DOI:
10.1039/D5DT01577D
(Paper)
Dalton Trans., 2025,
54, 17531-17545
Re(I)–quinolinate–pyridyl complexes with disubstituted benzimidazole pharmacophores as anticancer agents
Received
5th July 2025
, Accepted 28th October 2025
First published on 30th October 2025
Abstract
Five new neutral heteroleptic complexes fac-[Re(CO)3(QN)(L-X)] (Re–F, Re–Cl, Re–Br, Re–Me, and Re–Np) comprising the fac-[Re(CO)3]+ core, 8-quinolinate (QN), and the 2-(4-pyridyl)-5,6-disubstituted benzimidazole derivative (L-X = 2-(4-pyridyl)-5,6-difluorobenzimidazole (L-F) or 2-(4-pyridyl)-5,6-dichlorobenzimidazole (L-Cl) or 2-(4-pyridyl)-5,6-dibromobenzimidazole (L-Br) or 2-(4-pyridyl)-5,6-dimethylbenzimidazole (L-Me)) or 2-(4-pyridyl)-naphthanoimidazole (L-Np) were prepared from Re2(CO)10, 8-hydroxyquinoline (H-QN), and L-X via a one-pot approach. These complexes were characterized by ATR-IR, ESI-TOF-MS, NMR, and elemental analysis. Molecular structures of these complexes were determined by single-crystal X-ray diffraction analysis. The cytotoxicity study of the synthesized complexes was performed against cancer cell lines (murine mammary carcinoma 4T1, human lung adenocarcinoma A549, and human cervical carcinoma HeLa) and non-carcinogenic model cell lines (murine fibroblast L929, mouse myoblast C2C12, murine fibroblasts NIH 3T3, and cardiomyoblast H9C2 cells). Notably, complex Re–F, featuring a difluorobenzimidazole motif, exhibited potent anticancer activity with good selectivity towards 4T1 cells, while complex Re–Br, with a dibromobenzimidazole motif, showed better selectivity towards HeLa cells. In vitro studies demonstrated that Re–F and Re–Br induced the generation of reactive oxygen species (ROS), exhibited antiproliferative effects, caused cellular morphological alterations, triggered DNA damage, and induced apoptotic cell death. These complexes exhibited potent tumor growth inhibition against multicellular tumor spheroids. Molecular docking and dynamics studies revealed good binding affinity and interactions of the complexes with B-DNA.
Introduction
Extensive research has been carried out to find new anti-cancer drug candidates to overcome the drawbacks of conventional platinum-based drugs.1,2 Among the other metal complexes, fac-[Re(CO)3]+ core-based coordination complexes have been emerging as among the leading candidates in this field.2 These complexes have many advantages, including kinetic inertness, thermal stability, 3D geometry, and rich photophysical properties.3–6 Notably, few of the rhenium complexes have entered clinical trials, highlighting their promising therapeutic potential, which encourages researchers to develop new potential fac-[Re(CO)3]+ core-based anti-cancer drug molecules.3–6 Apart from the metal center, ligands play a pivotal role in tuning the therapeutic properties of the complexes. Organic molecules such as 8-hydroxyquinoline (H-QN) and its derivatives are also well-known in medicinal chemistry for their antibacterial, antifungal, and anticancer properties.7 Several metal complexes of 8-quinolinate (QN) and its derivatives are also known to exhibit significant potential applications in medicinal fields, especially as potent anticancer agents.8 Recently, rhenium complexes containing 8-quinolinate and 1,3,5-triaza-7-phosphaadamantane were synthesized and evaluated for their anticancer activity against cervical (HeLa) cancer cell lines.9a Very recently, similar rhenium-quinolinate complexes containing 3,5-dimethylpyrazole, 3-phenylpyrazole, imidazole, 2-methylimidazole, and triphenylphosphine were reported. These complexes showed anticancer activities against leukemia (HL-60), ovarian (SKOV-3), prostate (PC-3), and breast (MCF-7) cancer cell lines.9b This indicated that tuning the neutral ligand motifs could also influence the anticancer properties of rhenium–quinolinate complexes. Moreover, benzimidazole and its derivatives are well-established pharmacophores in cancer research due to their ability to interact with a variety of biological targets through π–π stacking, hydrogen bonding contacts, and coordination with metal ions.10 However, reports on the synthesis and cytotoxicity evaluation of fine new Re(I)–quinolinate complexes bearing a free benzimidazole unit are scarce. The presence of a free benzimidazole unit in the complex is expected to enhance interactions with DNA.10g We envision that tuning either the benzimidazole motif by substituting the 5th and 6th positions with functional atoms such as halogen (F, Cl, Br) and a methyl group or extended π-conjugation as a naphthanoimidazole motif may also be capable of interacting with DNA, inducing cancer cell death. Herein, we have synthesized five new rhenium(I) tricarbonyl mononuclear acyclic complexes in combination with 5,6-disubstituted pyridylbenzimidazole fac-[Re(CO)3(QN)(L-X)] (Re–F, (X = F); Re–Cl, (X = Cl); Re–Br, (X = Br); Re–Me, (X = Me)) and the pyridyl naphthanoimidazole motif (Re–Np) (Scheme 1) and studied their cytotoxicity against cancer cell lines (murine mammary carcinoma 4T1, human lung adenocarcinoma A549, and human cervical carcinoma HeLa) and non-carcinogenic model cell lines (murine fibroblast L929, mouse myoblast C2C12, murine fibroblasts NIH 3T3, and cardiomyoblast H9C2 cells). In-depth studies of Re–F on 4T1 cells and Re–Br on HeLa cells were conducted to investigate their ability to generate ROS, exhibit antimetastatic properties, and induce nucleosomal fragmentation. The cytotoxic efficacy of Re–Br and Re–F complexes was evaluated using 3D spheroid models of the HeLa and 4T1 cell lines. The binding affinity and interactions between the metal complexes and various DNA sequences were investigated through molecular docking studies. Molecular dynamics simulation of the metal complexes was carried out for 600 ns to understand and compare the interaction stability of the complexes with DNA.
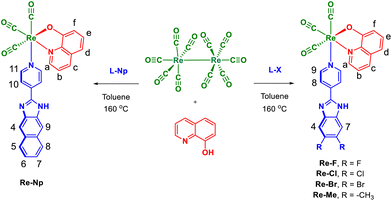 |
| | Scheme 1 Synthesis of complexes Re–F, Re–Cl, Re–Br, Re–Me, and Re–Np. L-X = 2-(4-pyridyl)-5,6-dihalobenzimidazole (or) 2-(4-pyridyl)-5,6-dimethylbenzimidazole. L-Np = 2-(4-pyridyl)-naphthanoimidazole. | |
Results and discussion
Synthesis and characterization of the complexes
2-(4-Pyridyl)-5,6-difluorobenzimidazole (L-F), 2-(4-pyridyl)-5,6-dichlorobenzimidazole (L-Cl), 2-(4-pyridyl)-5,6-dibromobenzimidazole (L-Br), 2-(4-pyridyl)-5,6-dimethylbenzimidazole (L-Me), and 2-(4-pyridyl)-naphthanoimidazole (L-Np) were synthesized by mixing 4,5-difluorobenzene-1,2-diamine/4,5-dichlorobenzene-1,2-diamine/4,5-dibromobenzene-1,2-diamine/4,5-dimethylbenzene-1,2-diamine/naphthalene-2,3-diamine, 4-pyridine carboxaldehyde, and sodium bisulfite in DMF. The content was heated to 80 °C for 5 h (SI).
The ligands were further utilized for the synthesis of five mononuclear acyclic rhenium(I) complexes, Re–F, Re–Cl, Re–Br, Re–Me, and Re–Np, which were obtained by treating Re2(CO)10 with 8-hydroxyquinoline, L-X (X = F/Cl/Br/Me) or L-Np in toluene at 160 °C using a one-pot solvothermal approach (Scheme 1). The complexes are air and moisture-stable. The ATR-IR spectra of the complexes displayed three strong bands in the range of 2015–1882 cm−1, characteristic of the fac-[Re(CO)3]+ core (Fig. S1 in the SI).6 ESI-MS spectra of the complexes displayed molecular ion peaks (m/z 647.0557 for [Re–F + H]+, m/z 678.2133 for [Re–Cl + H]+, m/z 768.8932 for [Re–Br + H]+, m/z 639.1037 for [Re–Me + H]+ and m/z 661.0894 for [Re–Np + H]+) (Fig. S2–S6 in the SI). The proton NMR spectra for these complexes were recorded in DMSO-d6 (Fig. 1A and Fig. S7–S11 in the SI). These complexes displayed well-resolved splitting patterns in comparison with their corresponding free ligands. The proton peaks of these complexes were assigned using the 1H–1H COSY NMR spectra (Fig. S12–S16 in the SI).
 |
| | Fig. 1 (A) Partial 1H NMR spectrum of Re–F indicating isomeric peaks in red and blue; and (B and C) DOSY and 1H–1H COSY spectra of Re–F in d6-DMSO (* = toluene solvent peak). | |
Since all the complexes differ only at the 5th and 6th positions of the benzimidazole motif, their corresponding spectra display similar patterns. The spectra showed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 proton ratio for both ligand motifs in the complexes. In all the complexes, the Ha proton exhibited a downfield shift relative to the free N,O-chelating quinolinate ligand. This shift was attributed to the loss of electron density upon Re metal coordination. Moreover, the 1H-NMR spectra of all complexes exhibited two distinct resonances for each proton, indicating chemically inequivalent environments and suggesting the presence of two solution-phase isomers in varying ratios. These isomers likely correspond to the monomeric and dimeric forms of the complex. This assignment is supported by DOSY NMR measurements, which display two distinct diffusion bands, with coefficients of 1.68 × 10–10 m2 s−1 and 2.32 × 10–10 m2 s−1, confirming the coexistence of two species in solution (Fig. S22).11 The faster diffusion coefficient (2.32 × 10–10 m2 s−1) was attributed to the monomeric species (red protons), while the slower coefficient (1.68 × 10–10 m2 s−1) corresponded to the larger dimeric species (blue protons). Furthermore, concentration-dependent 1H-NMR experiments demonstrated a dynamic equilibrium, with the dimeric species predominating at higher concentrations (15.5 mM) and the monomeric species favoured at lower concentrations (5.5 mM). The above observation clearly reveals the stability of two isomers in the solution. Furthermore, the time-dependent 1H NMR spectra of all complexes in DMSO-d6 confirmed their solution-phase stability for at least 48 hours (Fig. S17–S21 in the SI).
