
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKey role of short-lived halogens on global atmospheric oxidation during historical periods†
Adriana
Bossolasco
 ab,
Rafael P.
Fernandez
cd,
Qinyi
Li
e,
Anoop S.
Mahajan
f,
Julián
Villamayor
a,
Javier A.
Barrera
gh,
Dwayne E.
Heard
i,
Carlos A.
Cuevas
a,
Cyril
Caram
j,
Sophie
Szopa
j and
Alfonso
Saiz-Lopez
*a
ab,
Rafael P.
Fernandez
cd,
Qinyi
Li
e,
Anoop S.
Mahajan
f,
Julián
Villamayor
a,
Javier A.
Barrera
gh,
Dwayne E.
Heard
i,
Carlos A.
Cuevas
a,
Cyril
Caram
j,
Sophie
Szopa
j and
Alfonso
Saiz-Lopez
*a
aDepartment of Atmospheric Chemistry and Climate, Institute of Physical Chemistry Blas Cabrera, CSIC, Madrid, Spain. E-mail: a.saiz@csic.es
bPhysics Institute of Northwest Argentina, National Research Council (INFINOA-CONICET), National University of Tucumán (UNT), Tucumán, Argentina
cInstitute for Interdisciplinary Science, National Research Council (ICB-CONICET), Mendoza, Argentina
dSchool of Natural Sciences (FCEN), National University of Cuyo (UNCuyo), Mendoza, Argentina
eEnvironment Research Institute, Shandong University, Qingdao, China
fIndian Institute of Tropical Meteorology, Ministry of Earth Sciences, Pune, India
gResearch Institute for Physical Chemistry of Córdoba, National Research Council (INFIQC-CONICET), Córdoba, Argentina
hDepartment of Physical Chemistry, School of Chemical Sciences, National University of Córdoba (UNC), Córdoba, Argentina
iSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK
jLaboratoire des Sciences du Climat et de l'Environnement, LSCE/IPSL, CEA-CNRS-UVSQ, Université Paris-Saclay, Gif-sur-Yvette, France
First published on 19th February 2025
Abstract
Atmospheric oxidation largely determines the abundance and lifetime of short-lived climate forcers like methane, ozone and aerosols, as well as the removal of pollutants from the atmosphere. Hydroxyl, nitrate and chlorine radicals (OH, NO3 and Cl), together with ozone (O3), are the main atmospheric oxidants. Short-lived halogens (SLH) affect the concentrations of these oxidants, either through direct chemical reactions or indirectly by perturbing their main sources and sinks. However, the effect of SLH on the combined abundance of global oxidants during historical periods remains unquantified and is not accounted for in air quality and climate models. Here, we employ a state-of-the-art chemistry–climate model to comprehensively assess the role of SLH on atmospheric oxidation under both pre-industrial (PI) and present-day (PD) conditions. Our results show a substantial reduction in present-day atmospheric oxidation caused by the SLH-driven combined reduction in the global boundary layer levels of OH (16%), NO3 (38%) and ozone (26%), which is not compensated by the pronounced increase in Cl (2632%). These global differences in atmospheric oxidants show large spatial heterogeneity due to the variability in SLH emissions and their nonlinear chemical interactions with anthropogenic pollution. Remarkably, we find that the effect of SLH was more pronounced in the pristine PI atmosphere, where a quarter (OH: −25%) and half (NO3: −49%) of the boundary layer concentration of the main daytime and nighttime atmospheric oxidants, respectively, were controlled by SLH chemistry. The lack of inclusion of the substantial SLH-mediated reduction in global atmospheric oxidation in models may lead to significant errors in calculations of atmospheric oxidation capacity, and the concentrations and trends of short-lived climate forcers and pollutants, both historically and at present.
Environmental significanceIn this work, we used a state-of-the-art chemistry–climate model to evaluate the effect of short-lived halogens (SLH) on the combined abundance of global atmospheric oxidants in the pre-industrial (PI) and present-day (PD) atmospheres. The results show a significant global reduction in the PD levels of hydroxyl (OH), nitrate (NO3), and ozone (O3) concentrations. The simulations also show that the role of halogens on atmospheric oxidation was more prominent in the pristine pre-industrial atmosphere where a quarter of OH and half of NO3 concentrations in the boundary layer are accounted for by SLH. We conclude that atmospheric oxidation, particularly in the preindustrial atmosphere, cannot be fully understood without consideration of SLH emissions and chemistry. |
1 Introduction
The global abundance of atmospheric oxidants determines the lifetime of natural and anthropogenic chemicals emitted into the atmosphere. Hence, understanding the burden and budget of atmospheric oxidants is critical for tackling air quality and climate change.1,2 Indeed, the interplay of key atmospheric oxidants and other important reactive compounds such as carbon monoxide (CO), methane (CH4), and volatile organic compounds (VOCs) determines a large fraction of the tropospheric composition.3,4 The hydroxyl radical (OH) is the principal oxidant in the atmosphere.5 However, other tropospheric oxidants like ozone (O3), nitrate (NO3), and chlorine radicals (Cl) also influence the concentrations of crucial atmospheric components.5–10 By improving our quantitative understanding of the chemical processes regulating the sources and sinks of the main atmospheric oxidants, uncertainties in long-term trends and interannual variability of other compounds should thus decrease.11,12Accurately estimating the levels of these oxidants remains a significant challenge due to their short lifetime and the many factors influencing their production and loss; indeed, measuring these oxidants directly is difficult at large scales.13 Therefore, chemistry–climate models (CCMs) are valuable tools for understanding their temporal trends and spatial variability, although several uncertainties and shortcomings remain.14–16 This highlights the need to improve our understanding of the main chemical processes affecting the levels of these oxidants in the atmosphere.11,17,18 One factor attracting growing attention is the complex role of short-lived halogens (SLH) emitted from both natural and anthropogenic sources, as they play a critical role in tropospheric chemistry.8,19–24 SLH change the lifetime of methane, the HOx (hydrogen oxides – OH and HO2) and NOx (nitrogen oxides – NO and NO2) ratios, atmospheric mercury oxidation, and aerosol formation.8,20,25,26 SLH also lead to the depletion of tropospheric O3,21,27 which consequently impacts the primary production of OH and NO3.28,29 SLH represent a large source of Cl radicals in the atmosphere.30 Consequently, SLH alter the levels of atmospheric oxidants either directly (for example, through catalytic destruction of O3) or indirectly (for example, by changing the primary and secondary production of OH). The effects of SLH can be highly variable due to the spatial heterogeneity in emissions and the complex nonlinear atmospheric chemistry of reactive halogens. For instance, they can reduce OH levels in clean marine environments31,32 but increase OH production in polluted regions with high NOx and VOC emissions.33,34 Indeed, previous studies have reported global decreases in OH and O3,23,35 while others suggest regional enhancements of OH, O3, and Cl,21,34,36 highlighting the importance of quantifying the counteracting effects of SLH on the production and loss pathways of the main oxidants.
Measurements of Arctic and Alpine ice cores, as well as tree rings in Tibet, show that the emissions of natural iodine-containing SLH have increased by a factor of two to three since pre-industrial times37–39 due to a positive feedback mechanism between anthropogenic pollution and oceanic SLH emissions.40–42 Following past climate changes, the emissions of SLH have also shown considerable variability on paleo-climate timescales43,44 and ice cores also show an increase in bromine and chlorine during the industrial era.45,46
While previous studies have highlighted the role of SLH in specific regions or processes, a quantitative global evaluation of the impact of SLH on the combined abundance of atmospheric oxidants during historical periods has not yet been performed, which adds to the uncertainty in model projections of past and present climate. To address this critical gap, this study employs a global CCM – the Community Earth System Model (CESM), with a state-of-the-art SLH chemistry mechanism implemented in the Community Atmospheric Model with Chemistry (CAM-Chem) (see Methods). We conducted global simulations for both pre-industrial (PI, representative of the year 1850) and present-day (PD, representative of the year 2000) conditions with (wSLH) and without (noSLH) halogen chemistry and emissions to assess the role played by halogens on altering atmospheric oxidants levels. The results reveal that SLH substantially reduce atmospheric oxidation at present and even more in the pristine pre-industrial atmosphere. The reduction in atmospheric oxidant levels in turn results in an increase in the lifetime and loading of key air quality and climate-relevant chemical compounds in the atmosphere. These findings underscore the importance of considering the impact of SLH on atmospheric oxidants, and their associated implications for past and present air quality and climate, both in pristine and polluted regions, on regional and global scales.
2 Methods
2.1 CESM (CAM-Chem) model set-up and experiments design
The Community Earth System Model (CESM) version 1.1.1, with the Community Atmospheric Model with interactive chemistry (CAM-Chem) version 4.0,47 was used to explore the combined impact of SLH on global atmospheric oxidation at pre-industrial (PI) times and present-day (PD). The model extends from the surface to approximately 40 km (3.5 hPa in the upper stratosphere), following a hybrid sigma pressure coordinate with 26 vertical levels and has a horizontal resolution of 1.9° latitude × 2.5° longitude (96 × 144 grid points, respectively). This version of CAM-Chem contains a detailed representation of the tropospheric and stratospheric chemistry, which includes 169 species with both gas-phase and heterogeneous reactions coupled to the radiation module.48 Updates for the chemical processing of SLH include a state-of-the-art chemical mechanism for halogens in the troposphere and the stratosphere, which has been described in detail in previous studies.20,42,49–51 Further, we updated CAM-Chem with an improved chlorine emission mechanism from the lofting of iron-bearing mineral dust aerosols from the Sahara with sea spray aerosols (van Herpen et al., 2023).30 When these dust aerosols descend into the marine boundary layer (MBL) over the Atlantic, they mix with sea spray aerosol to form Mineral Dust-Sea Spray Aerosols (MDSA). Within MDSA, iron transforms into Fe(III) ions under specific atmospheric conditions, which then react with abundant chloride from sea spray to form Fe(III) 30 chlorides. These Fe(III) chlorides can be activated by sunlight, releasing Cl atoms that can combine to form Cl2 gas. Additionally, the iron component is regenerated, making the process catalytic.The MDSA mechanism was included in our model configuration with an extended vertical range beyond 900 hPa (van Herpen et al., 2023).30 Our model showed a MDSA-induced Cl2 production peaking around 850–900 hPa, with 2.2 × 10−13 kg s−1, decreasing significantly (<1.5 × 10−13 kg s−1) above 800 hPa. This indicates that the MDSA mechanism is most active in the lower troposphere, and its contribution to Cl2 production decreases significantly at higher altitudes.
