
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peroxy acetyl nitric anhydride (PAN) and peroxy acetic acid (PAA) over the Atlantic west of Africa during CAFE-Africa and the influence of biomass-burning†
John N.
Crowley
 *a,
Raphael
Dörich
a,
Philipp
Eger
a,
Frank
Helleis
a,
Ivan
Tadic
a,
Horst
Fischer
a,
Jonathan
Williams
a,
Achim
Edtbauer
a,
Nijing
Wang
a,
Bruna A.
Holanda
b,
Mira
Poehlker‡
b,
Ulrich
Pöschl
b,
Andrea
Pozzer
a and
Jos
Lelieveld
a
*a,
Raphael
Dörich
a,
Philipp
Eger
a,
Frank
Helleis
a,
Ivan
Tadic
a,
Horst
Fischer
a,
Jonathan
Williams
a,
Achim
Edtbauer
a,
Nijing
Wang
a,
Bruna A.
Holanda
b,
Mira
Poehlker‡
b,
Ulrich
Pöschl
b,
Andrea
Pozzer
a and
Jos
Lelieveld
a
aAtmospheric Chemistry Department, Max-Planck-Institute for Chemistry, 55128-Mainz, Germany. E-mail: john.crowleympic.de
bMultiphase Chemistry Department, Max-Planck-Institute for Chemistry, 55128-Mainz, Germany
First published on 14th April 2025
Abstract
PAN (CH3C(O)O2NO2) is often the most important chemical reservoir of reactive nitrogen compounds throughout the free- and upper troposphere and provides a means of transport of reactive nitrogen from source regions to more remote locations. Both PAN and PAA (peroxy acetic acid, CH3C(O)OOH) are formed exclusively via reactions of the CH3C(O)O2 radical, with PAA favoured under low NOX conditions. We present airborne measurements of PAN and PAA taken with a chemical-ionisation mass spectrometer on board the High Altitude-Long range (HALO) aircraft over the North and tropical Atlantic Ocean west of Africa in August–September 2018. Our observations showed that mixing ratios of PAN and PAA are enhanced in biomass-burning impacted air masses and we determined molar enhancement ratios for both trace gases relative to CO and CH3CN. The PAA-to-PAN ratio was enhanced in biomass-burning impacted air masses compared to background air, which may reflect the continued photochemical formation of PAA in such plumes even after NOX has been largely depleted. This was confirmed by the large ratio of PAN/(PAN + NOX), which was on average ≈0.8 at 7–8 km altitude and approached unity in biomass burning impacted air masses. Although no measurements of total reactive nitrogen species (NOy) or HNO3 were available, a major fraction of NOX was likely sequestered in the form of PAN in this region, especially in air masses that had been impacted by biomass burning.
Environmental significanceThis work deals with one of the most important atmospheric trace gases, PAN and the influence of biomass burning on its abundance over the North- and tropical Atlantic Ocean west of Africa in August September. Along with PAN, airborne measurements of peroxy acetic acid (PAA) provide insight into the processing of reactive nitrogen in biomass burning impacted air masses (from both African and Boreal fires) as they travel away from source regions. |
1 Introduction
Our analysis focusses on two trace gases, peroxy acetic acid (CH3C(O)OOH, PAA) and peroxy acetyl nitric anhydride (CH3C(O)OONO2, PAN), which have a common precursor, the peroxy acetyl radical, CH3C(O)O2.| CH3C(O)O2 + HO2 → CH3C(O)OOH (PAA) + O2 | (R1a) |
| → CH3C(O)OH + O3 | (R1b) |
| → OH + CH3CO2 + O2 | (R1c) |
| CH3C(O)O2 + NO2 + M → CH3C(O)OONO2 (PAN) + M | (R2) |
| CH3C(O)O2NO2 + M → CH3C(O)OO + NO2 + M | (R3) |
Globally, the main source of CH3C(O)O2 is the OH-initiated degradation of acetaldehyde, with important contributions from the photolysis of acetone and methyl-glyoxal.1 The conversion rate of CH3C(O)O2 to either PAN or PAA depends on the relative abundance of HO2 and NO2 (ref. 2) and the rate of thermal decomposition of PAN, which is highly temperature dependent.
The central role of PAN in atmospheric chemistry has been established for decades3 and is documented in numerous ground-level, airborne and remote-sensing measurements of its abundance and in several model studies of, e.g. its global distribution and role as a supplier (via long-range transport) of NOX to remote regions.1,4,5 The lifetime of PAN can be as short as 10 minutes in the warm boundary layer but can increase to weeks or months in the much colder free and upper troposphere where it is the principal reservoir of NOX.1 PAN mixing ratios can vary from a few pptv in remote regions to several ppbv in heavily polluted air masses.1 As well as sequestering NOX, the formation of PAN converts an organic peroxy radical (CH3C(O)O2) to a long-lived reservoir, thus affecting photochemical ozone formation through NO to NO2 conversion.
Ambient measurements of PAA in the gas phase are scarce, though its observation in the boundary layer2,6–11 and at higher altitudes12,13 indicate that it is present throughout the troposphere. Recent airborne measurements of PAA indicate mixing ratios of 50–100 ppt for the Northern (winter) and Southern (summer and winter) Hemispheres with little variation with altitudes up to 10 km. In contrast, in the Northern Hemisphere in summer, up to 300 ppt were measured with the largest mixing ratios generally found in the lowermost 4 km.14 PAA contributes to the formation and ageing of secondary organic aerosol,15 but unlike other organic acids, the direct emission of PAA by the biosphere has not been documented.
Until recently, the lifetime of PAA was believed to be dominated by reaction with the OH radical for which a rate coefficient of ∼10−11 cm3 per molecule per s had been measured.16 However, new kinetic data has shown that PAA reacts orders of magnitude more slowly (the rate coefficient is ∼4 × 10−14 cm3 per molecule per s at 298 K) and has a lifetime with respect to reaction with OH of ∼1 year. The reduction in the rate coefficient for OH + PAA results in a substantial increase in its global, modelled concentration.17 Above the boundary layer, where deposition is important,2,6 the dominant loss process for PAA is thought to be photolysis, with a noon-time J-value at mid-latitudes and cloud-free sky of ∼7 × 10−7 s−1, which (assuming equal length of day and night) results in a diel averaged J-value of ∼2.2 × 10−7 s−1 or a lifetime of several weeks. Later, when analysing PAA datasets, we provide a more detailed analysis of its sinks.
Biomass-burning contributes to the global budgets of many trace gases18,19 and can result in elevated mixing ratios thereof over vast regions of the Indian and Pacific Oceans as well as the tropical Atlantic Ocean. The main source regions for biomass burning emissions are Central Africa during the dry-season (November to March in the northern hemisphere, July to October in the Southern Hemisphere), South America (from August to October) and, to a lesser extent, the boreal forest which has a short biomass burning season centred on July.20 Along with many volatile organic compounds, nitrogen oxides are formed/released during biomass burning, the photochemical transformation of which results in the formation of O3,21–23 which is an important greenhouse gas at higher altitudes and respiratory irritant in the boundary layer, as well as particles that may affect air quality, cloud formation and precipitation. Observations indicate that PAN is formed rapidly in the presence of fire-generated NOx and non-methane hydrocarbons,24–35 with ∼40% of initially fire-generated NOX partitioned to PAN.
Although PAA has been less frequently linked with biomass burning, there are reports of PAA downwind of de-forestation fires in biomass-burning plumes sampled a few hours after emission36 and in air masses over the boreal forest that were impacted by biomass burning several days prior to the measurement.2,6,37 PAA resulting from biomass burning has also been observed following residual crop burning in China38 and in hour-old biomass-burning plumes from Californian fires.39
In this work, we use airborne measurements of PAN and PAA to derive relative enrichment ratios (compared to CO and CH3CN) to assess to what extent their abundance throughout the North Atlantic troposphere is impacted by biomass burning and to examine the extent to which NOX has been converted to PAN in such air masses.
2 Methods
In August–September 2018 the High Altitude and Long-range research aircraft (HALO) provided the platform to perform the “CAFE-Africa” campaign (Chemistry of the Atmosphere: Field Experiment in Africa). A total of 16 measurement flights were conducted, mainly over the Tropical Atlantic west of Africa.40 Sal Airport (Cape Verde Islands) was the mission base.In this paper, we focus on 4 flights in which the aircraft intercepted biomass-burning impacted air masses, as evidenced by clearly enhanced levels of black carbon, CO and CH3CN. These were flights F4, F10, F12 and F13, which were conducted on the 10th, 24th, 29th and 31st of August, respectively. The flight tracks, colour-coded by black-carbon, CO and CH3CN levels are displayed in Fig. S1 of the ESI.† The altitudes at which the biomass-burning plumes were intercepted (i.e., the points on the flight tracks with enhanced black-carbon, CO and CH3N) and the associated 10-day air-mass back-trajectories are described in Section 3 (see also Fig. S1†).
2.1 Measurement of PAN and PAA
The I-CIMS configured for airborne operation during CAFE-Africa has been described recently in detail.41 Briefly, I− primary ions were generated by passing CH3I through a 210Po ion source before reacting with atmospheric trace gases in an ion-molecule reactor (IMR) held at a constant pressure of 24 mbar. PAN was detected as CH3CO2− (m/z 59), which is formed in the reaction between I− and CH3C(O)O2, the latter generated during the thermal dissociation of PAN in a heated inlet (∼170 °C, 100 mbar). PAA was also detected as CH3CO2−, in this case formed by the direct reaction of PAA with I−. Clusters of I- with PAA or with the CH3C(O)O2 radical are not observed with this CIMS even when declustering is switched off and is related to collisional declustering at the gas-expansion between CDC and octople-chamber.41In order to separate the signals of PAN and PAA, NO was periodically added to the heated inlet to remove CH3C(O)O2 and thus eliminate sensitivity to PAN. A constant amount of H2O was added to the IMR to reduce fluctuations in sensitivity to PAN and PAA caused by air masses with different water content. However, as the I-CIMS sensitivity to PAA is strongly dependent on the ambient relative humidity2 corrections to PAA mixing ratios based on in-flight changes in the ratios of signals at m/z = 127 (I−) and m/z = 145 (I−(H2O)) were applied.
The forward-facing trace gas inlet (TGI, located on top of the aircraft) was configured to force RAM-air through ¼ inch (OD) PFA tubing sampling at an angle of 90° to the flight direction. The ¼ inch tubing was connected to a ½ inch (OD) PFA tube attached to an exhaust plate at the underside of the aircraft to create a fast “bypass” flow. The bypass flow was sub-sampled (again at 90° and by ¼ inch PFA tubing heated to 40 °C) by the ∼1.3 L (STP) min−1 flow into the I-CIMS. Prior to take-off, the inlet line and TGI were flushed with nitrogen to prevent contamination by the high levels of pollutant trace gases at the airport. With this setup, PAA and PAN data points (m/z 59 integrated over 0.8 s) were taken every ∼10 s.