:1 proton ratio for both ligand motifs in the complexes. In all the complexes, the Ha proton exhibited a downfield shift relative to the free N,O-chelating quinolinate ligand. This shift was attributed to the loss of electron density upon Re metal coordination. Moreover, the 1H-NMR spectra of all complexes exhibited two distinct resonances for each proton, indicating chemically inequivalent environments and suggesting the presence of two solution-phase isomers in varying ratios. These isomers likely correspond to the monomeric and dimeric forms of the complex. This assignment is supported by DOSY NMR measurements, which display two distinct diffusion bands, with coefficients of 1.68 × 10–10 m2 s−1 and 2.32 × 10–10 m2 s−1, confirming the coexistence of two species in solution (Fig. S22).11 The faster diffusion coefficient (2.32 × 10–10 m2 s−1) was attributed to the monomeric species (red protons), while the slower coefficient (1.68 × 10–10 m2 s−1) corresponded to the larger dimeric species (blue protons). Furthermore, concentration-dependent 1H-NMR experiments demonstrated a dynamic equilibrium, with the dimeric species predominating at higher concentrations (15.5 mM) and the monomeric species favoured at lower concentrations (5.5 mM). The above observation clearly reveals the stability of two isomers in the solution. Furthermore, the time-dependent 1H NMR spectra of all complexes in DMSO-d6 confirmed their solution-phase stability for at least 48 hours (Fig. S17–S21 in the SI).
Molecular structures of SCCs
Single crystals of the complexes suitable for X-ray diffraction analysis were obtained directly from the solvothermal reaction vessels upon cooling the reaction mixture.
ORTEP diagrams of all the complexes are shown in Fig. 2. The crystallographic details, such as data collection, solution, refinement, and selected bond lengths, are given in Tables S1 and S2 in the SI. Complexes Re–F, Re–Me and Re–Np crystallize in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) while Re–Cl and Re–Br crystallize in P21/c. The asymmetric unit of Re–F, Re–Me, and Re–Np contains one disordered toluene molecule. The complexes consist of one fac-[Re(CO)3]+ core, 8-quinolinate, and a 5,6-disubstituted benzimidazole-pyridyl/naphthanoimidazole pyridyl motif. The Re(I) center exhibits distorted octahedral geometry bound to the nitrogen atom of the pyridylbenzimidazole monodentate ligand, nitrogen and oxygen atoms of the quinolinate bidentate ligand, and three carbon atoms of three facially arranged carbonyl (CO) ligands. Both ligand motifs in the complex are arranged orthogonally to each other, forming an L-shaped framework. The bonding parameters of Re–N4, Re–O4 and Re–N3 are found within the expected range (Table S3 in the SI).6d All the complexes show out-of-plane bending of the neutral pyridyl-benzimidazole motif from the plane through the Re-1, N1, C13 and C17 with dihedral angles of ∼3.5°, 14.7°, 17.2°, 17.3° and 20.9°, respectively. As the size of the substituents at the 5th and 6th positions of the benzimidazole group increases, the degree of bending exhibits a corresponding increase with the following trend: Re–F > Re–Cl > Re–Br > Re–Me > Re–Np. The crystal structures are stabilized by various intermolecular non-covalent interactions. In complex Re–F, two benzimidazole motifs are aligned anti-cofacially, exhibiting π⋯π interactions with a distance of 3.365 Å. The fluorine atom interacts with two CO molecules via intermolecular F⋯O contacts (2.88/2.98 Å).6c Additionally, a C–H group of the pyridyl ring interacts with an adjacent carbonyl unit (C–H⋯O: 2.67 Å and 126°). Displaced π⋯π interactions between two pyridyl units are also observed. Furthermore, the oxygen atom of the quinolinate interacts with the neighboring NH unit (N–H⋯O: 2.035 Å and 166°). In complex Re–Cl, two neighbouring molecules contact each other through lone pair⋯π contacts (Cl⋯CQN/pyridyl = 3.44/3.42 Å). In addition, a strong hydrogen bonding interaction between NH2 and the oxygen atom of quinolinate is also present (N–H⋯O = 2.056 and 179°). A similar type of dimeric structure was observed in the Re–Br complex. However, in the dimeric structure of Re–Br, the bromine atom contacts with the carbon atom of the pyridyl unit. No close contacts were observed between Br and the QN motif. Nitrogen and oxygen donor atoms of quinolinate form a five-membered ring with rhenium and are in plane with two CO groups. This leads to pyridylbenzimidazole and CO groups occupying axial positions.
while Re–Cl and Re–Br crystallize in P21/c. The asymmetric unit of Re–F, Re–Me, and Re–Np contains one disordered toluene molecule. The complexes consist of one fac-[Re(CO)3]+ core, 8-quinolinate, and a 5,6-disubstituted benzimidazole-pyridyl/naphthanoimidazole pyridyl motif. The Re(I) center exhibits distorted octahedral geometry bound to the nitrogen atom of the pyridylbenzimidazole monodentate ligand, nitrogen and oxygen atoms of the quinolinate bidentate ligand, and three carbon atoms of three facially arranged carbonyl (CO) ligands. Both ligand motifs in the complex are arranged orthogonally to each other, forming an L-shaped framework. The bonding parameters of Re–N4, Re–O4 and Re–N3 are found within the expected range (Table S3 in the SI).6d All the complexes show out-of-plane bending of the neutral pyridyl-benzimidazole motif from the plane through the Re-1, N1, C13 and C17 with dihedral angles of ∼3.5°, 14.7°, 17.2°, 17.3° and 20.9°, respectively. As the size of the substituents at the 5th and 6th positions of the benzimidazole group increases, the degree of bending exhibits a corresponding increase with the following trend: Re–F > Re–Cl > Re–Br > Re–Me > Re–Np. The crystal structures are stabilized by various intermolecular non-covalent interactions. In complex Re–F, two benzimidazole motifs are aligned anti-cofacially, exhibiting π⋯π interactions with a distance of 3.365 Å. The fluorine atom interacts with two CO molecules via intermolecular F⋯O contacts (2.88/2.98 Å).6c Additionally, a C–H group of the pyridyl ring interacts with an adjacent carbonyl unit (C–H⋯O: 2.67 Å and 126°). Displaced π⋯π interactions between two pyridyl units are also observed. Furthermore, the oxygen atom of the quinolinate interacts with the neighboring NH unit (N–H⋯O: 2.035 Å and 166°). In complex Re–Cl, two neighbouring molecules contact each other through lone pair⋯π contacts (Cl⋯CQN/pyridyl = 3.44/3.42 Å). In addition, a strong hydrogen bonding interaction between NH2 and the oxygen atom of quinolinate is also present (N–H⋯O = 2.056 and 179°). A similar type of dimeric structure was observed in the Re–Br complex. However, in the dimeric structure of Re–Br, the bromine atom contacts with the carbon atom of the pyridyl unit. No close contacts were observed between Br and the QN motif. Nitrogen and oxygen donor atoms of quinolinate form a five-membered ring with rhenium and are in plane with two CO groups. This leads to pyridylbenzimidazole and CO groups occupying axial positions.
 |
| | Fig. 2 ORTEP diagrams of the molecular structures of the complexes drawn at the 50% probability level. Solvent molecules were removed for clarity. | |
Biological studies
Lipophilicity.
Lipophilicity of a drug molecule is a fundamental parameter to determine biological processes such as membrane penetration capability, absorption, metabolism, and distribution.12 The lipophilicity of the complexes was assessed by calculating the octanol–water partition coefficient (logKo/w) using the shake flask method. The logKo/w values for Re–F and Re–Br were 0.70 and 1.50, respectively, indicating their hydrophobic/lipophilic nature.
Biocompatibility.
Prior to assessing in vitro anticancer efficacy, the cytocompatibility of rhenium(I) tricarbonyl complexes (Re–F, Re–Cl, Re–Br, Re–Me, and Re–Np) was systematically evaluated in non-cancerous cell lines: L929 fibroblasts, NIH 3T3 murine fibroblasts, H9C2 cardiomyoblasts, and C2C12 myoblasts to establish preliminary therapeutic indices. Cells were exposed to a concentration gradient of 1–250 μM (1, 2, 5, 10, 25, 50, 100, and 250) of each complex for 24 hours under standard culture conditions. Dose–response analyses revealed negligible cytotoxicity (cell viability ≥80%) at concentrations ≤25 μM across all cell types (Table 1 and Fig. S23 in the SI), with IC50 values exceeding 50 μM, indicating favourable biocompatibility within therapeutically relevant dosage ranges. This preliminary profiling in diverse normal cell models supports the suitability of these complexes for subsequent anticancer screening while highlighting their potential selectivity toward malignant versus non-malignant tissues.
Table 1 IC50 values of the complexes determined using the MTT assay towards cancer cell lines (4T1, A549, and HeLa) and non-cancer cell lines (L929, NIH 3T3, H9C2 and C2C12)
| IC50 (µM) |
| Cell lines |
Re–F
|
Re–Cl
|
Re–Br
|
Re–Me
|
Re–Np
|
| 4T1 |
16.25 ± 1.63 |
22.92 ± 2.29 |
23.01 ± 2.30 |
18.34 ± 1.83 |
18.59 ± 1.86 |
| A549 |
23.30 ± 2.33 |
19.71 ± 1.97 |
12.87 ± 1.29 |
18.85 ± 1.88 |
24.50 ± 2.45 |
| HeLa |
25.01 ± 2.50 |
20.40 ± 2.04 |
12.27 ± 1.23 |
19.03 ± 1.90 |
26.04 ± 2.60 |
| L929 |
30 |
37.5 |
30 |
40 |
37.5 |
| NIH 3T3 |
30 |
37.5 |
21 |
30 |
37.5 |
| H9C2 |
50 |
50 |
60 |
50 |
50 |
| C2C12 |
70 |
17.5 |
25 |
50 |
37.5 |
Cytotoxicity.
The cytotoxic profiles of rhenium-based complexes Re–F, Re–Cl, Re–Br, Re–Me, and Re–Np were evaluated against 4T1 (murine mammary carcinoma), A549 (human lung adenocarcinoma), and HeLa (human cervical carcinoma) cell lines via an MTT assay. Dose-dependent cytotoxicity analyses revealed notable sensitivity differences: Re–Br exhibited the highest IC50 potency in HeLa cells (12.27 ± 1.23 µM), while Re–F demonstrated optimal IC50 activity in 4T1 cells (16.25 ± 1.63 μM) (Table 1). Untreated cells served as negative controls, with viability normalized to 100%.