Globally, our model results show a boundary layer (BL, 1000–850 hPa) MDSA-induced Cl burden of 15 Tg per year, which is comparable to the 13 Tg per year reported by van Herpen et al. (2023).30 This agreement suggests that our extended MDSA parameterization provides a reasonable estimate of global Cl production from this mechanism. While the MDSA mechanism can potentially operate at higher altitudes, the limited availability of observational data and the complexities of atmospheric chemistry at these altitudes limit our ability to accurately parameterize the process. Future research on atmospheric halogens should include improving the representation of MDSA in chemistry climate models.
The natural biogenic sources include nine halocarbons (CHBr3, CH2Br2, CH2BrCl, CHBr2Cl, CHBrCl2, CH3I, CH2I2, CH2IBr and CH2ICl) which are the result of micro- and macro-algae as well as phytoplankton metabolism coupled to photochemistry at the ocean's surface.50 Inorganic iodine (HOI and I2) is directly emitted from the ocean surface following O3 deposition on seawater and its reactions with aqueous iodide.40–42 The current chemical scheme includes the heterogeneous recycling of inorganic halogens reservoirs on SSA (sea-salt aerosols), for example, HOX and XONO2 (X = I, Br, Cl), which constitute a net source of chlorine and bromine.
Anthropogenic SLH sources are included following an emission inventory of the two dominant organic chlorine species (CH2Cl2 and C2Cl4),52 complemented by lower boundary conditions of other anthropogenic chlorinated substances (CHCl3, C2H4Cl2 and C2HCl3). In the PI simulation set-up, long-lived halocarbon lower boundary conditions (LBC) are zeroed, except for CH3Cl and CH3Br for which we assume the same natural contribution as for PD. Further we utilized a contemporary anthropogenic global emission inventory of reactive inorganic halogen species for 2014, adjusted to present-day conditions. This inventory encompassed inorganic chlorine (HCl and particulate chloride) from coal, biomass, and waste combustion, as well as inorganic bromine (HBr and Br2) and iodine (HI and I2) from coal combustion, following the methodology established in Saiz-Lopez et al., 2023.20
The standard anthropogenic emissions and biomass burning emissions for pollutants developed for the Chemistry–Climate Intercomparison Project (CCMI) (IPCC 2021) have been used here following Tilmes et al.48 The aircraft emissions of black carbon and nitrogen dioxide, as well as volcanic emissions of sulfur and sulfate, are vertically distributed. For the present-day CH4 emission, we have included an emissions-driven approach for CH4 instead of applying the standard lower boundary surface mixing ratios for long-lived species. The main CH4 sources include agriculture, landfill, fossil fuel industry, biomass and biofuel burning, and natural emissions from wetlands (see Li et al., 2022 (ref. 25) for further details) representative of year 2020. Biogenic emissions are calculated online throughout the land module using the Model of Emissions of Gases and Aerosols from Nature (MEGAN) version 2.1.53
The CAM-Chem simulations were conducted using a Specified Dynamics (SD) configuration,47 nudging the model towards high-frequency meteorological fields from a common year (nominally 2000) cyclically repeated across 20 years (obtained from a previous simulation that omitted the contribution of SLH54), ensuring consistent meteorological physics (e.g., transport, hydrology and BL fluxes) between the PI and PD simulations. This approach allows us to isolate the impact of changes in atmospheric composition and chemistry, particularly SLHs, while minimizing the influence of meteorological variability. In addition, the PI and PD simulations include different SST and sea ice conditions representative of each time period, in order to allow the model estimate changes in surface emissions that depend on these parameters. These conditions, also modulate MDSA-induced chlorine (Cl) production and the chemical partitioning between reactive and reservoir chlorine species within the online MDSA mechanism. Despite this, the resulting MDSA-induced Cl production is comparable in both the PI (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 079 Gg) and PD (15444 Gg) periods.
079 Gg) and PD (15444 Gg) periods.
All experiments were initialized from a previous simulation after allowing 40 years of spin-up to ensure all chemical species, particularly CH4, were stabilized. In both PI and PD conditions the long-lived halogens species (e.g., halons, CFCs, etc.) are included as LBC based on the A1 halogens scenario from the Scientific Assessment on Ozone Depletion Report.55
We conduct four sets of simulations with and without SLH for both periods PI and PD, Table 1 shows the configuration for each case together with the changes in surface flux emissions for the SLHCl, SLHI, SLHBr and VOCs, CH4 and CO. It is important to note that the anthropogenic and biogenic emissions other than SLH are identical within the noSLH, therefore the changes in atmospheric composition (CH4, NO3, O3, OH, etc.) between wSLH and noSLH represent the effect of SLH. All the simulations are running for a period of 5 years, 1850–1854 for PI and 2000–2004 for PD. We then used the average values of these 5 years for the analyses of our results. To isolate the effect of SLH emissions between PD and PI, we have used the same chemical mechanisms in both scenarios, therefore the observed differences in the results are primarily attributed to variations in SLH emission and other pollutant (like NOx) emissions in the different periods, which lead to distinct distributions and transformations of halogenated compounds in each scenario, such as he stronger impact of SLH in PI due to the lower NOx concentration than in PD.
| Cases | SLH emissions (Gg per year) | VOCs emission fluxs (Tg per year) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Surface flux halocarbons | Sea salt recycling* inorganic halogens | Oceanic emissions | Acid-displacement* | Cl from MDSA* | VOCs | CH4 | CO | ||||
| SLHCL | SLHBr | SLHI | SLHCl | SLHBr | I2/HOI | SLHCl | Cl2 | ||||
| a noSLH_PI, standard chemical scheme without SLH sources. b wSLH_PI, only natural SLH emissions for pre-industrial. c noSLH_PD standard chemical scheme without SLH sources. d wSLH_PD, natural plus anthropogenic SLH sources from present-day conditions. | |||||||||||
| noSLH_PIa | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 782 | 189 | 459.5 |
| wSLH_PIb | 60.9 | 595.7 | 586.4 | 2483 | 1578 | 898 | 2090 | 15079 |
782 | 189 | 459.5 |
| noSLH_PDc | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 828 | 499 | 1142 |
| wSLH_PDd | 849.7 | 616.9 | 591.7 | 2136 | 2750 | 1875 | 14214 |
15444 |
828 | 499 | 1142 |
The percentage of changes of each scenario (wSLH_PI or wSLH_PD) is presented relatively to the corresponding noSLH scenario as follow:
| % Δ(wSLH–noSLH) = ((wSLH_X − noSLH_X)/noSLH_X) × 100%. |
The global tropospheric OH, O3, NO3 and Cl burden from the different sources and sinks is computed from the surface to the tropopause considering all latitudes (90° N–90° S) and longitudes (0°–360°).
The global annual mean surface flux (SF) source strength for SLH is computed as follows: SLHCl = CH2Cl2 + C2Cl4 + CH2BrCl + CH2ICl + CHBrCl2 + CHBr2Cl; SLHBr = CHBr3 + CH2Br2 + CH2BrCl + CH2IBr + CHBrCl2 + CHBr2Cl; SLHI = CH3I + CH2I2 + CH2ICl + CH2IBr. Oceanic emission of inorganic iodine (HOI/I2) are computed online and depends on near-surface O3 concentration, wind speed and SST (Prados-Roman et al., 2015 (ref. 42)). Surface flux for the VOCs = ISOPRENE + C10H16 + CH3OH + C2H5OH + CH2O+ CH3CHO + CH3COOH + CH3COCH3 + HCOOH + C2H2 + C2H4 + C2H6 + C3H8 + C3H6 + BIGALK + BIGENE + MEK + TOLUENE, including it corresponding biogenic emissions form MEGAN for ISOPRENE + C10H16 + CH3OH + C2H5OH + CH2O+ CH3CHO + CH3COOH + CH3COCH3 + HCOOH + CO + C2H4 + C2H6 + C3H8 + C3H6 + BIGALK + BIGENE + MEK + TOLUENE.
• Halogen heterogeneous recycling at the boundary layer on Seas Salt Aerosols (SSA) are calculated from the following reactions:
| BrONO2 → 0.65 × Br2 + 0.35 × BrCl | (R1) |
| BrNO2 → 0.65 × BR2 + 0.35 × BrCl | (R2) |
| HOBr → 0.65 × BR2 + 0.35 × BrCl | (R3) |
| ClONO2 → 1 × Cl2 | (R4) |
| ClNO2 → 1 × Cl2 | (R5) |
| HOCl → 1 × Cl2 | (R6) |
| IONO2 → 0.5 × IBr + 0.5 × ICl | (R7) |
| INO2 → 0.5 × IBr + 0.5 × ICl | (R8) |
| HOI → 0.5 × IBr + 0.5 × ICl | (R9) |
• Acid-displacement heterogeneous reactions on SSA:
| HNO3 → HCl | (R10) |
| N2O5 → ClNO2 + HNO3 + N2O5 | (R11) |
The total mass of inorganic halogens emitted by each source is computed by considering the halogen atomicity of each species and the halogen mass.
