
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the role of anthropogenic terpenoids in urban secondary pollution under summer conditions by a box modeling approach†
M.
Farhat
ab,
L.
Pailler
a,
M.
Camredon
c,
A.
Maison‡
d,
K.
Sartelet
 e,
L.
Patryl
f,
P.
Armand
f,
C.
Afif
bg,
A.
Borbon
*a and
L.
Deguillaume
*ah
e,
L.
Patryl
f,
P.
Armand
f,
C.
Afif
bg,
A.
Borbon
*a and
L.
Deguillaume
*ah
aUniversité Clermont Auvergne, Laboratoire de Météorologie Physique, OPGC/CNRS UMR 6016, Clermont-Ferrand, France. E-mail: laurent.deguillaume@uca.fr
bEMMA Research Group, Center for Analysis and Research, Faculty of Sciences, Saint Joseph University of Beirut, Beirut, Lebanon
cUniv Paris Est Creteil and Université Paris Cité, CNRS, LISA, Créteil, F-94010, France
dUniversité Paris-Saclay, INRAE, AgroParisTech, UMR EcoSys, Palaiseau, 91120, France
eCEREA, École nationale des ponts et chaussées, EdF R & D, IPSL, Marne la Vallée, France
fCEA, DAM, DIF, Arpajon, 91297, France
gClimate & Atmosphere Research Centre (CARE-C), The Cyprus Institute, Nicosia, Cyprus
hUniversité Clermont Auvergne, Observatoire de Physique du Globe de Clermont-Ferrand, UAR 833, Clermont-Ferrand, France. E-mail: agnes.borbon@uca.fr
First published on 4th April 2025
Abstract
Terpenoids, including isoprene and monoterpenes, are highly reactive volatile organic compounds (VOCs) that play an essential role in atmospheric chemistry, contributing to the formation of ozone and secondary organic aerosols (SOAs). While known for decades for their biogenic origin, their anthropogenic origin is now well established in urban areas worldwide. Nevertheless, there is still a lack of clarity regarding the relative significance of these emissions and their impact on secondary pollution at the urban scale where biogenic and anthropogenic emissions coexist. The objective of this study is to evaluate the role of anthropogenic terpenoids in secondary pollution over the megacity of Paris, a typical northern mid-latitude urban area, using a box model. The model employs the Master Chemical Mechanism (MCM v3.3.1) to describe the gaseous reactivity. A physico-chemical scenario was developed to reproduce a typical summertime environment built upon in situ observations collected during the EU-MEGAPOLI campaign in Paris. Emission ratios of anthropogenic VOCs over carbon monoxide were used to parametrize the primary emissions of more than 60 species (including anthropogenic terpenoids). The comparison between in situ observations and modelled trace gas concentrations demonstrated the model's capacity to reproduce the levels and their temporal variability. Two sensitivity tests were conducted to quantify the impact of terpenoid emissions on ozone formation and their potential to form SOA mass concentration according to two simulations modulating anthropogenic and biogenic emissions of terpenoids based on the uncertainties associated with their estimation. Ozone concentration slightly increases by 1 (±0.5)% when increasing anthropogenic terpenoid emissions and by 3 (±2)% when increasing biogenic terpenoid emissions; the increase of O3 with increasing VOCs is consistent with the high-NOx chemical regime. Looking at the potential terpenoid derived SOA production, isoprene and limonene dominate. The estimated total mass concentration of SOAs produced over a 24 h period is 0.53 μg m−3, with a maximum hourly produced mass concentration of 0.045 μg m−3 observed in the morning. This modelling study suggests that the production of SOAs through the oxidation of terpenoids emitted from anthropogenic sources is competitive with that derived from their biogenic sources and remains significant at night.
Environmental significanceTerpenoids are crucial in atmospheric chemistry, given their high reactivity and involvement in the production of secondary pollutants. Recent studies have demonstrated their anthropogenic origin but have given little consideration to their contribution to secondary pollution in urban environments where anthropogenic and biogenic emissions coexist. Using a modelling approach, we aim to evaluate the role of anthropogenic terpenoids in secondary pollution at the urban scale. We simulate an urban scenario representative of a northern mid-latitude megalopolis: Paris. We used emission ratios of anthropogenic VOCs over CO to parametrize the primary emissions of 60 species, including anthropogenic terpenoids. This study suggests that the SOA production through the oxidation of terpenoids from anthropogenic sources is competitive with that derived from biogenic sources. |
1. Introduction
Among Volatile Organic Compounds (VOCs), terpenoids, including isoprene and monoterpenes with the (C5H8)n formula, have at least one double bond, making them extremely reactive. Thus, they play a key role in atmospheric chemistry by being involved in the ozone cycle and in the formation of a range of organic compounds contributing to the complex composition of secondary organic aerosols (SOAs).1–3 SOAs and O3 are key compounds in the evaluation of air quality4 and belong to short-lived climate forcers. Given the important role of terpenoids in secondary air pollution, several studies have focused on their quantification and their source identification. Terpenoids have been known for a long time to be emitted from biogenic sources; their biogenic emissions are estimated to be 347.7 Tg year−1 for isoprene and 184.4 Tg year−1 for monoterpenes on a global scale.5However, few studies have also focused on the anthropogenic origin of terpenoids from multiple sources. Since the work of Rouvière et al.6 in the Alpine valleys, several studies have shown that wood heating is one of the anthropogenic sources of terpenoids.7 Recently, studies in North American cities have highlighted terpenoid emissions from Volatile Chemical Product (VCP) use; they demonstrated that 50% of monoterpenes result from VCP volatilization.8–10 Since terpenoids are associated with the use of solvents, several industrial activities have been identified as emitters of these compounds. For example, they account for 8.2 tons per year of monoterpenes in the Dunkirk urban area, North of France.11 It is in the same order of magnitude as the non-industrial anthropogenic emissions of monoterpenes determined in Paris by Borbon et al.,12 which were estimated to be 11.8 tons per year. Moreover, Dominutti et al.13 demonstrated that traffic could be a source of terpenoid emissions, where up to 40% of monoterpenes were attributed to road transport in cities around the world, extending the seminal work by Borbon et al.14 on isoprene. Nevertheless, there is still uncertainty on the relative importance of these emissions and their effect on secondary pollution. Global modeling studies have estimated that SOA production from isoprene oxidation ranges from 6 to 20 Tg year−115–17 and that from monoterpene oxidation accounts for 13 to 40 Tg year−1.18,19 While the role of anthropogenic VOCs as precursors in SOA formation has been extensively studied, the impact of anthropogenic terpenoids on air pollution remains an area of active investigation in cities worldwide.
The objective of this study is to evaluate the impact of anthropogenic terpenoids on secondary pollution at the urban scale under summer conditions. To do so, we optimized an explicit gas phase box model to assess how terpenoids from biogenic and anthropogenic origins participate in the secondary production of oxidants such as ozone and to the VOC multiphasic chemistry over a northern mid-latitude urban area. This model considers the gas-phase Master Chemical Mechanism (MCM v3.3.1) and has been developed to enhance its capability to capture both local-scale dynamics and biogenic and anthropogenic sources to simulate the atmospheric processes governing terpenoid concentrations and their effect on atmospheric chemistry. To simulate realistic concentrations, we constrained and evaluated the model with in situ observations that were collected over the Paris metropolitan area during the EU-MEGAPOLI field campaign (megacities: emissions, urban, regional, and global atmospheric pollution and climate effects, and integrated tools for assessment and mitigation) conducted in July 2009.20
We first present the developments performed on the model to reproduce the chemical environment over the Paris area. Diel emissions of NOx, SO2, and CO follow the emission database of the “association for Air Quality Monitoring Network in the Paris region” (AIRPARIF), and emission ratios of anthropogenic VOCs over carbon monoxide were used to parametrize their primary emissions. Biogenic emissions were also considered in the model based on recent modeling work over the Paris region during the STREET project (impact of STress on uRban trEEs on ciTy air quality). The temporal variation of the boundary layer height is also considered to modulate the intensity of emissions/depositions and the concentration of chemical compounds through dilution/entrainment. Dilution and advection by the wind have also been implemented in the model to represent sources/sinks induced by atmospheric transport. These developments were evaluated by comparing the concentrations of the reference simulation to the observations from the MEGAPOLI campaign. Then, the second part of this work is devoted to sensitivity studies to quantify the effect of terpenoid emissions on atmospheric chemistry (i.e., the secondary production of ozone and SOAs). In this framework, we also performed sensitivity analysis by modulating the relative proportion of their anthropogenic and biogenic emissions based on the uncertainties associated with their quantification.
