteries (AZIBs) represent a promising frontier in energy storage technology, characterized by its cost-effectiveness, safety, and environmental sustainability. However, the dendrite formation and corrosion on the zinc metal anode cause performance degradation and early cell failure in AZIBs. Herein, a novel tellurium complex treatment has been developed for a highly reversible zinc anode. The HTeCl5 complex (tellurium complex) rapidly forms uniform tellurium nanoparticles on the zinc metal surface in a short period of time, creating a hydrophilic surface that promotes uniform zinc-ion flux. These beneficial effects effectively suppress zinc dendrite growth and side reactions, resulting in significant cycling stability for 2500 h at 1 mA cm−2 for 1 mA h cm−2 and for 300 h with 50% depth of discharge (DOD) under symmetric cell conditions. In particular, the performance of full cells incorporating LiMn2O4 (LMO) and NaV3O8 (NVO) as cathodes exhibited notable enhancement in capacity retention, a result of employing a zinc anode treated with the tellurium complex. This study provides a practical path for realizing long-term stable AZIBs.
Broader contextThe limitations of lithium-ion batteries (LIBs), such as the toxicity of their organic electrolytes and the potential for explosion hazards, have led to a growing demand for the development of next-generation battery systems. Aqueous zinc-ion batteries (AZIBs) have emerged as promising alternatives due to their cost-effectiveness and environmental benefits. However, challenges such as dendrite formation and zinc anode corrosion have hindered their widespread adoption. A novel approach involving treatment with a tellurium complex effectively mitigates these issues by forming uniform tellurium nanoparticles on zinc surfaces. This treatment promotes a hydrophilic environment that facilitates uniform zinc-ion flux and suppresses dendrite growth and side reactions. These effects result in impressive cycling stability and improved capacity retention, particularly notable in cells with various cathodes. The simple and rapid tellurium treatment offers promising prospects for improving the performance and stability of AZIBs. |
Introduction
Over the past decades, the application of lithium-ion batteries (LIBs) in mobile devices and electric vehicles has been realized. However, the limitations of LIBs, such as the toxicity of their organic electrolytes and explosion hazards, have generated an increasing demand for next-generation battery systems. To overcome these safety issues, battery systems using nonflammable aqueous electrolytes are receiving increasing attention. Aqueous zinc-ion batteries (AZIBs) are promising options for next-generation energy storage systems due to their low cost, abundant resources, and environmental friendliness.1,2 AZIBs with their zinc anode also have other advantages of high theoretical capacity (820 mA h g−1) and low redox potential (−0.76 V vs. SHE).3 Nonetheless, there are severe side reactions such as the hydrogen evolution reaction (HER) and corrosion of the zinc metal anode.4–6 Moreover, the formation of dendrites on the zinc anode degrades the lifespan of AZIBs.7,8Numerous research endeavors are underway to address the mentioned limitations. Key areas of investigation include the use of electrolyte additives involving salts and organic molecules, separator modification, three-dimensional (3D) structural anodes, and zinc anode surface modification.9–17 Among them, the zinc surface modification has a distinct advantage over the former methods. The artificial layer formed by surface modification plays a role similar to that of the solid electrolyte interphase (SEI) of the zinc anode, which not only regulates the electrochemical environment but also acts as a physical protective layer.18,19
Improving the performance of AZIBs by surface modification of zinc anodes has been attempted in several ways. In particular, many modification methods with chalcogens have been reported due to their stable physical properties.20 For example, modification with zinc sulfide, which was adapted as a protective layer for zinc anodes, is described in one of the pioneering works.21 Aside from the zinc sulfide layer, selenium exhibits its beneficial effects on stabilizing zinc anodes.22,23 In addition, zinc telluride has received considerable attention as an alternative anode for AZIBs due to its zincophilic characteristics.24,25 Moreover, tellurium, known for having the highest conductivity among nonmetallic materials, including chalcogen elements, is recognized as a promising material capable of enabling rapid reaction kinetics.26,27 Tellurium has been hybridized with a metal organic framework (MOF), which was then applied as a protective layer on a zinc anode.28 Although chalcogens have a tremendous synergetic effect with zinc anodes, complicated processing methods (e.g. high-energy ball milling or chemical vapor deposition) are inevitable for most of the surface modifications with chalcogens. In particular, among chalcogens, tellurium has the lowest reactivity with zinc metal, resulting in limited research into its coating applications.
Herein, we propose a novel surface treatment method using a tellurium complex. The tellurium complex reacts rapidly with zinc, whereupon tellurium nanoparticles are uniformly formed on the zinc anode surface. Previously reported surface modifications of zinc anodes using chalcogens noted that they required long reaction times and high energy (heat and pressure) consumption. In contrast, this tellurium complex treatment is carried out in a very fast, facile, and simple manner, completely overcoming the complicated processes of previous studies. This simple process is considered the most powerful strength of this treatment. The tellurium layer enhanced the hydrophilicity of the zinc anode and inhibited the formation of zinc hydroxide sulfate hydrate (ZHS) by preventing side reactions. Furthermore, it also promoted homogeneous and dense zinc deposition by generating even zinc-ion flux across the zinc surface, which effectively suppressed the formation of dendrites. These effects resulted in excellent cycling stability over 2500 h at 1 mA cm−2 for 1 mA h cm−2 and over 300 h with 50% depth of discharge (DOD) under symmetric cell conditions. Furthermore, the cycling stability over 1000 cycles at 1 A g−1 was significantly improved on that of LiMn2O4 (LMO) under full-cell conditions. For the NaV3O8 (NVO) full cell, when galvanostatic charge/discharge was performed at 5 A g−1, it showed high performance with 81% capacity retention after 1000 cycles.
Results and discussion
The tellurium surface treatment on zinc metal is carried out when considering that tellurium with its 4+ oxidation state easily forms a complex with halide ions. The HTeCl5 complex reacts with zinc metal to form a thin tellurium layer on the metal surface (Fig. 1a and eqn (1) and (2)):29| TeO2 + 5H+ + 5Cl− ↔ HTeCl5 + 2H2O | (1) |
| 2HTeCl5 + 5Zn ↔ 2Te + 5ZnCl2 + H2 | (2) |
 | ||
| Fig. 1 Illustration of the novel tellurium complex treatment and characterization of the tellurium layer. (a) Schematic illustration of the tellurium complex treatment. (b) Solubility of TeO2 in various solvents. (c) Photographs of bare Zn, Zn-HA and Zn-TC. SEM images of the surface of (d) bare Zn, (e) Zn-HA and (f) Zn-TC. (g) Cross-sectional SEM image of Zn-TC. (h) XRD patterns of bare Zn, Zn-HA and Zn-TC. (i and j) HR-TEM images of tellurium nanoparticles of Zn-TC (the inset in (i) shows the SAED pattern of tellurium particles). Contact angle analysis of (k) bare Zn, (l) Zn-HA and (m) Zn-TC. | ||
Exploring in depth the formation of the tellurium complex, reagent addition sequence was varied in several ways (Fig. 1b). TeO2 has a very low solubility in deionized (DI) water, and if hydrochloric acid (HCl) was not preferentially added, the complex could not be formed and TeO2 would remain insoluble. On the other hand, when TeO2 was added to concentrated HCl, it dissolved into a clear yellow solution by complexation. This complex was well preserved even when diluted with DI water, which suggests that diluting the solution after the formation of the tellurium complex is necessary to prevent hydration. When zinc foil is immersed in the complex solution, it immediately begins to form a tellurium layer and the tellurium complex treatment is completed in a few seconds. The distinctive appearances of the zinc foil under no reaction conditions (referred to as bare Zn), the zinc foil when reacted with diluted HCl lacking TeO2 (Zn-HA), and the zinc foil treated with the tellurium complex (Zn-TC) are shown in Fig. 1c. Notably, it can be seen that the yellowish tellurium layer is uniformly coated on the Zn foil without cracking and detachment, even when using a large piece of zinc foil (Fig. S1, ESI†).