Selectivity indices (SI) were calculated to assess therapeutic specificity using the formula: SI = (IC50 value in normal cells)/(IC50 value in cancer cells).13
Thresholds define SI as follows: SI < 1 (normal cell toxicity), SI = 1 (equivalent toxicity), SI > 1 (cancer cell selectivity), and SI ≫ 1 (high specificity). At a threshold of SI ≥ 4, Re–Br showed an SI of 4.88 in HeLa relative to H9C2 cardiomyoblasts, while Re–F achieved an SI of 4.3 in 4T1 compared to C2C12 myoblasts (Table 2 and Fig. S24 in the SI). These values indicate preferential cytotoxicity toward malignant cells, underscoring their potential as selective chemotherapeutic agents with an expanded therapeutic window.
Table 2 Selectivity index values of the complexes determined using the MTT assay towards cancer cell lines (4T1, A549 and HeLa) and non-cancer cell lines (L929, NIH 3T3, H9C2 and C2C12)
| Selectivity index |
Re–Cl
|
Re–Br
|
Re–F
|
Re–Me
|
Re–Np
|
| L929/4T1 |
1.63 |
1.3 |
1.84 |
2.185 |
1.97 |
| L929/A549 |
1.9 |
2.3 |
1.3 |
2.12 |
1.53 |
| L929/HeLa |
1.83 |
2.4 |
1.2 |
2.1 |
1.44 |
| |
| NIH3T3/4T1 |
1.63 |
0.7 |
1.84 |
1.64 |
1.97 |
| NIH3T3/A549 |
1.9 |
1.61 |
1.3 |
1.59 |
1.53 |
| NIH3T3/HeLa |
1.83 |
1.7 |
1.2 |
1.57 |
1.44 |
| |
| H9C2/4T1 |
2.17 |
2.6 |
3.07 |
2.73 |
2 |
| H9C2/A549 |
2.53 |
4.63 |
2.17 |
2.66 |
2.04 |
| H9C2/HeLa |
2.45 |
4.88 |
2 |
2.63 |
1.93 |
| |
| C2C12/4T1 |
0.76 |
1.8 |
4.3 |
2.73 |
1.97 |
| C2C12/A549 |
0.88 |
1.92 |
3.04 |
2.66 |
1.53 |
| C2C12/HeLa |
0.86 |
2.03 |
2.8 |
2.63 |
1.44 |
Live/dead assay
The anticancer efficacy of complexes Re–Br and Re–F was qualitatively assessed via FDA/PI (fluorescein diacetate/propidium iodide) dual staining, a fluorescence-based live/dead assay. Following 24-hour exposure to IC50 concentrations, HeLa and 4T1 cells were incubated with FDA (5 μg mL−1) and PI (2 μg mL−1) for 15 minutes. FDA was hydrolyzed by viable cells to emit green fluorescence (ex/em: 488/530 nm), indicating metabolic activity, while PI selectively stained the nuclei of membrane-compromised cells with red fluorescence (ex/em: 535/617 nm).
Fluorescence microscopy revealed a pronounced increase in PI-positive cells (bright red fluorescence) in Re–Br treated HeLa and Re–F treated 4T1 cultures compared to untreated controls and other treatment groups (Fig. 3). The untreated groups exhibited predominantly green fluorescence, consistent with >90% viability, whereas cisplatin (positive control) showed intermediate PI staining. These observations correlate with prior cytotoxicity data, confirming the selective induction of cell death by Re–Br and Re–F in a cell line-dependent manner.
 |
| | Fig. 3 Live–dead cell images and their quantification analysis of 4T1 cells treated with Re–F/cisplatin (A and B) and HeLa cells treated with Re–Br/cisplatin (C and D). Scale bar: 100 μm. Untreated cells (negative control) and cisplatin (positive control). FDA (fluorescein diacetate) and PI (propidium iodide) dyes stain the live and dead cells, respectively. | |
Reactive oxygen species (ROS) generation
The intracellular ROS generation mediated by complexes Re–Br and Re–F in HeLa and 4T1 cancer cells, respectively, was quantified via the 2′,7′-dichlorofluorescein diacetate (DCFDA) assay, a widely employed fluorometric method for detecting oxidative stress.14 Following 24-hour exposure to the complexes, cells were incubated with 10 µM DCFDA in serum-free medium, allowing the cell-permeable probe to undergo de-esterification by intracellular esterases to non-fluorescent DCFH, which is subsequently oxidized by ROS to fluorescent dichlorofluorescein (DCF). Fluorescence microscopy revealed a marked elevation in DCF-derived green fluorescence intensity (ex/em: 488/530 nm) in treated cells compared to untreated controls and other treatment groups (Fig. 4). HeLa cells treated with Re–Br exhibited a prominent increase in fluorescence signal relative to controls, while Re–F induced a bright green fluorescence in 4T1 cells, indicating ROS accumulation in a cell line- and complex-dependent manner. Untreated cells demonstrated minimal fluorescence, consistent with basal ROS levels maintained by endogenous antioxidant systems. These findings validate the pro-oxidant efficacy of Re–Br and Re–F in destabilizing redox homeostasis, correlating with their observed cytotoxic effects.
 |
| | Fig. 4 Images showing ROS generation (green fluorescence) with (A) 4T1 cells treated with Re–F/cisplatin and (B) HeLa cells treated with Re–Br/cisplatin (scale bar: 100 μm). Untreated cells (negative control) and cisplatin (positive control). | |
Antimigration evaluation
The antimetastatic efficacy of complexes Re–F and Re–Br was evaluated using an in vitro scratch assay to quantify migration inhibition in HeLa and 4T1 cells.15 Confluent monolayers were established in 12-well plates, and uniform scratches were introduced via mechanical disruption with a sterile pipette tip. Cells were treated with IC50 concentrations of Re–Br (HeLa) or Re–F (4T1), while untreated and cisplatin-treated groups served as negative and positive controls. Phase-contrast microscopy images captured at 0, 6, 12, and 24-hour intervals revealed a complete closure of the scratch for the untreated groups. Quantitative analysis demonstrated that only 10% of the scratch area was repopulated by Re–Br and Re–F treated cells after 24 hours, compared to 90–95% closure in untreated controls (Fig. 5 and Fig. S25 in the SI). This 9-fold decrease in migration aligns with prior studies showing that the rhenium complexes disrupt cytoskeletal dynamics and extracellular matrix remodeling through ROS-mediated pathways. The pronounced attenuation of motility underscores the dose-dependent antimetastatic potential of these complexes, likely mediated by their interference with metalloproteinase activity. These findings reveal that complexes Re–Br and Re–F act as promising candidates for inhibiting cancer cell dissemination, a critical target in overcoming therapeutic resistance.
 |
| | Fig. 5 (A) Evaluation of the antimetastatic effects of Re–F on 4T1 cells using the scratch assay, untreated cells (negative control), and cisplatin (positive control) (scale bar: 100 μm). (B) Percentage of scratch closure at 0, 6, 12 and 24 h of treatment. | |
Apoptosis detection
The pro-apoptotic effects of complexes Re–Br and Re–F in HeLa and 4T1 cells were investigated via acridine orange/ethidium bromide (AO/EtBr) dual fluorescence staining to delineate ROS-mediated cell death mechanisms. Post-treatment with IC50 concentrations, cells were co-stained with AO (2 μg mL−1) and EtBr (2 μg mL−1) for 10 minutes, enabling differential visualization of nuclear morphology and membrane integrity. Fluorescence microscopy revealed a significant increase in apoptotic cells (EtBr+/AO+, orange-red nuclei with condensed chromatin) in Re–Br treated HeLa (40 ± 2.8%) and Re–F treated 4T1 (14 ± 1%) populations compared to untreated controls (<5%) (Fig. 6). Untreated cells exhibited uniform green fluorescence. Cisplatin, a positive control, showed intermediate apoptosis (∼9%), corroborating the superior efficacy of rhenium complexes in inducing ROS-dependent apoptotic cascades. These findings align with prior DCFDA data, substantiating the hypothesis that ROS overproduction underlies the selective cytotoxicity of Re–Br and Re–F in malignant cells.
 |
| | Fig. 6 (A and D) Detection of apoptosis using acridine orange (AO) and ethidium bromide (EtBr) on HeLa cells in the presence of Re–Br and 4T1 cells in the presence of Re–F. Living cells are stained green, while apoptotic cells are stained red. Cells without treatment were used as a negative control and cells treated with cisplatin served as a positive control. Scale bar: 50 μm. Quantification data of the fluorescence intensity of AO (B and E) and EtBr (C and F). | |
DNA fragmentation assay
To evaluate the effects of Re–Br and Re–F on the nuclear material, 4,6-diamidino-2-phenylindole (DAPI) staining was utilized.16 DAPI, a fluorescent dye, specifically stains DNA in both viable and non-viable cells; however, it exhibits diminished fluorescence intensity and membrane permeability in viable cells. Untreated HeLa and 4T1 cells (control) displayed preserved nuclear morphology, distinguished by a centrally located blue-fluorescent nucleus. In comparison with the control group, cells treated with Re–Br and Re–F demonstrated higher fluorescence intensity and displayed lobular formations, accompanied by the dispersion or leakage of nuclear fragments, signifying DNA damage (Fig. 7 and Fig. S26 in the SI).
 |
| | Fig. 7 (A) Detection of DNA damage after being treated with Re–Br and cisplatin. Control cells (untreated HeLa cells) were used as a negative control and cells treated with cisplatin were used as a positive control (scale bar: 20 µm). (B) Quantification data of fluorescence intensity. | |
3D spheroid model
The 3D spheroid model experiment provides more accurate mimics of architecture, microenvironment, and physiological conditions of solid tumours compared to traditional two-dimensional (2D) cell cultures.17a 3D spheroid replicates key tumor features including cell–cell and cell–matrix interactions, oxygen and nutrient gradients, and the presence of proliferating, quiescent, and necrotic cell populations, closely resembling the complexity of in vivo tumors.17b,c Thus, the 3D spheroid study reveals tumor biology, drug penetration, and resistance mechanisms and provides predictive and clinically relevant data for evaluating chemotherapeutic efficacy and toxicity. As a result, 3D spheroid cultures serve as a valuable bridge between conventional cell culture and animal models, enhancing the reliability of preclinical drug screening and reducing the need for animal testing.17d Therefore, the cytotoxic efficacy of Re–Br and Re–F complexes was evaluated using 3D spheroid models of the HeLa and 4T1 cell lines. Following spheroid maturation, the constructs were treated with the respective complexes, and viability was assessed 24 hours post-treatment through qualitative live/dead imaging utilizing FDA and PI dual staining. This staining methodology enabled simultaneous visualization of viable cells (fluorescein-positive) and necrotic cellular regions (PI-positive nuclei) within the spheroid architecture. Qualitative analysis revealed significantly reduced cellular viability in treated spheroids compared to untreated controls and other control groups, as evidenced by increased PI penetration and diminished fluorescein signal intensity (Fig. 8). The observed cytotoxic response in 3D cultures correlated with previous findings from 2D monolayer assays, demonstrating consistent compound efficacy across both experimental models. This concordance suggests preserved drug penetration capabilities and biological activity within the spheroid microenvironment, despite the enhanced physiological relevance of 3D systems compared to traditional monolayer cultures.