• Cl production from Mineral Dust-Sea Spray Aerosols (MDSA) at the boundary layer.
Note that MDSA and acid displacement only release chlorine. In addition, during PD, values from Cl Acid-displacement are equivalent to values from MDSA. During PI, acid displacement is smaller and MDSA Cl production becomes more important.
2.2 OH recycling probability
The so-called recycling probability “r” represents the probability that OH, once formed, is recycled. The calculation of r follows the definition of Lelieveld et al., 2002:56 | (1) |
In eqn (1), the sum of primary (P) and secondary (S) sources of OH (see Table 2) is the “oxidation power”, or the time rate at which OH is produced (gross OH formation (G)) P and S (Tmol per year) was calculated as follows:
 | (2) |
| Sources (Tmol per year) | noSLH_PD | wSLH_PD | noSLH_PI | wSLH_PI |
|---|---|---|---|---|
| Primary | ||||
| O1D + H2O | 87 (47%) | 71 (41%) | 53 (50%) | 41 (43%) |
|
||||
| Secondary | ||||
| NOx + HO2 | 59 (31%) | 56 (32%) | 31 (29%) | 28 (29%) |
| O3 + HO2 | 22 (12%) | 17 (10%) | 11 (11%) | 8 (8.5%) |
| H2O2 + hν | 14 (8%) | 14 (8%) | 8 (8%) | 8 (8%) |
| VOCs, ROOH + hν | 5.0 (2.7%) | 5.3 (3.0%) | 3.4 (3.1%) | 3.5 (3.6%) |
| HOX + hν | — | 11.0 (6%) | — | 7.5 (8%) |
| Others | 0.18 (0.1%) | 0.2 (0.09%) | 0.08 (0.07%) | 0.07 (0.07%) |
| Total secondary | 100 (53%) | 103 (59%) | 54 (50%) | 56 (57%) |
| G (P + S) | 187.2 | 173.7 | 107.3 | 97.02 |
| Recycling probability, r (%) | 53 | 59 | 50 | 57 |
2.3 CESM model evaluation
Our model configuration is based on the well-established CESM1.2 (CAM-Chem) with SLH framework, which has been extensively validated against a wide range of ground, ship, aircraft and satellite-based observations over the past 20 years, particularly in the context of halogen chemistry and key atmospheric species such as O3, CH4, and HCl. This is revised in Saiz-Lopez et al. (2023)20 based on numerous previous studies of the group.21,22,49,50,57,58 Specifically, our model configuration is based in the configuration of Saiz-Lopez et al., 2023 (ref. 20) and incorporates the latest advancements in halogen chemistry, including the MDSA mechanism from van Herpen et al., 2023.30 This allows us to provide a more comprehensive representation of halogen-mediated atmospheric processes and their impact on climate.Briefly, Ordóñez et al. 2012,50 validate the natural oceanic sources of short-lived halocarbons within CESM1 (CAM-Chem) with data collected from near-surface and aircraft campaigns conducted in extra-polar regions, incorporating additional observations of reactive short-lived halogens and ozone in tropical regions to further validate the model. Furthermore, Fernandez et al., 2014,49 Fernandez et al., 2017 and Saiz-Lopez and Fernandez 2016 (ref. 57 and 59) demonstrated the model's accurate representation of bromine transport and its improvement in the total O3 column and the Antarctic ozone hole. Barrera et al., 2020 (ref. 54) showed the model's ability to reproduce observed stratospheric halogen levels. Additionally, the implementation of iodine chemistry in CESM (CAM-Chem)51 allowed reproducing aircraft observations in the tropical upper tropopause, suggesting the occurrence of iodine-driven stratospheric O3 depletion, which was later confirmed by Koenig et al., 2020.60 Prados Roman et al., 2015 (ref. 42) demonstrated the need to consider an inorganic iodine source from the ocean surface to accurately reproduce the observed iodine oxide mixing ratios over the open ocean. Cuevas et al., 2022 (ref. 58) reported the improved model performance of stratospheric O3 by including reactive iodine chemistry into the CESM/WACCM4-SD configuration. Furthermore, Li et al., 2022 (ref. 25) validated the model's representation of key atmospheric species, including ozone, methane, as well as for the global sea-salt aerosol abundance in the marine boundary layer compared with global observational results. To summarize, these studies, provide strong evidence for the model's ability to accurately simulate halogen chemistry.
In addition to these core validations, our model incorporates the latest advancements in halogen chemistry, including the Cl atoms' photochemical production from Sahara dust (MDSA mechanism) from van Herpen et al., 2023.30 This mechanism has been initially validated using 13C depletion in CO in air samples from Barbados.61 In the ESI,† we added more details about the contribution of Cl production from MDSA and comparison to observation (see Section 1 in the ESI†).
Here we provide further evaluation of the model results for OH burdens production (Table S4†). OH is a short-lived species, making its direct measurement challenging. However, our model simulations of OH burden are in the range of the reported values using other model simulations.9,56 Indeed, our simulated global tropospheric OH concentration in the noSLH_PD and wSLH_PD cases are 1.06 × 106 molec. per cm3 and 9.28 × 105 molec. per cm3, respectively, and are in agreement with the averaged value reported by observation and previous modeling studies ∼1.0 × 106 molec. per cm3 for the 21st century.9,12,18 This agreement provides confidence in the model's ability to represent the complex interactions between OH and other atmospheric species. ESI Table S5† shows that our tropospheric O3 and CH4 budgets in PI and PD with and without reactive halogens are in agreement with the values reported by other model studies.27,62
3 Results and discussion
3.1 Changes in global atmospheric oxidants due to halogens
SLH lead to the release of halogen radicals (i.e., Cl, Br and I atoms and ClO, BrO and IO oxides) on photolysis, which can react with numerous species in the atmosphere, leading to a change in the O3, OH, NO3 and Cl burdens and their distribution, which in turn affect the lifetime of many key atmospheric constituents. Bromine and iodine predominantly lead to the destruction of O3, and this reduction is more pronounced over the oceans, which are the largest bromine- and iodine-SLH sources. Chlorine can also lead to ozone destruction; however, in the presence of VOCs and NOx, it can also lead to the formation of O3. Our results show that SLH reduce tropospheric O3 by 20% in PD and 21% in PI, respectively (Fig. 1a). The simulations show a larger reduction in ozone in the PD (20%) compared to previous studies22,25,27 (12–16%). This difference results from the recent inclusion of additional chlorine sources from dust30 in our model (see Methods for details). During PD, the net integrated tropospheric O3 burden is reduced from 330 Tg (noSLH) to 260 Tg (wSLH). Despite the total ozone burden being much lower in the PI period (noSLH: 230 Tg), the relative reduction due to SLH chemistry (wSLH: 180 Tg, 21%) is similar to PD. | ||
| Fig. 1 Global integrated tropospheric burden of main oxidants (OH, O3, NO3, and Cl) and their relative changes due to the inclusion of SLH in the present-day (PD) and pre-industrial (PI) atmospheres. Filled bars represent the wSLH case, hatched bars the noSLH case, and crosshatch bars the absolute differences between them. The relative percentage differences for PD and PI times are indicated above the bars in bold and are calculated as follow: % Δoxidant = [oxidant]wSLH − [oxidant]noSLH/[oxidant]noSLH × 100%. (a) Changes for O3 (Tg), (b) changes for OH (Mg), (c) changes for NO3 (Gg) and (d) changes for Cl (Mg). | ||
O3 photolysis is the main source of OH in the troposphere56 and hence the SLH-driven ozone destruction indirectly reduces the tropospheric OH burden by 13% in the PD (wSLH: 181 Mg and noSLH: 210 Mg; Fig. 1b). This value is larger than the 8.2% decrease reported by Sherwen et al.28 and the 4.5% decrease reported by Stone et al.35 This is because these studies did not account for sea-salt debromination and dust sources of Cl, which represent a significant fraction of the tropospheric halogen burden (see Table 1). Additionally, the study by Stone et al.35 did not include iodine chemistry which is the dominant halogen leading to tropospheric O3 reduction.23 The reduction in global OH due to SLH increases from 13% during PD to 17% during PI (i.e., reducing the tropospheric burden from 213 Mg (noSLH) to 177 Mg (wSLH)). The production and loss pathways controlling the OH levels in PI and PD are detailed in the next section.
NO3 radicals, the main atmospheric oxidant at night, are mainly formed by reaction of ozone with NO2, therefore SLH reduce NO3 formation primarily by depleting ozone and titrating NO2 to form reservoir halogen nitrates and nitrites.8 Similar to O3 and OH, NO3 reduction caused by SLH is more pronounced during the PI than in the PD (Fig. 1c), primarily due to slightly larger ozone depletion and lower NO2 abundance in the pristine preindustrial atmosphere, reducing the titration of reactive SLH to reservoir species. During PD, halogen chemistry reduces the net integrated tropospheric NO3 burden from 6.78 Gg (noSLH) to 4.33 Gg (wSLH; 36% reduction). While the NO3 levels are much lower in the PI period (noSLH: 2.44 Gg), as expected due to the absence of anthropogenic NOx emissions, the relative reduction due to SLH chemistry is larger (wSLH: 1.4 Gg; 43% reduction).