The changes in the morphology of each piece of zinc metal foil were demonstrated by surface and cross-sectional analysis under a scanning electron microscope (SEM) (Fig. 1d–g). Unlike the bare Zn, as shown in Fig. 1d, a coarsely etched surface on Zn-HA can be observed (Fig. 1e), while nano-sized particles are uniformly formed on the surface of Zn-TC (Fig. 1f). This uniform distribution of tellurium was demonstrated by energy dispersive spectroscopy (EDS) (Fig. S2, ESI†). A cross-sectional SEM image revealed the formation of the nanoscale tellurium layer with a thickness of approximately 110 nm on the zinc metal surface (Fig. 1g). The cross-sectional EDS mapping showed a uniform distribution of tellurium within the top layer of the zinc metal (Fig. S3, ESI†). This confirms that the thin and uniform tellurium layer acts as a protective coating for the zinc metal surface.
X-ray diffraction (XRD) analysis was performed to examine the composition of each Zn foil (Fig. 1h and Fig. S4, ESI†). Upon comparing the XRD patterns in Fig. 1h, it can be observed that Zn-HA exhibits a pattern resembling that of bare Zn, suggesting that the HCl treatment does not change the surface composition of zinc metal. On the other hand, Zn-TC shows diffraction peaks of tellurium (PDF#36-1452) at 23.0 and 27.5°, confirming the formation of a tellurium layer without any impurities. To further investigate the tellurium layer of Zn-TC, high-resolution transmission electron microscopy (HR-TEM) analysis was performed. Tellurium nanoparticles were observed, and the selected area electron diffraction (SAED) pattern showed the formation of polycrystalline tellurium (Fig. 1i). Furthermore, the lattice spacing of 0.39 nm corresponded to the (100) plane of tellurium, which proved that the particles from the surface of Zn-TC in the TEM image were tellurium (Fig. 1j).30 In addition, EDS mapping showed that the tellurium particles were uniformly distributed (Fig. S5, ESI†). As a result, the aforementioned results confirmed that the tellurium complex treatment effectively forms uniform polycrystalline tellurium nanoparticles on the zinc surface. The uniformly distributed tellurium nanoparticles act as zincophilic sites, accelerating a uniform zinc-ion flux and thereby guiding uniform zinc deposition.22,23,31–33 Moreover, crystalline tellurium, with its smaller lateral dimension compared to bulk tellurium, enhances contact between the surface and electrolytes. This, along with the large surface area and uniform distribution of the nanoparticles and the inherently high conductivity of tellurium, maximizes these advantages. Consequently, it is expected that the kinetics and ion flux on the zinc metal surface will be improved.24,34
To investigate the hydrophilicity of each zinc foil, contact angle analysis was performed with 2 M ZnSO4 aqueous electrolyte (Fig. 1k–m). The difference between bare Zn (Fig. 1k) and Zn-HA (Fig. 1l) was not noticeable. On the other hand, the contact angle of the Zn-TC was significantly smaller, suggesting that the hydrophilicity of the zinc foil was increased by the tellurium complex treatment (Fig. 1m). The enhanced hydrophilicity can reduce the interfacial free energy between the zinc electrode and the aqueous electrolyte. Therefore, the uniform flux of zinc ions is promoted, resulting in dense zinc deposition and nucleation without dendrite growth.35–38
To analyze zinc deposition behavior, chronoamperometry (CA) was conducted on a symmetric cell at a constant overpotential (Fig. S6, ESI†). Fig. 2a shows the change of current density with time for the three types of electrodes. From this, the changes in the morphology of the zinc deposition can be deduced. An increase in current density implies an enlarged surface area and two-dimensional (2D) diffusion. During 2D diffusion, zinc ions laterally diffuse toward sites with low surface energy, and rampant 2D diffusion tends to promote dendrite formation. This leads to the so-called “tip-effect”, where zinc grows predominantly at the particle tip.39 In contrast, 3D diffusion effectively constrains 2D diffusion, preventing ion concentration in specific areas and enabling zinc to grow uniformly and densely. It can be inferred that the current density of the bare Zn cell continuously increases, leading to continuous 2D diffusion and the formation of zinc dendrites. For the Zn-HA cell, the increase in current density was less than that of bare Zn, but 2D diffusion continued until the end of the experiment. In contrast, the Zn-TC cell exhibited 2D diffusion of very short duration and soon maintained a constant current density. This confirms that the formation of dendrites with continuously increasing surface area is inhibited in the Zn-TC electrode and 3D diffusion becomes dominant, which indicates the prevention of local charge accumulation and the promotion of uniform zinc deposition.40 After the CA test, these three electrodes were observed by using an optical microscope (OM) and the differences in zinc deposition were determined (Fig. 2b–d). Bare Zn and Zn-HA electrodes showed inhomogeneous surfaces and severe dendrite formation (Fig. 2b and c). On the other hand, uniform and dense zinc deposition could be observed on the Zn-TC electrode, which substantiates the results shown in the CA test (Fig. 2d).
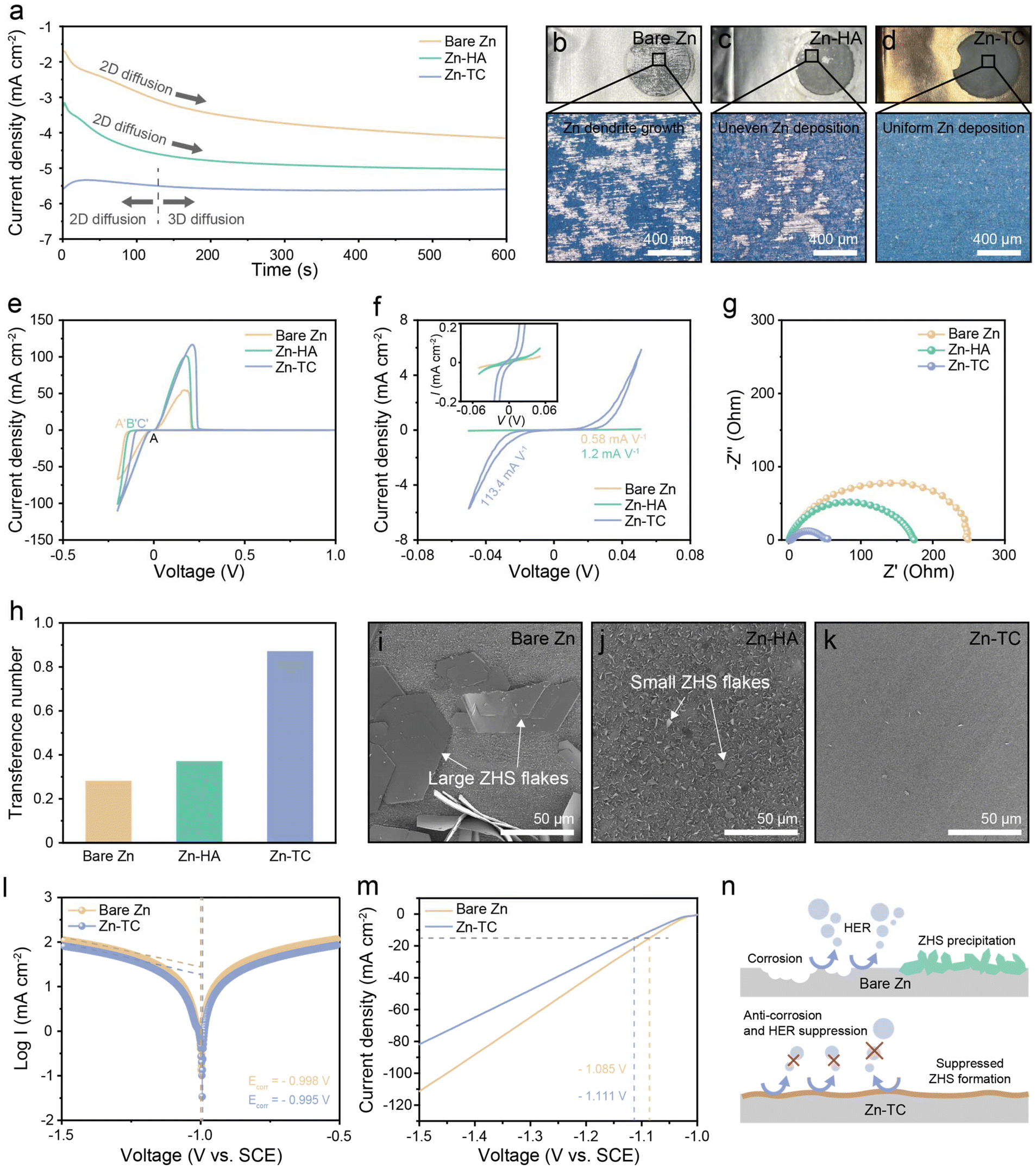 | ||
| Fig. 2 Electrochemical analyses of the tellurium complex layer. (a) Chronoamperograms of bare Zn, Zn-HA and Zn-TC. Photographs and optical microscope images of (b) bare Zn, (c) Zn-HA, and (d) Zn-TC. (e) CV curves of asymmetric cells and (f) CV curves of symmetric cells of bare Zn, Zn-HA, and Zn-TC. (g) EIS curves and (h) transference numbers of bare Zn, Zn-HA, and Zn-TC. Surface SEM images of (i) bare Zn, (j) Zn-HA, and (k) Zn-TC after immersing in 2 M ZnSO4 electrolyte for 7 days. (l) Tafel plots of bare Zn and Zn-TC cells at 5 mV s−1. (m) LSV curves of bare Zn and Zn-TC cells at 5 mV s−1. (n) Schematic illustration of side reactions at bare Zn and Zn-TC anodes. | ||