 |
| | Fig. 8 Cell images of (A) 3-D spheroids of 4T1 cells after 24 h of treatment with 4T1 cells treated with Re–F and (B) HeLa cells treated with Re–Br. Untreated cells (negative control) and cisplatin (positive control). FDA (fluorescein diacetate) and PI (propidium iodide) dyes stain the live and dead cells, respectively. The merged image represents the co-localization of live and dead cells. Scale bar: 100 μm. | |
Molecular docking
The DNA fragmentation assay suggests that the prepared metal complexes cleave the DNA in a specific manner. To investigate the interactions between DNA and the prepared metal complexes, molecular docking simulations were performed. As reported earlier, ligand and DNA can interact via grooving (major or minor groove), inter-crossing, and intercalation.18a Hence, various DNA models such as 1BNA, 1BWG, 1 AU5, 3FT6, and 1Z3F were considered to assess the possible interaction with the metal complexes.18b–e The metal complexes favour minor groove binding with 1BNA and 1BWG and intercalate with 3FT6 and 1Z3F DNA models. The expected covalent cross-linking in the 1 AU5 model, which did not occur as expected, indicates the least binding stability of the metal complexes with DNA. The binding energy of metal complexes with DNA models is given in Table S5. The results show that metal complexes prefer the minor groove and intercalation modes of binding with DNA. For minor groove binding (1BNA) and intercalation (1Z3F), results are considered based on their higher binding energies and interactions. The best binding conformer positions of the complexes with 1BNA and 1Z3F are shown in Fig. 9. All the complexes bind towards the continuous part of the helix (minor groove) with 1BNA. In contrast, the metal complexes are intercalated with 1Z3F in the CG region, particularly the 5,6-disubstituted benzimidazole-pyridyl/naphthanoimidazole pyridyl motif.
 |
| | Fig. 9 The best conformer of metal complexes into DNA is represented in 3D view and 2D interaction of complexes with 1BNA (black box) and 1Z3F (red box) DNA models. (A) Re–F, (B) Re–Cl, (C) Re–Br, (D) Re–Me, and (E) Re–Np complexes (DG – deoxyguanosine, DA – deoxyadenosine, DT – deoxythymidine, and DC – deoxycytidine nucleotides). The metal complexes are colored in blue for better visualization in the 3D representation. In 2D interactions, atom colors are as follows: grey – C, black – H, blue – N, red – O, cyan – Re, pale blue – F, green – Cl, and brown – Br. The interaction distance is specified in Å units, and bond distances are in the range of their respective domain interactions. | |
The individual metal complex–DNA interactions are explored further, and the 2D interactions are presented in Fig. 9. The 2D interaction representation shows hydrogen bonding and other interactions between the DNA and the metal complex. The T-shaped π⋯π non-covalent interaction is recorded in Re–F, Re–Cl, and Re–Br complexes. Re–Me exhibits only an H-bond and Re–Np has π⋯σ interaction with 1BNA. The more unfavourable acceptor–acceptor interaction made between the complex (Re–F and Re–Np) and B-DNA becomes repulsive, which can affect the binding affinity. The binding energies of Re–F, Re–Cl, Re–Br, Re–Me, and Re–Np with 1BNA are found to be −8.69 kcal mol−1, −9.75 kcal mol−1, −10.21 kcal mol−1, −9.52 kcal mol−1, and −10.50 kcal mol−1, respectively. The interaction of metal complexes and 1Z3F-DNA is represented in Fig. 9. The 2D interaction diagram reveals that the complexes form hydrogen bonding, π⋯π non-covalent interaction, π⋯π stacked interaction, and π⋯alkyl interactions with DNA. The binding energies of Re–F, Re–Cl, Re–Br, Re–Me, and Re–Np complexes are found to be −7.17 kcal mol−1, −8.30 kcal mol−1, −8.44 kcal mol−1, −8.59 kcal mol−1, and −8.41 kcal mol−1, respectively. This indicates that the metal complexes exhibit a strong binding affinity towards DNA. Molecular docking analyses indicated that all rhenium complexes bind selectively to the minor groove of B-DNA (1BNA), suggesting a capacity to interfere with DNA integrity. This computational prediction is substantiated by DAPI-based DNA fragmentation assays wherein DAPI preferentially binds to the minor groove of A-T-rich regions in the B-DNA. The results revealed lobular nuclear formations and leakage of DNA fragments, characteristic of apoptotic cell death. The close agreement between the structural binding data from docking and the nuclear damage visualized experimentally underscores that minor groove binding is the mechanistic trigger driving the cytotoxic effects of these complexes.
MD simulations
Molecular dynamics simulation has been carried out to understand and compare the binding stability of metal complexes with DNA (1BNA and 1Z3F) over a period of 600 ns in a water environment.19–25 The root mean square deviation (RMSD) plot for 1BNA and 1Z3F is shown in Fig. 10 and Fig. S27, respectively. The plot shows an unstable RMSD with significant fluctuations during the simulation, which is attributed to the metal complexes starting to move out of the intercalated position with the DNA, suggesting that the interaction is unstable over a period of time. This is not observed in the Re–Cl complex; here, the DNA unwinds initially; however, the Re–Cl complex binds to DNA with a weak binding energy compared to minor groove binding. In contrast, the minor groove binding mode in the metal complexes shows good stability for a longer period (Fig. 10). The complexes, Re–F, Re–Cl, Re–Br, and Re–Me, have a stable RMSD throughout the molecular dynamics simulation, whereas Re–Np shows an RMSD with more fluctuations due to the unstable minor groove binding of the Re–Np complex with 1BNA, which leads to separation of the metal complex and DNA. The other complexes bind well with 1BNA and are stable for up to 600 ns in water. To compare the binding stability of the complexes and the interaction of the complex with 1BNA (Table 3) and 1Z3F (Table S6), the Gibbs free energy of binding (ΔG) is calculated using the molecular mechanics Poisson–Boltzmann surface area (MMPBSA) method after molecular dynamics simulations. The complexes interact with 1BNA through strong van der Waals interactions and have higher attractive electrostatic energy. Re–Br is the most stable complex (−40.0 kcal mol−1) and Re–Np (−15.9 kcal mol−1) is the least stable complex under solvent conditions. From molecular dynamics simulations, based on the binding energy, it is evident that the metal complexes show stable binding with DNA models, particularly those with halogen-substituted metal complexes, compared to metal complexes with Me and Np substitutions.
 |
| | Fig. 10 Time evolution of RMSD of metal complexes–1BNA (A) Re–F, (B) Re–Cl, (C) Re–Br, (D) Re–Me, and (E) Re–Np. The metal complexes are colored in blue for better visualization in the 3D representation. The inserted snapshot in the plot is taken at 0 ns, 300 ns, and 600 ns, respectively. The atoms’ colors are represented as follows: grey – C, black – H, blue – N, red – O, cyan – Re, pale blue – F, green – Cl, and brown – Br. | |
Table 3 MMPBSA calculations of Re–F, Re–Cl, Re–Br, Re–Me, and Re–Np with 1BNA
| Components |
Re–F
|
Re–Cl
|
Re–Br
|
Re–Me
|
Re–Np
|
| All values are reported in kcal mol−1. |
| VDWAALS |
−47.2 |
−51.3 |
−51.8 |
−47.7 |
−23.6 |
| EEL |
−21.6 |
−19.4 |
−20.2 |
−18.5 |
−14.3 |
| EGB |
38.4 |
−36.1 |
36.2 |
35.8 |
23.9 |
| ESURF |
−4.2 |
−4.2 |
−4.2 |
−4.1 |
−2.0 |
| ΔG (gas) |
−68.8 |
−70.6 |
−72.0 |
−66.2 |
−37.8 |
| ΔG (solv) |
34.1 |
31.9 |
32.0 |
31.7 |
22.0 |
| ΔG (binding) |
−34.7 |
−38.8 |
−40.0 |
−34.5 |
−15.9 |
Compared to minor groove binding, the metal complex intercalation shows the least negative binding energy. Based on the MD simulation, minor groove binding is more stable than the intercalation mode of binding with DNA models over a period of 600 ns.
Conclusion
A series of five new Re(I)–quinolinate–pyridyl complexes featuring 5,6-disubstituted benzimidazole or naphthanoimidazole motifs were synthesized and characterized. These complexes exhibited anticancer properties against 4T1, A549, and HeLa cell lines. Notably, Re–F (5,6-difluorobenzimidazole-based) and Re–Br (5,6-dibromobenzimidazole-based) complexes displayed potent activity against 4T1 and HeLa cells, respectively. The incorporation of fluorine and bromine atoms significantly enhanced their anticancer activity. Both complexes demonstrated antimigratory properties, induced ROS production, and triggered apoptosis via nucleosomal fragmentation. Furthermore, they exhibited deep tumor tissue penetration in 3D spheroids. The complexes exhibited stable binding to B-DNA primarily through minor grooves via non-covalent interactions involving the 5,6-dihalobenzimidazole motif, with Re–Br showing higher binding affinity. Molecular simulation studies corroborated the in vitro findings, indicating stable interactions of halogen-substituted complexes with B-DNA over 600 ns. Collectively, these findings suggest that fluorine- and bromine-substituted benzimidazole-based complexes hold promise as anti-cancer agents for breast cancer and cervical cancer, respectively. Notably, this pioneering study explores the potential of fac-[Re(CO)3]+ core-based complexes with halogen-substituted benzimidazole motifs, opening new avenues for investigating their biological activity.