2. Methodology
2.1 In situ measurements during the summer MEGAPOLI campaign
The EU-MEGAPOLI project included two one-month field campaigns in Paris and its surrounding areas. The first one took place in July 2009, while the second began on January 15, 2010, and finished in February 15, 2010. This project aimed to provide a comprehensive, coherent, and more quantitative description of the impact of megacities on air quality, tropospheric chemistry, and climate. To this purpose, the objective was to document and analyze primary and secondary gases and particulates in the Paris area, including their concentrations, their spatial and temporal variations, their emission sources, and origins (local or long-range advected).21,22 In this study, the summertime field campaign was privileged to leverage the increased photochemical activity that occurs due to high temperatures and long daylight hours. It relies on the measurements performed at the urban background site of the Laboratoire d’Hygiène de la Ville de Paris (LHVP) (i.e., meteorological parameters, VOCs, oxygenated VOCs, peroxy acyl nitrates, CO, NO, NO2, and O3). The site is representative of Paris background pollution for its location on the southern outskirts of Paris in a large public garden 150 m away from a major street.23,24 Measurements performed at the Site Instrumental de Recherche par Télédétection Atmosphérique (SIRTA) (i.e., OH and O3) are also used to support the analysis featured in this paper. The SIRTA site is qualified as suburban for its location 14 km southwest of Paris, surrounded by wooded areas, fields, houses, and industries.25,26 During this campaign, a wide variety of measurement instruments were used and are listed in Table S1.† All measurements were subject to strict QA/QC protocols (e.g., calibration against certified standards and regular cross-validation with reference methods); the uncertainties were evaluated at ±5–20%. All the above-mentioned observations will be used in the model assessment for its capacity to simulate the urban environment.2.2 Box model description and developments
The 0D box model CLEPS (Cloud Explicit Physico-Chemical Scheme) has been used in this study to numerically describe the chemical and physical atmospheric processes as a function of time.27,28 In the following sections, the main developments of the model to reproduce an adequate urban environment are presented. This model is based on the Dynamically Simple Model for Atmospheric Chemical Complexity (DSMACC),29 using the Kinetic Pre-Processor (KPP).30 This box model was developed to cover the full range of chemical reactions in both the gaseous phase and aqueous phase.27 The box model CLEPS previously considered a reduced version of the gaseous MCM (Master Chemical Mechanism) v3.3.1 (i.e., 2049 reactions including 678 species) adapted to reproduce remote environments influenced by biogenic emissions.31 In this work, the entire MCM was implemented in the model (i.e., 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 707 reactions and 5837 species). The actinic flux and the subsequent photolysis rates are calculated using the radiative transfer model TUV version 4.5, while only the gaseous phase chemistry is activated in the model. In this section, we will present the physical and chemical scenario that we worked on, and then the development of the model adapted to the urban area in Paris. The model allows performing a chemical budget to quantify the sources and sinks of a chemical compound over the duration of the simulation.
707 reactions and 5837 species). The actinic flux and the subsequent photolysis rates are calculated using the radiative transfer model TUV version 4.5, while only the gaseous phase chemistry is activated in the model. In this section, we will present the physical and chemical scenario that we worked on, and then the development of the model adapted to the urban area in Paris. The model allows performing a chemical budget to quantify the sources and sinks of a chemical compound over the duration of the simulation.
 | ||
| Fig. 1 Description of the scenario implemented in the box model to reproduce the urban environment: emissions, deposition, boundary layer height, advection/dilution, and meteorological parameters (T, P, RH, and wind speed). | ||
For biogenic emissions, VOCs from biogenic origin were also described in the model. Biogenic emissions in urban environments are usually poorly or incompletely entered in inventories despite their presence in urban atmospheres and their potential reactivity.13 A recent modeling study focused on the description of biogenic emissions in Paris in the framework of the STREET (impact of STress on uRban trEEs on ciTy air quality) project.39 This work described the development of an emission inventory to model biogenic emissions in Paris during the months of June and July 2022 using the tree database called “ARBRE” from Paris city, allometric relations to determine tree characteristics and meteorological parameters estimated using the WRF model. Biogenic emissions were computed for each tree based on their leaf dry biomass, tree-species dependent emission factors and activity factors representing the effects of light and temperature. Here, gaseous emissions of 8 chemical compounds were estimated using the mean hourly emissions from the study of Maison et al.39 Three emission profiles were implemented in the model based on the mean diel profile of the eight compounds: the first one for isoprene and CO, the second one for α-pinene, limonene, and methanol, and the third one for β-pinene, acetone, and NO (Fig. 1④). All details and profiles are available in the ESI (Section 2.b.†).
All deposition rates were adapted from Michoud et al.20 and are reported in Table S6.† As explained above, a variable temporal BLH profile was also implemented in the model (Fig. 1⑤ and S1(d)†). Thus, deposition and emission rates are a function of the BLH variation according to the equations below:
 | (1) |
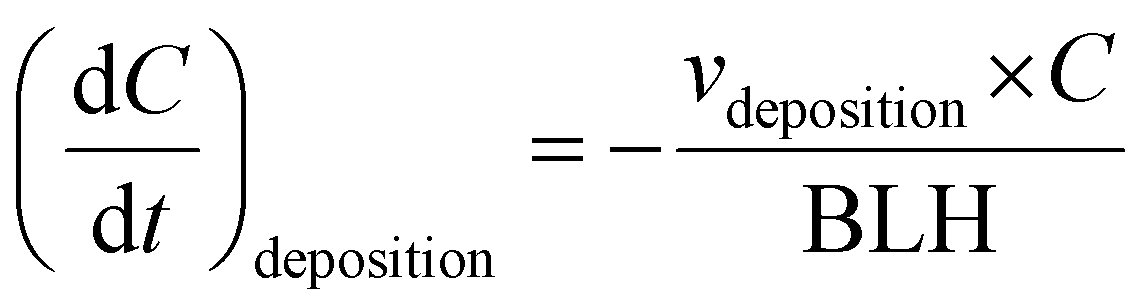 | (2) |
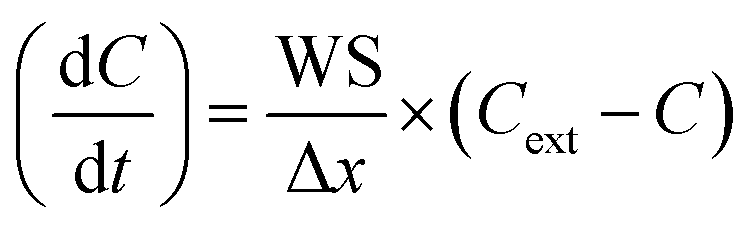 | (3) |
In this study, all compounds are subject to dilution, and only ozone is advected. No dominant wind direction was observed during the campaign. The ozone concentration outside Paris Intramuros was defined as a mean diel profile based on measurement from the peri-urban SIRTA site (Fig. 1⑦). The profile was determined from data obtained during the MEGAPOLI summer campaign (Fig. S1(e)†).
Vertical dilution and entrainment were also implemented in the model. This defines the dilution of all compounds and the exchange between the residual layer and the boundary layer when the BLH is increasing (Fig. 1⑤)  The following equation describes these processes:
The following equation describes these processes:
 | (4) |
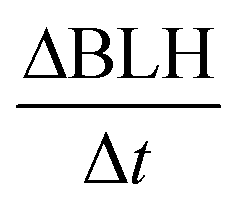 is the BLH variation (m s−1). In this study, all compounds are diluted vertically (Cresidual = 0); only ozone entrainment is considered (Cresidual ≈ 40 ppb).40
is the BLH variation (m s−1). In this study, all compounds are diluted vertically (Cresidual = 0); only ozone entrainment is considered (Cresidual ≈ 40 ppb).40
3. Results and discussion
3.1 Evaluation of the reference simulation towards in situ observations
Fig. 2 shows the temporal variability of the modeled concentrations (continuous curve) obtained on the last simulation day (i.e., at a daily stationary state) that was compared to the diel variability of measurements over one month (July 2009). This comparison is conducted for all compounds measured in this study, except for those having more than 75% of the measured values below the detection limit (see Section 3 in the ESI†). | ||
| Fig. 2 Comparison of a selection of simulated concentration (continuous line) vs. observations (box plots) during the MEGAPOLI field campaign. Measurements performed during the month of July 2009 are presented as hourly boxplots showing the average (markers “+”), the median, and the inter-quartile range in addition to the range of in situ data from the minimum to the maximum values highlighting outliers that could represent “unusual” events (* on figure legend: measured by PTR-MS). | ||
In practice, we confronted the hourly modeled concentrations for the last day with the 5th, 25th, 50th, 75th, and 95th percentiles of concentration measured during the MEGAPOLI campaign. This comparison is reported in the †SI (Comparison_Obs_Model.xls file).†
3.2 The O3/NOx chemical sub-system
Generally, the model appears to perform properly with regard to the temporal trend of NO concentrations emitted throughout the day (Fig. 2). As expected, at night, NO levels are low (below 1 ppb), since the traffic sources are reduced. However, the model overestimates the morning concentrations by a factor of 2.5 with a maximum value of 16.5 ppb at 8 AM (vs. observation at 6.3 ppb – median value). Fig. 2 also shows the considerable variability of NO2 observations, suggesting that at different times, the NO2 levels fluctuate significantly, which is common in urban areas due to varying traffic patterns and industrial activities.41 Nevertheless, the observed NO2 data show considerable variability and sharper peaks than the modeled NO2 concentration during the morning hours, but the simulated concentration is within the right order of magnitude. NOx simulated concentration is in line with observations demonstrating that the conversion of NO into NO2 may not be efficient enough in our simulation. This latter reaction represents the essential source of NO2 and sink for NO according to the chemical budget, while the key source of NO is the photolysis reaction of NO2, and the sink of NO2 is the reaction triggered by photolysis.The O3 diel pattern is well represented by the model. The analysis of the chemical budget of ozone shows that the O3 sink is controlled by its reaction with NO (titration) to give NO2 and by its photolysis. These two sinks are almost completely counterbalanced by the reaction of monoatomic O with O2 leading to O3. The additional source represented by the advection process together with the dilution allows the reproduction of a realistic O3 concentration. In the simulation, the contribution of horizontal advection of O3 by the wind is dominant in comparison to the advection of O3 by entrainment. With low levels of VOCs, the generated peroxyl radical (ROx) through VOC oxidation is probably not enough to contribute to the ozone production explaining the need of implementing ozone advection in the model (Fig. S4†).