The nucleation stage is another important factor that determines the zinc deposition behavior. Fig. 2e shows the cyclic voltammograms (CVs) of zinc plating/stripping on a titanium substrate at a scan rate of 1 mV s−1. The nucleation overpotential (NOP) is defined as the potential difference between the intersection point (A) and zinc nucleation point (A′, B′, or C′). The NOP has values of 124, 106, and 79 mV for bare Zn, Zn-HA, and Zn-TC cell, respectively. The lower NOP of the Zn-TC cell indicates a lower barrier for cathodic polarization and for zinc nucleation and deposition.41 Furthermore, a CV test was performed with a zinc symmetric cell at a scan rate of 1 mV s−1 (Fig. 2f). The CV curve of bare Zn, Zn-HA, and Zn-TC cell showed a current density of −0.03, −0.06, and −5.67 mA cm−2 at −50 mV, respectively. Thus, the gradient of the Zn-TC cell was 113.4 mA V−1, indicating much-improved electrochemical kinetics behavior compared to that of the bare Zn (0.58 mA V−1) and Zn-HC (1.2 mA V−1) cells.42 The lower polarization of the Zn-TC cell in the asymmetric CV test can be further investigated by electrochemical impedance spectroscopy (EIS) (Fig. 2g). The charge transfer resistance (Rct) tends to increase in the order of Zn-TC, Zn-HA, and bare Zn cell. The drastic decrease in Rct of the Zn-TC cell indicates that zinc-ion transport occurs much more efficiently here than in the bare Zn cell. This improved zinc-ion transport can be attributed to the uniformly distributed tellurium nanoparticles.26,43 The hydrophilic surface of Zn-TC contributes to reductions of the overpotential-like activation energy barrier and charge transfer resistance by accelerating uniform zinc-ion flux. To further verify the extent of zinc-ion flux of each electrode, the transference number was calculated. The change in current for each sample during polarization and the recording of the EIS curves are shown in Fig. S7 (ESI†). The transference numbers are 0.28, 0.37, and 0.87 for bare Zn, Zn-HA, and Zn-TC cells, respectively, further confirming that the tellurium layer can facilitate uniform zinc-ion flux by mitigating the strong electrostatic forces on the surface of the zinc metal anode due to the large atomic radius of tellurium (Fig. 2h).44
Zinc anodes are subjected to severe corrosion reactions when using mildly acidic ZnSO4 electrolyte, which forms a large deposit of ZHS as a byproduct. To determine the extent of the corrosion reaction, each zinc electrode was immersed in the electrolyte for 7 days. Large ZHS flakes were formed on the bare Zn due to a severe corrosion reaction (Fig. 2i). On the Zn-HA electrode, the ZHS did not grow significantly, but small ZHS flakes covered the entire surface of the electrode (Fig. 2j). In contrast, the Zn-TC electrode was well preserved without ZHS flakes, demonstrating the anti-corrosive effect of the tellurium layer (Fig. 2k). Additionally, the corrosion potential of Zn-TC as revealed by the Tafel plot increased from −0.998 V to −0.995 V, further confirming its anti-corrosion effect (Fig. 2l). Besides, linear sweep voltammetry (LSV) was conducted to investigate the impact of the tellurium layer on the HER (Fig. 2m). The onset potential of the HER for Zn-TC and bare Zn were −1.111 V and −1.085 V, respectively, with Zn-TC showing a value that was lower by approximately 26 mV. Additionally, the current response of the HER for Zn-TC decreased compared to that for bare Zn. These results demonstrated that the tellurium layer can effectively prevent the HER in mildly acidic electrolytes. As depicted in Fig. 2n, the tellurium layer effectively suppressed corrosion, the HER and the formation of ZHS byproducts.
Several electrochemical and surface analyses confirmed that the tellurium layer promotes a uniform zinc-ion flux, which inhibited the formation of zinc dendrites and effectively prevented the corrosion reaction of zinc metal. To verify the effectiveness of the tellurium layer in real-time during battery operation, operando OM imaging was performed. A symmetric cell was constructed and observed during zinc deposition. The zinc deposition behavior of the three types of electrodes is depicted in Fig. 3a–c. By operando optical surface observations, it was confirmed that the zinc deposition nuclei of the bare Zn electrode were locally concentrated, where deposition continued, resulting in severe zinc dendrite growth (Fig. 3a and d). Inhomogeneous zinc deposition was still observed on the Zn-HA electrode (Fig. 3b and e). In contrast, the Zn-TC electrode showed that simultaneous zinc nuclei occurred over the entire surface, and were manifested as homogeneous and dense zinc deposition (Fig. 3c and f). The cross-sectional operando OM images also demonstrate the dendrite suppression effect of the tellurium layer (Fig. S8, ESI†). While mossy zinc deposition persisted on the bare Zn electrode, the Zn-TC electrode exhibited much finer and more uniform growth. Notably, the cross-sectional images reveal that, unlike bare Zn, zinc deposition occurred uniformly across the entire electrode in the case of Zn-TC. This indicates that the uniformly distributed tellurium nanoparticles on the electrode facilitate zinc growth, thereby mitigating localized zinc growth. From the results of operando OM imaging, it was demonstrated that tellurium treatment significantly inhibited the formation of zinc dendrites during the actual operation process.