Considering that SLH are a direct source of Cl atoms, an increase in the Cl burden is expected in both PD and PI (Fig. 1d). Integrated throughout the troposphere, the Cl burden increases from 0.21 Mg (noSLH) to 1.87 Mg (wSLH; 778% increase) in the PD, while the increase is much higher in PI going from 0.20 Mg (noSLH) to 2.38 Mg (wSLH; 1070% increase). Note that the small fractions of chlorine present in the noSLH arise from the slow degradation of long-lived chlorocarbons, as some of them such as methyl chloride have natural sources. However, if we consider the total chlorine family (Cly), which includes all reactive and reservoir chlorine species (see Methods), the Cly tropospheric abundance is larger during PD (from 32 Gg (noSLH) to 302 Gg (wSLH); 833% increase) compared to PI (from 17 Gg (noSLH) to 200 Gg (wSLH); 1047% increase). While both Cl and Cly exhibit significant increases in both periods, the relative increase in Cl is larger in the PI compared to the PD simulation. This is due to the coupled interplay between direct emissions of SLHs and the chemical partitioning of chlorine species. This shift in partitioning, driven by higher NOx and HOx levels in the PD from anthropogenic emissions54 reducing the X/Xy ratios (X = Cl, Br, I) (Table S1†), leads to a greater fraction of chlorine being stored in unreactive reservoir species, reducing the available Cl atoms. Thus, despite the larger increase in total Cl emissions in the PD compared to PI (see Table 1), the net effect on Cl abundance is more significant in the PI simulation.
Note that the SLH-mediated percentage changes in oxidant burdens within the BL (1000–850 hPa) are higher than in the integrated troposphere (Fig. S2†). The reductions in oxidants in the BL are OH: −16% and −25%; O3: −26% and −32%; NO3: −38% and −49% for PD and PI, respectively, while an increase of Cl radicals of 2632% and 3114%, while an increase of Cl radicals of 2632% and 3114% are observed in the PD and PI, respectively.
3.2 Spatial heterogeneity in the influence of SLH on oxidants
The regional impact of SLH on atmospheric oxidants is highly dependent on natural and anthropogenic emissions and their chemical processing, which depend on the local environment. SLH can lead to both the lowering of OH and O3 in remote, clean environments and an increase in OH production in polluted continental regions by increasing O3 formation through the oxidation of VOCs and forming OH through the photolysis of hypohalous acids (HOX; X = Cl, Br, I). At a global scale, SLH lead to large decreases in OH, O3 and NO3 over the oceans (Fig. 2). Present-day reductions of up to 40% in OH are observed over the Atlantic Ocean and in the Southern Ocean boundary layer. The average OH change over the global oceans is −19% at present. Comparatively, some locations over land show increased OH production due to the SLH reaction with pollutants and HOX photolysis. Therefore, although OH is globally reduced due to the catalytic loss of O3 by SLH, the decrease is smaller integrated over the land (PD: −7%; Table S2†). This is clearly observed in the northern mid-latitudes, where in some regions, anthropogenic sources of SLH also cause an increase in the secondary production of OH, leading to an increase in OH over the continents (Fig. 2a–c). | ||
| Fig. 2 Spatial distribution of relative changes in oxidant concentrations (OH, O3, NO3, and Cl) due to the inclusion of SLH in the boundary layer (1000–850 hPa) for the present-day (PD) and pre-industrial (PI) times. The color scale indicates the percentage change in each oxidant relative to the noSLH case. (a)–(c) Show the relative differences for OH in the PD, PI and the changes from PI to PD (wSLH scenarios); (d)–(f) show the changes for O3 in the PD, PI and the changes from PI to PD (wSLH scenarios); (g)–(i) show the relative changes for NO3 in the PD, PI and the changes from PI to PD (wSLH scenarios); and (j)–(l) show the relative changes for Cl radicals in the PD, PI due to SLH and the changes from PI to PD (wSLH scenarios), respectively; note that the color scale for the change in Cl is different due to the percentage changes for the Cl radical are positive and greater than 100%. (Panel l) Shows that the change from PI to PD are negative, indicating that Cl radicals are higher in pre-industrial periods. | ||
The effect of SLH on OH is even more pronounced in the pre-industrial atmosphere. The overall reduction in the global BL over oceans is −29% in the PI (compared to −19% in the PD). This is also true over the land, with a change of −13% (−7% in the PD). SLH lead to the catalytic loss of O3 throughout the global BL. O3 losses of up to 45% are observed over the northern Atlantic Ocean in the PD and are more than 50% in certain areas during the PI period (Fig. 2). Averaged in the global BL, the O3 reduction over the oceans (PD: −29% and PI: −34%) is larger as compared to over the land (PD: −20% and PI: −28%) (Table S2†). The smaller reduction in O3 over land compared to the ocean can be attributed to several factors, such as higher NOx emissions, specially over land areas. These relatively higher NOx levels can titrate reactive halogen species, limiting their potential to deplete ozone. On the other hand, as discussed in Barrera et al., 2023,27 the iodine-ozone feedback mechanism, particularly in the Western Pacific Warm Pool, plays a relevant role in oceanic ozone depletion. This mechanism is less prominent over land due to much lower iodine emissions.
The open, clean ocean regions also show the largest NO3 relative changes (up to 90%). The reduction is more pronounced in the northern Atlantic and the Southern Oceans in PD, while during the PI period, the largest changes are seen in the Southern, Atlantic and Northern Oceans. The change over the oceans (PD: −44% and PI: −59%) is larger than over the land (PD: −22% and PI: −38%) (Table S2†), similar to OH and O3. These results show that the levels and distribution of global NO3 radicals, cannot be understood without consideration of the crucial impact of SLH chemistry.
In the case of Cl, as expected, the inclusion of SLHs leads to significant increases in Cl concentrations in both the PI and PD simulations, particularly in regions with abundant dust and sea salt aerosols, such as the Atlantic Ocean. Continental regions also exhibit an increase in Cl radical, which is attributed to anthropogenic SLHCl emissions (Fig. 2j and k). However, while Cl concentrations increase in both the PI and PD periods, the relative increase of atomic Cl is more pronounced in PI. This behavior is primarily attributed to the interplay between SLH emissions and NOx concentrations. In the PI simulation, lower NOx levels favor the formation of reactive chlorine species, leading to a more pronounced Cl increase than in PD. Conversely, higher NOx levels in the PD simulation promote the formation of reservoir species like ClNO2 and ClONO2, reducing the active Cl budget. Consequently, despite the potential for increased Cl production due to SLH emissions, the dominant NOx-driven titration reactions ultimately result in lower Cl concentrations in the PD compared to the PI (Table S1†).
Vertically, the effect of SLH in OH, O3 and NO3 is highest in the BL and reduces gradually up to 300 hPa (Fig. 3). Large effects are seen in the polar regions, where emissions of SLH peak, particularly in springtime. Above 300 hPa, the effects of SLH are more marked in the equatorial regions, where large-scale convective systems transport SLH to the upper troposphere (Fig. 3). The effect of SLH on Cl differs from OH, O3 and NO3, peaking in the lower tropical troposphere, which arise mainly from dust-induced Cl production,30 aerosol recycling,63 and biomass burning.64,65 In the upper troposphere, Cl is elevated by the photodegradation of relatively longer-lived anthropogenic SLHCl, like dichloromethane and chloroform24,52 (Fig. 3).
 | ||
| Fig. 3 Vertical distribution of relative changes in oxidant concentrations (OH, O3, NO3, and Cl) due to the inclusion of SLH in the present-day (PD) and pre-industrial (PI) times. The color scale indicates the percentage change in each oxidant relative to the noSLH case. Panels from top to bottom shows the relative change for OH, O3, NO3 and Cl. Decreases are observed for OH, O3 and NO3, but Cl radicals show a large increase throughout the vertical column. Note that the color scale applied for ΔCl (%) is different. The vertical profile Δglobal average (%) for the last panel is calculate as the difference between the global average in the PDwSLH–noSLH and PIwSLH–noSLH. | ||
Due to the large spatial heterogeneity in the effect of SLH on oxidants, the resulting atmospheric oxidation capacity shows differences on regional scales. Li et al.34 have reported an increase in atmospheric oxidation due to SLH over China, particularly during the daytime due to enhanced OH and Cl radical production. This is also consistent with Dai et al.,66 who found that OH reactions with VOCs and CO account for most daytime oxidation capacity and the study by Chen et al.,67 who reported an increase in oxidation due to halogens in the Yangtze River Delta, with OH increasing by up to 16% and O3 increasing by 2%. Our results are consistent with this spatial heterogeneity while showing a global decrease in the combined concentration of atmospheric oxidants.
These findings underscore the spatial heterogeneity in the halogen-induced oxidation changes, showing that while regional models in polluted areas consistently indicate oxidation enhancements, the effects can be contrasting between different pristine, semi-polluted and polluted environments. This highlights the complexity and the need to incorporate detailed halogen emissions and chemistry in climate and air quality models to more accurately capture atmospheric oxidation from the regional to the global scale.
3.3 Influence of halogens on the chemical pathways for OH formation
Given OH dominates the oxidation of VOCs and pollutants in the troposphere, now we turn to quantify the SLH-driven changes on its major sources and sinks. The photolysis of O3 at wavelengths lower than 330 nm leads to the formation of O1D, which then reacts with water vapour to form OH. This is known as the primary production of OH (P). In addition, several reactions lead to the formation of OH through subsequent reactions involving VOCs and NOx, known as secondary sources of OH (S).9 Secondary production can in turn be subdivided into contributions by the so-called NOx mechanism (NOx + HO2), the Ox mechanism (O3 + HO2 and H2O2 + hv), the recycling of OH by the chemistry of VOCs, OVOCs and ROOH, as well as the OH production due to the photolysis of HOX (Table 2).In the absence of SLH, present-day P accounts for 87 Tmol per year (noSLH) of the OH production, similar to Lelieveld et al.9 (84 Tmol per year). In the absence of anthropogenic ozone pollution, P accounts for 53 Tmol per year (noSLH) in the PI atmosphere. When SLH are included, tropospheric P decreases by 18% in the PD and 22% in the PI, while total S remains almost identical (+3% and +4%, respectively; see Table 2).