Regarding OH, the simulated data are realistic in comparison with the observations, even if some discrepancies can be highlighted, particularly when photolysis conditions are at their maximum (Fig. 2) with a significant production of OH through O3 photolysis (20% of produced OH). The simulated OH concentrations are slightly higher (+25% for the maximal values) during these hours with respect to the measurements, with a slight time shift to the right (approx. 1 h), which is explained by the temporal profile of O3.
3.3 VOC chemical sub-system
Alkanes up to C12 (dodecane) follow the same temporal pattern as butane (Fig. 2). Similar to aromatics, the modelled concentrations of alkanes are mainly driven by the emission and dilution processes, with a slight underestimation between 5 PM and 5 AM, most probably due to low emissions in the model. For alkenes, the model simulates concentrations comparable to observations.
However, the most significant biases in aromatic, alkene, and alkane concentrations are observed for compounds presenting ERs calculated from Los Angeles,37,38 namely 2-methyl propene, 3-methyl-hexane, and o-ethyltoluene, demonstrating the possible specificity of Paris emissions and the need to reconsider these ratios for these species.
Modelled isoprene showed morning and evening peaks with a decrease between 9 AM and 2 PM. A delay in the morning peak exists between isoprene and the anthropogenic compounds. Indeed, the isoprene concentration is mainly governed by the biogenic emissions that are 4.5 times more important than anthropogenic emissions during daytime. The isoprene morning peak is the result of biogenic emission controlled by the BLH with higher concentrations in the model (maximum concentration of 0.54 ppb for modelled concentration vs. 0.25 ppb for the observed value). This urban cycle of isoprene is governed by traffic-related emissions, temperature, and light dependent biogenic emissions during the day, which counterbalance its consumption by the OH radical.14 In our simulation, this trend is not properly reproduced due to underestimation of biogenic emissions and the additional effect of local biogenic emissions from the trees of the nearby public garden affecting the observed concentrations.
In comparison with the study conducted by Guo et al.,43 the diel variability of α-pinene concentrations does not show a typical biogenic diel cycle (Fig. 2) as already shown in other urban areas such as Beirut;44 it appears to be similar to the urban anthropogenic diel cycle demonstrating the significant anthropogenic sources contributing to α-pinene concentrations in Paris.45 Indeed, the anthropogenic emission intensity is greater than the biogenic emissions by a factor of 1.3 on average. The model seems to capture the overall evolution of the observations but fails to reproduce the mean concentration with a significant underestimation, probably due to the modelled emissions.
Observations of certain compounds such as acetaldehyde and methanol show a pronounced cycle similar to the model but with generally higher values probably due to emissions or advection issues. This seems to indicate that the model is not correctly considering emissions of these compounds and/or that they need to be advected in the box model. These compounds could be transported at a regional/continental scale as underlined in a previous study analyzing VOC data measured in Paris.23
The PTR-MS sum of MEK + butanal + methylglyoxal (m/z 73) shows acceptable concentrations from 8 AM to 8 PM when compared to observations. In the model, MEK and butanal are primarily emitted in the morning and in the afternoon, and methylglyoxal is efficiently produced, which explains why concentration levels remained stable during the afternoon. However, the model simulates a lower concentration during nighttime, which is also potentially explained by the lack of advection of MEK and butanal, for example.
3.4 Sensitivity tests on terpenoid emissions
Looking at the emissions of terpenoids in the model, Fig. 3 presents the temporal variability of anthropogenic and biogenic emissions in terms of injected concentrations in the model (molec cm−3 s−1). These values result from multiplying the prescribed emissions by the BLH temporal variability. For isoprene, as underlined before, the biogenic sources dominate the diel emissions with maximum intensity a factor of 8.7 higher than its anthropogenic emissions; the opposite is observed for β-pinene and limonene, where anthropogenic emissions can be up to an order of magnitude higher than the biogenic emissions on an hourly basis. For α-pinene, diel emissions are comparable, with slightly higher anthropogenic emissions (maximum hourly factor of 1.7) for maximum emission values.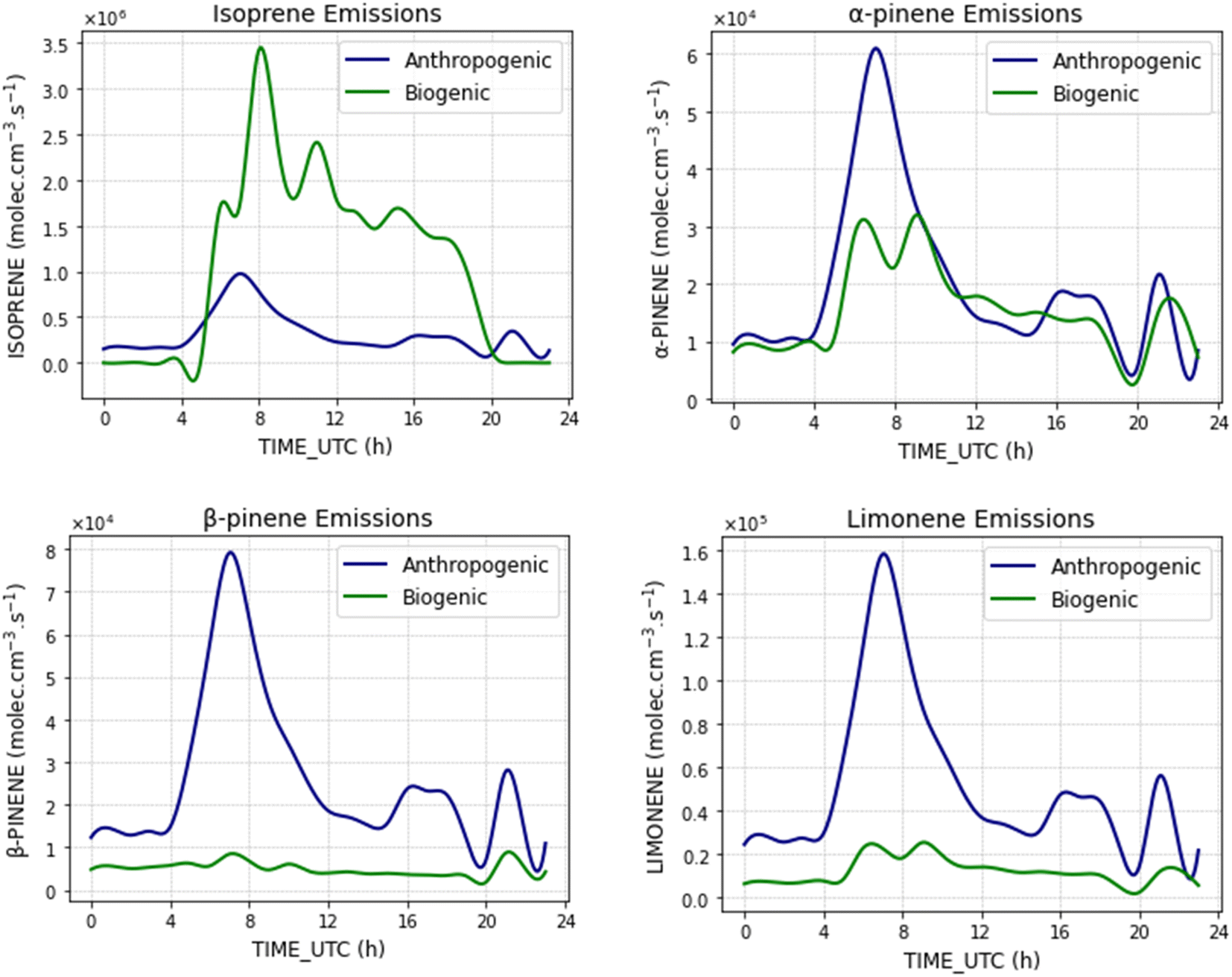 | ||
| Fig. 3 Comparison of anthropogenic and biogenic emissions of terpenoids implemented in the model for the reference simulation. Emissions are presented in terms of injected concentration (molec cm−3 s−1) controlled by the boundary layer height that evolves during the day. | ||
Evaluating the role of anthropogenic terpenoid emissions in secondary pollution relies on sensitivity tests by varying both anthropogenic and biogenic emissions of terpenoids. As for other VOCs (see the previous section), the emission ratio of anthropogenic isoprene, α-pinene, β-pinene, and limonene to CO during nighttime was quantified by implementing the regression fit method on the observed data. The scatterplots of α-pinene, β-pinene, and limonene to CO are illustrated in Fig. 4(a). The mean emission ratio is derived from the value of the linear fit (Table S3†).
 | ||
| Fig. 4 (a) Terpenoid scatter plots as a function of CO at nighttime, (b) and (c) comparison between the reference simulation and the “Anthro_high” test simulation on the one hand, and the “Bio_high” test simulation on the other hand. | ||
The scatterplots of isoprene, β-pinene and limonene are dispersed with an R2 between 0.1 and 0.4 but still show a linear relationship with CO. α-Pinene shows a much-scattered plot (R2 = 0.1) partly explained by the high amount of data below the detection limit (58%). This dispersed data behavior suggests either the presence of potent non-combustion and biogenic terpenoid sources or the nighttime reactivity with ozone and the nitrate radical. These might include terpenoid emissions from VCPs as demonstrated in some recent studies in North American urban cities where it was revealed that more than 50% of urban monoterpene loads are explained by VCP-dominated emissions.8,10,46 To consider the additional contribution of non-combustion emissions in the simulation, we derived a linear fit along the data point delimiting the upper part of the scatterplot (in dark blue). This value represents an estimate of the maximum emission ratio of terpenoids from anthropogenic origin. Fig. 4(b) shows the increase in terpenoid emissions considered in the model for this sensitivity test (“Anthro_high”). Emission ratios of isoprene, α-pinene, β-pinene and limonene to CO have increased by a factor of 4, 15, 2, and 3, respectively. The “Anthro_high” scenario for α-pinene shows the largest difference from the reference one because of the high dispersion of the scatterplot.