 | ||
| Fig. 3 Operando optical surface observations of zinc deposition behavior. Schematic illustration of zinc deposition behavior on (a) bare Zn, (b) Zn-HA, and (c) Zn-TC. Operando optical microscopy images of (d) bare Zn, (e) Zn-HA, and (f) Zn-TC. | ||
In order to verify how the tellurium treatment affects the electrochemical performance, a galvanostatic cycling test was conducted under symmetric zinc cell conditions for each of the electrodes. The overpotential of the Zn-TC cell was only 16.9 mV at 1 mA cm−2 for 1 mA h cm−2 (Fig. 4a). On the other hand, those overpotentials of the bare Zn and Zn-HA cells showed values of 58.9 and 54.6 mV, respectively. To examine whether there were any changes in the crystal structure of tellurium during the cycling test, the XRD patterns of the pristine Zn-TC electrode, the plated Zn-TC electrode, and the stripped Zn-TC electrode were compared (Fig. S9, ESI†). All three samples exhibited distinct tellurium peaks, indicating that the tellurium layer was maintained even during the zinc plating/stripping process. This demonstrates the characteristics of a protective layer that maintains its initial structure throughout battery operation, which can be considered another advantage of the tellurium layer.45 The long-term cycling test was performed under the same conditions, and the Zn-TC cell showed remarkable cycling stability for 2500 h (Fig. 4b). The short-circuiting of the bare Zn and Zn-HA cells occurred after 90 and 160 h, respectively. It can be assumed that the short-circuit was caused by zinc dendrites piercing the separator and directly contacting both electrodes. Notably, the Zn-TC cell showed the lowest voltage hysteresis throughout the test (Fig. S10, ESI†). It can be inferred that the significantly lower nucleation overpotential and voltage hysteresis of the Zn-TC cell is due to the hydrophilic tellurium layer, resulting in a uniform ion flux. This uniform ion flux was also confirmed by comparing the surface and cross-sections of the electrodes using SEM images obtained after 50 cycles (Fig. 4c–f and Fig. S11, ESI†). Prior to SEM analysis, the photographs showed severe zinc dendrites on bare Zn and Zn-HA electrodes, while uniform zinc deposition was observed on the Zn-TC electrode (Fig. S12, ESI†). The cross-sectional SEM image of the bare Zn electrode shows that large zinc dendrites grew vertically and adhered to the separator. Moreover, pits grew deep and wide on the electrode (Fig. 4c). On the Zn-HA electrode, the pit formation is somewhat alleviated, but large zinc dendrite formation is still observed (Fig. 4d). The surface SEM images also show that dendrite growth on bare Zn and Zn-HA electrodes is present (Fig. S11a and b, ESI†). Furthermore, the separator became entangled in the dendrites, further demonstrating the overall adhesion of the glass fibers to the surface. In contrast, the Zn-TC electrode showed significant dendrite suppression in both cross-sectional and surface SEM images (Fig. 4e and Fig. S11c, ESI†). The uniform zinc plating/stripping demonstrated by SEM can be the underlying reason for the superior and stable behavior observed in the previous electrochemical tests.
 | ||
| Fig. 4 Electrochemical performances of the Zn//Zn symmetric cells. (a) Voltage profiles of zinc deposition at 1 mA cm−2 for 1 mA h cm−2 during the first plating. (b) Long-term galvanostatic cycling at 1 mA cm−2 for 1 mA h cm−2 in the symmetric cell. SEM images of (c) bare Zn, (d) Zn-HA, and (e and f) Zn-TC electrodes after 50 cycles. (g) XRD patterns of bare Zn, Zn-HA, and Zn-TC after 50 cycles. (h) The ratio of the (100) and (002) zinc crystal planes. (i) Shelving recovery test for the symmetric cell. (j) Galvanostatic cycles at 3 mA cm−2 for 3 mA h cm−2. (k) DOD test at 8.6 mA cm−2 for 8.6 mA h cm−2 in symmetric cells. (l) Rate performance of symmetric cells at current densities from 1 to 5 mA cm−2. | ||
In the cross-sectional analysis, the surface of the Zn-TC electrode shows that zinc deposition proceeded uniformly along the (002) plane direction (Fig. 4f). It is well known that the (002) zinc plane has an in-plane direction relative to the zinc substrate. When zinc is preferentially deposited along the (002) direction, a compact and smooth layer is formed to prevent the corrosion reaction. Meanwhile, dominant zinc growth on the (100) plane, which corresponds to the direction perpendicular to the substrate, intensifies dendrite formation.46 Therefore, to elucidate the difference in zinc growth, the electrodes were examined by XRD analysis after 50 cycles. The intensity of the (002) peak was the lowest when compared to the intensity of the (100) peak in bare Zn. The intensity of the (002) peak was slightly increased in the Zn-HA electrode. In the Zn-TC electrode, the (002) plane became much more dominant than the (100) plane (Fig. 4g). Comparing the difference of the intensity ratio between the (002) and (100) planes, the effect of uniform zinc deposition by the tellurium treatment is apparent (Fig. 4h). In addition, the peak of the ZHS (PDF#44-0673), a product of the corrosion reaction, was not identified in the XRD pattern of the Zn-TC electrode. The previous analysis confirmed that the tellurium layer prevented corrosion of the zinc metal. Furthermore, it can be inferred that the tellurium layer promoted zinc deposition on the (002) plane, thereby maintaining its anti-corrosion effect during battery operation.47–49 It has been shown that this effect can be maintained for 50 cycles of zinc plating/stripping. Moreover, the characterization of the tellurium layer by XRD analysis (Fig. 1h) showed that the tellurium layer is well preserved even after 50 cycles (Fig. S13, ESI†). In the shelving recovery test, the Zn-TC symmetric cell shows remarkable reversibility and stability over 7 repetitions of each shelving cycle (600 h), while the bare Zn and Zn-HA cells failed during the second shelving period (Fig. 4i).50 To determine the effect of tellurium treatment under harsh conditions, galvanostatic charging and discharging was performed at 3 mA cm−2 for 3 mA h cm−2. Similar to its extended performance under a lower current density (Fig. 4b), the Zn-TC cell showed superior cycling stability for 600 h in these harsher conditions and low voltage hysteresis throughout the cycling test (Fig. 4j and Fig. S14, ESI†). The bare Zn and Zn-HA cells failed after 62 and 68 h, respectively. To assess practical applicability, the DOD test was performed under symmetric cell conditions. To verify the reversibility of the tellurium layer under extreme conditions, we conducted a 50% DOD test. This experiment corresponds to a plating/stripping process at a high current density value of approximately 8.6 mA cm−2 with a substantial capacity of 8.6 mA h cm−2. The Zn-TC cell maintained a significantly lower voltage hysteresis for 300 h without voltage fluctuation, while short-circuiting of the bare Zn and Zn-HA cells occurred within 30 h (Fig. 4k and Fig. S15, ESI†). The improved electrochemical performance of the Zn-TC cell was also evident in the rate performance test. When the current density was increased from 1 to 5 mA cm−2, the Zn-TC cell maintained very low voltage hysteresis even at 5 mA cm−2. On the other hand, the bare Zn and Zn-HA cells showed a sharp increase in operating voltage at 3 mA cm−2, which indicates that the polarization of the symmetric cells became severe at higher current density values (Fig. 4l). The Zn-TC cell demonstrated exceptional cycling stability, surpassing the performance of electrodes in most of the reported studies on zinc surface modification, as summarized in Table S1 (ESI†).
These findings confirmed that the uniform zinc-ion flux with the tellurium layer significantly stabilized the zinc electrodes and enhanced the reversibility of the symmetric cell. To verify if a tellurium layer on a copper electrode maintains its high reversibility of zinc plating/stripping, a tellurium complex treatment was applied to a copper foil (Fig. S16, ESI†). Similar to the Zn-TC foil, tellurium nanoparticles were uniformly coated without any areas of the copper surface being exposed (Fig. S17, ESI†). The galvanostatic cycling of the Zn//Cu-TC cell showed an average coulombic efficiency (CE) of 99.2%, confirming the high reversibility of zinc metal due to the tellurium layer (Fig. S18, ESI†). The lower initial CE of the Cu-TC electrode is attributed to its increased surface area from uniformly distributed tellurium nanoparticles, leading to greater SEI formation during initial plating, while subsequent rapid CE improvement suggests the tellurium layer enhances long-term zinc plating/stripping reversibility. In the voltage profile at the 10th cycle, the polarization decreased from 32.8 mV to 21.3 mV in the Zn//Cu-TC cell (Fig. S19, ESI†). The SEM images of the bare Cu surface revealed severe zinc dendrite formation on the bare electrode, whereas uniform zinc deposition was observed on the Cu-TC electrode (Fig. S20, ESI†). Such results demonstrated the improved kinetics of the Zn//Cu-TC asymmetric cell.