Most flights were of ∼8 hours duration, during which an hourly, in situ PAN calibration was performed whereby PAN was generated in a photochemical source42,43 and added to scrubbed ambient air. Based on in situ calibrations from all flights, the average sensitivity for PAN, normalised to 1 MHz signal at m/z = 127 from I−, was 1.01 ± 0.2 cts per pptv. PAA was calibrated once during the campaign by simultaneously sampling the air flow over a diffusion tube containing liquid PAA in acetic acid into the I-CIMS and into a modified commercial instrument “HypHop” (Model AL2021, Aero-Laser GmbH, Garmisch-Partenkirchen, Germany) based on the horseradish peroxidase/catalase/p-hydroxyphenyl acetic acid wet chemical fluorescence measurement technique.44,45 The sensitivity for PAA under dry conditions (i.e. with H2O added only to the IMR as described above), normalised to 1 MHz signal at m/z = 127 from I−, was 1.64 cts per pptv.
The PAN measurements have an uncertainty of 25%, which considers calibration reproducibility, potential systematic bias in the mixing ratio of NO used to generate PAN, uncertainty in the flows, uncertainty in the conversion factor for NO to PAN (0.9 ± 0.05), the necessary subtraction of the PAA signal and its interpolation and the potential loss of signal at m/z 59 due to reaction of CH3CO2− with other gases exiting the calibration source.46 The detection limit for PAN (derived from 3σ standard deviations of the zero-signal) is ∼10 pptv, resulting in a total measurement uncertainty of 25% + 10 pptv. However, large fluctuations in PAA concentrations (measured at the same mass as PAN) can result in a worsening of the PAN detection limit owing to interpolation uncertainty.
The uncertainty associated with the PAA measurement is related to the accuracy of the internal HypHop calibration (using a H2O2 standard), the scavenging efficiency of PAA in the scrubber (90 ± 5%), the non-tested assumption that PAA behaves like other acidic peroxides regarding the addition of peroxidase and the fact that no in-flight calibrations were possible, resulting in an estimated uncertainty of 50%. The limit of detection (based on 3σ standard deviations of the zero-signal obtained during scrubbing) was 10 pptv, resulting in an overall measurement uncertainty of 50% + 10 pptv.
2.2 Measurement of NOX, CO, CH3CN and black-carbon
NO was measured using a two-channel chemiluminescence detector CLD 790 SR (ECO Physics AG, Dürnten, Switzerland) that has been modified for in situ field measurements.40,47 The total measurement uncertainty for NO was 6% which includes uncertainty in the NO-standard (NO in N2) used for calibration. As NO2 measurements using the CLD are likely to be contaminated by thermally labile peroxy-nitrates in the upper troposphere,48 NO2 mixing ratios were calculated from measurements of NO, O3, the NO2 photolysis-frequency and assuming photo-stationary steady-state.40CO was measured by mid-infrared, quantum-cascade laser absorption spectroscopy (QCLAS) with “TRISTAR”, a multichannel spectrometer.49,50 The time resolution was 1 Hz and the measurements were associated with a total uncertainty of 4.3%. CH3CN was measured at m/z 42.0339 using a customized Proton Transfer Reaction Time of Flight mass spectrometer (PTR-TOF-MS 8000, Ionicon Analytik GmbH Innsbruck, Austria) with a limit of detection (3 times standard deviation) of 27 ppt and an uncertainty of 25–29% (1 s resolution). Black-carbon concentrations were measured at 1 s time resolution with a single particle soot photometer (SP2) manufactured by DMT (https://www.dropletmeasurement.com/). The SP2 is sensitive to refractory black-carbon-free particles in the optical size range of about 180 < Dp < 400 nm and to refractory black-carbon cores in the size range of about 80 < dMEV < 500 nm, with a counting efficiency close to unity.51
2.3 EMAC model calculations
We used the global, atmospheric chemistry-transport model EMAC to calculate the temperature, photolysis frequencies (for PAN and PAA), and OH, HO2, NO and NO2 levels along air-mass trajectories calculated by HYSPLIT52,53 using 1° meteorological data (GDAS) extending 10 days back from the point and time of interception of the biomass-burning impacted air mass by the HALO-aircraft.EMAC (ECHAM-MESSy) uses the 5th generation European Centre Hamburg general circulation model (ECHAM5 (ref. 54)) as the core atmospheric general circulation model.55,56 In this study, we used EMAC (ECHAM5 version 5.3.02, MESSy version 2.55.0) at T63L47MA-resolution, i.e. with a spherical truncation of T63 (∼1.8 by 1.8° in latitude and longitude) with 47 vertical hybrid terrain following-pressure levels up to 0.01 hPa. The model was weakly nudged in spectral space, applying Newtonian relaxation of the parameters temperature, vorticity, divergence and surface pressure to meteorological reanalysis data.57 The model set-up is identical to the simulation RED (reduced emissions due to lockdown, without cloud–aerosol interaction)58 where the model was evaluated against an aircraft campaign over Europe. In addition, the model has been evaluated on many occasions.59–61 For additional references, see http://www.messy-interface.org. EMAC was used as a chemical-transport model without feedbacks between photochemistry, radiation and atmospheric dynamics.62,63
3 Flights impacted by biomass-burning
We used measurements of CH3CN, CO and black carbon to identify periods in each flight where the air masses were clearly impacted by biomass-burning. CH3CN is produced almost exclusively by biomass burning,64,65 which makes it an excellent tracer for air masses that have been impacted by fires, though it may also be formed during industrial and vehicular combustion.66 CH3CN reacts only slowly with the OH radical (k(240 K) ∼1 × 10−14 cm3 per molecule per s) and thus has a lifetime in the free troposphere of the order of years and can be used to identify air masses that were impacted by biomass-burning emissions several days (or weeks) prior to its detection.CO, often used as a biomass-burning tracer,67 is formed with high yield in vegetation fires. Combining the rate coefficient (at 240 K) of ∼2 × 10−13 cm3 per molecule per s for the reaction of CO with OH and an approximate, diel averaged OH concentration of 1 × 106 molecule per cm3 results in a lifetime of ∼58 days and thus, on time-scales of a few days, CO may be considered a conservative marker of biomass-burning emissions. However, CO is also formed in the subsequent photochemical degradation of many organic trace gases that are co-emitted during biomass burning and transported through the atmosphere. As both CH3CN and CO are relatively long-lived, they acquire high background levels that can make identifying aged biomass-burning plumes difficult. For this reason, our primary (qualitative) identifier of fresh biomass-burning emissions is black carbon, which has a lifetime of ∼1 week and generally very low background levels compared to air masses recently impacted by biomass burning. Black-carbon is also emitted by industrial burning, however the black-carbon signal in these regions over the Atlantic Ocean originates from biomass-burning.51,68,69
In Fig. 1 we present binned data (red and blue data points, 1 min averages) and campaign averaged vertical profiles (black data points) of CH3CN, CO, PAN and PAA for flights F04–F15 (Fig. 1b–e, black data points) as well as black-carbon particle concentrations (Fig. 1a). The data covers a large region of the tropical Atlantic west of and above the African coast (see Fig. S1†).
 | ||
| Fig. 1 Vertical profiles of black carbon (a), CO (b), CH3CN (c), PAN (d) and PAA (e). The datapoints (1-minute averages) are colour-coded according to latitude, with blue and red datapoints obtained north and south of 10°N (the approximate location of the ITCZ during the campaign). The solid black circles are mean mixing ratios in 1 km bins. The open blue circles highlight elevated CO, CH3CN, PAN, PAA and black carbon north of the ITCZ when the aircraft intercepted a biomass-burning plume from Canada/N.W. USA. | ||
During CAFE-Africa, the Inter-Tropical Convergence Zone (ITCZ) was located at roughly 5–15°N for longitudes between 0 and 25° east,70 and we accordingly delineate the data as having been acquired either south (red data points) or north (blue data points) of 10°N. In all three biomass-burning tracers, black carbon, CH3CN and CO, the mixing ratios/concentrations obtained at low altitudes south of the ITCZ are greatly enhanced relative to those obtained at latitudes >10°N. The south–north gradient reflects both the location of biomass burning at this time of the year (African continent, south of the ITCZ as shown in Fig. 1) and also efficient in-cloud scavenging of black carbon (for which the North–South differences are most obvious) during transport through the ICTZ. This data indicates that the air masses most strongly influenced by biomass-burning emissions are located at low altitudes (up to ∼4 km) south of the ITCZ.71 Between 5 and 14 km, the north–south gradient is less pronounced but still apparent, with mixing ratios of CO and CH3CN obtained south of the ITCZ generally above the mean at most altitudes. A similar picture emerges for both PAN and PAA, with the largest mixing ratios mostly obtained at low altitudes south of the ITCZ. Exceptions to this (encircled in blue) are the data from flight 10, where elevated PAN mixing ratios of up to ∼700 pptv, CO mixing ratios of almost 200 ppb and PAA mixing ratios of up to 75 ppt were observed at altitudes of ∼6–7 km north of the ITCZ. As shown below, the air mass intercepted at this location was impacted by biomass burning emissions originating from boreal forest fires in the Northern US and Canada and enhancements in black carbon and CH3CN, although less obvious, are still apparent.
At altitudes above ∼5 km the differences in PAN (or PAA) when comparing measurements north of the ITCZ to those south of the ITCZ are no longer significant. Biomass burning clearly has a significant impact on the chemical composition of the atmosphere throughout much of the free troposphere in the region interrogated by the HALO flights during CAFE-Africa, especially at altitudes below ≈4 km south of the ITCZ.
For our analysis of the impact of biomass burning, we now focus on four flights (F04, F10, F12 and F13, see Fig. S1† for flight tracks and levels of black-carbon, CH3CN and CO) in which black-carbon particle concentration and the CH3CN and CO mixing ratios provided unambiguous evidence of biomass-burning influence for extended periods. Three biomass-burning plumes were intercepted south of the ITCZ, and one (F10) was intercepted at higher latitudes (25–30°N). Time series of the altitude as well as CH3CN, CO, PAN and PAA mixing ratios from these flights are displayed in Fig S2.†
Fig. S3† confirms the expected correlation between black-carbon and CH3CN for biomass-burning impacted air masses. Similar slopes (∼1800 ppbv−1) for black-carbon particle concentration per ppbv of CH3CN are observed for the biomass-burning plumes originating over the African Savanna in flights FL4, FL12 and FL13. In contrast, the air mass impacted by biomass burning in the US/Canada (FL10) was depleted in black-carbon with a slope of a factor of ten lower. This difference is likely to reflect the slightly longer time of transport from the fire and the influence of rain-out. However, for this analysis, we only wish to indicate that elevated black-carbon and CH3CN mixing ratios are (as expected) associated with biomass burning.