We also varied the biogenic emissions of terpenoids by relying on the work by Maison et al.39 over the Paris region. For this, we used this work where the effect of abiotic parameters on biogenic emissions was evaluated. In particular, they simulated the effect of micrometeorology at the street level with reflection and shading effects on the leaf temperature of trees, which is a more relevant variable of biogenic emissions compared to ambient temperature. They showed that the leaf temperature was on average +1.2 °C higher than the air temperature, resulting in an increase in isoprene and monoterpene emissions. Based on these results, we performed “Bio_high” sensitivity test where the biogenic emissions of isoprene and monoterpenes have been increased by a percentage of 36% and 25%, respectively, with respect to the reference simulation (Fig. 4(c)). This 1.2 °C temperature modulation is consistent with the temperature variability measured during the MEGAPOLI campaign, with standard deviations between 2 and 4 °C.
As expected, since concentrations of terpenoids are mostly controlled by the emission rates, modulating biogenic or anthropogenic emissions induces a change in their concentrations. We decided to present only in Table 1 the two tests where the biogenic and anthropogenic emissions of terpenoids have been increased. Under the “Anthro_high” scenario, the biogenic emissions of isoprene still dominate, while the anthropogenic ones of α- and β-pinenes and limonene prevail even more. With increasing anthropogenic emissions, the effect on concentrations is highest for limonene (123% of increase), and this leads to a significant rise for the 3 other targeted terpenoids (47% for α-pinene, 80% for β-pinene, and 103% for isoprene in average). The increase in biogenic emissions leads to a much more limited rise in concentrations, with the highest value observed for isoprene (87%).
| Min–max | Mean (±SD) | |||
|---|---|---|---|---|
| Sensitivity tests | Anthro_high | Bio_high | Anthro_high | Bio_high |
| Terpenoids | ||||
| Isoprene | 22–282 | 5–125 | 103 (±87) | 87 (±42) |
| α-Pinene | 35–59 | 37–58 | 47 (±7) | 45 (±5) |
| β-Pinene | 71–91 | 9–32 | 80 (±7) | 19 (±8) |
| Limonene | 106–138 | 12–34 | 123 (±10) | 19 (±5) |
|
||||
| Oxidation products | ||||
| MVK | 7–35 | 4–112 | 20 (±7) | 61 (±39) |
| MACR | 18–47 | 12–123 | 30 (±6) | 81 (±39) |
| Methylglyoxal | 11–33 | 11–58 | 18 (±7) | 43 (±16) |
| Glyoxal | 7–23 | 5–31 | 12 (±5) | 24 (±9) |
| Glycolaldehyde | 22–42 | 24–107 | 29 (±6) | 88 (±26) |
| Hydroxyacetone | 30–77 | 88–156 | 44 (±13) | 141 (±20) |
| Nopinone | 71–98 | 17–30 | 83 (±9) | 24 (±4) |
| Pinonaldehyde | 37–60 | 44–54 | 46 (±6) | 50 (±3) |
| Pinonic acid | 38–58 | 41–53 | 48 (±5) | 48 (±3) |
| Limonaldehyde | 108–142 | 17–24 | 121 (±9) | 22 (±2) |
|
||||
| Oxidants | ||||
| OH | 3–8 | 3–21 | 4 (±1) | 10 (±5) |
| Ozone | 0.3–2 | 0.3–6 | 1 (±0.5) | 3 (±2) |
Hence, as anticipated, these sensitivity tests not only impact the concentrations of the precursors but also those of their oxidation products. The latter, which are produced by chemical reactivity, are modified in almost the same order of magnitude as their emitted precursors (Table 1). MVK and MACR, the two main first generation oxidation products of isoprene, present concentrations affected by the increase in anthropogenic isoprene emissions by almost half of those affected by the increase in biogenic isoprene emissions. This is related to the fact that increasing biogenic emissions results in isoprene concentration almost double the isoprene concentration level reached by increasing anthropogenic emissions. The same conclusions can be drawn for other first- and second-generation products of isoprene such as methylglyoxal, glyoxal, glycolaldehyde and hydroxyacetone. However, nopinone and limonaldehyde produced through the oxidation of β-pinene and limonene show the highest concentrations for the “Anthro_high” scenario. Nevertheless, the relative increase in concentration of pinonaldehyde and pinonic acid produced by the oxidation of α-pinene is similar for both anthropogenic and biogenic sensitivity tests.
Concerning oxidants, increasing anthropogenic terpenoid emissions over 24 hours results in OH and ozone concentration changes of 4 (±1)% and 1 (±0.5)%, respectively, while increasing the biogenic terpenoid emissions impacts the two species by 10 (±5)% and 3 (±2)%, respectively. The increase in ozone with increasing VOC is consistent with the high-NOx chemical regimes.
In addition, the scenarios for varying terpenoid emissions have no significant effect on the concentrations of alkanes, aromatics or alkenes.
3.5 Secondary organic aerosols (SOAs) estimated from modeled terpenoid levels
The reactivity of terpenoids can contribute to the generation of SOAs, a key process in the evaluation of air quality. This last section evaluates the ability of terpenoids to form SOAs under the Paris summertime conditions and the respective role of their biogenic and anthropogenic emissions. SOAs are formed by the condensation or reactive uptake of oxidation products of VOC; the SOA formation potential (i.e., yield) through VOC oxidation depends mainly on their structures (carbon chain length, number and type of functional groups, etc.) and environmental conditions such as temperature, relative humidity, oxidant, organic aerosol, and NOx concentrations.47Formation yield “Y” expresses the capacity of a precursor to form SOAs. Defined by Pandis et al.,48Y corresponds to the quantity of a precursor gaseous organic compound that is converted into a SOA, i.e. (eqn (5)):
 | (5) |
These yields are determined in simulation chambers and/or in field experiments. These yields are usually quantified under different conditions such as high or low NOx concentrations.49 All these yield estimates were performed on a selection of precursor gases for specific oxidants (OH, O3, and NO3). In the work of Ait-Helal et al.,21 the potential SOA mass production was estimated from the measured concentrations of its anthropogenic gaseous precursors. They used a time resolved approach adapted from a previous study from Sjostedt et al.50 to quantify the relative contribution of measured anthropogenic gaseous precursors to SOA formation, namely aromatics and long-chain alkanes. In this work, we followed this approach to evaluate SOA production from simulated concentrations of terpenoids.
By assuming that these VOC precursors only form SOAs through their oxidation and SOAs are not subject to loss by deposition,50 we can estimate the potential SOA mass concentration C(SOA) produced on an hourly basis Δt by each terpenoid oxidized by a specific oxidant.
The potential produced SOA mass concentrations for a compound i C(SOA)estimated(i)(t) have been calculated with the model over the simulation time (t). For this, we are using the outputs of the model kTerpi(t), [Ox(t)], and kTerpi(t) as follows:
| Δ[Terpi](t) = kTerpi(t) × [Terpi(t)] × [Ox(t)] × Δt | (6) |
- kTerpi(t) is the rate constant of terpenoid i with an oxidant (Ox) (cm3 molec−1 s−1) during the simulation time (t);
- [Ox(t)] is the concentration of the oxidant (molecule cm−3) (Ox = OH, O3, NO3) during the simulation time (t);
- [Terpi(t)] is the concentration of a given terpenoid (isoprene, α-pinene, β-pinene, or limonene) during the simulation time (t);
- Δt corresponds to the time step of the model (3600 seconds in our case).