To determine how the thickness of the tellurium layer affects the performance of the zinc anode, we conducted further analyses and experiments. The tellurium treatment was carried out for 2, 5, 10, 20 and 30 s to determine the optimal time for forming a uniform tellurium layer. In the SEM images, the zinc metal surface seems to be incompletely formed over the whole surface of the electrodes during the first 2 to 5 s (Fig. S21a and b, ESI†). On the other hand, a uniform tellurium layer covered the entire surface of the zinc metal after 10 s (Fig. S21c, ESI†). From 20 s, the tellurium layer exhibits a remarkably different morphology with thin dendrites growing due to excessive tellurium treatment (Fig. S21d, ESI†). These tellurium dendrites became thicker after 30 s of the tellurium complex treatment. To compare the effect of the morphology of the tellurium layer, we further performed galvanostatic cycling at a current density of 1 mA cm−2 for 1 mA h cm−2 using electrodes that were treated for 2 s (Zn-TC (thin)) and 30 s (Zn-TC (thick)), respectively. Both cells showed a shortened lifespan of zinc plating/stripping compared to the Zn-TC cell, which can be attributed to the formation of the inhomogeneous tellurium layer. With the thin Zn-TC cell, short-circuiting occurred after 1600 h due to the exposure of the bare Zn foil. The thick Zn-TC cell failed before 1500 h as the zinc-ion flux was concentrated in the tellurium dendrites (Fig. S22, ESI†). Therefore, it is confirmed that the tellurium complex treatment for 10 s is optimal for the zinc anode.
Since practical applicability is an important factor in AZIBs, a full-cell test was conducted using LMO as the cathode (Fig. S23, ESI†). The CV test was performed from 1.4 V to 2.05 V at a scan rate of 0.2 mV s−1. Bare Zn//LMO and Zn-TC//LMO cells showed identical redox processes as they displayed two similar pairs of redox peaks. However, the Zn-TC//LMO cell demonstrated a higher current response than the bare Zn//LMO cell, which indicated the faster kinetics driven by tellurium treatment. Furthermore, the potential difference between the oxidation/reduction peaks (ΔEp) of the Zn-TC//LMO cell is lower than that of the bare Zn//LMO cell. The ΔEp is 219 mV and 284 mV for the Zn-TC//LMO cell and the bare Zn//LMO cell, respectively (Fig. 5a). This lower polarization of the Zn-TC//LMO cell is consistent with the reduced voltage hysteresis of the Zn-TC//LMO cell in the galvanostatic charge/discharge curve, where the discharge capacity of the Zn-TC//LMO cell during the first cycle was 104.1 mA h g−1 (the energy density being 184.2 W h kg−1) (Fig. 5b). These behaviors are attributed to the hydrophilic tellurium layer, which promotes even zinc-ion flux and suppresses dendrite formation. This difference was also observed during galvanostatic cycling. In rate performance tests when gradually increasing the current density from 1 A g−1 to 5 A g−1, the specific capacity of the bare Zn//LMO cell decreased sharply at the current density of 2 A g−1 (68.8 mA h g−1). At the current density of 5 A g−1, the specific capacity was much lower (13.3 mA h g−1) than that of the Zn-TC//LMO cell (26.0 mA h g−1). In contrast, the Zn-TC//LMO cell well-maintained its specific capacity at current density values from 1 (101.9 mA h g−1) to 5 A g−1 (26.0 mA h g−1) (Fig. 5c). To further demonstrate the improvement in cycling performance with the tellurium layer, a long-term cycling test was carried out at a current density of 1 A g−1. The initial specific capacity was similar for both bare Zn//LMO and Zn-TC//LMO cells (101.8 and 104.7 mA h g−1, respectively). However, the bare Zn//LMO cell exhibits only capacity retention of 32.3% after 1000 cycles, while the Zn-TC//LMO cell shows 67.4% capacity retention, demonstrating improved cycling stability (Fig. 5d). The Zn-TC//LMO cell shows significantly extended performances over cells in previously reported studies (Table S2, ESI†). The photographs of zinc anodes after 200 cycles reveal that the dendrite formation on the bare Zn electrode was much more severe. On the other hand, the surface of the Zn-TC electrode shows uniform deposition (Fig. S24, ESI†). This difference is even more evident in the SEM images. Similar to the symmetric cell conditions, the separator was adhered to the zinc anode due to dendrite formation on the bare Zn anode, and pits were formed on the surface owing to uneven ion flux. In contrast, the Zn-TC electrode maintained a uniform surface even after 200 cycles, demonstrating the performance improvement due to dendrite suppression by the tellurium layer (Fig. S25, ESI†). Meanwhile, XRD analysis of the Zn-TC electrode after 200 cycles revealed that the peak for tellurium was observed, indicating that the tellurium layer was well-preserved even after long-term cycling of the full cell. This understanding suggests that the beneficial effects persisted due to the preserved tellurium layer (Fig. S26, ESI†). In the full-cell test with the LMO cathode, the performance improvement due to the tellurium layer was confirmed. This high performance was still observed when we further performed electrochemical tests with the NVO full cell (Fig. S27, ESI†). In the CV test from 0.2 to 1.6 V at a scan rate of 0.2 mV s−1, two pairs of redox peaks were maintained in both the bare Zn//NVO and Zn-TC//NVO cells. The ΔEp value of the Zn-TC//NVO cell (155 mV) was lower than that of the bare Zn//NVO cell (226 mV), demonstrating the reduction of overpotential by the tellurium layer (Fig. 5e). When the galvanostatic cycling test was performed at a current density of 5 A g−1, the capacity during the first cycle was 163.6 mA h g−1 (the energy density being 183.2 W h kg−1). The capacity retention of the Zn-TC//NVO cell after 1000 cycles was about 80.6% showing a much higher stability than that of the bare Zn//NVO cell (46.8%) (Fig. 5f). In addition to uniform zinc deposition, corrosion suppression was reported to prevent vanadium dissolution, suggesting that the corrosion-inhibiting effect of the tellurium layer also contributed to the performance of the full cell.51 Through various electrochemical tests, it has been confirmed that the tellurium layer is beneficial and compatible for both LMO and NVO full cells.