Experimental details
Materials and instruments
Re2(CO)10 was purchased from Sigma-Aldrich. 8-Hydroxyquinoline, 4-pyridine carboxaldehyde, 4,5-difluorobenzene-1,2-diamine, 4,5-dichlorobenzene-1,2-diamine, 4,5-dibromobenzene-1,2-diamine, 4,5-dimethylbenzene-1,2-diamine, and 2,3-diaminonaphthalene were procured from BLD Pharma. Sodium bisulfite and DMF were obtained from commercial sources and used as received. Hexane and toluene were distilled using conventional procedures. All the complexes analysed for anticancer activity were dissolved in dimethylsulfoxide (DMSO, Merck). Elemental analysis was performed on a Vario MICRO Superuser V4.0.14. ESI-TOF-MS spectra were recorded on a Bruker maXis mass spectrometer. ATR-IR spectra were recorded on a Nicolet iS5 ATR-spectrometer in the 400–4000 cm−1 range. The 1H NMR spectra in DMSO-d6 were recorded on a Bruker Avance III 500 MHz spectrometer. The NMR spectra were referenced to the internal reference of tetramethylsilane.
Synthesis of fac-[Re(CO)3(QN)(L-F)] = Re–F·(C7H8)0.5.
A mixture of Re2(CO)10 (50.23 mg, 0.077 mmol), L-F (35.42 mg, 0.153 mmol), 8-hydroxyquinoline (22.3 mg, 0.154 mmol), and toluene (5 mL) was sealed in a Teflon-lined stainless steel bomb and heated at 160 °C for 24 h. Upon cooling the reaction vessel to room temperature, yellow crystals were obtained. The crystals were washed with hexane and air-dried. Yield: 77% (78 mg). Anal. calcd for C24H13F2N4O4Re·(C7H8)0.5: C, 49.75; H, 2.48; N, 7.10. Found: C, 49.82; H, 2.01; N, 7.07. 1H NMR (500 MHz, DMSO-d6): a mixture of two isomers with a 1:0.5 ratio without the solvent toluene molecule: δ 9.18 (d, J = 4.69 Hz, Ha), 8.95 (d, J = 4.57 Hz, Ha), 8.81 (d, J = 4.72 Hz, H9), 8.76 (d, J = 5.97 Hz, H9), 8.53 (d, J = 8.51 Hz, Hc), 8.45 (d, J = 8.10 Hz, Hc), 8.15 (d, J = 5.13 Hz, H8), 8.04 (d, J = 5.85 Hz, H8), 7.77–7.68 (m, H4–7), 7.65–7.60 (m, Hb), 7.48 (t, J = 7.92 Hz, He), 7.39 (t, J = 8.06 Hz, He), 7.09 (d, J = 8.06 Hz, Hf), 6.98 (d, J = 7.92 Hz, Hf), 6.94 (d, J = 7.92 Hz, Hd), 6.91 (d, J = 7.94 Hz, Hd). ESI (HR-MS). Calcd for C24H14F2N4O4Re [M + H]+: m/z 647.054. Found: m/z 647.0557. ATR-IR (cm−1): 2015 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O), 1914 (CO), and 1882 (CO).
O), 1914 (CO), and 1882 (CO).
Synthesis of fac-[Re(CO)3(QN)(L-Cl)] = Re–Cl.
By following the procedure similar to Re–F, yellow crystals were obtained using a mixture of Re2(CO)10 (51.04 mg, 0.078 mmol), L-Cl (41.12 mg, 0.156 mmol), 8-hydroxyquinoline (22.43 mg, 0.154 mmol), and toluene (5 mL). The crystals were washed with hexane and air-dried. Yield: 85% (89 mg). Anal. calcd for C24H13Cl2N4O4Re: C, 45.49; H, 1.93; N, 8.26. Found: C, 45.02; H, 1.33; N, 8.42. 1H NMR (500 MHz, DMSO-d6): a mixture of two isomers with a 1:0.5 ratio without the solvent toluene molecule δ 9.18 (d, J = 5.01 Hz, Ha), 8.95 (d, J = 4.77 Hz, Ha), 8.41 (s, H9), 8.77 (d, J = 6.46 Hz, H9), 8.54 (d, J = 8.16 Hz, Hc), 8.45 (d, J = 8.16 Hz, Hc), 8.19 (d, J = 5.46 Hz, H8), 8.03 (d, J = 6.56 Hz, H8), 7.95 (s, H4), 7.87 (s, H7), 7.65–7.58 (m, Hb), 7.47 (t, J = 7.81 Hz, He), 7.38 (t, J = 7.89 Hz, He), 7.09 (d, J = 8.00 Hz, Hf), 6.98 (d, J = 8.00 Hz, Hf), 6.94 (d, J = 7.92 Hz, H9), 6.91 (d, J = 7.92 Hz, Hd). ESI (HR-MS). Calcd for C24H14Cl2N4O4Re [M + H]+: m/z 678.9949. Found: m/z 678.2133. ATR-IR (cm−1): 2011 (CO), 1899 (CO), and 1880 (CO).
Synthesis of fac-[Re(CO)3(QN)(L-Br)] = Re–Br.
By following the procedure similar to Re–F, yellow crystals were obtained using a mixture of Re2(CO)10 (50.33 mg, 0.078 mmol), L-Br (54.1 mg, 0.214 mmol), 8-hydroxyquinoline (22.12 mg, 0.152 mmol), and toluene (5 mL). The crystals were washed with hexane and air-dried. Yield: 69% (81 mg). Anal. calcd for C24H13Br2N4O4Re: C, 39.56; H, 1.71; N, 7.30. Found: C, 39.71; H, 1.18; N, 7.19. 1H NMR (500 MHz, DMSO-d6): a mixture of two isomers with a 1:0.5 ratio without the solvent toluene molecule δ 9.18 (d, J = 4.42 Hz, Ha), 8.95 (d, J = 4.17 Hz, Ha), 8.83 (s, H9), 8.77 (d, J = 6.49 Hz, H9), 8.54 (d, J = 8.40 Hz, Hc), 8.45 (d, J = 8.21 Hz, Hc), 8.15 (d, J = 4.90 Hz, H8), 8.07 (s, H4), 8.04 (d, J = 8.02 Hz, H8), 8.00 (s, H7), 7.64–7.58 (m, Hb), 7.47 (t, J = 7.92 Hz, He), 7.38 (t, J = 8.02 Hz, He), 7.09 (d, J = 8.02 Hz, He), 6.98 (d, J = 7.92 Hz, Hf), 6.94 (d, J = 7.93 Hz, Hd), 6.90 (d, J = 7.88 Hz, Hd). ESI (HR-MS). Calcd for C24H14Br2N4O4Re [M + H]+: m/z 768.8940. Found: m/z 768.8932. ATR-IR (cm−1): 2011 (CO), 1901 (CO), and 1880 (CO).
Synthesis of fac-[Re(CO)3(QN)(L-Me)]·(C7H8) = Re–Me·(C7H8).
By following the procedure similar to Re–F, yellow crystals were obtained using a mixture of Re2(CO)10 (49.89 mg, 0.076 mmol), L-Me (34.56 mg, 0.153 mmol), 8-hydroxyquinoline (22.30 mg, 0.154 mmol), and toluene (5 mL). The crystals were washed with hexane and air-dried. Yield: 76% (74 mg). Anal. calcd for C33H27N4O4Re: C, 54.31; H, 3.73; N, 7.68. Found: C, 54.59; H, 3.67; N, 7.36. 1H NMR (500 MHz, DMSO-d6): a mixture of two isomers with a 1:0.5 ratio without the solvent toluene molecule δ 9.18 (d, J = 4.78 Hz, Ha), 8.95 (d, J = 4.72 Hz, Ha), 8.73 (d, J = 6.07 Hz, H9), 8.70 (d, J = 6.68 Hz, H9), 8.54 (d, J = 8.45 Hz, Hc), 8.45 (d, J = 8.45 Hz, Hc), 8.06 (d, J = 6.12 Hz, H8), 8.00 (d, J = 6.73 Hz, H8), 7.64–7.59 (m, Hb), 7.47 (t, J = 7.88 Hz, He), 7.42–7.36 (m, He,4,7), 7.09 (d, J = 7.92 Hz, Hf), 6.98 (d, J = 7.92 Hz, Hf), 6.94 (d, J = 7.83 Hz, Hd), 6.91 (d, J = 7.83 Hz, Hd), 2.34 (s, –CH3), 2.29 (s, –CH3). ESI (HR-MS). Calcd for C26H20N4O4Re [M + H]+: m/z 639.1042. Found: m/z 639.1037. ATR-IR (cm−1): 2012 (CO), 1905 (CO), and 1882 (CO).
Synthesis of fac-[Re(CO)3(QN)(L-Np)]·(C7H8) = Re–Np·(C7H8).
By following the procedure similar to Re–F, yellow crystals were obtained using a mixture of Re2(CO)10 (50.28 mg, 0.077 mmol), L-Np (38.52 mg, 0.157 mmol), 8-hydroxyquinoline (22.10 mg, 0.152 mmol), and toluene (5 mL). The crystals were washed with hexane and air-dried. Yield: 85% (85 mg). Anal. calcd for C35H25N4O4Re: C, 55.92; H, 3.35; N, 7.45. Found: C, 55.55; H, 3.32; N, 7.45. 1H NMR (500 MHz, DMSO-d6): a mixture of two isomers with a 1:0.7 ratio without the solvent toluene molecule δ 9.20 (d, J = 4.77 Hz, Ha), 8.95 (d, J = 4.83 Hz, Ha), 8.85 (d, J = 4.85 Hz, H11), 8.81 (d, J = 6.66 Hz, H11), 8.54 (d, J = 8.47 Hz, Hc), 8.46 (d, J = 8.31 Hz, Hc), 8.23 (d, J = 5.94 Hz, H10), 8.20 (s, H9) 8.16 (d, J = 6.51 Hz, H4,10), 8.05–8.03 (m, H5), 8.01–7.99 (m, H8), 7.66–7.63 (m, Hb), 7.62–7.59 (m, Hb), 7.47 (t, J = 8.01 Hz, Hf), 7.42–7.37 (m, H6,7) 7.09 (d, J = 7.97 Hz, Hf), 6.99 (d, J = 8.06 Hz, Hf), 6.96 (d, J = 7.99 Hz, Hd), 6.91 (d, J = 7.80 Hz, Hd), ESI (HR-MS). Calcd for C28H18N4O4Re [M + H]+: m/z 661.0881. Found: m/z 661.0894. ATR-IR (cm−1): 2014 (CO), 1907 (CO), and 1883 (CO).