3.1 Flight 04 on the 10th August 2018
During flight F04, the aircraft headed out in a south–south-easterly direction, crossing the ITCZ and entering the Southern Hemisphere at ∼12:30 UTC at an altitude of ∼12 km. As shown in Fig S2,† greatly enhanced levels of black carbon (>500 particles per cm3) were measured between 13:40 and 15:10 following a descent to altitudes of 2–3 km. Two distinct maxima in black carbon particle concentration were observed at 13:52 (∼930 cm−3 at 2 km, 291 K) and 14:37 (∼1200 cm−3 at 3 km, 284 K) and coincided with double maxima in the mixing ratios of CO (∼270 and 330 ppbv) and CH3CN (∼0.59 and 0.64 ppbv). Both maxima were also present in the PAA dataset (∼76 and 90 pptv), but only the second maximum (190 pptv) was clearly defined for PAN. The different peak shape of the PAN time series compared to CH3CN, CO and PAA is readily understood in terms of the strong dependence of the PAN lifetime (with respect to thermal dissociation) on temperature and, thus, altitude.Fig. 2 displays a 10-day back-trajectory calculation using HYSPLIT52,53 of the air mass intercepted at 14:00. The back-trajectory indicates that the air had passed over active fires 4–6 days previously at an altitude of between 3 and 5 km. Active fire data (red data points) were obtained from the Fire Information for Resource Management System (FIRMS) available through NASA's Earth Observing System Data and Information System and were recorded by the Moderate Resolution Imaging Spectroradiometer (MODIS) instruments on the satellites TERRA and AQUA. Only fires detected with 100% certainty during the time the back-trajectory crossed the continent are plotted.
 | ||
| Fig. 2 HYSPLIT back-trajectories (10 days, coloured by altitude) starting at the time of interception of the biomass-burning plume during flights 04, 10, 12 and 13. The black stars along the back-trajectories are separated by 24 hours. The red dots are Terra-MODIS fires detected during the first three weeks of August 2018. Only fires detected with 100% certainty are plotted. Back-trajectories obtained at 1 min intervals 5 min prior to and after those shown are plotted in Fig. S6.† | ||
3.2 Flight 10 on the 24th August 2018
During flight 10, the aircraft flew in a north–west-westerly direction before encountering (at ∼13:30–15:00) biomass-burning impacted air over a range of altitudes (4–8 km) between 25 and 30 °N over the central Atlantic. Compared to flight 04, black carbon number densities were low (<50 particles per cm3), but CO levels were similar (∼200 ppbv), and PAN levels were as high as 500 pptv. A HYSPLIT back-trajectory (Fig. 2) ending at the aircraft position at 14:00 indicates that the air mass had passed over active fires in Western USA and British Columbia (Canada) about 9 days prior to the measurement. In 2018, over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 wild-fires were registered in the Western US states with >20000 km2 (ref. 35) burnt and on 16th August 2018, >500 wildfires were active in British Columbia, which in 2018 experienced its most intense fire season on record with over 13000 km2 burnt.72 The longer transport time for this plume will have been partially responsible for the reduced concentration of black carbon particles which have a lifetime of ∼5–10 days, though other factors (e.g. in-cloud scavenging) may have contributed.
000 wild-fires were registered in the Western US states with >20000 km2 (ref. 35) burnt and on 16th August 2018, >500 wildfires were active in British Columbia, which in 2018 experienced its most intense fire season on record with over 13000 km2 burnt.72 The longer transport time for this plume will have been partially responsible for the reduced concentration of black carbon particles which have a lifetime of ∼5–10 days, though other factors (e.g. in-cloud scavenging) may have contributed.
3.3 Flight 12 on the 29th August 2018
Flight 12 intercepted a biomass-burning plume at an altitude of ∼3.9 km slightly south of the equator about 20° west of the African coast. The black carbon concentration at the peak of the plume (at 13:53, see Fig S2†) was ∼700 particles per cm3. The biomass-burning tracers, black carbon, CH3CN and CO were highly variable over ∼1 hour while flying at constant altitude with 2nd maxima (0.57 ppbv CH3CN and 240 ppbv CO) at ∼14:28. Both maxima are also observed in the PAA dataset (∼200 and 180 pptv) but not in the PAN measurements. A HYSPLIT back-trajectory (Fig. 2) ending at the aircraft position at 13:53 indicates that ∼6 days prior to the measurement, the air mass had passed over regions with active fires close to the West African coast.3.4 Flight 13 on the 31st August 2018
Flight 13 was a stacked flight on a roughly north–south transect designed to explore vertical gradients on both sides of the ITCZ. Typically, the aircraft stayed on a constant height level for 20–40 minutes before climbing/diving to a new altitude. Biomass burning impacted air masses with high black carbon particle number densities were intercepted on the outward leg, with peaks in black-carbon concentration at 12:12 (310 cm−3 at 2.2 km) and at 13:50 (∼500 cm−3 at 3.2 km). These maxima were also observed in the CO (∼160 and 240 ppbv), CH3CN (∼0.25 and 0.35 ppbv) and PAA (∼130 and 170 pptv) datasets. High levels of CO and CH3CN were also observed between 14:00 and 14:25 at an altitude of 7.7 km, during which the black carbon concentration was <10 particle per cm3, but not zero. The observation of enhanced CH3CN and CO at higher altitudes may indicate rapid convective transport from burning regions with wash-out of black carbon.73 The back-trajectory (ending at 14:21 and an altitude of 7.7 km, see Fig. 2) indicates that the air mass passed over biomass-burning regions ∼5–7 days prior to the measurement.4 PAA, PAN, CO and CH3CN correlations and molar enhancement ratios
The biomass-burning-induced molar enhancement ratio (MER) of a pair of trace gases can be derived from measurements of the difference in mixing ratio of each species within a biomass-burning plume compared to background levels or, more readily (if high-time-resolution data are available) by correlating mixing ratios and taking the slope of a linear regression.74 In the following, we differentiate between molar enhancement ratios in aged biomass-burning plumes (MER), those close to fires (MER0) and molar emission factors for trace gases directly generated in the fire (MEF).As both CO and CH3CN are known tracers of biomass burning, and trace-gas enhancement ratios are often reported relative to CO, we first examined the correlation between CO and CH3CN during the biomass-burning impacted periods of flights 04, 10, 12 and 13. The results, using 1-minute averaged data, are displayed in Fig. 3. Note that in the figure legend, the flights are subdivided into two or three parts reflecting differences in, e.g., flight altitude or different features in the time series (see Fig. S2† and text below). On average, using data from all 4 flights, we derive MER(CO/CH3CN) = 598 ± 20 (FL04), 763 ± 20 (FL10), 571 ± 75 (FL12) and 718 ± 42 (FL13).
 | ||
| Fig. 3 Correlation between the biomass burning tracers CH3CN and CO. The linear regressions are the molar enhancement ratios MER(CO/CH3CN) and are equal to 598 ± 20 (FL04), 763 ± 20 (FL10), 571 ± 75 (FL12) and 718 ± 42 (FL13). The labels refer to different flight segments. For example, BB4a refers to biomass-burning impacted periods in flight 4 between 13:45 and 14:00. | ||
By comparison, relative (mol-based) emission factors of MEF(CO/CH3CN) = 594 ± 299 for the burning of Savanna/grassland-biomass and 571 ± 288 for the burning of the boreal forest have been evaluated,67 whereby the uncertainties reflect the quoted standard deviation of the biomass burning events assessed. A more recent assessment66 reports a value of 498 ± 40. Our values of MER(CO/CH3CN) (which do not account for photochemical CO loss or formation during transport from the fire to the interception of the plume by HALO) thus lie within the range of previous observations of MEF(CO/CH3CN).
Using the same approach, we now use the measurement data to calculate the MER for PAN and PAA relative to both CO and CH3CN. The correlation plots are displayed in Fig. 4 and the slopes and associated flight altitude at the time of interception of the plume are listed in Table 1. Literature MER values are summarised in Table 2.
 | ||
| Fig. 4 Correlation between PAN and PAA and the biomass-burning tracers CH3CN and CO. The labels refer to different flight segments. For example, BB4a refers to biomass-burning impacted periods in flight 4 between 13:45 and 14:00. For the plot of PAN versus CH3CN and both PAA plot, the error bars are not shown to preserve clarity. | ||
| Alta | TT | MER(PAN/CO) (×10−4) | MER(PAN/CH3CN) | MER(PAA/CO) (×10−5) | MER(PAA/CH3CN) | FS (PAN) | FS (PAA) | FS (CO) | MER0(PAA/CO) (×10−5) | MER0 (PAA/CH3CN) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Flight altitude (km) at point of interception of plume by HALO. TT = approximate transport time (days) from biomass-burning source region. FS = fraction surviving transport, MER = molar enhancement ratio, MER0 = molar enhancement ratio corrected for loss of PAA and CO during transport, NLC = no linear correlation owing to lack of variation of one parameter, ID = insufficient data. FL04a (13:45–14:00), FL04b (14:06–15:07), FL10a (13:26–13:35). FL10b (13:36–14:21), FL10c (14:25–15:00), FL12a (13:47–14:02), FL12b (14:10–14:30), FL13a (12:10–12:54), FL13b (12:59–13:53). | |||||||||||
| FL04 (a) | 1.94 | 4.3 ± 0.3 | 0.62 ± 0.07 | ID | ID | ||||||
| FL04 (b) | 3.22 | 5.0 ± 0.3 | 0.35 ± 0.03 | 28 ± 3.3 | 0.22 ± 0.03 | ||||||
| MER | 5 | 4.7 ± 0.3 | 0.49 ± 0.15 | 28 ± 3.3 | 0.22 ± 0.03 | 1 | 86 | 87 | 28 | 0.26 | |
| FL10 (a) | 7.32 | 36 ± 11 | 2.0 ± 0.7 | ID | ID | ||||||
| FL10 (b) | 5.5 | 49 ± 3 | 3.2 ± 0.4 | ID | ID | ||||||
| FL10 (c) | 4.3 | 39 ± 5 | 3.9 ± 0.8 | ID | ID | ||||||
| MER | 8 | 39 ± 1 | 2.9 ± 0.08 | 39 ± 2 | 0.31 ± 0.02 | 62 | 75 | 81 | 42 | 0.41 | |
| FL12 (a) | 3.9 | NLC | NLC | ID | ID | ||||||
| FL12 (b) | 4.0 | 18 ± 3 | 0.9 ± 0.1 | ID | ID | ||||||
| MER | 6 | 18 ± 3 | 0.9 ± 0.1 | 54 ± 8 | 0.62 ± 0.15 | 18 | 78 | 81 | 56 | 0.79 | |
| FL13 (a) | 2.2 | NLC | NLC | 129 ± 12 | 0.60 ± 0.13 | ||||||
| FL13 (b) | 3.2 | NLC | NLC | 108 ± 9 | 0.70 ± 0.09 | ||||||
| MER | 7 | — | — | 110 ± 6 | 0.71 ± 0.07 | 82 | 70 | 86 | 135 | 1.01 | |
| Reference | MER(PAN/CO) (×10−4) | Plume age | Location |
|---|---|---|---|
| Holzinger et al., 2005 (ref. 75) | 65 | “Aged” | Eastern Mediterranean |
| Mauzerall et al., 1998 (ref. 26) | 53–75 | “Varying” | Tropical S. Atlantic |
| Singh et al.,2004 (ref. 76) | 38–41 | — | Pacific |
| Real et al., 2010 (ref. 21) | 32 | — | Central Africa |
| Fiedler et al., 2011 (ref. 73) | 21 | — | Africal Savanna |
| Alvarado et al., 2010 (ref. 27) | 11–87 | “Aged” | Boreal forest |
| Wolfe et al., 2021 (ref. 31) | 25–50 | 2–12 h | California |
| Juncosa Calahorrano et al., 2021 (ref. 35) | 200–500 | <1 day to >3 days | Western US |
| This work | 5–49 | 5–9 days | North/Tropical Atlantic |
| Reference | MER(PAN/CH3CN) | Plume age | Location |
|---|---|---|---|
| Holzinger et al., 2005 (ref. 75) | 3.25 | “Aged” | Eastern Mediterranean |
| Reference | MER(PAA/CH3CN) | Plume age | Location |
|---|---|---|---|
| This work | 0.2–0.7 | 5–9 days | North/Tropical Atlantic |
4.1 PAN: molar enhancement ratios (MER) relative to CO and CH3CN
Based on the slopes of the correlation plots displayed in Fig. 4 (top left), we derive molar enhancement ratios for PAN relative to CO, MER(PAN/CO) of 4–5 × 10−4 (FL04), 36–49 × 10−4 (FL10) and 18 × 10−4 (FL12). The values of MER(PAN/CO) are thus highly variable from flight to flight, which reflects variations in the PAN loss rate (and also its photochemical formation) during transport. The larger values of MER(PAN/CO) (flights F10 and F12) are consistent with previous reports of PAN/CO relative enhancement ratios derived from analysis of PAN and CO in aged biomass-burning plumes, including 65 × 10−4 for biomass-burning plumes of unknown origin observed over the Eastern Mediterranean,75 53–75 × 10−4 for biomass-burning plumes of varying age over the tropical South Atlantic,26 38 and 41 × 10−4 for biomass-burning plumes over the tropical Pacific at heights of between 3–10 km and 0–1 km, respectively,76,77 32 × 10−4 for Central African biomass-burning21 and 21 × 10−4 for aged biomass-burning plumes originating over the Southern Hemispheric African Savanna.73For biomass-burning emissions originating from the boreal forest, values of MER(PAN/CO) of 11–87 × 10−4 have been reported for aged plumes,27 which is consistent with our values for FL10 during which we intercepted a biomass-burning plume originating from the Western-US/Canada. In situ-measurements of Californian biomass-burning plumes reported MER values of 25–50 × 10−4,39 which are roughly independent of plume age (2–12 hours) and broadly consistent with those observed in this work. Values of MER(PAN/CO) derived from satellite measurements of wildfires in the Western US in 2018 vary from >500 × 10−4 for plumes less than 1 h old to ∼200 × 10−4 for plumes >3 h old.35
Mol-based enhancement ratios for PAN relative to CH3CN measured during the HALO mission (Fig. 4, top right) are MER(PAN/CH3CN) = 0.5 (FL04), 2.9 (FL10) and 0.9 (FL12). The larger value obtained in FL10 is thus comparable to the only previously reported value75 from measurements taken over the Eastern Mediterranean whereby MER(PAN/CO) = 0.0065 and MER(CH3CN/CO) = 0.002 convert to MER(PAN/CH3CN) = 3.25.