Regionally, the most substantial reductions in P due to SLH occur in the tropics, where P dominates the total OH production (Fig. 4a and c). P is also the dominant OH source up to approximately 500 hPa in the absence of SLH. When SLH are included, P still remains dominant up to 600 hPa, beyond which S becomes more important (Fig. S3†). Fig. S4† presents an expanded analysis of changes in P and S across the selected regions (Polluted Regions: China (20–40° N, 100–125° E); Semi-Polluted Regions: South America (10–30° S, 70–40° W); Clean Regions: Open Ocean (0–30° N, 180–120° W)), showing that in clean environments, the reaction O1D + H2O account for ∼74% of the OH formation.
 | ||
| Fig. 4 Spatial distribution of changes in primary (P) and secondary (S) OH production (molecules cm−3 s−1) due to the inclusion of SLH in the boundary layer (1000–850 hPa) for the present-day (PD) and pre-industrial (PI) times. (a) and (b) Shows the changes in the primary and secondary OH production due to SLH for present day, (c) and (d) shows the changes in the primary and secondary OH production due to SLH for pre-industrial times. PwSLH–noSLH = P(wSLH)PD/PI − P(noSLH)PD/PI. SwSLH–noSLH = S(wSLH)PD/PI − S(noSLH)PD/PI. Note that SLH lead to a decrease in primary OH production in both PD and PI, with more pronounced effects in the tropical and subtropical regions, while increase secondary OH production, particularly through the photolysis of HOX. | ||
S slightly increases from 100 Tmol per year (noSLH) to 103 Tmol per year (wSLH) in the PD, presenting an almost identical vertical profile for the different sensitivities (Fig. S2†). The photolysis of HOX contributes approximately 6% to boundary layer S in the PD (Table 2), increasing to 14% in the upper troposphere (Fig. S5†). S values in the noSLH case (100 Tmol per year) are in agreement with a previous study by Lelieveld et al.56 (96 Tmol per year) but lower than in a later study by Lelieveld et al.9 (167.2 Tmol per year), who used higher VOC emissions and a faster VOC degradation mechanism (Table S2†). Fig. S4† shows that in these clean regions, SLHs contribute 12% to total secondary OH sources. In polluted regions like China, with high NOx concentrations, the impact of SLHs on secondary OH production, and therefore on OH recycling, is less pronounced. The primary mechanism contributing to OH formation is NOx + HO2. Because NOx can titrate reactive halogen species, their contribution to OH production is limited in these regions. However, SLHs can still modulate OH recycling by influencing the balance between primary and secondary OH sources.
In the pre-industrial atmosphere, S increases from 54 Tmol per year (noSLH) to 56 Tmol per year, with a similar contribution from HOX photolysis of 8%. Note that several models with different levels of complexity have been used to study how halogen chemistry influences HOx. For instance, Whalley et al.68 showed that HOI and HOBr photolysis contributed roughly 13% to instantaneous OH formation in the tropical Atlantic BL, while Sommariva et al.69 calculated an increase of up to 15% in OH due to HOI photolysis, which are comparable to our simulations.
The spatial differences in P and S due to SLH in the boundary layer show that P mostly decreases in the tropical and extra-tropical marine regions. At the same time, increases in S are seen predominantly over the continents (Fig. 4b and d). The differences due to the inclusion of SLH are larger in the PD, showing that inclusion of halogen chemistry reduces the dependence of OH abundance on primary OH production, increasing the relative contribution of secondary sources. At present, S consistently exceeds P near the surface in the presence of SLH, but in the pre-industrial atmosphere, S shows lower values due to lower NOx and lack of anthropogenic SLH emissions (Fig. S3†).
Overall, our results highlight the complex interplay between SLHs, NOx, and other atmospheric constituents in shaping the oxidizing capacity of the atmosphere, which presents different behaviors between polluted and pristine environments. By understanding the regional variations in these interactions, we can better assess the impact of SLHs on climate and air quality.
3.4 Role of halogen chemistry on OH recycling in the atmosphere
The OH recycling probability (r)56 refers to the ability of OH to be regenerated from secondary sources. It is calculated as r = (1 − P/(P + S)) × 100. When secondary production exceeds primary production (S > P), the recycling probability surpasses 50%. Lelieveld et al.9 suggested that the atmospheric chemical system is buffered when r approaches 60%. Our calculations show that SLH enhance tropospheric OH recycling, with global r increasing from 53% to 59% at present (50% to 57% in PI) (Table 2).Fig. 5 shows that substantial regional differences in r are observed, with the tropical BL showing values of r < 50%, as this region strongly depends on P (more sensitive to O3 photolysis and water vapour) (see also Fig. S2 and S4† for vertical distribution). When SLH are incorporated, r increases (due to a significant reduction of P). In the northern mid-latitudes, halogens increase r values > 60% near the surface (Fig. S6†), with r values reaching up to 60% over the North Atlantic Ocean, where the photocatalytic chlorine production from Saharan dust is highest (Fig. 2). Southern mid-latitudes have lower r values due to lower NOx and SLH levels. Consequently, r values are below 60% (Fig. 5 and S6†), with both P and S lower as compared to the Northern Hemisphere (Fig. S3†).
 | ||
| Fig. 5 Spatial distribution of the recycling probability (r, %) for OH average at the boundary layer (BL, 1000–850 hPa) in both scenarios wSLH and noSLH for present-day (PD) and pre-industrial (PI) times. r was calculated as (1 − P/P + S) × 100%, where P and S refers to the primary and secondary OH production. (a) r values for wSLH case at PD, (b) r values for noSLH case at PD (c) shows the differences between panel (a) and (b), i.e. the increase in r due to SLH. panel (d)–(f) are equivalent to panel (a)–(c) but for PI. | ||
SLH significantly enhance the global OH recycling from 4–6% in continental region to 12–16% in the open ocean (Fig. 5). Pre-industrial r over oceans increases by 14% when SLH are included (Fig. S7†).
The impact of SLH on OH recycling is more pronounced in the PI, especially in the tropics (Fig. 5) due to the lower contribution of other secondary sources besides the HOX production channel (Table 2). This demonstrates the significant but largely heterogenous role of SLH in modulating OH recycling efficiency and, therefore, modulating the chemical pathways for OH formation in the atmosphere.
Our model results show that the reduction in P, as well as the corresponding enhancement of S are more pronounced in PD than in PI, as a consequence of nonlinear chemical interactions with anthropogenic pollution. Therefore, SLH not only reduce OH levels but also increase the r, and as a consequence SLH not only reduces the oxidation capacity of the global atmosphere but also represents an additional coupling route between different oxidants that buffer the response of main oxidants to the changes in the background composition of the troposphere. In other words, SLH increase the natural resilience of the atmosphere to external perturbations affecting the chemical composition of the atmosphere.
3.5 Impact of SLH on the abundance of key organic compounds
The overall SLH-driven reduction of oxidant levels has important implications for the atmospheric degradation of many organic compounds. While SLH generally decreases OH, it also increases Cl. Therefore, since many VOCs have different reaction rates towards oxidation by OH and Cl atoms, we investigated how the addition of SLH along with the difference in the reaction rates between VOCs with OH and Cl leads to positive/negative changes in the burdens of different VOCs.Our results show that the general reduction in OH levels caused by SLH increases the boundary layer burden of CO by 14% and 21%, CH4 by 9% and 13%, isoprene by 7% and 9%, propene (C3H6) by 4% and 17% and limonene (C10H16) by 11 and 13%, during PD and PI, respectively (Fig. 6a and S6a†). These changes in organic compound burdens have important implications for air quality and climate. For instance, the increased burden of methane, a potent greenhouse gas, contributes to enhanced radiative forcing. Additionally, the increased burden of isoprene can lead to increased secondary organic aerosol formation, affecting air quality and climate.