Materials & methodology
Thiazolyl blue tetrazolium bromide (MTT) was purchased from Sigma Aldrich, USA. Roswell Park Memorial Institute (RPMI), Dulbecco's Modified Eagle's Medium (DMEM), Fetal Bovine Serum (FBS), Phosphate-Buffered Saline (PBS), and Pen/Strep were purchased from HiMedia Chemicals, Mumbai, India. Propidium iodide (PI), fluorescein diacetate (FDA), and 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) were obtained from Sigma Aldrich, USA. The reagents utilized in the experiments were of analytical grade. For biological experiments, the complexes were initially dissolved in 1% dimethyl sulfoxide (DMSO) to prepare the primary stock solution. Prior to experimental application, working concentrations were further diluted in cell culture medium, ensuring the final DMSO concentration remained below 0.5% to minimize potential cytotoxic effects.
Cell lines and maintenance
The mouse fibroblast cell line (L929), mouse embryonic fibroblast cell line (NIH 3T3), embryonic cardiac muscle cell line (H9C2), mouse myoblast cell line (C2C12), murine mammary carcinoma cell line (4T1), human lung adenocarcinoma cell line (A549), and human cervical cancer cell line (HeLa) were obtained from the National Centre for Cell Sciences (NCCS), Pune, India. The cell lines were cultured in DMEM/RPMI medium supplemented with 10% (v/v) fetal bovine serum (FBS), 1% L-glutamine, and 100 U mL−1 penicillin/streptomycin and were maintained at 37 °C under a humidified atmosphere containing 5% CO2 under sterile conditions.
Lipophilicity
A 0.1 mM stock solution of Re–F and Re–Br in DMSO was prepared and diluted with 10 mL of 1:100 pH 7.4 PBS buffer. After recording the absorbance of this, 10 mL of octanol was added to 10 mL of the above aqueous buffer solution. The two layers were separated after the resulting solution was kept for 24 h on a mechanical stirrer. The aqueous layer was separated, and the absorbance was recorded. The partition coefficient was calculated using the formula Ko/w = [{(AiDFi − AfDFf)·Vwater}/AfDFf·Voctanol] in which Ai and Af are the absorbances of the aqueous layer before and after extraction, Vwater and Voctanol are the volumes of the water and octanol phases, and DFi and DFf are the dilution factors used to measure Ai and Af, respectively.
Biocompatibility studies
The biocompatibility of complexes (Re–F, Re–Cl, Re–Br, Re–Me, and Re–Np) was evaluated using four murine cell models: L929 mouse fibroblasts, NIH 3T3 mouse embryonic fibroblasts, H9C2 embryonic cardiac muscle cells, and C2C12 mouse myoblasts.26,27 Cells were seeded in 96-well plates at a density of 1 × 104 cells per well and allowed to adhere during a 24-hour pre-incubation period under standard culture conditions (37 °C, 5% CO2). Test compounds were serially diluted in complete growth medium to various concentrations (1, 2, 5, 10, 25, 50, 100, and 250 μM) and then incubated with adherent cell monolayers for 24 hours. Post-treatment viability was quantified using the MTT assay, where mitochondrial dehydrogenase activity reduces tetrazolium salts to formazan crystals proportional to metabolic activity.27 This standardized protocol allows systematic comparison of compound cytotoxicity across diverse cell types relevant to connective tissue, cardiac, and musculoskeletal systems.26
Cytotoxicity studies
The cytotoxicity of rhenium-based metallic complexes was evaluated in vitro using the murine mammary carcinoma (4T1), human lung adenocarcinoma (A549), and human cervical cancer (HeLa) cell lines using the MTT assay. Cells were seeded in 96-well plates at a density of 1 × 104 cells per well and incubated at 37 °C with 5% CO2 for 24 hours to promote cell adhesion. Subsequently, the cells were treated with varying concentrations (0.05, 0.5, 5, 50, and 250 μM) of the test compounds (Re–F, Re–Cl, Re–Br, Re–Me, and Re–Np). Untreated cells served as the negative control. After 24 hours of compound exposure, the medium was replaced with fresh DMEM or RPMI containing the MTT reagent (5 mg mL−1), and the cells were incubated for an additional 3 hours to allow for formazan crystal formation. The resulting crystals were dissolved in DMSO, and absorbance was measured at 570 nm and 630 nm using a multimode plate reader to assess cell viability.3e,27
Live/dead assay
Cells (HeLa and 4T1) were pre-treated with complexes (Re–Br and Re–F, respectively) and incubated for 24 hours. Following the treatment, the cells were incubated with both FDA and PI, typically by adding the dyes directly to the cell culture medium and incubating for 10 minutes at room temperature, protected from light. After incubation, the cells were immediately analyzed using a fluorescence microscope (Zoe Microscope). Live cells fluoresce green, while dead cells fluoresce red, allowing for clear two-color discrimination and qualitative or quantitative assessment of cell viability within the complexes.28
DCFDA analysis
The generation of ROS by rhenium-based metallic complexes was assessed using the DCFDA (2′,7′-dichlorodihydrofluorescein diacetate) staining assay. Following the seeding and attachment of HeLa and 4T1 cells in well plates, the cells were exposed to the synthesized complexes (Re–Br and Re–F, respectively). After a 24-hour incubation period, 10 µM DCFDA was introduced to each well, and fluorescence imaging was performed using a fluorescence microscope. Untreated cells served as the negative control, while cisplatin-treated cells were included as a positive control for ROS generation analysis.
Antimigration evaluation
The antimigratory effects of rhenium-based complexes (Re–Br and Re–F) were evaluated using an in vitro scratch assay.14d Briefly, HeLa and 4T1 cells were seeded into 12-well plates at a density of 1 × 105 cells per well and incubated for 24 hours under standard culture conditions (5% CO2, 37 °C) to establish confluent monolayers. Mechanical scratches were introduced into the adherent cell layers using a sterile 200 μL pipette tip, followed by gentle rinsing with PBS to remove dislodged cellular debris. The cells were subsequently exposed to IC50 concentrations of Re–Br and Re–F, derived from prior cytotoxicity assays, to assess migration inhibition. Phase-contrast microscopy images of the scratch regions were acquired immediately post-scratch (0 hour) and at 6, 12, and 24-hour intervals while maintaining incubation conditions. Scratch closure percentages were calculated by quantifying the reduction in the scratch area relative to the initial wound width (t = 0 hour) using image analysis software, enabling comparative evaluation of migration kinetics across treatment groups.
Apoptosis determination assay
The apoptotic activity was evaluated through acridine orange/ethidium bromide (AO/EtBr) dual fluorescence staining, a method distinguishing viable and apoptotic cells based on membrane integrity.29 HeLa and 4T1 cell lines (1 × 104 cells per well) were plated in 96-well plates and allowed to adhere under standard culture conditions. Following attachment, the cells were exposed to the respective rhenium complexes for 12 hours, with untreated cultures serving as negative controls and cisplatin-treated cells as positive controls. Post-incubation, the cells were stained with AO/EtBr (1:1 v/v), enabling differential fluorescence: AO intercalates into the DNA of viable cells (green emission), while EtBr permeates membrane-compromised apoptotic cells (red emission). Fluorescent micrographs were acquired using a Zoe fluorescence microscope to quantify apoptotic indices across treatment groups.
DNA fragmentation assay
HeLa cells (5 × 104 cells per well) and 4T1 cells (5 × 104 cells per well) were cultured in 6-well plates with coverslips and subjected to treatment with 12.27 μM Re–Br for HeLa cells and 16.25 μM Re–F for 4T1 cells, together with cisplatin for 48 hours, followed by fixation with 4% paraformaldehyde for 10 minutes. The cells were rinsed with 1× PBS, subsequently permeabilized with 0.1% Triton X-100 for 10 minutes, washed again with 1× PBS, and stained with DAPI (4,6-diamidino-2-phenylindole) for 5 minutes. The cells were assessed using a fluorescence microscope (Olympus Corp., Tokyo, Japan).
3D spheroid model
3D spheroids of HeLa and 4T1 cells were generated using ultra-low attachment (ULA) 96-well plates, which employ hydrophilic, non-adhesive surfaces to enforce scaffold-free self-assembly. Cells were seeded at densities of 3–4 × 103 cells per well and incubated for 5 days under standard culture conditions (5% CO2, 37 °C), enabling the formation of compact, hypoxic-core spheroids that recapitulate solid tumor physiology. Post-assembly, spheroids were treated for 24 hours with rhenium(I) tricarbonyl complexes (Re–Br and Re–F), which demonstrated antineoplastic activity in 3D models. The cell viability was assessed 24 hours post-treatment via dual FDA/PI staining. FDA (5 μg mL−1) was hydrolysed by intracellular esterases in viable cells to emit green fluorescence, indicating metabolic activity, while PI (2 μg mL−1) penetrated membrane-compromised cells, binding nuclear DNA and emitting red fluorescence. Fluorescence imaging was captured using a Zoe microscope, and viability indices were quantified as the ratio of FDA+/PI− cells to total cells, normalized against untreated controls. This methodology enabled evaluation of chemotherapeutic efficacy while preserving spheroid structural integrity throughout the assay.
Molecular docking and dynamics simulation
The metal complexes binding to DNA models are generated using AutoDock.19a The DNA structures are acquired from the Protein Data Bank.20–22 Initially, the undesirable water molecules and ligands are eliminated from the structure and then polar hydrogens are added to it. The complex–DNA interaction was explored with a grid box (1BNA – 60 Å × 58 Å × 94 Å, 1BWG – 48 Å × 50 Å × 90 Å, 1 AU5 –40 Å × 52 Å × 40 Å, 3FT6 – 40 Å × 40 Å × 50 Å, and 1Z3F – 48 Å × 44 Å × 52 Å) and 0.5 grid resolution. For Re–F docking with 1Z3F, a grid box of 52 Å × 40 Å × 46 Å with 0.375 grid resolution is preferred to achieve the intercalation mode of binding. The complexes have two torsional degrees of freedom, except for the Re–Me complex (torsion = 1). Docking is analyzed with 500 conformers and a population size of 1500. Genetic algorithms are preferred with parameters such as the number of generations (27000), the mutation rate (0.02) and the crossover rate (0.8). Additionally, each calculation is repeated five times to verify the consistency of the obtained results with the conformer. Finally, the best conformer is selected based on the highest binding energy and the total number of hydrogen bonds and the binding energy values are given in Table S5. Furthermore, the BIOVIA Discovery Studio Visualizer is used for visualizing the best interactive conformer to the DNA.19 The MD simulations for the complex with 1BNA and 1Z3F are executed in Amber2022.22 The metal complex is parameterized by easyPARM (S33). For DNA, hydrogens are added using web-server H++. Then, the metal complex and DNA were added together to obtain the topology and coordinate files using tleap with the DNA.bsc1 force field.23 The TIP3P solvation model (comparison of simple potential functions for simulating liquid water) induces water molecules around the complex and DNA with a buffer distance of 10 Å. To neutralize the system, Na+ ions are added. First, minimization calculation is carried out under restrained constant volume periodic boundary conditions with 10000 steps and a cut-off distance of 10.0 Å. The system is fixed to have 500 kcal mol−1 force constant in the minimization calculation. Then, the second minimization is calculated under unrestrained constant volume periodic boundary conditions with 2500 steps with the same cut off distance as previous minimization. Next, the system is heated to 300 K over a period of 20 ps in 100000 steps. Also, the DNA and metal complex is restrained with constant volume boundary conditions, 10 kcal mol−1 force constant and a similar cut off distance. The MD simulation was performed for all the complexes at 300 K, including Langevin damping of 1 ps−1. The simulation is carried out for 100 ps, 10 ns and finally for 600 ns with 50000, 50000000, and 300000000 steps respectively. The time step is set to be 2 fs without any constraints. Trajectory is analyzed by Cpptraj (AMBER) and Visual Molecular Dynamics software is used for visualization.24 The binding of the metal complex with DNA has been assessed using the MM-PBSA method in Amber22.25
Crystallography
The single crystal X-ray structural study of Re–F, Re–Cl, Re–Br, Re–Me, and Re–Np was performed on a Bruker D8 QUEST diffractometer [λ(Mo Kα) = 0.71073 A]. The molecular structures were refined using the SHELXL-2018/3 program (within the WinGX program package) and solved by direct methods using SHELXS-97 (Sheldrick 2008).30 All non-hydrogen atoms were refined anisotropically (CCDC: 2451107–2451111).