For both MER(PAN/CO) and MER(PAN/CH3CN), the lower enhancement ratios observed during FL04 are likely to have been caused by the efficient loss of PAN through thermal decomposition during transport at low altitudes. Our EMAC model results (see below) suggest that, for both FL04 and FL12, practically all of the PAN formed early on during biomass burning would have thermally decomposed (R3) during transport. The fact that, despite the likely loss of all initially present PAN during transport, a correlation between PAN and the biomass-burning markers CO and CH3CN still exists, indicates that PAN was (re)generated during transport and the rate of production was proportional to the concentration of organic and reactive nitrogen precursors (and thus CH3CN).
During FL13, the losses of PAN derived from EMAC J-values and temperatures are much lower, which reflects transport at higher altitudes and a slower rate of thermal decomposition. Such effects will also contribute to the range of slopes and non-linear correlation between PAN and CO or CH3CN during FL12. The fact that MER(PAN/CO) is highly dependent on the transport altitude is also reflected in the lack of agreement in modelled molar enhancement ratios between models that are driven by different meteorology whereby MER(PAN/CO) values between 18.7 × 10−4 and 70 × 10−4 have been reported.29
4.2 PAA: molar enhancement ratios (MER) relative to CO and CH3CN
Based on the slopes of the correlation plots displayed in Fig. 4 (bottom left), we derive molar enhancement ratios for PAA relative to CO, MER(PAA/CO), of ∼28 × 10−5 (FL04), 40 × 10−5 (FL10), 54 × 10−5 (FL12) and 110 × 10−5 (FL13). MER(PAA/CO) values are thus somewhat less variable than those of MER(PAN/CO), reflecting the fact that losses of PAA during transport from the biomass-burning region to the point of interception are much smaller than for PAN (see below) and, in contrast to PAN, are not strongly influenced by the altitude at which transport took place.Measurements of PAA in biomass-burning impacted air masses are limited, but simultaneous measurements of PAA and CO close to crop-residue and deforestation fires in the Yucatan have been used to derive initial emission ratios, MER0(PAA/CO) from smoke that was only a few minutes old, of 20 ± 10 × 10−5 (range 13–26 × 10−5) from three different fires.36 Observed an increase (over 1.4 hours) in the PAA mixing ratios in aging plumes, which is consistent with the higher value of 72 × 10−5 reported for more aged plumes originating from crop burning in the North China Plain.38 An increase in MER(PAA/CO) with time was also recorded for smoke from Californian fires39 with MER0(PAA/CO) ∼20 × 10−5 in plumes a few minutes old (and thus consistent with the observation of Yokelson et al.36) increasing to 150 × 10−5 for ∼12 h old plumes.
As highlighted by Wolfe et al.,39 the evolution of the PAA/CO ratio in biomass-burning plumes is a complex function of the organic material available for combustion, NO levels and the formation of reactive radicals (e.g. OH) that can sustain oxidation of primary emitted VOCs and thus the formation of radicals required to generate PAA. The evolution of CH3CHO is of critical importance to PAA (and PAN) formation as it represents an important source of the CH3C(O)O2 radical precursor. Wolfe et al.39 have shown that CH3CHO levels peaked at ∼2 hours after emission and then decreased steadily (by a factor of ≈2) over the next 10 hours.
From the four HALO flights, we derive MER(PAA/CH3CN) values of 0.22 ± 0.03 (FL04), 0.31 ± 0.03 (FL10), 0.62 ± 0.15 (FL12) and 0.71 ± 0.07 (FL13). As far as we are aware, there are no previous values for MER(PAA/CH3CN) with which to compare our data.
As mentioned above, PAN (and to a lesser extent PAA) that was rapidly generated during biomass burning can be lost during transport away from the fire, so that the MER is expected to be time-dependent. In order to estimate the fractional losses of PAN and PAA during transport from the biomass-burning region to the point of interception by the aircraft, we used the EMAC model to calculate time-dependent J-values (for PAA and PAN) and OH concentrations along the HYSPLIT back trajectories for each of the major biomass burning events observed in flights 04, 10, 12 and 13.
In the calculation of the fractional loss, PAN was removed by thermal decomposition (R3) and photolysis (R4a) and (R4b).
| CH3C(O)O2NO2 + hν → CH3C(O)O2 + NO2 | (R4a) |
| → CH3CO2 + NO3 | (R4b) |
Not all PAN that thermally decomposed or is photolysed in (R4a) is lost as some fraction of the CH3C(O)O2 radical thus formed can react with NO2 to reform PAN (via(R2)). The fraction of CH3C(O)O2 that reforms PAN depends on the relative concentrations of NO2, NO and HO2 (also taken from EMAC) and associated rate coefficients for their reaction with CH3C(O)O2. Reaction with NO leads to the formation of CH3O2 and thus always results in a net loss of PAN, while reaction with NO2 reforms PAN, so that it is conserved. For reaction with HO2, the only product that can regenerate CH3C(O)O2 is PAA (formed at a yield of ∼25% (R1a)). However, as the regeneration of CH3C(O)O2 requires that PAA reacts with OH (rather than be photolysed), this can be neglected (see below) and, to a very good approximation, the reaction of CH3C(O)O2 with HO2 will not lead to reformation of PAN. In calculating an effective PAN loss rate, we have taken its reformation into account by multiplying the thermal loss rate coefficient or the photolysis frequency for (R4a) by (1 − f), where f is a term that describes the fractional conversion of CH3C(O)O2 to PAN (see below). We see that despite thermal dissociation to CH3C(O)O2 and NO2, the nighttime losses of PAN are negligible as levels of radicals and NO (which compete with NO2 for reaction with CH3C(O)O2) are very low (and f is thus close to 1).
Fig. 5 presents the time-dependent, fractional losses of PAN along the back-trajectory for FL04. In this case, almost none of the PAN (red curve) formed early in the plume would have survived transport from the biomass-burning region as the plume travelled part of the time at low altitudes (i.e., at high temperatures). PAA may be lost via reaction with OH and photolysis:
| CH3C(O)OOH + OH → CH3C(O)O2 + H2O | (R5) |
| CH3C(O)OOH + hν → CH3CO2 + OH | (R6) |
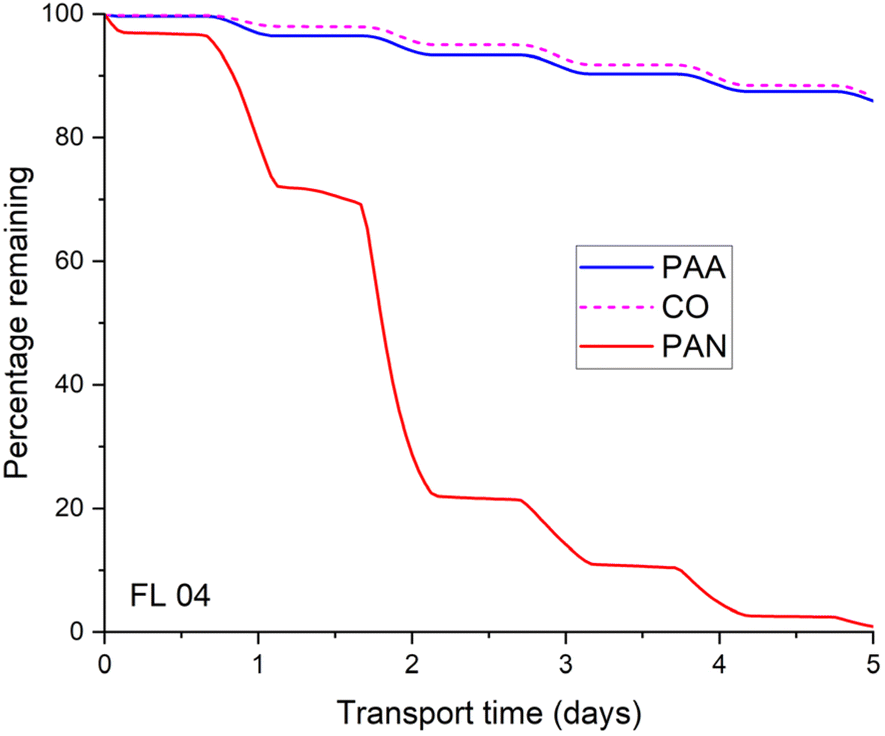 | ||
| Fig. 5 Calculated percentage of CO, PAA and PAN remaining after transport from the biomass-burning region to the point of interception during flights 04, 10, 12 and 13. The losses of CO, PAN and PAA were calculated from modelled (EMAC) temperatures, pressures, photolysis frequencies and OH-fields along the back-trajectories using rate expressions described in the text. The PAN loss frequency via its thermal decomposition or photolysis (to reform CH3C(O)O2) was modified to take its reformation through CH3C(O)O2 + NO2 into account (i.e. the rate coefficient or photolysis frequency of the channel leading to CH3C(O)O2 is multiplied by (1 − f)). | ||
Using the recently determined rate coefficient for (R5) (k5 ∼ 4 × 10−14 cm3 per molecule per s) and J-values based on evaluated PAA cross-sections and a photodissociation quantum yield of unity, we calculate that circa 86% of PAA formed early in biomass-burning plumes would survive transport to the point of measurement, with photolysis being by far the most import loss process. Note this does not consider wash-out of PAA in, e.g., rain-generating convection.