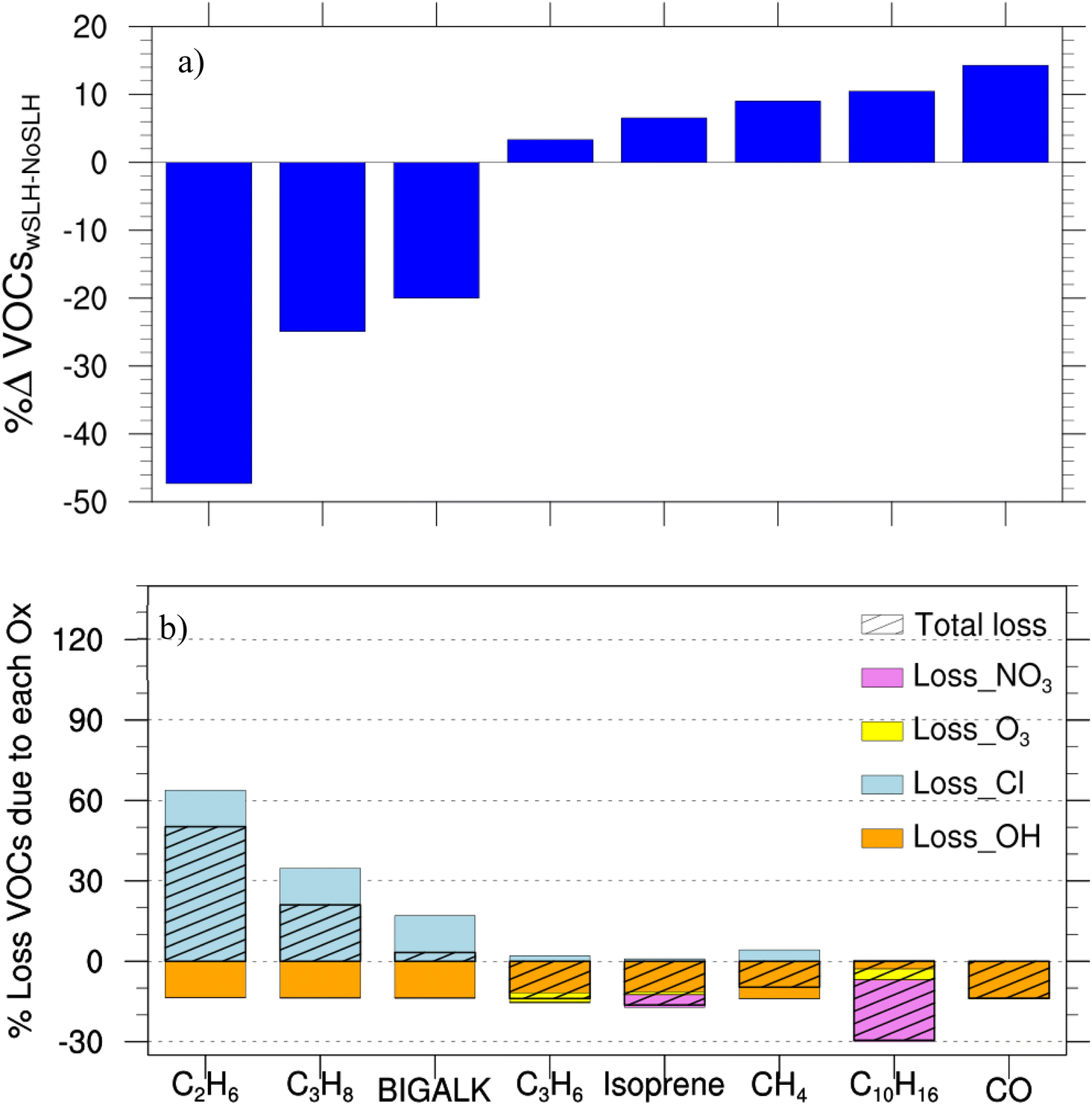 | ||
| Fig. 6 Percentage of changes in the concentration of some principal VOCs due to the inclusion of SLH for present-day (PD) and the percentage contribution of each oxidant to VOC loss at the boundary layer (1000–850 hPa). (a) Percentage changes in VOC concentrations due to SLH. (b) The stacked bar chart shows the percentage contribution of each oxidant (OH, Cl, O3 and NO3) due to SLH to the total loss of VOCs. The calculations are based on the rate constants for the reaction of each VOC with the different oxidants and its relative change due to SLH, and was calculated as follows. | ||
The halogen-mediated increase in CH4 burden is primarily driven by the indirect effect of reduced OH concentrations, which outweighs the direct impact of Cl-atom oxidation. This finding is consistent with previous studies,22,25 which have also highlighted the importance of halogen chemistry in regulating atmospheric methane levels. Our results further quantify the magnitude of this effect, showing that wSLHs increasing CH4 burden by approximately 350 Tg (9%) in the PD and 200 Tg (13%) in the PI, respectively (ESI Table S5†), which leads to increasing the CH4 lifetime (∼6–9% (Li et al. 2022)25).
By contrast, SLH reduce the boundary layer burden of ethane (C2H6) by 47% and 49%, propane (C3H8) by 25% and 23% and larger alkanes (BIGALK > C5) by 20% and 18%, during PD and PI, respectively (Fig. 6a and S6a†). This contrasting effect of SLH on the abundance and lifetime of VOCs results from the faster oxidation of the longer-chained VOCs by Cl than by OH (Fig. 6b). Indeed, in the presence of SLH, reaction with Cl is the main loss pathway for C2H6, C3H8 and big-alkanes and, as a consequence, their burden decreases in the presence of halogen chemistry, despite the decrease in OH levels (Fig. 6a).


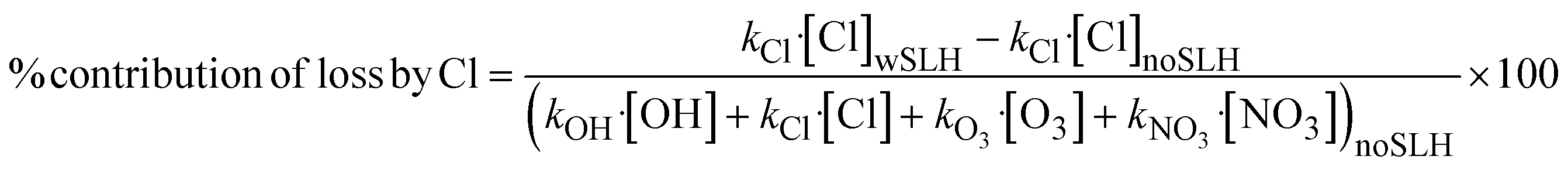

where kOH is the rate constant for the loss of VOCs with OH and kCl is the rate constant for the loss of VOCs with Cl. In the case of Isoprene, C3H6 and C10H16 we have also taken into account the additional loss due to O3 and NO3.

4 Summary and conclusions
This study presents the complex interplay between SLH and atmospheric oxidants. We show that in the present-day atmosphere, SLH substantially decrease atmospheric oxidation through a reduction effect on the combined concentration of the main atmospheric oxidants. Importantly, the effect of SLH on atmospheric oxidation has changed over time due to SLH emission changes and their chemical interaction with anthropogenic pollution, and is expected to vary further due to changing climate, therefore their effects should not be assumed constant in models. Remarkably, we find that the role of halogens on atmospheric oxidation was more prominent in the pristine pre-industrial atmosphere where a quarter of OH and half of NO3 concentrations in the boundary layer are accounted for by SLH. This result implies that the pre-industrial atmospheric oxidation capacity cannot be understood without the consideration of SLH emissions and chemistry. Critically, our analysis shows that while MDSA represents a significant source of chlorine in the boundary layer, its impact on global tropospheric OH reduction is relatively modest, increasing the total OH decrease by approximately 3% and 8% during the present day (PD) and pre-industrial (PI) periods, respectively, compared to simulations without MDSA-derived Cl. This limited impact underscores the dominant role of iodine and bromine in regulating tropospheric O3 and, consequently, OH.The results also show, particularly for the pre-industrial atmosphere, that SLH reduces the OH burden dependence on primary production and, in turn, increases OH recycling efficiency, which enhances the natural resilience of the atmosphere to external perturbations altering the OH budget. Furthermore, the interplay between oxidants, SLH, and VOCs ultimately results in differing but substantially changes in the burden of key atmospheric organic compounds, with important implications for air quality and aerosol formation.
Our findings reveal that the complex and multidirectional chemical interactions between SLH and atmospheric oxidants, currently unaccounted for in air quality and climate assessments, substantially reduce global atmospheric oxidation across the pre-industrial and present-day atmospheres, and thus influence the concentrations and trends of pollutants and short-lived climate forcers. These reductions exhibit spatial heterogeneity, with the most significant impacts observed over oceanic regions, which can be considered pristine environments. In contrast, the influence of SLHs on OH production is less pronounced in polluted continental regions, such as China, where high NOx concentrations limit the impact of halogen chemistry on overall OH production, although SLHs can still modulate OH recycling. Semi-polluted regions, such as South America, represent an intermediate case. This regional variation underscores the importance of considering local environmental conditions, including pollution levels, when assessing the impact of SLHs.
Our results reveal the dominant role of SLHs in the global oxidation state of the pre-industrial atmosphere. We therefore suggest incorporating detailed SLH emissions and chemistry into air quality and climate models to improve their predictive skills on atmospheric oxidation in past, present, and future climates.
Data availability
The CESM software used in this work is publicly available for download at https://www.cesm.ucar.edu/models/. Simulations outputs for each scenario analyzed in this manuscript are available in https://zenodo.org/records/13933571.Author contributions
A. S.-L. devised the research. A. B. performed the CESM simulations with the help of R. P. F. and Q. L. All authors discussed the findings and commented on the manuscript. A. B., A. S. M., and A. S.-L. wrote the manuscript with contributions from all authors.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This study has received funding from the European Research Council Executive Agency under the European Union's Horizon 2020 Research and Innovation Programme (Project ERC-2016-COG 726349 CLIMAHAL). The CESM project, supported primarily by the NSF (Nationa Science Foundation), is led by the National Center for Atmospheric Research (NCAR), a major facility sponsored by the NSF under cooperative agreement 1852977. The Climate Simulation Laboratory at NCAR's Computational and Information Systems Laboratory, sponsored by the NSF, provided computing resources, support and data storage.References
- IPCC, Climate Change 2021: the Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, 2021 Search PubMed.
- A. M. Fiore, L. J. Mickley, Q. Zhu and C. B. Baublitz, Climate and Tropospheric Oxidizing Capacity, Annu. Rev. Earth Planet. Sci., 2024, 52, 321–349 CrossRef CAS . Available from: https://www.annualreviews.org/content/journals/10.1146/annurev-earth-032320-090307.
- D. J. Jacob, Introduction to Atmospheric Chemistry, Princeton University Press, 1999 Search PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts, Atmospheric Chemistry: Fundamentals and Experimental Techniques, John Wiley, New York, 1986 Search PubMed.