Statistical analysis
The experiments were performed in triplicate and calculated as mean ± SD. A one-way ANOVA analysis was used to differentiate between research groups (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Conflicts of interest
There are no conflicts to declare.
Data availability
Supplementary information: spectroscopic data-IR, ESI-MS, NMR spectra, molecular docking and MD simulations studies. See DOI: https://doi.org/10.1039/d5dt01577d.
CCDC 2451107–2451111 contain the supplementary crystallographic data for this paper.31a–e
Acknowledgements
AKR thanks ICMR – CoE, FIW/2024/01/208, IIRPIG-2024-01-02351, SUPRA (SPR/2022/000230), and MOE-STARS2/2023/640 for funding, RLB, CLP, and NB thank UGC, MHRD, and IIT-H for the fellowship, respectively. RS acknowledges VIT for the SEED grant support with the reference number. RGEMS/SG20220030. MP thanks CO2RGTC for computational facilities and VIT management for the fellowship. RLB and BB thank Dr B. Shankar (Dept. of Chemistry, Thiagarajar College of Engineering, Madurai), Dr P. Kalimuthu (Dept. of Chemistry, The Gandhigram Rural Institute, Deemed to be University), and Dr M. Sathiyendiran (School of Chemistry, University of Hyderabad) for scientific support.
References
-
(a) L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584, DOI:10.1038/nrc2167;
(b) T. Zhong, J. Yu, Y. Pan, N. Zhang, Y. Qi and Y. Huang, Adv. Healthcare Mater., 2023, 12, e2300253, DOI:10.1002/adhm.202300253.
-
(a) R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653, 10.1039/c8dt00838h;
(b) R. L. Lucaciu, A. C. Hangan, B. Sevastre and L. S. Oprean, Molecules, 2022, 27, 6485, DOI:10.3390/molecules27196485;
(c) R. Paprocka, M. Wiese-Szadkowska, S. Janciauskiene, T. Kosmalski, M. Kulik and A. Helmin-Basa, Coord. Chem. Rev., 2022, 452, 214307, DOI:10.1016/j.ccr.2021.214307;
(d) S. Abdolmaleki, A. Aliabadi and S. Khaksar, Coord. Chem. Rev., 2024, 501, 215579, DOI:10.1016/j.ccr.2023.215579;
(e) A. Pothig and A. Casini, Theranostics, 2019, 9, 3150–3169, DOI:10.7150/thno.31828.
-
(a) K. M. Knopf, B. L. Murphy, S. N. MacMillan, J. M. Baskin, M. P. Barr, E. Boros and J. J. Wilson, J. Am. Chem. Soc., 2017, 139, 14302–14314, DOI:10.1021/jacs.7b08640;
(b) R. Kushwaha, A. Upadhyay, S. Peters, A. K. Yadav, A. Misha, A. Bera, T. Sadhukhan and S. Banerjee, Langmuir, 2024, 40, 12226–12238, DOI:10.1021/acs.langmuir.4c01296;
(c) A. Leonidova and G. Gasser, ACS Chem. Biol., 2014, 9, 2180–2193, DOI:10.1021/cb500528c;
(d) K. Schindler and F. Zobi, Molecules, 2022, 27, 539, DOI:10.3390/molecules27020539;
(e) E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kühn, Coord. Chem. Rev., 2019, 393, 79–117, DOI:10.1016/j.ccr.2019.04.014;
(f) L. E. Enslin, K. Purkait, M. D. Pozza, B. Saubamea, P. Mesdom, H. G. Visser, G. Gasser and M. Schutte-Smith, Inorg. Chem., 2023, 62, 12237–12251, DOI:10.1021/acs.inorgchem.3c00730;
(g) U. Das, B. Kar, S. Pete and P. Paira, Dalton Trans., 2021, 50, 11259–11290, 10.1039/D1DT01326B;
(h) D. Gupta and M. Sathiyendiran, ChemistrySelect, 2018, 3, 7439–7458, DOI:10.1002/slct.201800904;
(i) A. Sharma, N. Vaibhavi, B. Kar, U. Das and P. Paira, RSC Adv., 2022, 12, 20264–20295, 10.1039/D2RA03434D;
(j) J. R. Dilworth, Coord. Chem. Rev., 2021, 436, 213822, DOI:10.1016/j.ccr.2021.213822;
(k) P. Thanasekaran, C. C. Lee and K. L. Lu, Acc. Chem. Res., 2012, 45, 1403–1418, DOI:10.1021/om4005065.
-
(a) Z. Huang and J. J. Wilson, Eur. J. Inorg. Chem., 2021, 2021, 1312–1324, DOI:10.1002/ejic.202100031;
(b) I. Kitanovic, S. Can, H. Alborzinia, A. Kitanovic, V. Pierroz, A. Leonidova, A. Pinto, B. Spingler, S. Ferrari, R. Molteni, A. Steffen, N. Metzler-Nolte, S. Wölfl and G. Gasser, Chem. – Eur. J., 2014, 20, 2496–2507, DOI:10.1002/chem.201304012.
-
(a) S. Imstepf, V. Pierroz, R. Rubbiani, M. Felber, T. Fox, G. Gasser and R. Alberto, Angew. Chem., 2016, 128, 2842–2845, DOI:10.1002/ange.201511432;
(b) F.-X. Wang, J.-H. Liang, H. Zhang, Z.-H. Wang, Q. Wan, C.-P. Tan, L.-N. Ji and Z.-W. Mao, ACS Appl. Mater. Interfaces, 2019, 11, 13123–13133, DOI:10.1021/acsami.9b01057;
(c) R.-R. Ye, C.-P. Tan, Y.-N. Lin, L.-N. Ji and Z.-W. Mao, Chem. Commun., 2015, 51, 8353–8356, 10.1039/C5CC02354H.
-
(a) N. Montesdeoca, R. L. Borkar, M. Sathiyendiran and J. Karges, Chem. – Eur. J., 2024, e202400217, DOI:10.1002/chem.202400217;
(b) M. Kedia, S. Khatun, U. Phukon, B. Shankar, A. K. Rengan and M. Sathiyendiran, Dalton Trans., 2023, 52, 14314–14318, 10.1039/D3DT02535G;
(c) R. L. Borkar, C. L. Putta, B. Shankar, B. Bhattacharjee, A. K. Rengan and M. Sathiyendiran, Chem. – Asian J., 2025, 20, e202401656, DOI:10.1002/asia.202401656;
(d) M. Priyatharsini, I. Mishra, B. Shankar, N. Srinivasan, R. V. Krishnakumar and M. Sathiyendiran, J. Organomet. Chem., 2021, 953, 122052, DOI:10.1016/j.jorganchem.2021.122052.
-
(a) R. Wykowski, A. M. Fuentefria and S. F. de Andrade, Arch. Microbiol., 2022, 204, 535, DOI:10.1007/s00203-022-03122-2;
(b) I. Chaaban, H. Hafez, I. AlZaim, C. Tannous, H. Ragab, A. Hazzaa, S. Ketat, A. Ghoneim, M. Katary, M. M. Abd-Alhaseeb, F. A. Zouein, A. Albohy, A. N. Amer, A. F. El-Yazbi and A. S. F. Belal, Bioorg. Chem., 2021, 113, 105035, DOI:10.1016/j.bioorg.2021.105035.
-
(a) A. Ali, S. Banerjee, S. Kamaal, M. Usman, N. Das, M. Afzal, A. Alarifi, N. Sepay, P. Roy and M. Ahmad, RSC Adv., 2021, 11, 14362–14373, 10.1039/D1RA00172H;
(b) M. Kubanik, H. Holtkamp, T. Sohnel, S. M. F. Jamieson and C. G. Hartinger, Organometallics, 2015, 34, 5658–5668, DOI:10.1021/acs.organomet.5b00868;
(c) K. Mudarmah, N. Abeydeera, G. Chen, W. Jogadi, J. A. Krause, J. M. Budzik and S. D. Huang, Dalton Trans., 2025, 54, 9975–9983, 10.1039/D4DT02895C;
(d) X. Zhou, J. Inorg. Biochem., 2023, 238, 112051, DOI:10.1016/j.jinorgbio.2022.112051 , and references therein.
-
(a) V. L. Gantsho, M. Dotou, M. Jakubaszek, B. Goud, G. Gasser, H. G. Visser and M. Schutte-Smith, Dalton Trans., 2020, 49, 35–46, 10.1039/C9DT04025K;
(b) K. Łyczko, A. Pogorzelska, U. Czescik, M. Koronkiewicz, J. E. Rode, E. Bednarek, R. Kawecki, K. Wegrzynska, A. Baraniak, M. Milczarek and J. Dobrowolski, RSC Adv., 2024, 14, 18080–18092, 10.1039/D4RA03141E.