Fig. 6 shows that For FL04, thermal decomposition dominated the loss of PAN whereas photolysis was the dominant loss process for PAA. Similar figures for flights FL10, FL12 and FL13 are found in the ESI (Fig. S4 and S5)† with the fraction surviving transport listed in Table 1. In contrast to FL04, we see that thermal losses for PAN were less important for e.g. FL13 where the biomass-burning plume was intercepted at a higher altitude.
 | ||
| Fig. 6 Details of the loss processes for PAA and PAN during transport (using Hysplit back-trajectories) for flight FL04. R-OH + PAA was calculated from the model OH concentration and the temperature dependent expression for the OH + CH3C(O)OOH rate coefficient. J-PAN1 and J-PAN2 are the photolysis frequencies PAN via(R4a) and (R4b), respectively. k-PAN is the pressure and temperature dependent thermal decomposition rate coefficient for PAN, corrected for its reformation via recombination of CH3C(O)O2 and NO2. The percentage loss of both PAA and PAN (red and blue lines) is the same as in Fig. 5. | ||
The only loss process considered for CO was reaction with OH, whereby between 75 and 90% of initially formed CO would have survived transport.
| CO + OH (+O2) → CO2 + HO2 | (R7) |
Neglecting the formation of PAA during transport away from the biomass-burning area, we use a correction factor based on the surviving fraction of each trace gas during transport to calculate approximate values of MER0 for PAA relative to CH3CN and CO: the values obtained are listed in Table 1. As PAA and CO have similar lifetimes MER0(PAA/CO) values are similar to MER(PAA/CO), with the values increasing by a maximum of 22% for flight FL13. As CH3CN is not considered to be reactively removed to any significant extent during transport over such short time periods, the correction to the MER0(PAA/CH3CN) values is related to loss of PAA only and close to a factor of 1.3 to 1.4 for all flights except for FL04 (where the factor is 1.16).
The value of MER0(PAA/CO) can be compared to the PAA and CO measurements made by Yokelson et al.36 in biomass-burning plumes that were a few minutes old. They report an average value over three flights of MER0(PAA/CO) = (19 ± 10) × 10−5 (range = 13–26 × 10−5), which is at the lower end of our flight-to-flight variation in this parameter (range = 28–135 × 10−5). Using their single reported value of MER0(CO/CH3CN) = 232, we can also derive MER0(PAA/CH3CN) ∼0.04 from their data, which appears to be much lower than our values that lie in the range 0.26–1. Notwithstanding that we are dealing with potentially very different fuel types, some of the discrepancies between the extrapolated values of MER0(PAA/CO) reported in this work and those measured by Yokelson et al.36 are likely to be related to photochemistry during the transport of the biomass-burning plume. The poor agreement between the values of MER0(PAA/CH3CN) may reflect that the photochemical formation of PAA during transport results in a time-dependent increase in this parameter, leading to larger values in older plumes. This is consistent with the observation of Yokelson et al.,36 that the PAA mixing ratio increased with plume age. Such effects may be difficult to detect in the case of MER0(PAA/CO) as both PAA are products of the photochemical degradation of organic precursor trace gases abundant in biomass-burning-plumes.
4.3 PAA to PAN ratios and controlling factors
The MERs presented above indicate that the relative PAN to PAA enhancements in biomass-burning plumes is quite variable. Several factors control the relative formation and loss rates of PAN and PAA, both close to and remote from the biomass-burning region. As both PAN and PAA are formed from reactions of the peroxyacetyl radical (CH3C(O)O2), the relative abundance of HO2, NO and NO2 as the plume initially cools, dilutes and chemically ages will strongly influence the PAA-to-PAN ratio. Biomass with a higher nitrogen content will modify the NOx/HO2 ratio in favour of PAN formation.Initially, very close to the fire, O3 levels are very low,78 the NO-to-NO2 ratio is large, and neither PAN or PAA are formed as the CH3C(O)O2 radical is converted to CH3O2 (and CO2). However, within 10–20 min away from the fire photochemical O3 production has been observed.32
The gradual oxidation of NO to NO2 (e.g., via reactions with peroxy radicals) enables PAN to be formed, which, however, will be short-lived and decompose thermally back to CH3C(O)O2, which can react with HO2 to form PAA. At larger distances from the fire, as the plume rises and cools, the PAN lifetime will increase. In addition, the photochemical degradation of hydrocarbons released in the fire results in the formation of precursors of CH3C(O)O2 (notably CH3CHO and di-carbonyls79) so that both PAN and PAA will be formed during transport and the relative formation rates will depend on the relative abundance of HO2 and NO2.
The rate of initial vertical transport of the fresh plume depends on the type of biomass burning and the phase (smouldering or flaming) of the fire and has a strong impact on the PAN-to-PAA ratio both close to and distant from the source. Clearly, in biomass-burning impacted air masses, the absolute mixing ratios of PAN and PAA and the PAN/PAA ratio depend both on chemical processes that generate and destroy these trace gases as well as on meteorological considerations.
Fig. 7 plots the PAA mixing ratios versus those of PAN for flights FL04–FL08 and FL10–FL15. The PAA and PAN mixing ratios are weakly correlated (R = 0.48) with a slope of 0.1 ± 0.01. The weak correlation reflects the potentially different lifetimes of PAN depending e.g. on temperature when averaging over many flights where the data were taken at different altitudes. As shown in Fig. S5† the maximum daytime loss frequency for PAA (mainly photolysis) was about 1–2.5 × 10−6 s−1 resulting in a lifetime of ∼5–12 days. In contrast, the total PAN loss term was much more variable (between ∼2 × 10−6 and 5 × 10−5 s−1) resulting in lifetimes of 6 days (at high altitudes where only photolysis is important) to 6 hours at altitudes of 2–4 km. Below, we examine a subset of the PAA/PAN ratio in airmasses that were impacted by biomass-burning.
 | ||
| Fig. 7 Correlation between PAA and PAN for all CAFE-Africa flights. The red data are associated with air masses influenced by biomass burning (nBC > 20 cm−3). The encircled data (at ∼700 ± 100 ppt PAN) were from flight 10, which sampled biomass burning from high latitudes. | ||
Fig. 7 colour-codes the data according to whether the black-carbon particle concentration was above a threshold value of 20 particles per cm3, which is our indicator for air masses recently (i.e., within a few days) impacted by biomass burning. For this subset of the data (red data points), there is a cluster of measurements around a PAA/PAN slope close to one, indicating that PAA is more enhanced than PAN (by a factor of ∼10) in aged biomass-burning plumes compared to the background. As we discuss below, in aged biomass-burning plumes, the PAN-to-NOX ratio can be very large (and NOX very low), so further chemical processing of the air mass will favour PAA rather than PAN formation. Flight 10 appears to represent an exception as the PAA/PAN ratio, both with and without biomass-burning influence, is close to 0.1. As discussed above, there are many potential reasons for variability in the PAA-to-PAN ratio in aged biomass-burning impacted air masses, including differences in the fuel type (reactive nitrogen content) and rate of vertical transport (Western US/Canadian fires in flight 10 rather than Savannah fires in flights 4, 12 and 13). Also, wash-out of PAA in FL10 during convective transport is likely to have played a major role.
Insight into the variability of the PAN-to-PAA ratio can be gained by examining the (in situ) photochemical processes that lead to the formation of the acetyl-peroxy radical (CH3C(O)O2) and the factors that control whether this radical reacts to generate PAN or PAA.
While not intended to be exhaustive, the reaction scheme below shows the major routes to PAN and PAA and the known photochemical sinks (i.e., excluding deposition) of both trace gases:
| OH + CH3CHO → CH3C(O)O2 + H2O | (R8) |
| CH3C(O)CH3 + hν (O2) → CH3C(O)O2 + CH3O2 | (R9) |
| CH3C(O)O2 + NO2 + M → PAN + M | (R2) |
| PAN + M → CH3C(O)O2 + NO2 + M | (R3) |
| CH3C(O)O2 + NO → CH3O2 + NO2 + CO2 | (R10) |
| CH3C(O)O2 + RO2 → products | (R11) |
| CH3C(O)O2 + HO2 → PAA + O2 | (R1a) |
| → CH3C(O)OH + O3 | (R1b) |
| → OH + CH3O2 + CO2 | (R1c) |
| OH + PAA → CH3C(O)O2 + H2O | (R5) |
| PAN + hν → CH3C(O)O2 + NO2 | (R4a) |
| PAN + hν → CH3O2 + NO2 + CO2 | (R4b) |
| PAA + hν → CH3O2 + OH + CO2 | (R6) |
Within this simplified scheme, the PAN production rate (P-PAN) is given by:
| P-PAN = ([OH][CH3CHO]k8 + [CH3C(O)CH3]J9) × f | (1) |
| f = k2[NO2]/(k2[NO2] + k10[NO] + k1[HO2] + k11[RO2]) | (2) |
The terms ki and Ji are reaction rate coefficients or photolysis frequencies, respectively for reaction (Ri).
In a similar manner, the production rate of PAA (P-PAA) can be approximated by:
| P-PAA = ([OH][CH3CHO]k8 + [CH3C(O)CH3]J9 + [PAN]k3 + [PAN]J4a) × g | (3) |
| g = k1[HO2]α/(k2[NO2] + k10[NO] + k1[HO2] + k11[RO2]) | (4) |
The large PAA-to-PAN ratios represented by the biomass-burning impacted (red) datapoints in Fig. 7 can thus be explained by (1) an increase in PAA production terms combined with a decrease in PAN production terms as the air-mass ages and NOX is converted to PAN, (2) by the loss of PAN (and formation of PAA) as PAN-rich/NOX-poor air-masses descend to lower (warmer altitudes) which favour the thermal decomposition of PAN, or a combination of both. While the PAA/PAN ratio is to some extent controlled by the relative production term, an analysis of this ratio in order to extract NO2/HO2 ratios or HO2 levels as performed previously2 is difficult for biomass-burning impacted air masses of different plume age. We expect high PAA/PAN ratios in very freshly generated plumes at low altitudes in warm air where PAN is unstable, which will switch to lower ratios at higher altitudes as long as NOX levels are still high and PAN is generated. As the plume ages and NOX depletes, PAA formation will gain in importance and the ratio will be increased in time as PAN is photolyzed or the air mass descends and PAN is thermally lost. Analysis of the 4 biomass-burning impacted datasets revealed two different pairs of PAA/PAN ratios. These were 0.58 ± 0.09 and 0.58 ± 0.07 for flights 04 and 13 respectively. Lower values (0.1. ± 0.01 and 0.12 ± 0.02) were obtained for flights 10 and 12, respectively. These data are displayed as Fig. S7.† The larger PAA/PAN ratios observed during flights 04 and 13 suggest either that the plumes were more aged (i.e. PAN production is slow relative to PAA) or that the initial fuel contained a larger hydrocarbon-to-nitrogen ratio.