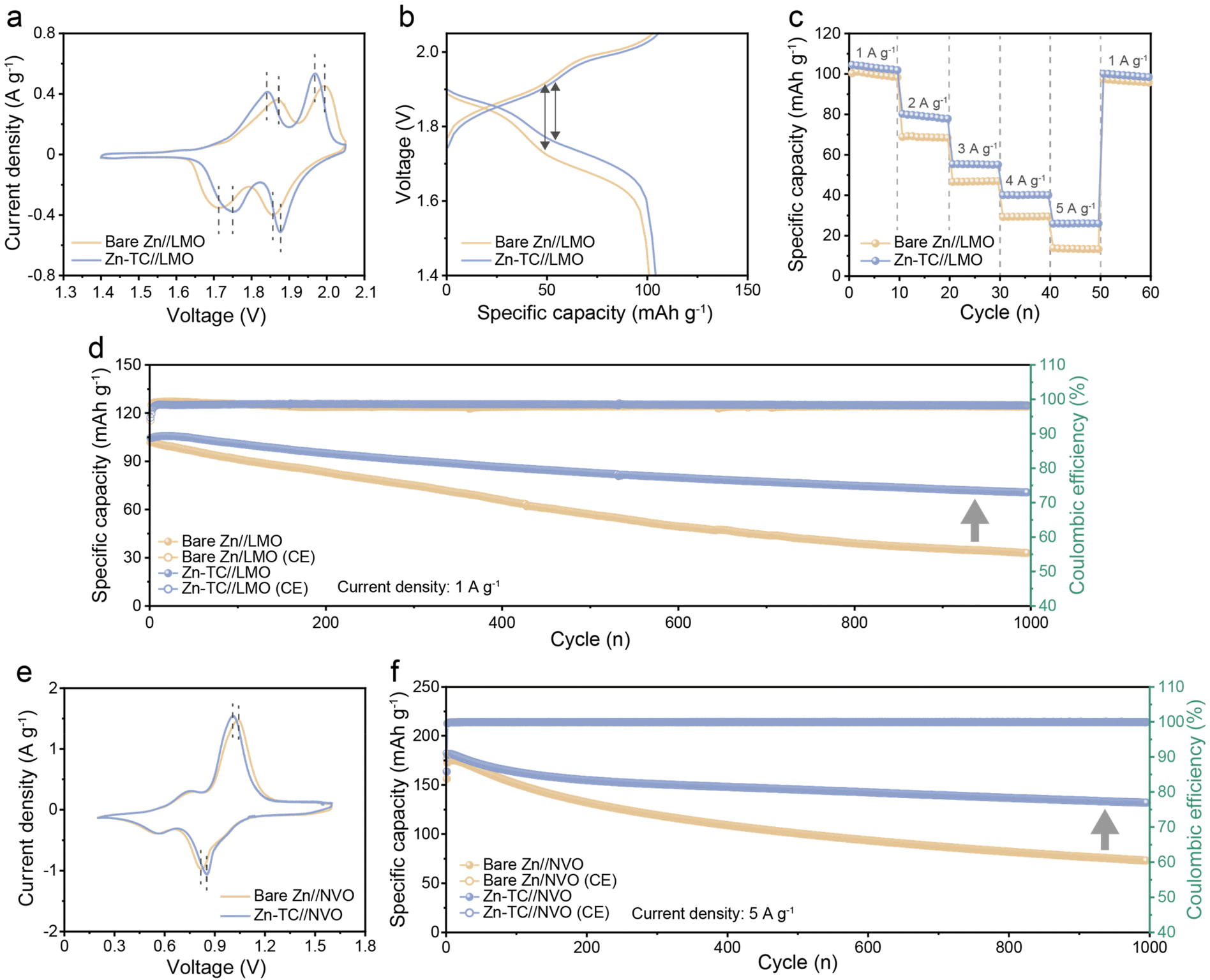 | ||
| Fig. 5 Electrochemical performances of the full cells. (a) Cyclic voltammograms at 0.2 mV s−1, (b) galvanostatic charge/discharge curves at 0.1 A g−1, (c) rate performances at various current density values and (d) long-term cycling stability test at 1 A g−1 of bare Zn//LMO and Zn-TC//LMO full cells. (e) Cyclic voltammograms at 0.2 mV s−1, (f) long-term cycling stability test at 5 A g−1 of bare Zn//NVO and Zn-TC//NVO full cells. | ||
The tellurium complex treatment is accomplished by simply immersing the electrode in the complex solution and it has a much faster deposition rate than other replacement reactions, facilitating scale-up (Fig. 6). The tellurium layer exhibits hydrophilic characteristics and enhances the diffusion kinetics of zinc ions, resulting in a uniform zinc-ion flux. These effects ultimately inhibit the corrosion reaction, induce preferential growth of zinc deposition on the (002) plane, and prevent dendrite formation. As a result, AZIBs demonstrating excellent stability and performance have been realized by this tellurium complex treatment.
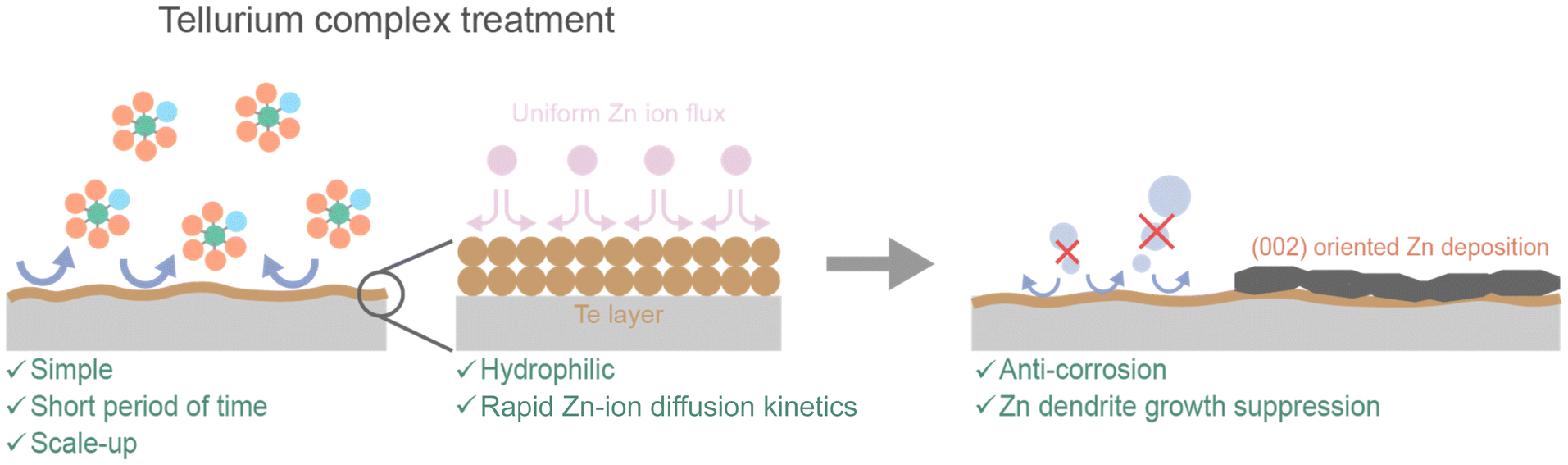 | ||
| Fig. 6 Schematic illustration of the tellurium complex treatment and its beneficial effects on AZIBs. | ||
Experimental
Electrode preparation
All chemicals were purchased from Sigma-Aldrich and used without further purification. Prior to use, zinc foil (Goodfellow, purity 99.99%, thickness 0.025 mm) and copper foil (Sigma-Aldrich, purity 99.8%, thickness 0.1 mm) were washed with acetone and dried. The solution for the Zn-HA foil was prepared by mixing 0.5 mL of HCl (37 wt%) in 100 mL of DI water. A clear yellow solution was obtained by stirring 0.05 g of TeO2 in 0.05 mL of hydrochloric acid for 30 minutes, and this solution was diluted in 100 mL of DI water to complete the solution for the Zn-TC foil. The zinc foil was immersed in each solution for 2, 10, and 30 s, washed with DI water and ethanol to remove residual solution, and dried at 70 °C in a vacuum oven for 6 h to obtain Zn-HA and Zn-TC. Cu-TC was fabricated using an analogous method.Sodium vanadate cathode synthesis
Firstly, 5.0 g of V2O5 bulk powder was combined with 50 mL of 2 M NaCl aqueous solution. The resulting mixture was stirred at 25 °C for 3 days, during which time the color of the solution changed from orange to reddish-orange. After the reaction period, the mixture was centrifuged, washed several times with DI water and ethanol, and dried at 70 °C for 24 h. Finally, a collection of reddish-orange colored NVO nanofibers in powder form was obtained.Materials characterization
The electrode morphologies were examined by SEM (SUPRA 55VP, Zeiss). The crystalline structures of the electrodes were identified by XRD (JP/D/MAX-2500H, Rigaku) using a Cu-Kα radiation source. HR-TEM (EM-2010, JEOL) was performed to characterize the morphology, elemental distribution and lattice spacing of the treated materials. A contact angle analyzer (Phoenix-MT, SEO Co.) was used to evaluate the hydrophilicity of the electrodes. Operando optical observations were performed to visualize the zinc deposition behavior on the zinc surface using custom-made operando electrochemical cells. The images of optical observations were captured using a DSFi3 CMOS camera connected to a Nikon Eclipse LV150N upright microscope. A potentiostat (PGSTAT302N, Autolab) was used to apply a current density of 1 mA cm−2 for zinc deposition.Electrochemical characterization studies
Aqueous 2 M ZnSO4 electrolyte was used for electrochemical characterization studies except for during the full-cell test with the LMO cathode. For the LMO full-cell test, aqueous 2 M Li2SO4 with 1 M ZnSO4 was used. CA measurement was performed with a symmetric cell consisting of zinc electrodes without a separator. The CA test was conducted at a constant voltage of −150 mV for 10 min. CV of symmetric and asymmetric cells was performed at a scan rate of 1 mV s−1. Symmetric cells were assembled from CR-2032 coin cells using zinc foil (bare Zn, Zn-HA, or Zn-TC) as both electrodes, 150 μL of 2 M ZnSO4 electrolyte, and glass fiber (Whatman GF/A) as the separator. The diameter of the zinc electrode and the glass fiber separator were 14 and 19 mm, respectively. For CV testing of asymmetric cells, titanium foil was used as the counter electrode, zinc foil was used as the working electrode, and Ag/AgCl (saturated KCl) was used as the reference electrode. Using an impedance analyzer (ZIVE SP1, Wonatech), EIS analysis was conducted at an amplitude of 10 mV at open circuit potential in the frequency range from 0.1 to 100 kHz. Transference numbers were determined in the symmetric cells combined with EIS plots before and after polarization with a constant voltage of 20 mV and calculated by the following equation (eqn (3)):52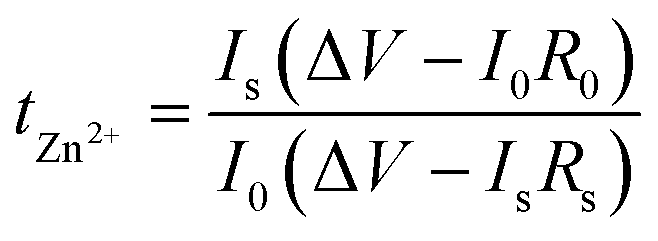 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 (wt%, LMO:Super P:PVDF). After casting the slurry on stainless foil, it was dried at 70 °C in a vacuum oven. Mass loading of LMO was ≈1.5 mg cm−2 and the diameter of the electrodes was 11 mm. Full cells were assembled in the same manner as the symmetric cells were constructed. The NVO full cell was fabricated using the same process. The NVO cathode underwent discharge first, whereas the LMO cathode was charged first. CV testing of full cells was performed at a scan rate of 0.2 mV s−1. Galvanostatic charge/discharge and cycling measurements were conducted with a battery cycler (WBCS3000L, Wonatech). The energy density E (W h kg−1) of the full cell was calculated based on the following equation (eqn (4)):