- R. P. Wayne, I. Barnes, P. Biggs, J. P. Burrows, C. E. Canosamas and J. Hjorth, et al., The Nitrate Radical – Physics, Chemistry, and the Atmosphere, Atmos. Environ. Part A Gen. Top., 1991, 25(1), 1–203 CrossRef.
- O. C. Zafiriou, Photochemistry of halogens in the marine atmosphere, J. Geophys. Res., 1974, 79(18), 2730–2732 CAS.
- R. von Glasow and P. J. Crutzen, Tropospheric halogen chemistry, in Treatise on Geochemistry, The Atmosphere, ed. R. Keeling, Elsevier-Pergamon, Oxford, 2007, pp. 1–76 Search PubMed.
- A. Saiz-Lopez and R. von Glasow, Reactive halogen chemistry in the troposphere, Chem. Soc. Rev., 2012, 41, 6448–6472 CAS.
- J. Lelieveld, S. Gromov, A. Pozzer and D. Taraborrelli, Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 2016, 16(19), 12477–12493 CrossRef CAS . Available from: https://acp.copernicus.org/articles/16/12477/2016/.
- J. Lelieveld, F. J. Dentener, W. Peters and M. C. Krol, On the role of hydroxyl radicals in the self-cleansing capacity of the troposphere, Atmos. Chem. Phys., 2004, 4(9/10), 2337–2344 CAS . Available from: https://acp.copernicus.org/articles/4/2337/2004/.
- C. D. Holmes, M. J. Prather, O. A. Søvde and G. Myhre, Future methane, hydroxyl, and their uncertainties: key climate and emission parameters for future predictions, Atmos. Chem. Phys., 2013, 13(1), 285–302 Search PubMed.
- D. Stone, L. K. Whalley and D. E. Heard, Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 2012, 41(19), 6348–6404, 10.1039/C2CS35140D.
- D. E. Heard, Field Masurements of Atmospheric Composition, in Analytical Techniques for Atmospheric Measurement, John Wiley & Sons, Ltd, 2006, pp. 1–71, DOI:10.1002/9780470988510.ch1.
- D. S. Stevenson, A. Zhao, V. Naik, F. M. O'Connor, S. Tilmes and G. Zeng, et al., Trends in global tropospheric hydroxyl radical and methane lifetime since 1850 from AerChemMIP, Atmos. Chem. Phys., 2020, 20(21), 12905–12920 CAS.
- V. Naik, A. Voulgarakis, A. M. Fiore, L. W. Horowitz, J. F. Lamarque and M. Lin, et al., Preindustrial to present-day changes in tropospheric hydroxyl radical and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 2013, 13(10), 5277–5298 Search PubMed.
- O. Wild, A. Voulgarakis, F. O'Connor, J. F. Lamarque, E. M. Ryan and L. Lee, Global sensitivity analysis of chemistry-climate model budgets of tropospheric ozone and OH: Exploring model diversity, Atmos. Chem. Phys., 2020, 20(7), 4047–4058 CrossRef CAS.
- G. Myhre, D. Shindel, F. M. Bréon, W. Collins, J. Fuglestvedt, J. Huang, et al., Anthropogenic and Natural Radiative Forcing, in Climate Change 2013: the Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013 Search PubMed.
- A. Voulgarakis, V. Naik, J. F. Lamarque, D. T. Shindell, P. J. Young and M. J. Prather, et al., Analysis of present day and future OH and methane lifetime in the ACCMIP simulations, Atmos. Chem. Phys., 2013, 13(5), 2563–2587 CrossRef.
- A. Badia, C. E. Reeves, A. R. Baker, A. Saiz-Lopez, R. Volkamer and T. K. Koenig, et al., Importance of reactive halogens in the tropical marine atmosphere: A regional modelling study using WRF-Chem, Atmos. Chem. Phys., 2019, 19, 3161–3189 CrossRef CAS.
- A. Saiz-Lopez, R. P. Fernandez, Q. Li, C. A. Cuevas, X. Fu and D. E. Kinnison, et al., Natural short-lived halogens exert an indirect cooling effect on climate, Nature, 2023, 618(7967), 967–973 CrossRef CAS PubMed.
- A. Saiz-Lopez, J. F. Lamarque, D. E. Kinnison, S. Tilmes, C. Ordóñez and J. J. Orlando, et al., Estimating the climate significance of halogen-driven ozone loss in the tropical marine troposphere, Atmos. Chem. Phys., 2012, 12, 3939–3949 CrossRef CAS.
- F. Iglesias-Suarez, A. Badia, R. P. Fernandez, C. A. Cuevas, D. E. Kinnison and S. Tilmes, et al., Natural halogens buffer tropospheric ozone in a changing climate, Nat. Clim. Change, 2020, 10(2), 147–154 CAS.
- T. Sherwen, J. A. Schmidt, M. J. Evans, L. J. Carpenter, K. Großmann and S. D. Eastham, et al., Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 2016, 16(18), 12239–12271 CAS . Available from: https://acp.copernicus.org/articles/16/12239/2016/.
- R. Hossaini, M. P. Chipperfield, S. A. Montzka, A. Rap, S. Dhomse and W. Feng, Efficiency of short-lived halogens at influencing climate through depletion of stratospheric ozone, Nat. Geosci., 2015, 8(3), 186–190, DOI:10.1038/ngeo2363.
- Q. Li, R. P. Fernandez, R. Hossaini, F. Iglesias-Suarez, C. A. Cuevas and E. C. Apel, et al., Reactive halogens increase the global methane lifetime and radiative forcing in the 21st century, Nat. Commun., 2022, 13(1), 2768 CAS.
- W. R. Simpson, S. S. Brown, A. Saiz-Lopez, J. A. Thornton and G. R. Von, Tropospheric Halogen Chemistry: Sources, Cycling, and Impacts, Chem. Rev., 2015, 115(10), 4035–4062 CrossRef CAS PubMed.
- J. A. Barrera, D. E. Kinnison, R. P. Fernandez, J. F. Lamarque, C. A. Cuevas and S. Tilmes, et al., Comparing the Effect of Anthropogenically Amplified Halogen Natural Emissions on Tropospheric Ozone Chemistry Between Pre-Industrial and Present-Day, J. Geophys. Res. Atmos., 2023, 128(14), e2022JD038283 CAS.
- T. Sherwen, M. J. J. Evans, L. J. J. Carpenter, S. J. J. Andrews, R. T. T. Lidster and B. Dix, et al., Iodine's impact on tropospheric oxidants: a global model study in GEOS-Chem, Atmos. Chem. Phys., 2016, 16(2), 1161–1186 CAS.
- W. J. Bloss, M. Camredon, J. D. Lee, D. E. Heard, J. M. C. Plane and A. Saiz-Lopez, et al., Coupling of HOx, NOx and halogen chemistry in the Antarctic boundary layer, Atmos. Chem. Phys., 2010, 10, 10187–10209 CAS.
- M. M. J. W. van Herpen, Q. Li, A. Saiz-Lopez, J. B. Liisberg, T. Röckmann and C. A. Cuevas, et al., Photocatalytic chlorine atom production on mineral dust–sea spray aerosols over the North Atlantic, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(31), e2303974120, DOI:10.1073/pnas.2303974120.
- K. A. Read, A. S. Mahajan, L. J. Carpenter, M. J. Evans, B. V. E. Faria and D. E. Heard, et al., Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean, Nature, 2008, 453(7199), 1232–1235 CAS.
- A. S. Mahajan, J. M. C. Plane, H. Oetjen, L. M. Mendes, R. W. Saunders and A. Saiz-Lopez, et al., Measurement and modelling of tropospheric reactive halogen species over the tropical Atlantic Ocean, Atmos. Chem. Phys., 2010, 10, 4611–4624 CAS.
- G. Sarwar, H. Simon, J. Xing and R. Mathur, Importance of tropospheric ClNO2 chemistry across the Northern Hemisphere, Geophys. Res. Lett., 2014, 41(11), 4050–4058 CAS.
- Q. Li, A. Badia, T. Wang, G. Sarwar, X. Fu and L. Zhang, et al., Potential Effect of Halogens on Atmospheric Oxidation and Air Quality in China, J. Geophys. Res. Atmos., 2020, 125(9), e2019JD032058 CAS.
- D. Stone, T. Sherwen, M. J. Evans, S. Vaughan, T. Ingham and L. K. Whalley, et al., Impacts of bromine and iodine chemistry on tropospheric OH and HO_2: comparing observations with box and global model perspectives, Atmos. Chem. Phys., 2018, 18(5), 3541–3561 CAS . Available from: https://acp.copernicus.org/articles/18/3541/2018/.
- Q. Li, R. Borge, G. Sarwar, D. de la Paz, B. Gantt and J. Domingo, et al., Impact of halogen chemistry on summertime air quality in coastal and continental Europe: application of the CMAQ model and implications for regulation, Atmos. Chem. Phys., 2019, 19(24), 15321–15337 CAS . Available from: https://acp.copernicus.org/articles/19/15321/2019/.
- C. A. Cuevas, N. Maffezzoli, J. P. Corella, A. Spolaor, P. Vallelonga and H. A. Kjær, et al., Rapid increase in atmospheric iodine levels in the North Atlantic since the mid-20th century, Nat. Commun., 2018, 9(1), 1452 CrossRef PubMed.
- M. Legrand, J. R. McConnell, S. Preunkert, M. Arienzo, N. Chellman and K. Gleason, et al., Alpine ice evidence of a three-fold increase in atmospheric iodine deposition since 1950 in Europe due to increasing oceanic emissions, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(48), 12136–12141 CAS.