4.4 Partitioning of NOX to PAN
As described above, chemically aged, biomass-burning impacted air masses are expected to have converted significant amounts of initially abundant NOX to PAN or other long-lived NOX reservoirs such as HNO3 during transport. Fig. 8 plots the ratio of PAN to (PAN + NOX) mixing ratios and indicates that at altitudes of 5–10 km, where PAN mixing ratios maximise, the average value of PAN/(PAN + NOX) is >0.7. In order to avoid values >1 (caused, e.g., by NOX mixing ratios lower than the instrumental LOD), only data where NOX > 10 pptv was used for the analysis. At the highest altitudes (e.g., at 13–14 km), PAN/(PAN + NOX) decreases to approximately 0.4 ± 0.1, which indicates either less chemically aged air masses, the effects of enhanced rates of PAN photolysis at high-altitudes and/or the input of fresh NOX from the stratosphere. Similarly, at lower altitudes, low values of PAN/(PAN + NOX) were observed, which may result from the thermal decomposition of PAN to NOX following descent from higher altitudes. In air masses impacted by biomass-burning (red data points) the PAN/(PAN + NOX) ratio in the 4–10 km altitude range is very close to one. Clearly, the photochemical aging of biomass-burning impacted air masses seen over the west coast of Africa efficiently converts a very large fraction of the initially available NOX into PAN, implying that such air masses are sufficiently hydrocarbon-rich to be in the NOX-limited regime with respect to, e.g., photochemical O3 formation. This also implies that the PAA to PAN ratio will continue to increase in aging (hydrocarbon rich) biomass-burning impacted air masses in which, to a large extent, NOX has already been converted to PAN, but in which PAA production will continue.36 The sequestering of high concentrations of biomass-burning generated NOX in the form of PAN has implications for the formation of ozone in remote regions.80 Following descent of PAN rich air at high altitudes to the warm boundary layer NO2 and CH3C(O)O2 will be formed by the thermal decomposition of PAN. NO2 photolysis is the primary source of photochemical O3 and the CH3C(O)O2 radical will enhance the pool of peroxy radicals (largely CH3O2 and HO2 in remote regions) that can convert NO to NO2. | ||
| Fig. 8 Conversion of NOX to PAN in chemically aged air masses. The grey datapoints are from all flights where NOX and PAN data were available. The black datapoints are mean values (+1σ) for all flights averaged over 1 km altitude bins; the red datapoints are those used for the analysis of MER in biomass-burning impacted air masses. | ||
5 Conclusions
Correlations between PAN, PAA and tracers of biomass-burning during four flights over the tropical to North Atlantic west of Africa showed that PAA (like PAN) is present at enhanced levels in biomass-burning impacted air masses. For PAN, a wide range of molar enhancement ratios of 4–39 × 10−4 (relative to CO) and 0.4–3.9 (relative to CH3CN) were observed, in broad agreement with previous measurements over different regions. For PAA, values of 28–110 × 10−5 (relative to CO) and 0.2–0.7 (relative to CH3CN) were obtained. The more compact ranges in molar enhancement ratio for PAA compared to PAN reflect differences in loss-process and lifetime, with estimates of the fractional survival of PAN during transport from biomass-burning region to its measurement varying between 1 and 82% compared to 70–86% for PAA. PAN was a significant reservoir of NOX for all air masses encountered between 2 and 10 km where PAN/(PAN + NOX) was >0.5. For air masses in which PAN was strongly correlated with biomass-burning tracers, this value approached unity, indicating that most NOX initially present in the biomass-burning plume had been converted to PAN during transport. This NOX sequestration limits O3 formation and the reformation (recycling) of OH radicals after reaction with CO and VOCs. In these biomass-burning impacted air masses, the PAA-to-PAN ratio was also enhanced, a result of continued photochemical production of PAA even after PAN production stopped due to the lack of NOX and also the conversion of PAN to PAA when air-masses descend and PAN dissociates thermally to form the CH3C(O)O2 radical. This suggests that, in terms of photochemical processing to form, e.g., O3 or PAN, the biomass-burning-influenced air masses encountered during CAFE-Africa were NOX limited. The sensitivity of the PAN-to-PAA ratio to the initial nitrogen/organic mix in biomass-fires, the levels of HOx and NOX during transport as well as the altitude and temperature at which transport takes place make simulation of this ratio an ultimate test for global chemical models.Data availability
Data for this article (time series in ascii format of mixing ratios of PAN, PAA, NO, NO2, CO, CH3CN and black-carbon as well as aircraft location for each flight) are available at the Max-Planck repository [EDMOND] at [https://doi.org/10.17617/3.2F0J4Z, Edmond, V1].Author contributions
Conceptualization: JL, HF, JW. Formal analysis: JC. Funding acquisition: JL. Investigation: RD, PE, JC, IT, HF, AE, NW, BH, AP. Methodology: FH. Writing – original draft: JC. Writing – review and editing: JL, HF, JW, MP, UP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the HALO pilots and ground crew for their indispensable participation in the Café-Africa mission.Notes and references
- E. V. Fischer, D. J. Jacob, R. M. Yantosca, M. P. Sulprizio, D. B. Millet, J. Mao, F. Paulot, H. B. Singh, A. Roiger, L. Ries, R. W. Talbot, K. Dzepina and S. P. Deolal, Atmos. Chem. Phys., 2014, 14, 2679 CrossRef CAS PubMed.
- G. J. Phillips, N. Pouvesle, J. Thieser, G. Schuster, R. Axinte, H. Fischer, J. Williams, J. Lelieveld and J. N. Crowley, Atmos. Chem. Phys., 2013, 13, 1129 Search PubMed.
- H. B. Singh and P. L. Hanst, Geophys. Res. Lett., 1981, 8, 941 CrossRef CAS.
- H. B. Singh, Environ. Sci. Technol., 1987, 21, 320 CrossRef CAS PubMed.
- J. M. Roberts, Atmos. Environ., Part A, 1990, 24, 243 CrossRef.
- J. N. Crowley, N. Pouvesle, G. J. Phillips, R. Axinte, H. Fischer, T. Petäjä, A. Nölscher, J. Williams, K. Hens, H. Harder, M. Martinez-Harder, A. Novelli, D. Kubistin, B. Bohn and J. Lelieveld, Atmos. Chem. Phys., 2018, 18, 13457 Search PubMed.
- S. Z. He, Z. M. Chen, X. Zhang, Y. Zhao, D. M. Huang, J. N. Zhao, T. Zhu, M. Hu and L. M. Zeng, J. Geophys. Res.: Atmos., 2010, 115, D17307 Search PubMed.
- X. Zhang, Z. M. Chen, S. Z. He, W. Hua, Y. Zhao and J. L. Li, Atmos. Chem. Phys., 2010, 10, 737 Search PubMed.
- H. Liang, Z. M. Chen, D. Huang, Y. Zhao and Z. Y. Li, Atmos. Chem. Phys., 2013, 13, 11259 Search PubMed.
- M. Fels and W. Junkermann, Geophys. Res. Lett., 1994, 21, 341 Search PubMed.
- S. J. Walker, M. J. Evans, A. V. Jackson, M. Steinbacher, C. Zellweger and J. B. McQuaid, Atmos. Chem. Phys., 2006, 6, 5525 CrossRef CAS.
- S. Y. Wang, R. S. Hornbrook, A. Hills, L. K. Emmons, S. Tilmes, J. F. Lamarque, J. L. Jimenez, P. Campuzano-Jost, E. A. Nault, J. D. Crounse, P. O. Wennberg, M. Kim, H. Allen, T. B. Ryerson, C. R. Thompson, J. Peischl, F. Moore, D. Nance, B. Hall, J. Elkins, D. Tanner, L. G. Huey, S. R. Hall, K. Ullmann, J. J. Orlando, G. S. Tyndall, F. M. Flocke, E. Ray, T. F. Hanisco, G. M. Wolfe, J. St Clair, R. Commane, B. Daube, B. Barletta, D. R. Blake, B. Weinzierl, M. Dollner, A. Conley, F. Vitt, S. C. Wofsy, D. D. Riemer and E. C. Apel, Geophys. Res. Lett., 2019, 46, 5601 CrossRef PubMed.
- J. D. Crounse, K. A. McKinney, A. J. Kwan and P. O. Wennberg, Anal. Chem., 2006, 78, 6726 CrossRef CAS PubMed.
- K. R. Travis, C. L. Heald, H. M. Allen, E. C. Apel, S. R. Arnold, D. R. Blake, W. H. Brune, X. Chen, R. Commane, J. D. Crounse, B. C. Daube, G. S. Diskin, J. W. Elkins, M. J. Evans, S. R. Hall, E. J. Hintsa, R. S. Hornbrook, P. S. Kasibhatla, M. J. Kim, G. Luo, K. McKain, D. B. Millet, F. L. Moore, J. Peischl, T. B. Ryerson, T. Sherwen, A. B. Thames, K. Ullmann, X. Wang, P. O. Wennberg, G. M. Wolfe and F. Yu, Atmos. Chem. Phys., 2020, 20, 7753 CrossRef CAS PubMed.
- K. S. Docherty, W. Wu, Y. B. Lim and P. J. Ziemann, Environ. Sci. Technol., 2005, 39, 4049 CrossRef CAS PubMed.
- H. Wu, Y. Wang, H. Li, L. Huang, D. Huang, H. Shen, Y. Xing and Z. Chen, Atmos. Environ., 2017, 164, 61 Search PubMed.
- K. H. Bates, M. J. Evans, B. H. Henderson and D. J. Jacob, Geosci. Model Dev., 2024, 17, 1511 Search PubMed.
- P. J. Crutzen and M. O. Andreae, Science, 1990, 250, 1669 CrossRef CAS PubMed.
- M. O. Andreae and P. Merlet, Global Biogeochem. Cycles, 2001, 15, 955 CrossRef CAS.
- L. Giglio, I. Csiszar and C. O. Justice, J. Geophys. Res.: Biogeosci., 2006, 111, G02016 Search PubMed.
- E. Real, E. Orlandi, K. S. Law, F. Fierli, D. Josset, F. Cairo, H. Schlager, S. Borrmann, D. Kunkel, C. M. Volk, J. B. McQuaid, D. J. Stewart, J. Lee, A. C. Lewis, J. R. Hopkins, F. Ravegnani, A. Ulanovski and C. Liousse, Atmos. Chem. Phys., 2010, 10, 3027 CrossRef CAS.
- J. Williams, J. M. Roberts, F. C. Fehsenfeld, S. B. Bertman, M. P. Buhr, P. D. Goldan, G. Hubler, W. C. Kuster, T. B. Ryerson, M. Trainer and V. Young, Geophys. Res. Lett., 1997, 24, 1099 CrossRef CAS.
- J. M. Roberts, J. Williams, K. Baumann, M. P. Buhr, P. D. Goldan, J. Holloway, G. Hübler, W. C. Kuster, S. A. McKeen, T. B. Ryerson, M. Trainer, E. J. Williams, F. C. Fehsenfeld, S. B. Bertman, G. Nouaime, C. Seaver, G. Grodzinsky, M. Rodgers and V. L. Young, J. Geophys. Res.: Atmos., 1998, 103, 22473 CrossRef CAS.
- H. B. Singh, D. Herlth, D. Ohara, K. Zahnle, J. D. Bradshaw, S. T. Sandholm, R. Talbot, G. L. Gregory, G. W. Sachse, D. R. Blake and S. C. Wofsy, J. Geophys. Res.: Atmos., 1994, 99, 1821 CrossRef CAS.
- H. B. Singh, D. Herlth, R. Kolyer, R. Chatfield, W. Viezee, L. J. Salas, Y. Chen, J. D. Bradshaw, S. T. Sandholm, R. Talbot, G. L. Gregory, B. Anderson, G. W. Sachse, E. Browell, A. S. Bachmeier, D. R. Blake, B. Heikes, D. Jacob and H. E. Fuelberg, J. Geophys. Res.: Atmos., 1996, 101, 24203 CrossRef CAS.