:2:1 (wt%, LMO:Super P:PVDF). After casting the slurry on stainless foil, it was dried at 70 °C in a vacuum oven. Mass loading of LMO was ≈1.5 mg cm−2 and the diameter of the electrodes was 11 mm. Full cells were assembled in the same manner as the symmetric cells were constructed. The NVO full cell was fabricated using the same process. The NVO cathode underwent discharge first, whereas the LMO cathode was charged first. CV testing of full cells was performed at a scan rate of 0.2 mV s−1. Galvanostatic charge/discharge and cycling measurements were conducted with a battery cycler (WBCS3000L, Wonatech). The energy density E (W h kg−1) of the full cell was calculated based on the following equation (eqn (4)):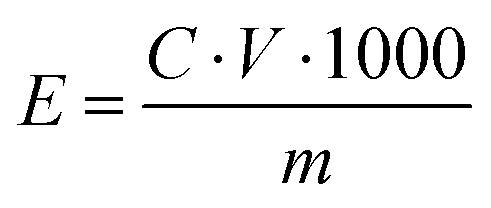 | (4) |
Conclusions
In this study, tellurium complex treatment was carried out to form a homogeneous tellurium layer on zinc foil in a short period of time. By various characterization methods, it was found that tellurium was homogeneously distributed on the zinc foil. This tellurium layer exhibited high hydrophilicity, which enhanced zinc-ion flux and effectively suppressed not only dendrite growth but also corrosion. This led us to demonstrate that zinc symmetric cells can be operated with low voltage hysteresis for up to 2500 h and showed stable operation during the shelving recovery test over 600 h. This excellent zinc plating/stripping capability was further demonstrated in a 50% DOD test, during which the cell stably cycled for 300 h. It was further confirmed that zinc dendrite growth was inhibited by promoting zinc growth on the (002) plane. Under the full-cell conditions, the Zn-TC//LMO cell showed significantly improved performance compared to the bare Zn//LMO cell. Also, the capacity retention of the Zn-TC//NVO cell was improved revealing a value of 80.6%, demonstrating good compatibility with and applicability of the tellurium treatment of AZIBs. Since the tellurium treatment process is effective, fast, and scalable, we believe this method will provide new insights into surface treatment techniques for zinc anodes. Furthermore, we hope to contribute to the development of high-performance AZIBs by introducing this simple treatment.Author contributions
Y.-H. Lee and E. Park conducted the methodology, investigation, formal analysis, and contributed to the original draft writing and visualization, while Y.-H. Lee conceptualized the work. Y. Jeoun and S.-H. Huh carried out the methodology, formal analysis, and visualization. K.-S. Ahn and Y.-E. Sung were responsible for project administration, funding acquisition, and supervision. S.-H. Yu handled project administration, funding acquisition, and supervision, and also contributed to the writing, reviewing, and editing of the manuscript.Data availability
All data generated or analyzed during this study are included in this article and the ESI.†Conflicts of interest
The authors declare no competing financial/commercial interests.Acknowledgements
This study was supported by the Institute for Basic Science (project code: IBS-R006-A2). This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (grant number 2018R1D1A3B05042787). This research was also supported by the National Research Foundation of Korea (NRF) funded by the Korea Government (MSIT) (NRF-RS-202400422387).References
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- J. Hao, X. Li, X. Zeng, D. Li, J. Mao and Z. Guo, Energy Environ. Sci., 2020, 13, 3917–3949 RSC.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- H. Liu, Q. Zhou, Q. Xia, Y. Lei, X. L. Huang, M. Tebyetekerwa and X. S. Zhao, J. Energy Chem., 2023, 77, 642–659 CrossRef.
- F. Tao, K. Feng, Y. Liu, J. Ren, Y. Xiong, C. Li and F. Ren, Nano Res., 2023, 16, 6789–6797 CrossRef.
- H. Li, L. Ma, C. Han, Z. Wang, Z. Liu, Z. Tang and C. Zhi, Nano Energy, 2019, 62, 550–587 CrossRef.
- H. J. Kim, S. Kim, K. Heo, J. H. Lim, H. Yashiro and S. T. Myung, Adv. Energy Mater., 2023, 13, 2203189 CrossRef.
- X. Zeng, J. Hao, Z. Wang, J. Mao and Z. Guo, Energy Storage Mater., 2019, 20, 410–437 CrossRef.
- P. Xiong, Y. Kang, N. Yao, X. Chen, H. Mao, W.-S. Jang, D. M. Halat, Z.-H. Fu, M.-H. Jung and H. Y. Jeong, ACS Energy Lett., 2023, 8, 1613–1625 CrossRef.
- H. Du, Y. Dong, Q. J. Li, R. Zhao, X. Qi, W. H. Kan, L. Suo, L. Qie, J. Li and Y. Huang, Adv. Mater., 2023, 35, 2210055 CrossRef PubMed.
- Z. Liu, R. Wang, Q. Ma, J. Wan, S. Zhang, L. Zhang, H. Li, Q. Luo, J. Wu and T. Zhou, Adv. Funct. Mater., 2023, 34, 2214538 CrossRef.
- P. Xiong, C. Lin, Y. Wei, J.-H. Kim, G. Jang, K. Dai, L. Zeng, S. Huang, S. J. Kwon and S.-Y. Lee, ACS Energy Lett., 2023, 8, 2718–2727 CrossRef.
- Z. Li, L. Ye, G. Zhou, W. Xu, K. Zhao, X. Zhang, S. Hong, T. Ma, M.-C. Li and C. Liu, Chem. Eng. J., 2023, 457, 141160 CrossRef.