Each hour, we can estimate the produced SOA concentration for each terpenoid i:
| C(SOA)estimated(i)(t) = ΔTrepi(t) × YSOA,i | (7) |
The 24 h SOA concentration produced for one specific terpenoid is therefore:
 | (8) |
Finally, the total concentration of SOAs for all terpenoids over 24 h of simulation is then calculated as follows:
 | (9) |
Table 2 lists the SOA yields used in this work from chamber experiments; the conditions of the chamber experiments are highly variable. We selected SOA yields with the 3 oxidants that were evaluated under conditions representative of those encountered during the MEGAPOLI field campaign (NOx, RH, T, …).
| Oxidant | Terpenoids | Isoprene | α-Pinene | β-Pinene | Limonene |
|---|---|---|---|---|---|
| a Ref. 51. b Ref. 52. c Ref. 53. d Ref. 54. e Ref. 55. f Ref. 56. g Ref. 56. h Ref. 57. i Ref. 58. | |||||
| OH | SOA yield (μg m−3 ppm−1) | 85a | 192b | 881c | 1186b |
| k(OH) (cm3 molec−1 s−1) | 1.0 × 10−10 | 5.3 × 10−11 | 7.9 × 10−11 | 1.7 × 10−10 | |
| O3 | SOA yield (μg m−3 ppm−1) | 20d | 1298e | 452f | 1807g |
| k(O3) (cm3 molec−1 s−1) | 1.2 × 10−17 | 9.3 × 10−17 | 1.9 × 10−17 | 2.1 × 10−16 | |
| NO3 | SOA yield (μg m−3 ppm−1) | 395h | 339b | 1863i | 1468b |
| k(NO3) (cm3 molec−1 s−1) | 6.9 × 10−13 | 6.3 × 10−12 | 2.5 × 10−12 | 1.2 × 10−11 | |
We used the model-simulated concentration data and model kinetic constants from the MCM mechanism to calculate over the last day of the simulation the potential daily total mass concentration of SOAs produced by the 4 studied terpenoids (isoprene, α- and β-pinene and limonene), for the reference case and the sensitivity scenarios “Anthro_high” and “Bio_high” (Fig. 5). SOA mass concentration was also analyzed by distinguishing the contribution of the oxidation by OH, O3 and NO3 for the 4 studied compounds (Fig. S6–S8†).
 | ||
| Fig. 5 Potential SOA mass concentrations produced from terpenoids predicted with the model: (a) “Reference” case; (b) “Anthro_high” case; (c) “Bio_high” case. (d): SOA mass concentration predicted over one day. | ||
For the reference case, the total estimated produced SOA mass concentration over 24 h is equal to 0.4 μg m−3 with a maximum hourly produced mass concentration of 0.032 μg m−3. When anthropogenic and biogenic terpenoid emissions are maximized, the SOA mass concentration level increases by a factor of 1.7 for both to reach 0.7 μg m−3 each, over 24 h (Fig. 5(d)). Regarding the reference case, the two main contributors to SOA mass formation are isoprene and limonene, representing 46% and 38% of the potential daily SOA mass production, respectively. During the day, the higher reactivity of isoprene and limonene compared to that of α and β-pinenes explains this statement; the contribution of O3 to SOA production is much lower than OH since it represents 28% of the diurnal SOA production (Fig. S6 and S7†). Moreover, limonene has the highest SOA formation yield compared to the other three terpenoids (Table 2) by one order of magnitude, explaining its major contribution even when its concentration is the lowest. At night, isoprene and limonene still contribute to the SOA production through their reactivity with NO3 (Fig. S8†). The nighttime production is significant since it represents 27% of the total SOA potential production through the reactivity of terpenoids with OH, O3, and NO3.
For the “Bio_high” scenario, isoprene and limonene are responsible for 90% of the SOA total mass production during one day. For isoprene, its relative contribution reached 65% on average, knowing that its emission intensity has been increased by 252% compared to a 195% increase for other terpenes. This increase is observed during daytime (Fig. 5(c)), resulting in a lower contribution of the nighttime production of SOAs (18%). For the “Anthro_high” case, the two main contributors to SOA mass formation are isoprene and limonene, representing 43% and 45% of the SOA production, respectively. The SOA production is enhanced during the 24 h of simulation; the nighttime production is, therefore, still important around 24% of the SOA production.
Among VOCs, BTEX (benzene, toluene, ethylbenzene, and xylenes) are also well-known SOA precursors.59 To answer the question of how terpenoids can compete with other anthropogenic compounds in SOA formation, we applied the same methodology using model outputs (see Fig. S9 and Table S8†). In the reference case, the estimated total concentration of SOAs produced over 24 h from BTEX is comparable to that from terpenoids. It is estimated to be 0.68 μg m−3 (vs. 0.4 μg m−3 for terpenoids). The maximum concentration at midday reaches a value of 0.094 μg m−3 for BTEX, which is significantly higher than the maximal SOA production from terpenoids. Daily production levels of SOAs from BTEX and terpenoids are comparable; as in the case of terpenoids, we were able to assess SOA production through their reactivity with other oxidants such as O3 or NO3, leading to significant SOA production at night. More specifically, toluene accounts for 91% of the SOA total mass production from BTEX over one day, despite the higher yield of benzene. This is attributed to the significantly higher simulated concentration of toluene compared to that of benzene.
This study is the first one attempting to estimate the contribution of anthropogenic and biogenic terpenoids to SOA production. While the terpenoid-derived SOA potential production cannot be directly compared to previous SOA estimations, our estimations bring new insights into the MEGAPOLI hypothesis. By combining two approaches derived from in situ observations, Ait-Helal et al.21 estimated that aromatics and intermediate VOC precursors (alkanes from C10 to C16) explained 11–36% and 2–7% of the measured SOA, respectively. However, 62 to 78% of the SOA remained unexplained. One reason was the exclusion of biogenic precursors, which was supported by the regional modelling60 that attributed 40% of SOAs to biogenic VOCs and long-range transport. By assimilating SOA mass concentration to its PMF-OOA factor derived from aerosol mass spectrometer measurements,61 the hourly mean mass concentration of SOAs is approximately 1 μg m−3 over the month of July and is consistent between the three Paris stationary sites, including LHVP and SIRTA. By applying the percentage contribution of aromatics and IVOCs to SOAs from Ait-Helal et al.,21 the anthropogenic SOA mass concentrations lie between 0.13 and 0.43 μg m−3. The terpenoid-derived SOA mass concentrations estimated here lie between 0.01 and 0.07 μg m−3 (Fig. 5): it is in the same order of magnitude as the IVOC-derived SOA mass concentrations (0.02 and 0.07 μg m−3), giving confidence to our estimations. However, almost half of the measured SOAs is not explained, and the contribution of terpenoids is not sufficient to fill the gap. While it is out of the scope of the paper, other hypotheses still need to be investigated including the ones already raised by Ait-Helal et al.21 and Zhang et al.60: the role of long range transport given the regional feature of SOAs in the Paris area, the underestimation of IVOC emissions for which the range of volatility was limited, the underestimation of the biogenic emissions of terpenoids, the non-representation of sesquiterpene biogenic emissions, the non-representation of VCP anthropogenic emissions in the model including terpenoids and other compounds,8 and the non-representation of the autooxidation processes of terpenes leading to highly oxygenated molecules (HOMs).
To go further, Fig. 6 represents the relative contribution of terpenoids to the SOA production by distinguishing their biogenic vs. anthropogenic origins for the reference and the two sensitivity tests. For the reference case, terpenoids from anthropogenic sources could exhibit 50% of the potential SOA formation during the 24 h of the simulation; their relative contribution can reach 80% at night and be as low as 28% during the day. For the “Anthro_high” case, this relative contribution reaches 71% over 24 h, whereas for the “Bio_high” case, the terpenoids from biogenic origin induce 70% of the SOA formation over 24 h. Therefore, the production of SOAs through the oxidation of terpenoids emitted from anthropogenic sources has been shown by this modeling study to be competitive with that derived from their biogenic sources.
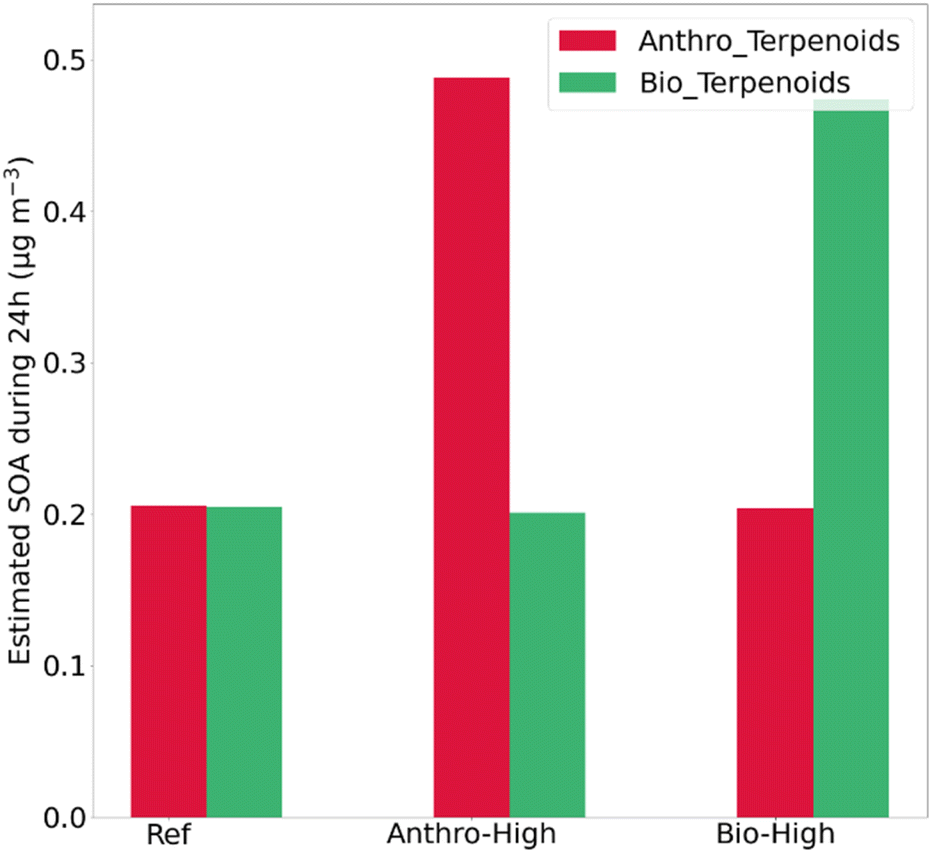 | ||
| Fig. 6 SOA mass concentration predicted over one day by separating the contribution of biogenic and anthropogenic sources of terpenoids. | ||
4. Conclusions
This study evaluated the role of anthropogenic and biogenic terpenoids in secondary pollution in the Paris megacity as representative of northern mid-latitude cities by a box modelling approach. A physico-chemical scenario typical of summertime conditions was defined based on the Paris MEGAPOLI-EU campaign of July 2009.First, the model was designed to reproduce the summertime physico-chemical environment by including the complete version of the Master Chemical Mechanism, anthropogenic VOC emissions by the application of VOC-to-CO emission ratios, and a new biogenic VOC emission inventory recently developed for Paris. The model demonstrated its ability to reproduce major oxidants and trace gas concentrations.