- X. Zhao, X. Hou and W. Zhou, Atmospheric Iodine (127I and 129I) Record in Spruce Tree Rings in the Northeast Qinghai-Tibet Plateau, Environ. Sci. Technol., 2019, 53(15), 8706–8714 CrossRef CAS PubMed.
- L. J. Carpenter, S. M. MacDonald, M. D. Shaw, R. Kumar, R. W. Saunders and R. Parthipan, et al., Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 2013, 6(2), 108–111 CrossRef CAS.
- S. M. MacDonald, J. C. Gómez Martín, R. Chance, S. Warriner, A. Saiz-Lopez and L. J. Carpenter, et al., A laboratory characterisation of inorganic iodine emissions from the sea surface: dependence on oceanic variables and parameterisation for global modelling, Atmos. Chem. Phys., 2014, 14(11), 5841–5852 CrossRef.
- C. Prados-Roman, C. a. Cuevas, R. P. Fernandez, D. E. Kinnison, J. F. Lamarque and A. Saiz-Lopez, A negative feedback between anthropogenic ozone pollution and enhanced ocean emissions of iodine, Atmos. Chem. Phys., 2015, 15(4), 2215–2224 CrossRef CAS.
- J. P. Corella, N. Maffezzoli, A. Spolaor, P. Vallelonga, C. A. Cuevas and F. Scoto, et al., Climate changes modulated the history of Arctic iodine during the Last Glacial Cycle, Nat. Commun., 2022, 13(1), 6–14 CrossRef PubMed.
- P. Vallelonga, N. Maffezzoli, A. Saiz-Lopez, F. Scoto, H. A. Kjær and A. Spolaor, Sea-ice reconstructions from bromine and iodine in ice cores, Quat. Sci. Rev., 2021, 269, 107133 CrossRef.
- S. Zhai, J. R. McConnell, N. Chellman, M. Legrand, T. Opel and H. Meyer, et al., Anthropogenic Influence on Tropospheric Reactive Bromine Since the Pre-industrial: Implications for Arctic Ice-Core Bromine Trends, Geophys. Res. Lett., 2024, 51(5), e2023GL107733 CrossRef CAS.
- S. Zhai, X. Wang, J. R. McConnell, L. Geng, J. Cole-Dai and M. Sigl, et al., Anthropogenic Impacts on Tropospheric Reactive Chlorine Since the Preindustrial, Geophys. Res. Lett., 2021, 48(14), 1–12 Search PubMed.
- J. F. Lamarque, L. K. Emmons, P. G. Hess, D. E. Kinnison, S. Tilmes and F. Vitt, et al., CAM-chem: description and evaluation of interactive atmospheric chemistry in the Community Earth System Model, Geosci. Model Dev., 2012, 5(2), 369–411 CrossRef . Available from: https://gmd.copernicus.org/articles/5/369/2012/.
- S. Tilmes, J. F. Lamarque, L. K. Emmons, D. E. Kinnison, D. Marsh and R. R. Garcia, et al., Representation of the Community Earth System Model (CESM1) CAM4-chem within the Chemistry-Climate Model Initiative (CCMI), Geosci. Model Dev., 2016, 9(5), 1853–1890 CrossRef CAS . Available from: https://gmd.copernicus.org/articles/9/1853/2016/.
- R. P. Fernandez, R. J. Salawitch, D. E. Kinnison, J. F. Lamarque and A. Saiz-Lopez, Bromine partitioning in the tropical tropopause layer: implications for stratospheric injection, Atmos. Chem. Phys., 2014, 14(24), 13391–13410 CrossRef . Available from: https://acp.copernicus.org/articles/14/13391/2014/.
- C. Ordóñez, J. F. Lamarque, S. Tilmes, D. E. Kinnison, E. L. Atlas and D. R. Blake, et al., Bromine and iodine chemistry in a global chemistry-climate model: description and evaluation of very short-lived oceanic sources, Atmos. Chem. Phys., 2012, 12(3), 1423–1447 CrossRef . Available from: https://acp.copernicus.org/articles/12/1423/2012/.
- A. Saiz-Lopez, R. P. Fernandez, C. Ordóñez, D. E. Kinnison, J. C. Gómez Martin and J. F. Lamarque, et al., Iodine chemistry in the troposphere and its effect on ozone, Atmos. Chem. Phys., 2014, 14(23), 13119–13143 CrossRef . Available from: https://acp.copernicus.org/articles/14/13119/2014/.
- T. Claxton, R. Hossaini, C. Wilson, S. A. Montzka, M. P. Chipperfield and O. Wild, et al., A Synthesis Inversion to Constrain Global Emissions of Two Very Short Lived Chlorocarbons: Dichloromethane, and Perchloroethylene, J. Geophys. Res. Atmos., 2020, 125(12), e2019JD031818, DOI:10.1029/2019JD031818.
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl and L. K. Emmons, et al., The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 2012, 5(6), 1471–1492 CrossRef . Available from: https://gmd.copernicus.org/articles/5/1471/2012/.
- J. A. Barrera, R. P. Fernandez, F. Iglesias-Suarez, C. A. Cuevas, J. F. Lamarque and A. Saiz-Lopez, Seasonal impact of biogenic very short-lived bromocarbons on lowermost stratospheric ozone between 60N and 60S during the 21st century, Atmos. Chem. Phys., 2020, 20(13), 8083–8102 CrossRef CAS . Available from: https://acp.copernicus.org/articles/20/8083/2020/.
- WMO, Scientific Assessment of Ozone Depletion: 2010 Global Ozone Research and Monitoring Project Report 52, WMO, 2011 Search PubMed.
- J. Lelieveld, W. Peters, F. J. Dentener and M. C. Krol, Stability of tropospheric hydroxyl chemistry, J. Geophys. Res. Atmos., 2002, 107(D23), 1–11, DOI:10.1029/2002JD002272.
- A. Saiz-Lopez and R. P. Fernandez, On the formation of tropical rings of atomic halogens: Causes and implications, Geophys. Res. Lett., 2016, 43(6), 2928–2935, DOI:10.1002/2015GL067608.
- C. A. Cuevas, R. P. Fernandez, D. E. Kinnison, Q. Li, J. F. Lamarque and T. Trabelsi, The influence of iodine on the Antarctic stratospheric ozone hole, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(7), e2110864119 CrossRef CAS PubMed.
- R. P. Fernandez, D. E. Kinnison, J. F. Lamarque, S. Tilmes and A. Saiz-Lopez, Impact of biogenic very short-lived bromine on the Antarctic ozone hole during the 21st century, Atmos. Chem. Phys., 2017, 17(3), 1673–1688 CrossRef CAS . Available from: https://acp.copernicus.org/articles/17/1673/2017/.
- T. K. Koenig, S. Baidar, P. Campuzano-Jost, C. A. Cuevas, B. Dix and R. P. Fernandez, et al., Quantitative detection of iodine in the stratosphere, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(4), 1860–1866, DOI:10.1073/pnas.1916828117.
- J. E. Mak, G. Kra, T. Sandomenico and P. Bergamaschi, The seasonally varying isotopic composition of the sources of carbon monoxide at Barbados, West Indies, J. Geophys. Res. Atmos., 2003, 108(D20), 4635, DOI:10.1029/2003JD003419.
- C. Caram, S. Szopa, A. Cozic, S. Bekki, C. A. Cuevas and A. Saiz-Lopez, Sensitivity of tropospheric ozone to halogen chemistry in the chemistry–climate model LMDZ-INCA vNMHC, Geosci. Model Dev., 2023, 16(14), 4041–4062 CrossRef CAS . Available from: https://gmd.copernicus.org/articles/16/4041/2023/.
- Y. J. Tham, X. C. He, Q. Li, C. A. Cuevas, J. Shen and J. Kalliokoski, et al., Direct field evidence of autocatalytic iodine release from atmospheric aerosol, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(4), 1–8 CrossRef PubMed.
- J. M. Roberts, H. D. Osthoff, S. S. Brown and A. R. Ravishankara, N2O5 oxidizes chloride to Cl2 in acidic atmospheric aerosol, Science, 2008, 321(5892), 1059 CrossRef CAS PubMed.
- S. Bleicher, J. C. Buxmann, R. Sander, T. P. Riedel, J. A. Thornton and U. Platt, et al., The influence of nitrogen oxides on the activation of bromide and chloride in salt aerosol, Atmos. Chem. Phys. Discuss., 2014, 14(7), 10135–10166 Search PubMed.
- J. Dai, G. P. Brasseur, M. Vrekoussis, M. Kanakidou, K. Qu and Y. Zhang, et al., The atmospheric oxidizing capacity in China-Part 1: Roles of different photochemical processes, Atmos. Chem. Phys., 2023, 23(22), 14127–14158 CrossRef CAS.
- H. Chen, P. Liu, Q. Wang, R. Huang and G. Sarwar, Impact and pathway of halogens on atmospheric oxidants in coastal city clusters in the Yangtze River Delta region in China, Atmos. Pollut. Res., 2024, 15(2), 101979 CrossRef CAS PubMed.
- L. K. Whalley, K. L. Furneaux, A. Goddard, J. D. Lee, A. S. Mahajan and H. Oetjen, et al., The chemistry of OH and HO2 radicals in the boundary layer over the tropical Atlantic Ocean, Atmos. Chem. Phys., 2010, 10(4), 1555–1576 CrossRef CAS.
- R. Sommariva, W. J. Bloss, N. Brough, N. Carslaw, M. Flynn and A. L. Haggerstone, et al., OH and HO2 chemistry during NAMBLEX: roles of oxygenates, halogen oxides and heterogeneous uptake, Atmos. Chem. Phys., 2006, 6, 1135–1153 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ea00141a |
| This journal is © The Royal Society of Chemistry 2025 |