- D. L. Mauzerall, J. A. Logan, D. J. Jacob, B. E. Anderson, D. R. Blake, J. D. Bradshaw, B. Heikes, G. W. Sachse, H. Singh and B. Talbot, J. Geophys. Res.: Atmos., 1998, 103, 8401 CrossRef CAS.
- M. J. Alvarado, J. A. Logan, J. Mao, E. Apel, D. Riemer, D. Blake, R. C. Cohen, K. E. Min, A. E. Perring, E. C. Browne, P. J. Wooldridge, G. S. Diskin, G. W. Sachse, H. Fuelberg, W. R. Sessions, D. L. Harrigan, G. Huey, J. Liao, A. Case-Hanks, J. L. Jimenez, M. J. Cubison, S. A. Vay, A. J. Weinheimer, D. J. Knapp, D. D. Montzka, F. M. Flocke, I. B. Pollack, P. O. Wennberg, A. Kurten, J. Crounse, J. M. St Clair, A. Wisthaler, T. Mikoviny, R. M. Yantosca, C. C. Carouge and P. Le Sager, Atmos. Chem. Phys., 2010, 10, 9739 CrossRef CAS.
- R. J. Pope, N. A. D. Richards, M. P. Chipperfield, D. P. Moore, S. A. Monks, S. R. Arnold, N. Glatthor, M. Kiefer, T. J. Breider, J. J. Harrison, J. J. Remedios, C. Warneke, J. M. Roberts, G. S. Diskin, L. G. Huey, A. Wisthaler, E. C. Apel, P. F. Bernath and W. Feng, Atmos. Chem. Phys., 2016, 16, 13541 Search PubMed.
- S. R. Arnold, L. K. Emmons, S. A. Monks, K. S. Law, D. A. Ridley, S. Turquety, S. Tilmes, J. L. Thomas, I. Bouarar, J. Flemming, V. Huijnen, J. Mao, B. N. Duncan, S. Steenrod, Y. Yoshida, J. Langner and Y. Long, Atmos. Chem. Phys., 2015, 15, 6047 Search PubMed.
- I. Bourgeois, J. Peischl, J. A. Neuman, S. S. Brown, H. M. Allen, P. Campuzano-Jost, M. M. Coggon, J. P. DiGangi, G. S. Diskin, J. B. Gilman, G. I. Gkatzelis, H. Guo, H. A. Halliday, T. F. Hanisco, C. D. Holmes, L. G. Huey, J. L. Jimenez, A. D. Lamplugh, Y. R. Lee, J. Lindaas, R. H. Moore, B. A. Nault, J. B. Nowak, D. Pagonis, P. S. Rickly, M. A. Robinson, A. W. Rollins, V. Selimovic, J. M. St. Clair, D. Tanner, K. T. Vasquez, P. R. Veres, C. Warneke, P. O. Wennberg, R. A. Washenfelder, E. B. Wiggins, C. C. Womack, L. Xu, K. J. Zarzana and T. B. Ryerson, Atmos. Meas. Tech., 2022, 15, 4901 CrossRef CAS.
- L. Xu, J. D. Crounse, K. T. Vasquez, H. Allen, P. O. Wennberg, I. Bourgeois, S. S. Brown, P. Campuzano-Jost, M. M. Coggon, J. H. Crawford, J. P. DiGangi, G. S. Diskin, A. Fried, E. M. Gargulinski, J. B. Gilman, G. I. Gkatzelis, H. Guo, J. W. Hair, S. R. Hall, H. A. Halliday, T. F. Hanisco, R. A. Hannun, C. D. Holmes, L. G. Huey, J. L. Jimenez, A. Lamplugh, Y. R. Lee, J. Liao, J. Lindaas, J. A. Neuman, J. B. Nowak, J. Peischl, D. A. Peterson, F. Piel, D. Richter, P. S. Rickly, M. A. Robinson, A. W. Rollins, T. B. Ryerson, K. Sekimoto, V. Selimovic, T. Shingler, A. J. Soja, J. M. St. Clair, D. J. Tanner, K. Ullmann, P. R. Veres, J. Walega, C. Warneke, R. A. Washenfelder, P. Weibring, A. Wisthaler, G. M. Wolfe, C. C. Womack and R. J. Yokelson, Sci. Adv., 2021, 7, eabl3648 CrossRef CAS PubMed.
- M. A. Robinson, Z. C. J. Decker, K. C. Barsanti, M. M. Coggon, F. M. Flocke, A. Franchin, C. D. Fredrickson, J. B. Gilman, G. I. Gkatzelis, C. D. Holmes, A. Lamplugh, A. Lavi, A. M. Middlebrook, D. M. Montzka, B. B. Palm, J. Peischl, B. Pierce, R. H. Schwantes, K. Sekimoto, V. Selimovic, G. S. Tyndall, J. A. Thornton, P. Van Rooy, C. Warneke, A. J. Weinheimer and S. S. Brown, Environ. Sci. Technol., 2021, 55, 10280 Search PubMed.
- G. I. Gkatzelis, M. M. Coggon, C. E. Stockwell, R. S. Hornbrook, H. Allen, E. C. Apel, M. M. Bela, D. R. Blake, I. Bourgeois, S. S. Brown, P. Campuzano-Jost, J. M. St. Clair, J. H. Crawford, J. D. Crounse, D. A. Day, J. P. DiGangi, G. S. Diskin, A. Fried, J. B. Gilman, H. Guo, J. W. Hair, H. S. Halliday, T. F. Hanisco, R. Hannun, A. Hills, L. G. Huey, J. L. Jimenez, J. M. Katich, A. Lamplugh, Y. R. Lee, J. Liao, J. Lindaas, S. A. McKeen, T. Mikoviny, B. A. Nault, J. A. Neuman, J. B. Nowak, D. Pagonis, J. Peischl, A. E. Perring, F. Piel, P. S. Rickly, M. A. Robinson, A. W. Rollins, T. B. Ryerson, M. K. Schueneman, R. H. Schwantes, J. P. Schwarz, K. Sekimoto, V. Selimovic, T. Shingler, D. J. Tanner, L. Tomsche, K. T. Vasquez, P. R. Veres, R. Washenfelder, P. Weibring, P. O. Wennberg, A. Wisthaler, G. M. Wolfe, C. C. Womack, L. Xu, K. Ball, R. J. Yokelson and C. Warneke, Atmos. Chem. Phys., 2024, 24, 929 CrossRef CAS.
- J. F. Juncosa Calahorrano, J. Lindaas, K. O'Dell, B. B. Palm, Q. Peng, F. Flocke, I. B. Pollack, L. A. Garofalo, D. K. Farmer, J. R. Pierce, J. L. Collett Jr, A. Weinheimer, T. Campos, R. S. Hornbrook, S. R. Hall, K. Ullmann, M. A. Pothier, E. C. Apel, W. Permar, L. Hu, A. J. Hills, D. Montzka, G. Tyndall, J. A. Thornton and E. V. Fischer, J. Geophys. Res.: Atmos., 2021, 126, e2020JD033484 Search PubMed.
- J. F. Juncosa Calahorrano, V. H. Payne, S. Kulawik, B. Ford, F. Flocke, T. Campos and E. V. Fischer, Geophys. Res. Lett., 2021, 48, e2021GL093405 Search PubMed.
- R. J. Yokelson, J. D. Crounse, P. F. DeCarlo, T. Karl, S. Urbanski, E. Atlas, T. Campos, Y. Shinozuka, V. Kapustin, A. D. Clarke, A. Weinheimer, D. J. Knapp, D. D. Montzka, J. Holloway, P. Weibring, F. Flocke, W. Zheng, D. Toohey, P. O. Wennberg, C. Wiedinmyer, L. Mauldin, A. Fried, D. Richter, J. Walega, J. L. Jimenez, K. Adachi, P. R. Buseck, S. R. Hall and R. Shetter, Atmos. Chem. Phys., 2009, 9, 5785 CrossRef CAS.
- J. Williams, J. Crowley, H. Fischer, H. Harder, M. Martinez, T. Petaja, J. Rinne, J. Back, M. Boy, M. Dal Maso, J. Hakala, M. Kajos, P. Keronen, P. Rantala, J. Aalto, H. Aaltonen, J. Paatero, T. Vesala, H. Hakola, J. Levula, T. Pohja, F. Herrmann, J. Auld, E. Mesarchaki, W. Song, N. Yassaa, A. Nolscher, A. M. Johnson, T. Custer, V. Sinha, J. Thieser, N. Pouvesle, D. Taraborrelli, M. J. Tang, H. Bozem, Z. Hosaynali-Beygi, R. Axinte, R. Oswald, A. Novelli, D. Kubistin, K. Hens, U. Javed, K. Trawny, C. Breitenberger, P. J. Hidalgo, C. J. Ebben, F. M. Geiger, A. L. Corrigan, L. M. Russell, H. G. Ouwersloot, J. V. G. de Arellano, L. Ganzeveld, A. Vogel, M. Beck, A. Bayerle, C. J. Kampf, M. Bertelmann, F. Kollner, T. Hoffmann, J. Valverde, D. Gonzalez, M. L. Riekkola, M. Kulmala and J. Lelieveld, Atmos. Chem. Phys., 2011, 11, 10599 CrossRef CAS.
- Y. Wang, Z. Chen, Q. Wu, H. Liang, L. Huang, H. Li, K. Lu, Y. Wu, H. Dong, L. Zeng and Y. Zhang, Atmos. Chem. Phys., 2016, 16, 10985 CrossRef CAS.
- G. M. Wolfe, T. F. Hanisco, H. L. Arkinson, D. R. Blake, A. Wisthaler, T. Mikoviny, T. B. Ryerson, I. Pollack, J. Peischl, P. O. Wennberg, J. D. Crounse, J. M. St. Clair, A. Teng, L. G. Huey, X. Liu, A. Fried, P. Weibring, D. Richter, J. Walega, S. R. Hall, K. Ullmann, J. L. Jimenez, P. Campuzano-Jost, T. P. Bui, G. Diskin, J. R. Podolske, G. Sachse and R. C. Cohen, Atmos. Chem. Phys. Discuss., 2021, 2021, 1 Search PubMed.
- I. Tadic, C. M. Nussbaumer, B. Bohn, H. Harder, D. Marno, M. Martinez, F. Obersteiner, U. Parchatka, A. Pozzer, R. Rohloff, M. Zöger, J. Lelieveld and H. Fischer, Atmos. Chem. Phys., 2021, 21, 8195 CrossRef CAS.
- R. Dörich, P. Eger, J. Lelieveld and J. N. Crowley, Atmos. Meas. Tech., 2021, 14, 5319 CrossRef.
- P. Warneck and T. Zerbach, Environ. Sci. Technol., 1992, 26, 74 CrossRef CAS.
- F. M. Flocke, A. J. Weinheimer, A. L. Swanson, J. M. Roberts, R. Schmitt and S. Shertz, J. Atmos. Chem., 2005, 52, 19 CrossRef CAS.
- A. L. Lazrus, G. L. Kok, J. A. Lind, S. N. Gitlin, B. G. Heikes and R. E. Shetter, Anal. Chem., 1986, 58, 594 CrossRef CAS.
- Z. Hamryszczak, A. Hartmann, D. Dienhart, S. Hafermann, B. Brendel, R. Königstedt, U. Parchatka, J. Lelieveld and H. Fischer, Atmos. Meas. Tech., 2023, 16, 4741 CrossRef CAS.
- L. H. Mielke and H. D. Osthoff, Int. J. Mass Spectrom., 2012, 310, 1 CrossRef CAS.
- Z. Hosaynali Beygi, H. Fischer, H. D. Harder, M. Martinez, R. Sander, J. Williams, D. M. Brookes, P. S. Monks and J. Lelieveld, Atmos. Chem. Phys., 2011, 11, 8497 Search PubMed.
- N. Wei, E. A. Marais, G. Lu, R. G. Ryan and B. Sauvage, Characterization of reactive nitrogen in the global upper troposphere using recent and historic commercial and research aircraft campaigns and GEOS-Chem, EGUsphere, 2024, vol. 2024, p. 1 Search PubMed.
- C. L. Schiller, H. Bozem, C. Gurk, U. Parchatka, R. Konigstedt, G. W. Harris, J. Lelieveld and H. Fischer, Appl. Phys. B: Lasers Opt., 2008, 92, 419 CrossRef CAS.
- I. Tadic, U. Parchatka, R. Konigstedt and H. Fischer, Appl. Phys. B, 2017, 123, 146 Search PubMed.
- B. A. Holanda, M. A. Franco, D. Walter, P. Artaxo, S. Carbone, Y. Cheng, S. Chowdhury, F. Ditas, M. Gysel-Beer, T. Klimach, L. A. Kremper, O. O. Krüger, J. V. Lavric, J. Lelieveld, C. Ma, L. A. T. Machado, R. L. Modini, F. G. Morais, A. Pozzer, J. Saturno, H. Su, M. Wendisch, S. Wolff, M. L. Pöhlker, M. O. Andreae, U. Pöschl and C. Pöhlker, Commun. Earth Environ., 2023, 4, 154 CrossRef.
- R. R. Draxler and G. D. Rolph, HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Trajectory) Model access via NOAA ARL READY Website, NOAA Air Resources Laboratory, Silver Spring, MD, 2011, http://ready.arl.noaa.gov/HYSPLIT.php Search PubMed.
- A. F. Stein, R. R. Draxler, G. D. Rolph, B. J. B. Stunder, M. D. Cohen and F. Ngan, Bull. Am. Meteorol. Soc., 2015, 96, 2059 CrossRef.
- E. Roeckner, R. Brokopf, M. Esch, M. Giorgetta, S. Hagemann, L. Kornblueh, E. Manzini, U. Schlese and U. Schulzweida, J. Clim., 2006, 19, 3771 CrossRef.
- P. Jöckel, H. Tost, A. Pozzer, C. Bruhl, J. Buchholz, L. Ganzeveld, P. Hoor, A. Kerkweg, M. G. Lawrence, R. Sander, B. Steil, G. Stiller, M. Tanarhte, D. Taraborrelli, J. Van Aardenne and J. Lelieveld, Atmos. Chem. Phys., 2006, 6, 5067 CrossRef.
- P. Jöckel, A. Kerkweg, A. Pozzer, R. Sander, H. Tost, H. Riede, A. Baumgaertner, S. Gromov and B. Kern, Geosci. Model Dev., 2010, 3, 717 CrossRef.
- A. B. M. Jeuken, P. C. Siegmund, L. C. Heijboer, J. Feichter and L. Bengtsson, J. Geophys. Res.: Atmos., 1996, 101, 16939 CrossRef.
- S. F. Reifenberg, A. Martin, M. Kohl, S. Bacer, Z. Hamryszczak, I. Tadic, L. Röder, D. J. Crowley, H. Fischer, K. Kaiser, J. Schneider, R. Dörich, J. N. Crowley, L. Tomsche, A. Marsing, C. Voigt, A. Zahn, C. Pöhlker, B. A. Holanda, O. Krüger, U. Pöschl, M. Pöhlker, P. Jöckel, M. Dorf, U. Schumann, J. Williams, B. Bohn, J. Curtius, H. Harder, H. Schlager, J. Lelieveld and A. Pozzer, Atmos. Chem. Phys., 2022, 22, 10901 CrossRef CAS.
- A. Pozzer, A. de Meij, K. J. Pringle, H. Tost, U. M. Doering, J. van Aardenne and J. Lelieveld, Atmos. Chem. Phys., 2012, 12, 961 CrossRef CAS.
- A. Pozzer, P. Zimmermann, U. M. Doering, J. van Aardenne, H. Tost, F. Dentener, G. Janssens-Maenhout and J. Lelieveld, Atmos. Chem. Phys., 2012, 12, 6915 CrossRef CAS.
- Y. Y. Yan, J. T. Lin, A. Pozzer, S. F. Kong and J. Lelieveld, Atmos. Environ., 2019, 213, 25 CrossRef CAS.
- S. F. Reifenberg, A. Martin, M. Kohl, Z. Hamryszczak, I. Tadic, L. Röder, D. J. Crowley, H. Fischer, K. Kaiser, J. Schneider, R. Dörich, J. N. Crowley, L. Tomsche, A. Marsing, C. Voigt, A. Zahn, C. Pöhlker, B. Holanda, O. O. Krüger, U. Pöschl, M. Pöhlker, P. Jöckel, M. Dorf, U. Schumann, J. Williams, J. Curtius, H. Harder, H. Schlager, J. Lelieveld and A. Pozzer, Atmos. Chem. Phys. Discuss., 2021, 2021, 1 Search PubMed.
- R. Deckert, P. Jockel, V. Grewe, K. D. Gottschaldt and P. Hoor, Geosci. Model Dev., 2011, 4, 195 CrossRef.
- J. A. de Gouw, C. Warneke, D. D. Parrish, J. S. Holloway, M. Trainer and F. C. Fehsenfeld, J. Geophys. Res.: Atmos., 2003, 108(D11), 4329 CrossRef.
- H. W. Bange and J. Williams, Atmos. Environ., 2000, 34, 4959 CrossRef.
- Y. Huangfu, B. Yuan, S. Wang, C. Wu, X. He, J. Qi, J. de Gouw, C. Warneke, J. B. Gilman, A. Wisthaler, T. Karl, M. Graus, B. T. Jobson and M. Shao, Geophys. Res. Lett., 2021, 48, e2020GL092322 CrossRef CAS.
- M. O. Andreae, Atmos. Chem. Phys., 2019, 19, 8523 CrossRef CAS.
- H. M. Royer, M. L. Pöhlker, O. Krüger, E. Blades, P. Sealy, N. N. Lata, Z. Cheng, S. China, A. P. Ault, P. K. Quinn, P. Zuidema, C. Pöhlker, U. Pöschl, M. Andreae and C. J. Gaston, Atmos. Chem. Phys., 2023, 23, 981 CrossRef CAS.
- B. A. Holanda, M. L. Pöhlker, D. Walter, J. Saturno, M. Sörgel, J. Ditas, F. Ditas, C. Schulz, M. A. Franco, Q. Wang, T. Donth, P. Artaxo, H. M. J. Barbosa, S. Borrmann, R. Braga, J. Brito, Y. Cheng, M. Dollner, J. W. Kaiser, T. Klimach, C. Knote, O. O. Krüger, D. Fütterer, J. V. Lavrič, N. Ma, L. A. T. Machado, J. Ming, F. G. Morais, H. Paulsen, D. Sauer, H. Schlager, J. Schneider, H. Su, B. Weinzierl, A. Walser, M. Wendisch, H. Ziereis, M. Zöger, U. Pöschl, M. O. Andreae and C. Pöhlker, Atmos. Chem. Phys., 2020, 20, 4757 CrossRef CAS.
- A. B. Collier and A. R. W. Hughes, J. Geophys. Res., 2011, 116, D05105 CrossRef.
- J. Williams, H. Fischer, S. Wong, P. J. Crutzen, M. P. Scheele and J. Lelieveld, J. Geophys. Res.: Atmos., 2002, 107, INX2–6 Search PubMed.
- C.-A. Papanikolaou, E. Giannakaki, A. Papayannis, M. Mylonaki and O. Soupiona, Sensors, 2020, 20(18), 5442 CrossRef CAS PubMed.
- V. Fiedler, F. Arnold, S. Ludmann, A. Minikin, T. Hamburger, L. Pirjola, A. Dornbrack and H. Schlager, Atmos. Chem. Phys., 2011, 11, 3211 CrossRef CAS.
- C. Jost, J. Trentmann, D. Sprung, M. O. Andreae, J. B. McQuaid and H. Barjat, J. Geophys. Res.: Atmos., 2003, 108(D13), 8482 CrossRef.
- R. Holzinger, J. Williams, G. Salisbury, T. Klüpfel, M. de Reus, M. Traub, P. J. Crutzen and J. Lelieveld, Atmos. Chem. Phys., 2005, 5, 39 CrossRef CAS.
- H. B. Singh, L. J. Salas, R. B. Chatfield, E. Czech, A. Fried, J. Walega, M. J. Evans, B. D. Field, D. J. Jacob, D. Blake, B. Heikes, R. Talbot, G. Sachse, J. H. Crawford, M. A. Avery, S. Sandholm and H. Fuelberg, J. Geophys. Res.: Atmos., 2004, 109, D15S07 Search PubMed.
- H. B. Singh, W. Viezee, Y. Chen, J. Bradshaw, S. Sandholm, D. Blake, N. Blake, B. Heikes, J. Snow, R. Talbot, E. Browell, G. Gregory, G. Sachse and S. Vay, Atmos. Environ., 2000, 34, 635 CrossRef CAS.
- P. V. Hobbs, P. Sinha, R. J. Yokelson, T. J. Christian, D. R. Blake, S. Gao, T. W. Kirchstetter, T. Novakov and P. Pilewskie, J. Geophys. Res.: Atmos., 2003, 108(D13), 8485 Search PubMed.
- K. J. Zarzana, K.-E. Min, R. A. Washenfelder, J. Kaiser, M. Krawiec-Thayer, J. Peischl, J. A. Neuman, J. B. Nowak, N. L. Wagner, W. P. Dubè, J. M. St Clair, G. M. Wolfe, T. F. Hanisco, F. N. Keutsch, T. B. Ryerson and S. S. Brown, Environ. Sci. Technol., 2017, 51, 11761 CrossRef CAS PubMed.
- I. Bourgeois, J. Peischl, J. A. Neuman, S. S. Brown, C. R. Thompson, K. C. Aikin, H. M. Allen, H. Angot, E. C. Apel, C. B. Baublitz, J. F. Brewer, P. Campuzano-Jost, R. Commane, J. D. Crounse, B. C. Daube, J. P. DiGangi, G. S. Diskin, L. K. Emmons, A. M. Fiore, G. I. Gkatzelis, A. Hills, R. S. Hornbrook, L. G. Huey, J. L. Jimenez, M. Kim, F. Lacey, K. McKain, L. T. Murray, B. A. Nault, D. D. Parrish, E. Ray, C. Sweeney, D. Tanner, S. C. Wofsy and T. B. Ryerson, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2109628118 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ea00006h |
| ‡ Now at: Leipzig Institute for Meteorology, Universität Leipzig, 04103 Leipzig, Germany and Experimental Aerosol and Cloud Microphysics Department, Leibniz Institute for Tropospheric Research, 04318 Leipzig, Germany.. |
| This journal is © The Royal Society of Chemistry 2025 |