The increase in ozone does not exceed 3% ± 2% in all the simulations (reference case and sensitivity tests modulating anthropogenic and biogenic emissions of terpenoids). However, terpenoid emissions result in a significant increase in total potential SOA mass concentration over a 24 h period, with isoprene and limonene identified as the primary contributors accounting for 42 and 41% of total SOA mass production (0.53 μg m−3). Moreover, terpenoids from anthropogenic sources induce 57% of the SOA formation during the 24 h of simulation; their relative contribution can be up to 86% at night and down to 34% during the day.
This work is the first modelling work evaluating the respective role of anthropogenic and biogenic terpenoids in secondary pollution at the urban scale. First, it suggests that the anthropogenic emissions of terpenoids are competitive with their biogenic emissions regarding SOA production. The systematic presence of anthropogenic emissions of terpenoids is now well established in contrasting cities worldwide,12 and therefore, there is an urgent need to accurately quantify those emissions in emission inventories. Hence, source apportionment studies of organic aerosol relying on the chemical tracer approach must incorporate the new findings. Second, implementing the box model to other contrasting urban environments in terms of climatic conditions and emissions would be useful to complete this work. Third, our study raises the role of terpenoids in nighttime chemistry and secondary pollutant production. It may therefore be useful for simulating concentrations during other field campaigns to use emission ratios determined with in situ measurements when emission inventories do not detail all the emissions of VOCs.
Data availability
The model used in this study is based on the DSMACC model available at https://github.com/barronh/DSMACC. The gaseous chemical mechanism is the Master Chemical Mechanism (v3.3.1) available at https://mcm.york.ac.uk/MCM/. VOC and trace gas concentrations measured at LHVP during the MEGAPOLI experiment can be retrieved from https://cds-espri.ipsl.upmc.fr/etherTypo/index.php?id=1450%26L=1.Author contributions
AB, CA and LD designed the project. LP, MF and LD were responsible for the modeling and performed the simulations and data analysis. MC participated actively in the model developments. CA, MF, LP, AB and LD conducted the scientific analyses. MF, LP, and LD prepared the manuscript, analyzed the data, and designed the figures, with contributions from all authors. MC, AM, KS, and CA revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Olivier Perrussel from AIRPARIF for fruitful discussions on the emission inventory. This work is part of the DATAbASE project (Do Anthropogenic emissions of Terpenoids mAtter in AtmoSpheric chEmistry ?) supported by the French National Program LEFE (“Les Enveloppes Fluides et l’Environement”). CEA (contract no. CEA CAJ_18-100/C34067 – CNRS 217485) funded the PhD contract of Lucas Pailler. LaMP acknowledges the Ecole Doctorale des Sciences Fondamentales (EDSF, Clemont-Ferrand University), the CIR 4/I-Site 2025 and the InVolc Graduate Track of Clermont Auvergne University for their financial contribution to Mariana Farhat's grant. EMMA also acknowledges the Research Council and the Faculty of Sciences at Saint Joseph University of Beirut – Lebanon for Mariana Farhat's scholarship.References
- G. Curci, P. I. Palmer, T. P. Kurosu, K. Chance and G. Visconti, Estimating European volatile organic compound emissions using satellite observations of formaldehyde from the Ozone Monitoring Instrument, Atmos. Chem. Phys., 2010, 10, 11501–11517 CrossRef CAS.
- C. L. Faiola, M. Wen and T. M. VanReken, Chemical characterization of biogenic secondary organic aerosol generated from plant emissions under baseline and stressed conditions: inter- and intra-species variability for six coniferous species, Atmos. Chem. Phys., 2015, 15, 3629–3646 CrossRef CAS.
- B. Nozière, M. Kalberer, M. Claeys, J. Allan, B. D'Anna, S. Decesari, E. Finessi, M. Glasius, I. Grgić, J. F. Hamilton, T. Hoffmann, Y. Iinuma, M. Jaoui, A. Kahnt, C. J. Kampf, I. Kourtchev, W. Maenhaut, N. Marsden, S. Saarikoski, J. Schnelle-Kreis, J. D. Surratt, S. Szidat, R. Szmigielski and A. Wisthaler, The molecular identification of organic compounds in the atmosphere: State of the art and challenges, Chem. Rev., 2015, 115, 3919–3983 CrossRef PubMed.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- H. Wang, X. Liu, C. Wu and G. Lin, Regional to global distributions, trends, and drivers of biogenic volatile organic compound emission from 2001 to 2020, Atmos. Chem. Phys., 2024, 24, 3309–3328 CrossRef CAS.
- A. Rouvière, G. Brulfert, P. Baussand and J.-P. Chollet, Monoterpene source emissions from Chamonix in the Alpine Valleys, Atmos. Environ., 2006, 40, 3613–3620 CrossRef.
- J. B. Gilman, B. M. Lerner, W. C. Kuster, P. D. Goldan, C. Warneke, P. R. Veres, J. M. Roberts, J. A. de Gouw, I. R. Burling and R. J. Yokelson, Biomass burning emissions and potential air quality impacts of volatile organic compounds and other trace gases from fuels common in the US, Atmos. Chem. Phys., 2015, 15, 13915–13938 CrossRef CAS.
- M. M. Coggon, G. I. Gkatzelis, B. C. McDonald, J. B. Gilman, R. H. Schwantes, N. Abuhassan, K. C. Aikin, M. F. Arend, T. A. Berkoff, S. S. Brown, T. L. Campos, R. R. Dickerson, G. Gronoff, J. F. Hurley, G. Isaacman-VanWertz, A. R. Koss, M. Li, S. A. McKeen, F. Moshary, J. Peischl, V. Pospisilova, X. Ren, A. Wilson, Y. Wu, M. Trainer and C. Warneke, Volatile chemical product emissions enhance ozone and modulate urban chemistry, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2026653118 CrossRef CAS PubMed.
- G. I. Gkatzelis, M. M. Coggon, B. C. McDonald, J. Peischl, J. B. Gilman, K. C. Aikin, M. A. Robinson, F. Canonaco, A. S. H. Prevot, M. Trainer and C. Warneke, Observations confirm that volatile chemical products are a major source of petrochemical emissions in U.S. cities, Environ. Sci. Technol., 2021, 55, 4332–4343 CrossRef CAS PubMed.
- B. C. McDonald, J. A. de Gouw, J. B. Gilman, S. H. Jathar, A. Akherati, C. D. Cappa, J. L. Jimenez, J. Lee-Taylor, P. L. Hayes, S. A. McKeen, Y. Y. Cui, S.-W. Kim, D. R. Gentner, G. Isaacman-VanWertz, A. H. Goldstein, R. A. Harley, G. J. Frost, J. M. Roberts, T. B. Ryerson and M. Trainer, Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Science, 2018, 359, 760–764 CrossRef CAS PubMed.
- M. Farhat, C. Afif, S. Zhang, S. Dusanter, H. Delbarre, V. Riffault, S. Sauvage and A. Borbon, Investigating the industrial origin of terpenoids in a coastal city in northern France: a source apportionment combining anthropogenic, biogenic, and oxygenated VOC, Sci. Total Environ., 2024, 928, 172098 CAS.
- A. Borbon, P. Dominutti, A. Panopoulou, V. Gros, S. Sauvage, M. Farhat, C. Afif, N. Elguindi, A. Fornaro, C. Granier, J. R. Hopkins, E. Liakakou, T. Nogueira, T. Corrêa dos Santos, T. Salameh, A. Armangaud, D. Piga and O. Perrussel, Ubiquity of anthropogenic terpenoids in cities worldwide: emission ratios, emission quantification and implications for urban atmospheric chemistry, J. Geophys. Res. Atmos., 2023, 128, e2022JD037566 CAS.
- P. A. Dominutti, B. T. P. Thera, A. Colomb and A. Borbon, Composition and chemical processing of volatile organic compounds in boundary layer polluted plumes: insights from an airborne Q-PTR-MS on-board the French ATR-42 aircraft, Sci. Total Environ., 2024, 941, 173311 CAS.
- A. Borbon, H. Fontaine, M. Veillerot, N. Locoge, J. C. Galloo and R. Guillermo, An investigation into the traffic-related fraction of isoprene at an urban location, Atmos. Environ., 2001, 35, 3749–3760 CAS.
- C. L. Heald, D. K. Henze, L. W. Horowitz, J. Feddema, J.-F. Lamarque, A. Guenther, P. G. Hess, F. Vitt, J. H. Seinfeld, A. H. Goldstein and I. Fung, Predicted change in global secondary organic aerosol concentrations in response to future climate, emissions, and land use change, J. Geophys. Res. Atmos., 2008, 113, D05211 Search PubMed.
- D. K. Henze and J. H. Seinfeld, Global secondary organic aerosol from isoprene oxidation, Geophys. Res. Lett., 2006, 33, L09812 Search PubMed.
- C. R. Hoyle, T. Berntsen, G. Myhre and I. S. A. Isaksen, Secondary organic aerosol in the global aerosol – chemical transport model Oslo CTM2, Atmos. Chem. Phys., 2007, 7, 5675–5694 CrossRef CAS.
- D. V. Spracklen, J. L. Jimenez, K. S. Carslaw, D. R. Worsnop, M. J. Evans, G. W. Mann, Q. Zhang, M. R. Canagaratna, J. Allan, H. Coe, G. McFiggans, A. Rap and P. Forster, Aerosol mass spectrometer constraint on the global secondary organic aerosol budget, Atmos. Chem. Phys., 2011, 11, 12109–12136 CrossRef CAS.
- K. Tsigaridis, N. Daskalakis, M. Kanakidou, P. J. Adams, P. Artaxo, R. Bahadur, Y. Balkanski, S. E. Bauer, N. Bellouin, A. Benedetti, T. Bergman, T. K. Berntsen, J. P. Beukes, H. Bian, K. S. Carslaw, M. Chin, G. Curci, T. Diehl, R. C. Easter, S. J. Ghan, S. L. Gong, A. Hodzic, C. R. Hoyle, T. Iversen, S. Jathar, J. L. Jimenez, J. W. Kaiser, A. Kirkevåg, D. Koch, H. Kokkola, Y. H. Lee, G. Lin, X. Liu, G. Luo, X. Ma, G. W. Mann, N. Mihalopoulos, J. J. Morcrette, J. F. Müller, G. Myhre, S. Myriokefalitakis, N. L. Ng, D. O'Donnell, J. E. Penner, L. Pozzoli, K. J. Pringle, L. M. Russell, M. Schulz, J. Sciare, Ø. Seland, D. T. Shindell, S. Sillman, R. B. Skeie, D. Spracklen, T. Stavrakou, S. D. Steenrod, T. Takemura, P. Tiitta, S. Tilmes, H. Tost, T. van Noije, P. G. van Zyl, K. von Salzen, F. Yu, Z. Wang, Z. Wang, R. A. Zaveri, H. Zhang, K. Zhang, Q. Zhang and X. Zhang, The AeroCom evaluation and intercomparison of organic aerosol in global models, Atmos. Chem. Phys., 2014, 14, 10845–10895 CrossRef.
- V. Michoud, A. Kukui, M. Camredon, A. Colomb, A. Borbon, K. Miet, B. Aumont, M. Beekmann, R. Durand-Jolibois, S. Perrier, P. Zapf, G. Siour, W. Ait-Helal, N. Locoge, S. Sauvage, C. Afif, V. Gros, M. Furger, G. Ancellet and J. F. Doussin, Radical budget analysis in a suburban European site during the MEGAPOLI summer field campaign, Atmos. Chem. Phys., 2012, 12, 11951–11974 CrossRef CAS.
- W. Ait-Helal, A. Borbon, S. Sauvage, J. A. de Gouw, A. Colomb, V. Gros, F. Freutel, M. Crippa, C. Afif, U. Baltensperger, M. Beekmann, J. F. Doussin, R. Durand-Jolibois, I. Fronval, N. Grand, T. Leonardis, M. Lopez, V. Michoud, K. Miet, S. Perrier, A. S. H. Prévôt, J. Schneider, G. Siour, P. Zapf and N. Locoge, Volatile and intermediate volatility organic compounds in suburban Paris: variability, origin and importance for SOA formation, Atmos. Chem. Phys., 2014, 14, 10439–10464 CrossRef.
- A. Baudic, V. Gros, S. Sauvage, N. Locoge, O. Sanchez, R. Sarda-Estève, C. Kalogridis, J. E. Petit, N. Bonnaire, D. Baisnée, O. Favez, A. Albinet, J. Sciare and B. Bonsang, Seasonal variability and source apportionment of volatile organic compounds (VOCs) in the Paris megacity (France), Atmos. Chem. Phys., 2016, 16, 11961–11989 CrossRef CAS.
- V. Gros, C. Gaimoz, F. Herrmann, T. Custer, J. Williams, B. Bonsang, S. Sauvage, N. Locoge, O. d'Argouges, R. Sarda-Estève and J. Sciare, Volatile organic compounds sources in Paris in spring 2007. Part I: qualitative analysis, Environ. Chem., 2011, 8, 74–90 CrossRef CAS.
- J. Sciare, O. d'Argouges, Q. J. Zhang, R. Sarda-Estève, C. Gaimoz, V. Gros, M. Beekmann and O. Sanchez, Comparison between simulated and observed chemical composition of fine aerosols in Paris (France) during springtime: contribution of regional versus continental emissions, Atmos. Chem. Phys., 2010, 10, 11987–12004 CrossRef CAS.
- M. Haeffelin, L. Barthès, O. Bock, C. Boitel, S. Bony, D. Bouniol, H. Chepfer, M. Chiriaco, J. Cuesta, J. Delanoë, P. Drobinski, J. L. Dufresne, C. Flamant, M. Grall, A. Hodzic, F. Hourdin, F. Lapouge, Y. Lemaître, A. Mathieu, Y. Morille, C. Naud, V. Noël, W. O'Hirok, J. Pelon, C. Pietras, A. Protat, B. Romand, G. Scialom and R. Vautard, SIRTA, a ground-based atmospheric observatory for cloud and aerosol research, Ann. Geophys., 2005, 23, 253–275 CrossRef.
- J. Sciare, O. d'Argouges, R. Sarda-Estève, C. Gaimoz, C. Dolgorouky, N. Bonnaire, O. Favez, B. Bonsang and V. Gros, Large contribution of water-insoluble secondary organic aerosols in the region of Paris (France) during wintertime, J. Geophys. Res. Atmos., 2011, 116 Search PubMed.
- C. Mouchel-Vallon, L. Deguillaume, A. Monod, H. Perroux, C. Rose, G. Ghigo, Y. Long, M. Leriche, B. Aumont, L. Patryl, P. Armand and N. Chaumerliac, CLEPS 1.0: a new protocol for cloud aqueous phase oxidation of VOC mechanisms, Geosci. Model Dev., 2017, 10, 1339–1362 CrossRef CAS.
- C. Rose, N. Chaumerliac, L. Deguillaume, H. Perroux, C. Mouchel-Vallon, M. Leriche, L. Patryl and P. Armand, Modeling the partitioning of organic chemical species in cloud phases with CLEPS (1.1), Atmos. Chem. Phys., 2018, 18, 2225–2242 CrossRef CAS.
- K. M. Emmerson and M. J. Evans, Comparison of tropospheric gas-phase chemistry schemes for use within global models, Atmos. Chem. Phys., 2009, 9, 1831–1845 CrossRef CAS.
- V. Damian, A. Sandu, M. Damian, F. Potra and G. R. Carmichael, The kinetic preprocessor KPP-a software environment for solving chemical kinetics, Comput. Chem. Eng., 2002, 26, 1567–1579 CrossRef CAS.
- M. E. Jenkin, J. C. Young and A. R. Rickard, The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 2015, 15, 11433–11459 CrossRef CAS.
- C. M. Nussbaumer and R. C. Cohen, The role of temperature and NOx in ozone trends in the Los Angeles basin, Environ. Sci. Technol., 2020, 54, 15652–15659 CrossRef CAS PubMed.
- A. Borbon, J. B. Gilman, W. C. Kuster, N. Grand, S. Chevaillier, A. Colomb, C. Dolgorouky, V. Gros, M. Lopez, R. Sarda-Esteve, J. Holloway, J. Stutz, H. Petetin, S. McKeen, M. Beekmann, C. Warneke, D. D. Parrish and J. A. de Gouw, Emission ratios of anthropogenic volatile organic compounds in northern mid-latitude megacities: observations versus emission inventories in Los Angeles and Paris, J. Geophys. Res. Atmos., 2013, 118, 2041–2057 Search PubMed.
- L. Menut, B. Bessagnet, D. Khvorostyanov, M. Beekmann, N. Blond, A. Colette, I. Coll, G. Curci, G. Foret, A. Hodzic, S. Mailler, F. Meleux, J. L. Monge, I. Pison, G. Siour, S. Turquety, M. Valari, R. Vautard and M. G. Vivanco, CHIMERE 2013: a model for regional atmospheric composition modelling, Geosci. Model Dev., 2013, 6, 981–1028 CrossRef.
- P. Dominutti, S. Keita, J. Bahino, A. Colomb, C. Liousse, V. Yoboué, C. Galy-Lacaux, E. Morris, L. Bouvier, S. Sauvage and A. Borbon, Anthropogenic VOCs in Abidjan, southern West Africa: from source quantification to atmospheric impacts, Atmos. Chem. Phys., 2019, 19, 11721–11741 CrossRef CAS.
- E. von Schneidemesser, B. C. McDonald, H. Denier van der Gon, M. Crippa, D. Guizzardi, A. Borbon, P. Dominutti, G. Huang, G. Jansens-Maenhout, M. Li, C.-F. Ou-Yang, S. Tisinai and J.-L. Wang, Comparing urban anthropogenic NMVOC measurements with representation in emission inventories—a global perspective, J. Geophys. Res. Atmos., 2023, 128, e2022JD037906 CrossRef CAS.
- J. A. de Gouw, J. B. Gilman, S.-W. Kim, S. L. Alvarez, S. Dusanter, M. Graus, S. M. Griffith, G. Isaacman-VanWertz, W. C. Kuster, B. L. Lefer, B. M. Lerner, B. C. McDonald, B. Rappenglück, J. M. Roberts, P. S. Stevens, J. Stutz, R. Thalman, P. R. Veres, R. Volkamer, C. Warneke, R. A. Washenfelder and C. J. Young, Chemistry of volatile organic compounds in the Los Angeles basin: Formation of oxygenated compounds and determination of emission ratios, J. Geophys. Res. Atmos., 2018, 123, 2298–2319 CrossRef CAS.
- J. A. de Gouw, J. B. Gilman, S.-W. Kim, B. M. Lerner, G. Isaacman-VanWertz, B. C. McDonald, C. Warneke, W. C. Kuster, B. L. Lefer, S. M. Griffith, S. Dusanter, P. S. Stevens and J. Stutz, Chemistry of volatile organic compounds in the Los Angeles basin: nighttime removal of alkenes and determination of emission ratios, J. Geophys. Res. Atmos., 2017, 122, 11843–11861 Search PubMed.
- A. Maison, L. Lugon, S. J. Park, A. Baudic, C. Cantrell, F. Couvidat, B. D'Anna, C. Di Biagio, A. Gratien, V. Gros, C. Kalalian, J. Kammer, V. Michoud, J. E. Petit, M. Shahin, L. Simon, M. Valari, J. Vigneron, A. Tuzet and K. Sartelet, Significant impact of urban tree biogenic emissions on air quality estimated by a bottom-up inventory and chemistry transport modeling, Atmos. Chem. Phys., 2024, 24, 6011–6046 Search PubMed.
- B. Thera, P. Dominutti, A. Colomb, V. Michoud, J. F. Doussin, M. Beekmann, F. Dulac, K. Sartelet and A. Borbon, O3–NOy photochemistry in boundary layer polluted plumes: insights from the MEGAPOLI (Paris), ChArMEx/SAFMED (North West Mediterranean) and DACCIWA (southern West Africa) aircraft campaigns, Environ. Sci.: Atmos., 2022, 2, 659–686 CAS.
- E. Penn and T. Holloway, Evaluating current satellite capability to observe diurnal change in nitrogen oxides in preparation for geostationary satellite missions, Environ. Res. Lett., 2020, 15, 034038 Search PubMed.
- T. Salameh, S. Sauvage, N. Locoge, J. Gauduin, O. Perrussel and A. Borbon, Spatial and temporal variability of BTEX in Paris megacity: two-wheelers as a major driver, Atmos. Environ. X, 2019, 1, 100003 CAS.
- P. Guo, Y. Su, X. Sun, C. Liu, B. Cui, X. Xu, Z. Ouyang and X. Wang, Urban–rural comparisons of biogenic volatile organic compounds and ground-level ozone in Beijing, Forests, 2024, 15(3), 508 CrossRef.
- A. Borbon, T. Salameh, S. Sauvage and C. Afif, Light oxygenated volatile organic compound concentrations in an Eastern Mediterranean urban atmosphere rivalling those in megacities, Environ. Pollut., 2024, 350, 123797 CrossRef CAS PubMed.
- A. Panopoulou, E. Liakakou, S. Sauvage, V. Gros, N. Locoge, I. Stavroulas, B. Bonsang, E. Gerasopoulos and N. Mihalopoulos, Yearlong measurements of monoterpenes and isoprene in a Mediterranean city (Athens): natural vs. anthropogenic origin, Atmos. Environ., 2020, 243, 117803 CrossRef CAS.
- A. M. Yeoman, M. Shaw, N. Carslaw, T. Murrells, N. Passant and A. C. Lewis, Simplified speciation and atmospheric volatile organic compound emission rates from non-aerosol personal care products, Indoor Air, 2020, 30, 459–472 CrossRef CAS PubMed.
- J. H. Kroll and J. H. Seinfeld, Chemistry of secondary organic aerosol: formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS.
- S. N. Pandis, R. A. Harley, G. R. Cass and J. H. Seinfeld, Secondary organic aerosol formation and transport, Atmos. Environ. Part A. General Top., 1992, 26, 2269–2282 CrossRef.
- L. Stirnweis, C. Marcolli, J. Dommen, P. Barmet, C. Frege, S. M. Platt, E. A. Bruns, M. Krapf, J. G. Slowik, R. Wolf, A. S. H. Prévôt, U. Baltensperger and I. El-Haddad, Assessing the influence of NOx concentrations and relative humidity on secondary organic aerosol yields from α-pinene photo-oxidation through smog chamber experiments and modelling calculations, Atmos. Chem. Phys., 2017, 17, 5035–5061 CrossRef CAS.
- S. J. Sjostedt, J. G. Slowik, J. R. Brook, R. Y. W. Chang, C. Mihele, C. A. Stroud, A. Vlasenko and J. P. D. Abbatt, Diurnally resolved particulate and VOC measurements at a rural site: indication of significant biogenic secondary organic aerosol formation, Atmos. Chem. Phys., 2011, 11, 5745–5760 CrossRef CAS.
- J. H. Kroll, N. L. Ng, S. M. Murphy, R. C. Flagan and J. H. Seinfeld, Secondary organic aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 2006, 40, 1869–1877 CrossRef CAS PubMed.
- A. Mutzel, Y. Zhang, O. Böge, M. Rodigast, A. Kolodziejczyk, X. Wang and H. Herrmann, Importance of secondary organic aerosol formation of α-pinene, limonene, and m-cresol comparing day- and nighttime radical chemistry, Atmos. Chem. Phys., 2021, 21, 8479–8498 CrossRef CAS.
- M. Sarrafzadeh, J. Wildt, I. Pullinen, M. Springer, E. Kleist, R. Tillmann, S. H. Schmitt, C. Wu, T. F. Mentel, D. Zhao, D. R. Hastie and A. Kiendler-Scharr, Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 2016, 16, 11237–11248 CrossRef CAS.
- K. Sato, S. Inomata, J.-H. Xing, T. Imamura, R. Uchida, S. Fukuda, K. Nakagawa, J. Hirokawa, M. Okumura and S. Tohno, Effect of OH radical scavengers on secondary organic aerosol formation from reactions of isoprene with ozone, Atmos. Environ., 2013, 79, 147–154 Search PubMed.
- T. Nah, R. C. McVay, X. Zhang, C. M. Boyd, J. H. Seinfeld and N. L. Ng, Influence of seed aerosol surface area and oxidation rate on vapor wall deposition and SOA mass yields: a case study with α-pinene ozonolysis, Atmos. Chem. Phys., 2016, 16, 9361–9379 CrossRef CAS.
- D. C. Draper, D. K. Farmer, Y. Desyaterik and J. L. Fry, A qualitative comparison of secondary organic aerosol yields and composition from ozonolysis of monoterpenes at varying concentrations of NO2, Atmos. Chem. Phys., 2015, 15, 12267–12281 CrossRef CAS.
- N. L. Ng, A. J. Kwan, J. D. Surratt, A. W. H. Chan, P. S. Chhabra, A. Sorooshian, H. O. T. Pye, J. D. Crounse, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 2008, 8, 4117–4140 CrossRef CAS.
- J. L. Fry, D. C. Draper, K. C. Barsanti, J. N. Smith, J. Ortega, P. M. Winkler, M. J. Lawler, S. S. Brown, P. M. Edwards, R. C. Cohen and L. Lee, Secondary organic aerosol formation and organic nitrate yield from NO3 oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 2014, 48, 11944–11953 CrossRef CAS PubMed.
- Q. Li, G. Su, C. Li, P. Liu, X. Zhao, C. Zhang, X. Sun, Y. Mu, M. Wu, Q. Wang and B. Sun, An investigation into the role of VOCs in SOA and ozone production in Beijing, China, Sci. Total Environ., 2020, 720, 137536 Search PubMed.
- Q. J. Zhang, M. Beekmann, E. Freney, K. Sellegri, J. M. Pichon, A. Schwarzenboeck, A. Colomb, T. Bourrianne, V. Michoud and A. Borbon, Formation of secondary organic aerosol in the Paris pollution plume and its impact on surrounding regions, Atmos. Chem. Phys., 2015, 15, 13973–13992 CrossRef CAS.
- F. Freutel, J. Schneider, F. Drewnick, S. L. von der Weiden-Reinmüller, M. Crippa, A. S. H. Prévôt, U. Baltensperger, L. Poulain, A. Wiedensohler, J. Sciare, R. Sarda-Estève, J. F. Burkhart, S. Eckhardt, A. Stohl, V. Gros, A. Colomb, V. Michoud, J. F. Doussin, A. Borbon, M. Haeffelin, Y. Morille, M. Beekmann and S. Borrmann, Aerosol particle measurements at three stationary sites in the megacity of Paris during summer 2009: meteorology and air mass origin dominate aerosol particle composition and size distribution, Atmos. Chem. Phys., 2013, 13, 933–959 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ea00112e |
| ‡ Current address: Laboratoire de Météorologie Dynamique-IPSL, CNRS/Sorbonne Université/École Normale Supérieure-PSL Université/École Polytechnique-Institut Polytechnique de Paris, Paris, 75005, France. |
| This journal is © The Royal Society of Chemistry 2025 |