-
(a) B. Pathare and T. Bansode, Results Chem., 2021, 3, 100200, DOI:10.1016/j.rechem.2021.100200;
(b) N. J. Basha, Polycyclic Aromat. Compd., 2023, 43, 6549–6569, DOI:10.1080/10406638.2022.2118334;
(c) R. S. Keri, A. Hiremathad, S. Budagumpi and B. M. Nagaraja, Chem. Biol. Drug Des., 2015, 86, 19–65, DOI:10.1111/cbdd.12462;
(d) S. R. Brishty, M. J. Hossain, M. U. Khandaker, M. R. I. Faruque, H. Osman and S. M. A. Rahman, Front. Pharmacol., 2021, 12, 762807, DOI:10.3389/fphar.2021.762807;
(e) B. Narasimhan, D. Sharma and P. Kumar, Med. Chem. Res., 2012, 21, 269–283, DOI:10.1007/s00044-010-9533-9;
(f) B. Farag, M. E. A. Zaki, D. A. Elsayed and S. M. Gomha, RSC Adv., 2025, 15, 18593–18647, 10.1039/D5RA01077B;
(g) K. Mahmood, Z. Akhter, F. Perveen, Aisha, M. Bibi, H. Ismail, N. Tabassum, S. Yousuf, A. R. Ashraf and M. A. Qayyum, RSC Adv., 2023, 13, 11982–11999, 10.1039/D3RA00982C.
- T. Yamanoi, Y. Oda and K. Katsuraya, Magnetochemistry, 2017, 3, 38, DOI:10.3390/magnetochemistry3040038.
- M. F. Harris and J. L. Logan, J. Chem. Educ., 2014, 91, 915–918, DOI:10.1021/ed400655b.
- T. Gokturk, E. S. Cetin, T. Hokelek, H. Pekel, O. Sensoy, E. N. Aksu and R. Gupta, ACS Omega, 2023, 8, 31839–31856, DOI:10.1021/acsomega.8b00582.
-
(a) E. Eruslanov and S. Kusmartsev, Methods Mol. Biol., 2010, 594, 57–72, DOI:10.1007/978-1-60761-411-1_4;
(b) D. Figueroa, M. Asaduzzaman and F. Young, J. Pharmacol. Toxicol. Methods, 2018, 94, 26–33, DOI:10.1016/j.vascn.2018.03.007;
(c) M. P. Murphy, H. Bayir, V. Belousov, C. J. Chang, K. J. A. Davies, M. J. Davies, T. P. Dick, T. Finkel, H. J. Forman, Y. Janssen-Heininger, D. Gems, V. E. Kagan, B. Kalyanaraman, N.-G. Larsson, G. L. Milne, T. Nyström, H. E. Poulsen, R. Radi, H. Van Remmen, P. T. Schumacker, P. J. Thornalley, S. Toyokuni, C. C. Winterbourn, H. Yin and B. Halliwell, Nat. Metab., 2022, 4, 651–662, DOI:10.1038/s42255-022-00591-z;
(d) M. Katerji, M. Filippova and P. Duerksen-Hughes, Oxid. Med. Cell. Longevity, 2019, 2019, 1–29, DOI:10.1155/2019/1279250;
(e) L. Wu, A. C. Sedgwick, X. Sun, S. D. Bull, X.-P. He and T. D. James, Acc. Chem. Res., 2019, 52, 2582–2597, DOI:10.1021/acs.accounts.9b00302;
(f) S. Escada-Rebelo, F. G. Mora, A. P. Sousa, T. Almeida-Santos, A. Paiva and J. Ramalho-Santos, Asian J. Androl., 2020, 22, 465–471, DOI:10.4103/aja.aja_132_19.
-
(a) X. Ma, J. Lu, P. Yang, B. Huang, R. Li and R. Ye, Front. Chem., 2022, 10, DOI:10.3389/fchem.2022.890925;
(b) P. Collery, D. Desmaele, A. Harikrishnan and V. Veena, Anticancer Res., 2025, 45, 73–79, DOI:10.21873/anticanres.17394;
(c) N. Shtemenko, C. Galiana-Rosello, A. Gil-Martínez, S. Blasco, J. Gonzalez-García, H. Velichko, O. Holichenko, O. Shtemenko and E. García-España, RSC Adv., 2024, 14, 19787–19793, 10.1039/d4ra02669a;
(d) C.-C. Liang, A. Y. Park and J.-L. Guan, Nat. Protoc., 2007, 2, 329–333, DOI:10.1038/nprot.2007.30;
(e) G. Cory, Methods Mol. Biol., 2011, 769, 25–30, DOI:10.1007/978-1-61779-207-6_2.
-
(a) M. Pebam, P. S. Rajalakshmi, M. Gangopadhyay, S. Thatikonda and A. K. Rengan, ACS Appl. Bio Mater., 2022, 5, 5333–5346, DOI:10.1021/acsabm.2c00724;
(b) J. Kapuscinski, Biotech. Histochem., 1995, 70, 220–233, DOI:10.3109/10520299509108199;
(c) P. Majtnerova, J. Capek, F. Petira, J. Handl and T. Rousar, Sci. Rep., 2021, 11, 1–13, DOI:10.1038/s41598-021-91380-3.
-
(a) M. Cavaco, P. Fraga, J. Valle, D. Andreu, M. A. R. B. Castanho and V. Neves, Pharmaceutics, 2021, 13, 1863, DOI:10.3390/pharmaceutics13111863;
(b) P. Nayak, V. Bentivoglio, M. Varani and A. Signore, Cancers, 2023, 15, 4846, DOI:10.3390/cancers15194846;
(c) M. E. Katt, A. L. Placone, A. D. Wong, Z. S. Xu and P. C. Searson, Front. Bioeng. Biotechnol., 2016, 4 DOI:10.3389/fbioe.2016.00012;
(d) S. Cordeiro, B. B. Oliveira, R. Valente, D. Ferreira, A. Luz, P. V. Baptista and A. R. Fernandes, Front. Cell Dev. Biol., 2024, 12, 1507388, DOI:10.3389/fcell.2024.1507388.
-
(a) Y. Gilad and H. Senderowitz, J. Chem. Inf. Model., 2014, 54, 96–107, DOI:10.1021/ci400352t;
(b) G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791, DOI:10.1002/jcc.21256;
(c) R. Masnikosa, M. M. Milutinović, I. Crnolatac, A. Tot, S. Veličković, Ž. Bojić-Trbojević and A. Rilak-Simović, J. Inorg. Biochem., 2020, 208, 111090, DOI:10.1016/j.jinorgbio.2020.111090;
(d) J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898, DOI:10.1021/acs.jcim.1c00203;
(e) M. Bhol, R. L. Borkar, B. Shankar, S. K. Panda, M. Wolff and M. Sathiyendiran, Inorg. Chem., 2023, 62, 11554–11569, DOI:10.1021/acs.inorgchem.3c01213.
-
(a) S. Kasibhatla, G. P. Amarante-Mendes, D. Finucane, T. Brunner, E. Bossy-Wetzel and D. R. Green, CSH Protoc., 2006, 2006 DOI:10.1101/pdb.prot4429;
(b) K. Liu, P.-c. Liu, R. Liu and X. Wu, Med. Sci. Monit. Basic Res., 2015, 21, 15–20, DOI:10.12659/MSMBR.893327.
- F. Ponte, S. Scoditti, P. Barretta and G. Mazzone, Inorg. Chem., 2023, 62, 8948–8959, DOI:10.1021/acs.inorgchem.3c00592.
- H. R. Drew, R. M. Wing, T. Takano, C. Broka, S. Tanaka, K. Itakura and R. E. Dickerson, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2179–2183, DOI:10.1073/pnas.78.4.2179.
-
D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao and P. A. Kollman, AMBER 2016, University of California, San Francisco, 2016 Search PubMed.
- I. Ivani, P. D. Dans, A. Noy, A. Perez, I. Faustino, A. Hospital, J. Walther, P. Andrio, R. Goni, A. Balaceanu, G. Portella, F. Battistini, J. L. Gelpi, C. A. Laughton, S. A. Harris, D. A. Case and M. Orozco, Nat. Methods, 2016, 13, 55–61, DOI:10.1038/nmeth.3658.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38, DOI:10.1016/0263-7855(96)00018-5.
- P. A. Kollman, I. Masova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheathan, Acc. Chem. Res., 2000, 33, 889–897, DOI:10.1021/ar000033j.
-
(a) C. Wiegand and U.-C. Hipler, Skin Pharmacol. Physiol., 2009, 22, 74–82, DOI:10.1159/000178866;
(b) C. Ma, X. Gao, Y. Yang, X. Bian, B. Wang, X. Liu, Y. Wang, D. Su, G. Zhang, L. Qu and N. Zhang, Front. Bioeng. Biotechnol., 2024, 11, 1329183, DOI:10.3389/fbioe.2023.1329183.
-
(a) P. Kumar, A. Nagarajan and P. D. Uchil, Cold Spring Harb. Protoc., 2018, 2018 DOI:10.1101/pdb.prot095505;
(b) C. L. Putta, H. S. Buddhiraju, D. N. Yadav, A. Basa and A. K. Rengan, ACS Appl. Bio Mater., 2025, 8, 661–675, DOI:10.1021/acsabm.4c01533.
- Y. Rai, R. Pathak, N. Kumari, D. K. Sah, S. Pandey, N. Kalra, R. Soni, B. S. Dwarakanath and A. N. Bhatt, Sci. Rep., 2018, 8, 1531, DOI:10.1038/s41598-018-19930-w.
-
(a) Y. Huang, Z. Guan, X. Dai, Y. Shen, Q. Wei, L. Ren, J. Jiang, Z. Xiao, Y. Jiang, D. Liu, Z. Huang, X. Xu, Y. Luo and C. Zhao, Nat. Commun., 2021, 12, 1–22, DOI:10.1038/s41467-021-24564-0;
(b) S. R. Motmaen, S. Tavakol, M. T. Joghataei and M. Barati, Int. Wound J., 2020, 17, 137–148, DOI:10.1111/iwj.13248;
(c) K. H. Jones and J. A. Senft, J. Histochem. Cytochem., 1985, 33, 77–79, DOI:10.1177/33.1.2578146.
-
(a)
G. M. Sheldrick, SHELXS-97: Program for Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122, DOI:10.1107/S0108767307038627;
(c) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S2053273314026370;
(d) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13, DOI:10.1107/S0021889802022112.
-
(a)
CCDC 2451107: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2n8kzh;
(b)
CCDC 2451108: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2n8l0k;
(c)
CCDC 2451109: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2n8l1l;
(d)
CCDC 2451110: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2n8l2m;
(e)
CCDC 2451111: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2n8l3n.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *c and
Aravind Kumar
Rengan
*c and
Aravind Kumar
Rengan