- X. Xie, S. Liang, J. Gao, S. Guo, J. Guo, C. Wang, G. Xu, X. Wu, G. Chen and J. Zhou, Energy Environ. Sci., 2020, 13, 503–510 RSC.
- G. Wang, H. Fu, J. Lu, S. Huang, C. Pei, D. Min, Q. Zhang and H. S. Park, Adv. Energy Mater., 2024, 14, 2303549 CrossRef.
- T. Huang, K. Xu, N. Jia, L. Yang, H. Liu, J. Zhu and Q. Yan, Adv. Mater., 2023, 35, 2205206 CrossRef CAS PubMed.
- F. Tao, Y. Liu, X. Ren, J. Wang, Y. Zhou, Y. Miao, F. Ren, S. Wei and J. Ma, J. Energy Chem., 2022, 66, 397–412 CrossRef CAS.
- B. Li, X. Zhang, T. Wang, Z. He, B. Lu, S. Liang and J. Zhou, Nano-Micro Lett., 2022, 14, 1–31 CrossRef CAS.
- A. Chen, C. Zhao, J. Gao, Z. Guo, X. Lu, J. Zhang, Z. Liu, M. Wang, N. Liu and L. Fan, Energy Environ. Sci., 2023, 16, 275–284 RSC.
- Q. Zhang, Y. Su, Z. Shi, X. Yang and J. Sun, Small, 2022, 18, 2203583 CrossRef CAS.
- J. Hao, B. Li, X. Li, X. Zeng, S. Zhang, F. Yang, S. Liu, D. Li, C. Wu and Z. Guo, Adv. Mater., 2020, 32, 2003021 CrossRef CAS.
- T. C. Li, Y. V. Lim, X. Xie, X. L. Li, G. Li, D. Fang, Y. Li, Y. S. Ang, L. K. Ang and H. Y. Yang, Small, 2021, 17, 2101728 CrossRef CAS.
- L. Zhang, B. Zhang, T. Zhang, T. Li, T. Shi, W. Li, T. Shen, X. Huang, J. Xu and X. Zhang, Adv. Funct. Mater., 2021, 31, 2100186 CrossRef CAS.
- Z. Chen, Q. Yang, F. Mo, N. Li, G. Liang, X. Li, Z. Huang, D. Wang, W. Huang and J. Fan, Adv. Mater., 2020, 32, 2001469 CrossRef CAS PubMed.
- M. H. Tsai, T. Y. Lin, T. S. Su, G. M. Chen, Y. C. Liu and Y. Z. Chen, Batteries Supercaps, 2023, 6, e202300107 CrossRef CAS.
- Z. Chen, Y. Zhao, F. Mo, Z. Huang, X. Li, D. Wang, G. Liang, Q. Yang, A. Chen and Q. Li, Small Struct., 2020, 1, 2000005 CrossRef.
- S. Dong, D. Yu, J. Yang, L. Jiang, J. Wang, L. Cheng, Y. Zhou, H. Yue, H. Wang and L. Guo, Adv. Mater., 2020, 32, 1908027 CrossRef CAS PubMed.
- Y.-C. Fu, T.-Y. Lin and Y.-Z. Chen, J. Colloid Interface Sci., 2023, 649, 471–480 CrossRef CAS PubMed.
- N. V. Sidgwick, The chemical elements and their compounds, Citeseer, 1950 Search PubMed.
- K.-W. Song, J.-H. Bae, H.-K. Kim, T.-H. Kim, M.-H. Park and C.-W. Yang, J. Nanosci. Nanotechnol., 2011, 11, 6559–6562 CrossRef CAS PubMed.
- L. Wu, Y. Zhang, P. Shang, Y. Dong and Z.-S. Wu, J. Mater. Chem. A, 2021, 9, 27408–27414 RSC.
- Q. Zhang, J. Luan, X. Huang, L. Zhu, Y. Tang, X. Ji and H. Wang, Small, 2020, 16, 2000929 CrossRef.
- Q. Hu, J. Hu, F. Ma, Y. Liu, L. Xu, L. Li, S. Zhang, X. Liu, J. Zhao and H. Pang, Energy Environ. Sci., 2024, 17, 2554–2565 RSC.
- Y. Liu, J. Wang, Y. Xu, Y. Zhu, D. Bigio and C. Wang, J. Mater. Chem. A, 2014, 2, 12201–12207 RSC.
- S. H. Park, S. Y. Byeon, J.-H. Park and C. Kim, ACS Energy Lett., 2021, 6, 3078–3085 CrossRef.
- X. Han, H. Leng, Y. Qi, P. Yang, J. Qiu, B. Zheng, J. Wu, S. Li and F. Huo, Chem. Eng. J., 2022, 431, 133931 CrossRef.
- H. Zhou, W. Zhang, L. Wang, X. Sui, P. Wang and Z. Wang, Chem. Eng. J., 2023, 474, 145689 CrossRef.
- M. Liu, L. Yang, H. Liu, A. Amine, Q. Zhao, Y. Song, J. Yang, K. Wang and F. Pan, ACS Appl. Mater. Interfaces, 2019, 11, 32046–32051 CrossRef.
- D. Xie, Z. W. Wang, Z. Y. Gu, W. Y. Diao, F. Y. Tao, C. Liu, H. Z. Sun, X. L. Wu, J. W. Wang and J. P. Zhang, Adv. Funct. Mater., 2022, 32, 2204066 CrossRef.
- Y.-M. Li, W.-H. Li, W.-Y. Diao, F.-Y. Tao, X.-L. Wu, X.-Y. Zhang and J.-P. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 23558–23569 CrossRef PubMed.
- Y. Zhang, P. Chen, M. Li, S. Li, Y. Yue, Y. Wang, S. Xie and W. Zhou, J. Mater. Chem. A, 2023, 11, 14333–14344 RSC.
- J. Ko, S. So, M. Kim, I. T. Kim, Y. N. Ahn and J. Hur, Chem. Eng. J., 2023, 462, 142308 CrossRef.
- C. Choi, J. B. Park, J. H. Park, S. Yu and D.-W. Kim, Chem. Eng. J., 2023, 456, 141015 CrossRef.
- Z. Chen, Q. Yang, D. Wang, A. Chen, X. Li, Z. Huang, G. Liang, Y. Wang and C. Zhi, ACS Nano, 2022, 16, 5349–5357 CrossRef CAS PubMed.
- G. Wang, Q. K. Zhang, X. Q. Zhang, J. Lu, C. Pei, D. Min, J. Q. Huang and H. S. Park, Adv. Energy Mater., 2024, 2304557 CrossRef.
- H. Zhang, Y. Zhong, J. Li, Y. Liao, J. Zeng, Y. Shen, L. Yuan, Z. Li and Y. Huang, Adv. Energy Mater., 2023, 13, 2203254 CrossRef CAS.
- R. Ramanauskas, P. Quintana, L. Maldonado, R. Pomés and M. Pech-Canul, Surf. Coat. Technol., 1997, 92, 16–21 CrossRef CAS.
- A. Gomes and M. da Silva Pereira, Electrochim. Acta, 2006, 51, 1342–1350 CrossRef CAS.
- Q. Li, L. Han, Q. Luo, X. Liu and J. Yi, Batteries Supercaps, 2022, 5, e202100417 CrossRef CAS.
- P. Wang, S. Liang, C. Chen, X. Xie, J. Chen, Z. Liu, Y. Tang, B. Lu and J. Zhou, Adv. Mater., 2022, 34, 2202733 CrossRef CAS.
- Y. Kim, Y. Park, M. Kim, J. Lee, K. J. Kim and J. W. Choi, Nat. Commun., 2022, 13, 2371 CrossRef CAS PubMed.
- H. Dong, X. Hu and G. He, Nanoscale, 2022, 14, 14544–14551 RSC.
- Y. Cui, Q. Zhao, X. Wu, Z. Wang, R. Qin, Y. Wang, M. Liu, Y. Song, G. Qian and Z. Song, Energy Storage Mater., 2020, 27, 1–8 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4eb00015c |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |