
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterisation, and magnetic properties of a permethylindenyl manganocene†
Alexander
Evans
 a,
Thomas A. Q.
Arnold
a,
Paul
Ransom
a,
Samantha C.
Binding
a,
William K.
Myers
b,
Agamemnon E.
Crumpton
a,
Zoë R.
Turner
a,
Jean-Charles
Buffet
a and
Dermot
O'Hare
*a
a,
Thomas A. Q.
Arnold
a,
Paul
Ransom
a,
Samantha C.
Binding
a,
William K.
Myers
b,
Agamemnon E.
Crumpton
a,
Zoë R.
Turner
a,
Jean-Charles
Buffet
a and
Dermot
O'Hare
*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, OX1 3TA Oxford, UK. E-mail: dermot.ohare@chem.ox.ac.uk
bCentre for Advanced Electron Spin Resonance (CAESR), Inorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK
First published on 11th July 2025
Abstract
The heptamethylindenyl ligand (C9Me7−; Ind*) has been used to synthesise the manganese complex, (η5-Ind*)2Mn, by lithiation of the proligand and subsequent salt metathesis with MnCl2. The solid state molecular structure indicates a low-spin (S = ½) ground state for the Mn2+ metal centre at 100 K. Solution EPR spectroscopy, solid state magnetic susceptibility measurements, and computational studies suggest the complex undergoes a thermally accessible spin-crossover to a high-spin (S = 5/2) state.
Introduction
Following the discovery of the prototypical metallocene complex, ferrocene,1–3 the cyclopentadienyl anion (Cp) has become ubiquitous in the isostructural transition metal metallocenes (η5-Cp2)M (e.g.: M = V–Ni),4–6 and further existing in complexes exhibiting a range of hapticities.7 Cp2M metallocene sandwich complexes, were later expanded to include s-block (Cp2Mg)8,9 and p-block (Cp2Sn and Cp2Pb) metals.10–12 More recently, homo- and heterobimetallic sandwich complexes have been reported in the literature from across the periodic table utilising Cp or sterically and electronically tuned derivatives.13–16 Additionally, increased steric bulk provided by Cp permethylation, Cp*, yields increased kinetic stability compared to their Cp analogues.17The indenyl anion (C9H7−; Ind), formally a benzannulated derivative of Cp, displays significantly altered chemistry from Cp, predominantly due to its flexibility in binding modes, and has become an important ligand framework in organometallic chemistry. Transition metal bis(η5-Ind) complexes are well documented, with Ind2Fe and Ind2Co first reported in 1954;18 however, it is notable that Ind2Mn has eluded characterisation as a non-THF adduct. To date η6-, η3-, η2- and η1-indenyl transition metal complexes have also been reported19,20 in addition to mixed complexation modes with Nesmeyanov et al. reporting the synthesis of a tungsten complex displaying both η3- and η5-bound Ind ligands to the same metal centre.21 An array of substituted manganocenes were reported by Hanusa and co-workers in 2010 and later by Maekawa et al.22,23 A spectrum of binding modes was reported, but five compounds (IndR2Mn, R = 2-SiMe3, 1,3-SiMe3, 1,3-iPr, 1,3-tBu and 1,3-Cy) displayed η5 binding. Magnetic and (DFT) studies clearly illustrate that the above species all contain high-spin manganese, mirroring the original discovery concerning Cp2Mn.
In many cases, indenyl metallocene, and indeed indenyl metallocenophane, complexes, exhibit significantly divergent reactivities compared to their Cp counterparts.24 This is reasoned to be caused by the ability of the indenyl group to undergo η5,η3-haptotropic ring slip while maintaining the aromaticity of the benzene ring. This phenomenon is commonly referred to as the “indenyl effect”.25 The η5–η3 ring slip is formally a two-electron oxidation of the metal, which becomes electrophilic and coordinatively unsaturated, allowing concomitant associative substitution processes not common for 18 electron complexes,26 leading to a several-thousand-fold increase in the rate of ligand substitution.27 This differing reactivity is classically exemplified in the SN2 (associative) substitution of PPh3 into the rhodium complex, (η5-Ind)Rh(CO)2, which proceeds 108 times faster than its Cp analogue.28
Similarly to Cp, permethylation confers kinetic stability to permethylindenyl (Ind*) complexes through increased steric bulk. This reduces the propensity for reactive collision, and the positive inductive effect of the methyl groups through  hyperconjugation generally increases the thermodynamic stability.29 The synthesis of the bulky Ind*H ligand precursor from (2E)-2-methylbut-2-enoic acid is well established,29 enabling its use in the preparation of Ind* metallocenes (Ind*2M) for a variety of d- and f-block elements through reactions with alkali metal Ind* salts and metal sources (such as Co(acac)2 or FeCl2·xTHF).30–36 The electronic properties of the Ind* ligand in Ind*2Fe have been studied by D. O'Hare et al. – a combined electrochemical and photoelectron spectroscopic approach showed quantitatively the electron donating nature of the Ind* ligand by a reduction in the first ionisation energies and redox potentials compared to Cp*2Fe.30
hyperconjugation generally increases the thermodynamic stability.29 The synthesis of the bulky Ind*H ligand precursor from (2E)-2-methylbut-2-enoic acid is well established,29 enabling its use in the preparation of Ind* metallocenes (Ind*2M) for a variety of d- and f-block elements through reactions with alkali metal Ind* salts and metal sources (such as Co(acac)2 or FeCl2·xTHF).30–36 The electronic properties of the Ind* ligand in Ind*2Fe have been studied by D. O'Hare et al. – a combined electrochemical and photoelectron spectroscopic approach showed quantitatively the electron donating nature of the Ind* ligand by a reduction in the first ionisation energies and redox potentials compared to Cp*2Fe.30
Manganocene, and its methyl-substituted derivatives, have been extensively studied due to their unique property of having two energetically accessible spin states (Fig. 1).37–41 These can be readily interconverted, induced by external stimuli such as light, pressure, or temperature, in a phenomenon known as spin-crossover.42 It has been found that while Cp2Mn is a high-spin (6A1g) species and its permethylated counterpart Cp*2Mn occupies an S = ½ (2E2g) ground state, whereas a singularly methylated Cp complex Cp′2Mn varies the occupation of the two spin states quite significantly with temperature.43–48 Work by Walter et al. has examined the high temperature spin-crossover behaviours of a variety of manganocenes using both EXAFS and solid-state magnetic measurements,49 whilst recent studies have investigated the low-spin d5 metallocene as a photoredox agent.50 Herein, we report the synthesis and characterisation of the manganese sandwich complex, Ind*2Mn – the first indenyl-containing manganocene to display thermally induced variation in the Mn2+ spin state.
 | ||
| Fig. 1 Mn d5 ground states in distorted Oh fields.37–41 | ||
Results and discussion
Synthesis and characterisation of Ind*2Mn
Two procedures for the preparation of proligand (C9Me7H; Ind*H) were used in this work: (1) methylation of hexamethylindene (C9Me6H2; Ind#H) by the action of MeI on the lithiated precursor (Ind#Li), and (2) an improved synthesis following the previously reported route, minimising reaction steps and optimising reagents (ESI†).13 In both cases Ind*H was subsequently deprotonated with nBuLi at −78 °C in THF and isolated as a solid powder. Synthesis of Ind*2Mn was achieved by adding two equivalents of Ind*Li to a stirred THF solution of MnCl2 at −78 °C, allowing to warm to room temperature overnight (Scheme 1). Work-up afforded Ind*2Mn as a purple solid. | ||
| Scheme 1 Synthesis of Ind*2Mn. | ||
The solution phase 1H NMR spectrum was paramagnetically broadened as expected for a d5 Mn2+ centre (Fig. S4†). On further investigation by variable temperature 1H NMR spectroscopy (Fig. 2), the manganocene's wing tip methyl (Me2) resonance demonstrated a dramatic response to temperature (Δδ = 9.8 ppm between 333 and 233 K) whilst other Me resonances varied slightly (Δδ = 0.18 ppm) over the same temperature range. The temperature dependence of the chemical shifts did not obey the Curie law (δ ∝ T−1) and we were unable to extract meaningful quantitative data (vide infra; Fig. S12†). This has been similarly observed by Ashley et al. for the paramagnetic bimetallic chromium permethylpentalene complex and Cloke and co-workers for an asymmetric manganese pentalene complex; where variation in ligand field and sterics confound solution phase thermodynamic interpretation.51–53
 | ||
| Fig. 2 Variable-temperature 1H NMR spectra of Ind*2Mn in toluene-d8 solution. | ||
Single crystals suitable for X-ray diffraction were obtained from a concentrated hexanes solution after cooling to −24 °C overnight. The molecular structure is depicted in Fig. 3. The molecule crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; alternate views are shown in Fig. S7,† and relevant bond lengths and angles are given in Table S2.† Scrutinising the molecular structure; the Ind* ligands are nearly coplanar, as shown by the low value of the fold angles about the C6–C5 ring junctions (1.91(6)° and 2.68(6)°) and a tilt angle = 4.80(5)°. Crystal structures previously reported for neutral Ind*2M complexes of the first row transition-metals are those of Ind*2Cr,31 and Ind*2Fe.54 The expected trend in decreasing average M–C distance is observed across the period (Cr = 2.182 Å, Mn = 2.1272(13) Å, Fe = 2.074 Å). The average Mn–C distance is significantly different from the values observed for all other structurally characterised alkyl manganocenes, IndR2Mn where R = 2-SiMe3, 1,3-SiMe3, 1,3-iPr, 1,3-tBu and 1,3-cyclohexyl (Table 1), where the reported average Mn–C distance is 2.41 Å.22,23 Furthermore, the average Mn–Cpcent distance is 1.7400(6) Å for Ind*2Mn compared to 2.05 Å for the other IndR2Mn complexes. By both measures, the Ind*2Mn distance is 0.3 Å shorter, clearly indicating a major change in the electronic structure.
; alternate views are shown in Fig. S7,† and relevant bond lengths and angles are given in Table S2.† Scrutinising the molecular structure; the Ind* ligands are nearly coplanar, as shown by the low value of the fold angles about the C6–C5 ring junctions (1.91(6)° and 2.68(6)°) and a tilt angle = 4.80(5)°. Crystal structures previously reported for neutral Ind*2M complexes of the first row transition-metals are those of Ind*2Cr,31 and Ind*2Fe.54 The expected trend in decreasing average M–C distance is observed across the period (Cr = 2.182 Å, Mn = 2.1272(13) Å, Fe = 2.074 Å). The average Mn–C distance is significantly different from the values observed for all other structurally characterised alkyl manganocenes, IndR2Mn where R = 2-SiMe3, 1,3-SiMe3, 1,3-iPr, 1,3-tBu and 1,3-cyclohexyl (Table 1), where the reported average Mn–C distance is 2.41 Å.22,23 Furthermore, the average Mn–Cpcent distance is 1.7400(6) Å for Ind*2Mn compared to 2.05 Å for the other IndR2Mn complexes. By both measures, the Ind*2Mn distance is 0.3 Å shorter, clearly indicating a major change in the electronic structure.
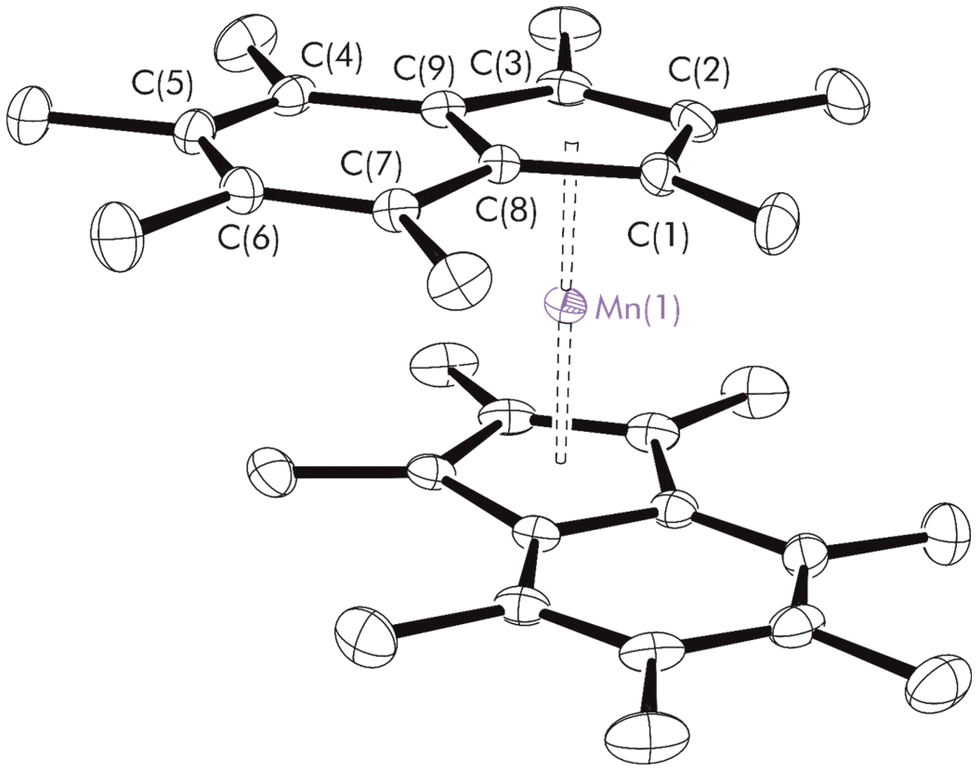 | ||
| Fig. 3 Molecular structure of Ind*2Mn. For clarity, hydrogens atoms were omitted (displacement ellipsoids are drawn at 30% probability). Selected distances (Å) and angles (°): Mn–Cpcent 1.7400(6), ΔM–C 0.073, ΔM–C 0.077, RA 85.2(2), C6–C5 1.443(2), C21–C22 1.441(2). Ring-slip parameter (ΔM–C) defined in eqn (S1).† Rotation angle (RA) based on adapted torsion angle, visually defined in Fig. S8.†55 | ||
| IndR2Mn | M–C avg. (Å) | Cpcent–M (Å) | Δ M–C (Å) | RAb (°) |
|---|---|---|---|---|
| a Ring-slip parameter (ΔM–C) defined in eqn (S1).† b Rotation angle (RA) based on adapted torsion angle, visually shown in Fig. S8.†55 | ||||
| R = 1,2,3,4,5,6,7-Me | 2.127 | 1.740 | 0.075 | 85.2 |
| R = 2-SiMe322 | 2.409 | 2.079 | 0.14 | 180 |
| R = 1,3-SiMe322 | 2.42 | 2.085 | 0.12 | 83.7 |
| R = 1,3-iPr22 | 2.4 | 2.04 | 0.07 | 176 |
| R = 1,3-tBu23 | 2.42 | 2.10 | 0.156 | 86.0 |
| R = 1,3-cyclohexyl23 | 2.383 | 1.96 | 0.148 | 91.35 |
In fact, the measured Mn–Cpcent distance much more closely matches the value for Cp*2Mn (1.734 Å), a low-spin manganocene.56 A DFT analysis conducted by Maekawa et al. on the effect of the spin state of Mn in their bis(indenyl) analogue suggested dramatically different values for the M–Cpcent distance in the low- and high-spin cases, encouraging us to investigate the effect of Ind* computationally.23 Interestingly, the rotation angle (RA) for Ind*2Mn is 85.2(2)°, whereas that for the chromium and iron species are 180° and 151° respectively; this value is similar to that found for the cationic cobalt and chromium species, and does not fit previous conclusions that the RA for permethylindenyl complexes fall broadly into two regions: 180° for the neutral species and 90° for the cations.31 However, it is in good agreement with the observed and calculated values for IndR2Mn, where R = 1,3-tBu and 1,3-SiMe3 are reported as 86.0° and 83.7° respectively.22,23 It was noted that the ring-slip parameter values (ΔM–C = 0.073 and 0.077 Å) were within the range of literature values (0.07–0.156 Å).
Electron paramagnetic resonance (EPR) studies
The temperature dependence of the X-band CW-EPR signal for Ind*2Mn was studied between 4.5–160 K in a toluene glass.Representative spectra are shown in Fig. 4. At 12 K, the signal is characteristic of low-spin Mn2+. As the temperature approaches 100 K, the unresolved outer manifold transitions of high-spin Mn2+, MS = ±5/2 ↔ MS = ±3/2 and MS = ±3/2 ↔ MS = ±½, are apparent on either side of the sextet of 55Mn hyperfine lines at g = 2 from the low-spin Mn2+ and MS = ±½ ↔ MS = ±½ of high-spin Mn2+. Double integration of the total EPR signal over the temperatures studied reveals an increasing amplitude with temperature as the high-spin state is populated. The low-spin signal was simulated, shown in Fig. S13,† providing g-values of g‖ = 2.56 and g⊥ = 1.975, while the 55Mn hyperfine simulations were A‖(55Mn) = 206 MHz and A⊥(55Mn) = 80 MHz. The theory of Maki and Berry has a separation of g‖ and g⊥ defined as follows:57
 | (1) |
 | ||
| Fig. 4 X-band CW-EPR of the Ind*2Mn complex at 12, 40 and 100 K. Conditions were a microwave power of 50 W, 100 kHz modulation amplitude of 0.8 mT, microwave frequency of 9.372(4) GHz and a time constant of 20.48 ms. | ||
According to eqn (1), the Ind* ligand provides a new extreme of an axial distortion, shown in Fig. S8.† Comparison of the g-values of Ind*2Mn with those of Cp2Mn, Cp*2Mn, and Cp′2Mn reveals a clear trend.48
The g-value of the lowest symmetry ligand, Cp′, is most similar to that of Ind*, followed by Cp* and Cp. On a lesser note, samples in frozen solution provide g-values that are closer than samples in magnetically-dilute solids.
Magnetic susceptibility measurements
The solid state magnetic susceptibility of Ind*2Mn has been measured between 6–300 K (Fig. 5). No difference was observed between the magnetic susceptibility data of a zero-field cooled (ZFC) compared to a field cooled (FC) sample over this temperature range. Attempts to fit the data to the Curie–Weiss law either gave very poor agreement or unrealistic values. For example, a least squares fit to the magnetic susceptibility data between 50 and 200 K gives a Weiss constant, θ = –76 K, and an effective magnetic moment, μeff = 4.18μB. Such a large value for the Weiss constant, implied antiferromagnetic behaviour, is not reasonable for a paramagnetic metallocene (Fig. S10†).58 | ||
| Fig. 5 Temperature dependence of reciprocal molar magnetic susceptibility (1/χmol) and effective magnetic moment (μeff) for Ind*2Mn. | ||
In the solid state, Ind*2Mn undergoes a thermally induced spin-crossover. At 6 K, μeff = 1.67μB which is close to the value predicted on the basis of the low temperature EPR (μeff = 1.89μB. S = ½; 〈g〉 = 2.19). Between 6 K and 300 K, the effective magnetic moment of the sample steadily increases, reaching a μeff = 3.85μB at 300 K, which is consistent with a thermally induced partial population of a high-spin, S = 5/2 state. Assuming an ideal high-spin excited state with a μeff = 5.92, the magnetic moment at 300 K is consistent with a 36% population of this high temperature form.47 A derived Arrhenius plot (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K vs. T−1) over the studied temperature range enabled reasonable extraction of thermodynamic parameters via a linear fit regression (Fig. S11;†R2 = 0.992, ΔH = 0.485 ± 0.007 kJ mol−1, and ΔS = –7.98 ± 0.17 J mol−1).49 Negligible enthalpic contributions in the solid state suggest an entropy driven process towards the thermally induced spin-crossover. Variable-temperature single-crystal X-ray diffraction was also employed to probe the system; all crystal structures exhibit largely the same unit cell parameters, with only minor variations attributed to thermal expansion. Notably, there were no significant changes to the RA, suggesting that any geometry induced spin-state transition does not manifest in the crystalline phase; however, the complex may behave differently in a toluene glass/solution due to reduced conformational rigidity. Consequently, we believe the magnetometry studies isolate the thermodynamic data associated with purely the spin-crossover phenomena, whilst the NMR spectroscopic data includes a second regime related to the ligand geometry, supported by DFT (vide infra), confounding the extraction of thermodynamic parameters.
K vs. T−1) over the studied temperature range enabled reasonable extraction of thermodynamic parameters via a linear fit regression (Fig. S11;†R2 = 0.992, ΔH = 0.485 ± 0.007 kJ mol−1, and ΔS = –7.98 ± 0.17 J mol−1).49 Negligible enthalpic contributions in the solid state suggest an entropy driven process towards the thermally induced spin-crossover. Variable-temperature single-crystal X-ray diffraction was also employed to probe the system; all crystal structures exhibit largely the same unit cell parameters, with only minor variations attributed to thermal expansion. Notably, there were no significant changes to the RA, suggesting that any geometry induced spin-state transition does not manifest in the crystalline phase; however, the complex may behave differently in a toluene glass/solution due to reduced conformational rigidity. Consequently, we believe the magnetometry studies isolate the thermodynamic data associated with purely the spin-crossover phenomena, whilst the NMR spectroscopic data includes a second regime related to the ligand geometry, supported by DFT (vide infra), confounding the extraction of thermodynamic parameters.
The solution phase effective magnetic moment was measured at 298 K using the Evans method59 and found to be 2.53 μB in a range of hydrocarbon solvents. Negligible variation of the moment over the accessible temperature range, 193–333 K, was observed. The existence of a low-spin configuration contrasts all previously reported manganese bis(indenyl) complexes which have high-spin, S = 5/2 ground states from 4–300 K: (IndR2)2Mn (R = iPr, tBu, or Cy), μeff = 5.25–5.9μB.4,5
The magnetic susceptibility behaviour of Ind*2Mn reflects the trend observed with bis(indenyl)chromium(II) complexes studied by Hanusa, Yee and co-workers, where the staggered and eclipsed conformations favour high-spin ground states, and the gauche conformations are generally associated with low-spin complexes.60 In addition, they also report a general correlation between increasing methylation and a propensity for low-spin ground states; this trend is mirrored in magnetic studies on the manganocenes Cp2Mn, Cp*2Mn, and Cp′2Mn.43–48 Extrapolating from these observations, Ind*2Mn would be expected to favour a low-spin configuration.
Computational studies
Magnetometry measurements at 300 K indicate that 36% of the complex exists in the high-spin state.47 At 130 K, EPR spectroscopy reveals the loss of hyperfine splitting, suggesting the presence of a high-spin complex. This observation is consistent with the expectation that a significant high-spin population would overwhelm the signal from the low-spin state and may likewise explain the lack of variation of the effective magnetic moment in solution using the Evans method. Given the discrepancies between observed spin-crossover behaviours in the NMR and EPR spectroscopic, and SQUID magnetometric measurements, the complex was computationally investigated in order to probe the ligand influence on the Mn spin-state.Accurate computational prediction of relative spin state energy levels for metal complexes are notoriously difficult to obtain.61,62 Screening a variety of DFT functionals from the crystallographically-defined coordinates of Ind*2Mn yielded a wide range of electronic energy difference between the low- and high-spin states (approximately 420 kJ mol−1 ΔEHL; Table S4†). Given the disparity in ΔEHL, similarly observed in the literature for manganese spin-crossover complexes,42 more accurate methods were utilised to ascertain an appropriate functional for the reactive surface. Consequently, complete active space self-consistent field with second-order N-electron valence state perturbation theory (CASSCF/NEVPT2) along with domain-based local pair natural orbital coupled-cluster theory (DLPNO-CCSD(T)) were selected due to their increased reliability in determining ΔEHL: CASSCF–NEVPT2 ΔEHL = 117 kJ mol−1 and CCSD(T) ΔEHL = 134 kJ mol−1.42 Reasonable agreement between the methods (ΔE = 17 kJ mol−1) prompted selection of the DFT functional MN15 (ΔE = 138 kJ mol−1), due to its alignment with CCSD(T).42 Consequently, optimised geometries were performed, with a scan across the RA for the optimised low- and high-spin structures where single point calculations were performed at DLPNO-CCSD(T) (Fig. 6). The potential energy surfaces of the high-spin and low-spin states were found to intersect, supporting the experimental observation that Ind*2Mn undergoes thermally induced spin-crossover.
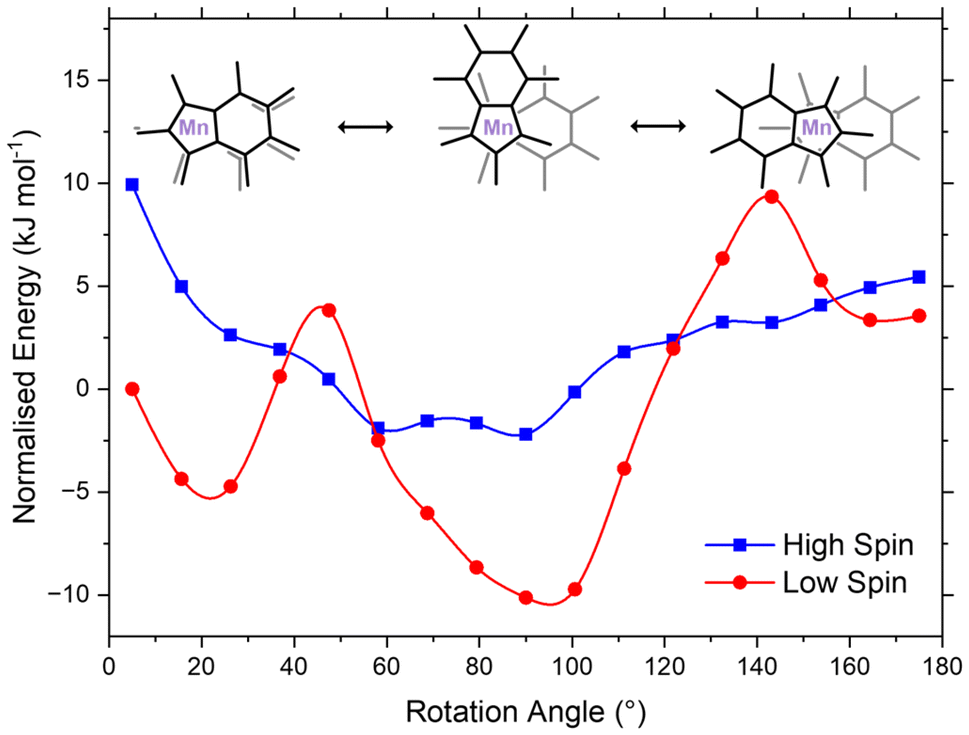 | ||
| Fig. 6 Single point calculations performed at CCSD(T) for both high- and low-spin Ind*2Mn, energies normalised to the low-spin complex at 5°. | ||
In order to probe the differences in the nature of the Cp* and Ind* ligand architectures, supplementary energy decomposition analysis (EDA), via sobEDA, of L2Mn (R = Ind* and Cp*) was carried out. The total energy interaction was found to be broadly similar (Table S7†). While both ligands stabilise the manganese centre through electrostatic and orbital (Eorb) interactions, the Ind* ligand contributes both larger Pauli repulsive (Erep) and orbital attractive forces, markedly counteracting one another. Increased Erep is reasonably explained by the imposed steric requirements of the benzannulated ring. Further decomposition of Eorb was undertaken using EDA-NOCV. The pairwise deformation densities reveal that, in the Ind* complex, the benzene ring contributes additional electron density to the Mn centre through its extended π-system (Fig. S16†).
Conclusions
The solid state structure and the low temperature magnetic responses of Ind*2Mn suggest that it is the first indenyl manganese complex with a low-spin ground state. At room temperature the solid state magnetic moment suggest that a high-spin (S = 5/2) configuration is significantly populated. Given the ubiquity of Cp*, certain circumstances likely demand increased steric constraint; consequently use of Ind* as an alternate ligand framework may provide attractive complex properties and we hope this puts into greater focus the work being done already within the group.63–65Experimental
Synthesis of Ind*H
7.40 g 1,2,4,5,6,7-hexamethyl-1H-indene (Ind#H, 36.9 mmol, 1 eq.) was dissolved in 150 mL THF and was cooled, in a 300 mL schlenk under N2, to −78 °C before fast dropwise addition of 23.1 mL n-butyllithium (1.6 M in hexanes; 36.93 mmol, 1 eq.). The reaction mixture was allowed to warm to room temperature over 16 h before cooling to −78 °C and 2.53 mL MeI (40.63 mmol, 1.1 eq.) added dropwise. The reaction mixture was allowed to warm to room temperature over 2 h. THF was removed in vacuo and the residue dissolved in DCM and transferred to a separating funnel containing brine solution. Product was extracted in 3 × 50 mL DCM, dried over MgSO4, DCM removed by rotary evaporation before placing the oil in a −4 °C freezer. Crystalline solid product was isolated by Buchner filtration and recrystallised from EtOH at −4 °C yielding 7.35 g (92%) yellow, crystalline 1,2,3,4,5,6,7-heptamethylindene (Ind*H). 1H NMR (Benzene-d6, 298 K, 400 MHz) δ (ppm): 3.03 (q, 1H, 3JHH = 7.5, CH), 2.44 (s, 3H, Ar–Me), 2.21 (s, 3H, Ar–Me), 2.18–2.13 (m, 9H, Ar–Me and Cp–Me), 1.82 (s, 3H, Cp–Me), 2.43 (d, 3H, 3JHH = 7.5, Cp–Me).Synthesis of Ind*2Mn
One equivalent of MnCl2 (0.27 g, 2.18 mmol) was stirred in an ampoule in 30 mL THF for 16 hours, yielding a white suspension. This mixture was cooled to −78 °C and a slurry of two equivalents of Ind*Li (1.00 g, 4.36 mmol) in 30 mL THF added. The yellow-brown mixture was allowed to warm to room temperature and stirred for 15 hours, affording a brown-orange solution. The solvent was removed under vacuum to give a brown-orange solid. Extraction with hexane gave a purple solution, which was filtered through Celite and dried in vacuo affording a purple solid in 60% yield (0.543 g, 1.30 mmol). 1H NMR (benzene-d6, 298 K, 499.9 MHz) δ (ppm): 9.06, 3.05, 2.44, 2.16, 1.82, 1.18 (all broad). 13C{1H} NMR (benzene-d6, 298 K, 499.9 MHz) δ (ppm): 145.3, 142.3, 141.2, 133.5, 132.7, 130.6, 128.6, 126.5, 46.6, 16.8, 16.4, 16.3, 16.3, 16.1, 15.5, 12.3. MS (EI): calc.: 481.2667 found: 481.2301. UV/Vis: λmax(n-hexanes) = 533.1 nm (ε = 5.30 × 102 mol−1 dm3 cm−1). FT-IR (KBr, cm−1): 2965 (s), 1616 (m), 1447 (m), 1385 (s), 1261 (s), 1093 (m), 1022 (m), 801 (s). CHN analysis (calculated, found; %): C = 79.79, 79.91; H = 8.80, 8.83.X-ray crystallography details
Colourless single crystals of Ind*H were isolated from concentrated pentane solution at room temperature. C16H22, Mr = 214.33, monoclinic, P21/c, a = 13.0353(4) Å, b = 10.9285(4) Å, c = 9.0453(3) Å, α = 90°, β = 91.765(3)°, γ = 90°, V = 1287.95(7) Å3, Z = 4, T = 150 K, block, colourless, 5161 independent reflections, R1 = 0.0470, wR2 = 0.1368 [I> = 2σ(I)]. CCDC deposition number: 2421030† Dark purple single crystals of Ind*2Mn were isolated from a concentrated solution (hexanes) after cooling to −24 °C overnight. C32H42Mn, Mr = 481.59, triclinic, P, a = 9.05120(10) Å, b = 10.43980(10) Å, c = 15.81240(10) Å, α = 73.2730(10)°, β = 84.7780(10)°, γ = 63.9150(10)°, V = 1284.09(2) Å3, Z = 2, T = 100°K, block, purple, 5238 independent reflections, R(int) = 0.0279, R1 = 0.0290, wR2 = 0.0801 [I > = 2σ(I)]. CCDC deposition number: 2421029.†
Author contributions
A. E. carried out the investigation and formal analysis unless otherwise stated. W. K. M. performed the investigation and formal EPR analysis. A. E. C performed the investigation and formal computational analysis. S. C. B. assisted the SQUID investigation. T. A. Q. A. developed the improved ligand synthesis. A. E. managed the project, and drafted the manuscript. All authors revised the manuscript. D. O'H supervised the research and acquired funding.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for Ind*H and Ind*2Mn has been deposited at the CCDC repository under 2421030 and 2421029.†Acknowledgements
The authors would like to thank SCG Chemicals PLC, (Thailand) and the University of Oxford for funding. Thanks to Louis J. Morris for crystallographic assistance with regards to the Ind*H data collection and structure refinement. Thanks to Stephen Boyer of London Metropolitan University for the elemental analysis. We gratefully acknowledge the EPSRC for a Strategic Equipment Grant (EP/V028995/1). We acknowledge with thanks the use of the University of Oxford Advanced Research Computing (ARC) facility in carrying out this work. https://doi.org/10.5281/zenodo.22558.References
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- S. A. Miller, J. A. Tebboth and J. F. Tremaine, J. Chem. Soc., 1952, 632–635 RSC.
- H. Werner, Angew. Chem., Int. Ed., 2012, 51, 6052–6058 CrossRef CAS PubMed.
- P. J. Chirik and J. E. Bercaw, Metallocenes: Synthesis Reactivity Applications, Wiley-VCH, Weinheim, 1998 Search PubMed.
- P. L. Pauson, Q. Rev., Chem. Soc., 1955, 9, 391–414 RSC.
- G. Wilkinson, P. L. Pauson and F. A. Cotton, J. Am. Chem. Soc., 1954, 76, 1970–1974 CrossRef CAS.
- J. M. O'Connor and C. P. Casey, Chem. Rev., 1987, 87, 307–318 CrossRef.
- E. O. Fischer and W. Hafner, Z. Naturforsch., B, 1954, 9, 503–505 CrossRef.
- G. Wilkinson and F. A. Cotton, Chem. Ind., 1954, 307 CAS.
- L. Wirtz and A. Schäfer, Chem. – Eur. J., 2021, 27, 1219–1230 CrossRef CAS PubMed.
- E. O. Fischer and U. Piesbergen, Z. Naturforsch., B, 1956, 11, 758–759 CrossRef.
- E. O. Fischer and H. Grubert, Z. Anorg. Allg. Chem., 1956, 286, 237–242 CrossRef CAS.
- I. Resa, E. Carmona, E. Gutierrez-Puebla and A. Monge, Science, 2004, 305, 1136–1138 CrossRef CAS PubMed.
- J. T. Boronski, A. E. Crumpton, L. L. Wales and S. Aldridge, Science, 2023, 380, 1147–1149 CrossRef CAS PubMed.
- I.-A. Bischoff, S. Danés, P. Thoni, B. Morgenstern, D. M. Andrada, C. Müller, J. Lambert, E. C. J. Gießelmann, M. Zimmer and A. Schäfer, Nat. Chem., 2024, 16, 1093–1100 CrossRef CAS PubMed.
- T. Piou, F. Romanov-Michailidis, M. Romanova-Michaelides, K. E. Jackson, N. Semakul, T. D. Taggart, B. S. Newell, C. D. Rithner, R. S. Paton and T. Rovis, J. Am. Chem. Soc., 2017, 139, 1296–1310 CrossRef CAS PubMed.
- D. C. Calabro, J. L. Hubbard, C. H. Blevins, A. C. Campbell and D. L. Lichtenberger, J. Am. Chem. Soc., 1981, 103, 6839–6846 CrossRef CAS.
- P. L. Pauson and G. Wilkinson, J. Am. Chem. Soc., 1954, 76, 2024–2026 CrossRef CAS.
- I. A. Lobanova and V. I. Zdanovich, Russ. Chem. Rev., 1988, 57, 967–980 CrossRef.
- M. Stradiotto and M. J. McGlinchey, Coord. Chem. Rev., 2001, 219–221, 311–378 CrossRef CAS.
- A. N. Nesmeyanov, N. A. Ustynyuk, L. G. Makarova, V. G. Andrianov, Y. T. Struchkov, S. Andrae, Y. A. Ustynyuk and S. G. Malyugina, J. Organomet. Chem., 1978, 159, 189–199 CrossRef CAS.
- J. A. Crisp, R. M. Meier, J. S. Overby, T. P. Hanusa, A. L. Rheingold and W. W. Brennessel, Organometallics, 2010, 29, 2322–2331 CrossRef CAS.
- M. Maekawa, C. G. Daniliuc, M. Freytag, P. G. Jones and M. D. Walter, Dalton Trans., 2012, 41, 10317–10327 RSC.
- V. Cadierno, J. Díez, M. Pilar Gamasa, J. Gimeno and E. Lastra, Coord. Chem. Rev., 1999, 193–195, 147–205 CrossRef CAS.
- E. O. Fischer, D. Seus and R. Jira, Z. Naturforsch., B, 1953, 8, 692–693 CrossRef.
- A. J. Hart-Davis and R. J. Mawby, J. Chem. Soc. A, 1969, 2403–2407 RSC.
- A. J. Hart-Davis, C. White and R. J. Mawby, Inorg. Chim. Acta, 1970, 4, 441–446 CrossRef CAS.
- M. E. Rerek, L.-N. Ji and F. Basolo, J. Chem. Soc., Chem. Commun., 1983, 1208–1209 RSC.
- T. K. Miyamoto, M. Tsutsui and L.-B. Chen, Chem. Lett., 1981, 10, 729–730 CrossRef.
- D. Ohare, J. C. Green, T. Marder, S. Collins, G. Stringer, A. K. Kakkar, N. Kaltsoyannis, A. Kuhn, R. Lewis, C. Mehnert, P. Scott, M. Kurmoo and S. Pugh, Organometallics, 1992, 11, 48–55 CrossRef CAS.
- D. O'Hare, V. J. Murphy and N. Kaltsoyannis, J. Chem. Soc., Dalton Trans., 1993, 383–392 RSC.
- T. M. Frankcom, J. C. Green, A. Nagy, A. K. Kakkar and T. B. Marder, Organometallics, 1993, 12, 3688–3697 CrossRef CAS.
- D. Ohare, V. Murphy, G. M. Diamond, P. Arnold and P. Mountford, Organometallics, 1994, 13, 4689–4694 CrossRef CAS.
- T. M. Trnka, J. B. Bonanno, B. M. Bridgewater and G. Parkin, Organometallics, 2001, 20, 3255–3264 CrossRef CAS.
- M. Tsutsui, L. B. Chen, D. E. Bergbreiter and T. K. Miyamoto, J. Am. Chem. Soc., 1982, 104, 855–856 CrossRef CAS.
- D. O'Hare, V. Murphy, G. M. Diamond, P. Arnold and P. Mountford, Organometallics, 1994, 13, 4689–4694 CrossRef.
- N. Hebendanz, F. H. Koehler, G. Mueller and J. Riede, J. Am. Chem. Soc., 1986, 108, 3281–3289 CrossRef CAS.
- D. Cozak and F. Gauvin, Organometallics, 1987, 6, 1912–1917 CrossRef CAS.
- J. H. Ammeter, R. Bucher and N. Oswald, J. Am. Chem. Soc., 1974, 96, 7833–7835 CrossRef CAS.
- S. Evans, M. L. H. Green, B. Jewitt, G. H. King and A. F. Orchard, J. Chem. Soc., Faraday Trans. 2, 1974, 70, 356–376 RSC.
- J. A. Bandy, F. G. N. Cloke, G. Copper, J. P. Day, R. B. Girling, R. G. Graham, J. C. Green, R. Grinter and R. N. Perutz, J. Am. Chem. Soc., 1988, 110, 5039–5050 CrossRef CAS.
- M. Drosou, C. A. Mitsopoulou and D. A. Pantazis, Polyhedron, 2021, 208, 115399 CrossRef CAS.
- F. Engelmann, Z. Naturforsch., B, 1953, 8, 775–776 CrossRef.
- E. O. Fischer and H. Leipfinger, Z. Naturforsch., B, 1955, 10, 353–355 CrossRef.
- L. T. Reynolds and G. Wilkinson, J. Inorg. Nucl. Chem., 1959, 9, 86–92 CrossRef.
- G. Wilkinson, F. A. Cotton and J. M. Birmingham, J. Inorg. Nucl. Chem., 1956, 2, 95–113 CrossRef CAS.
- M. E. Switzer, R. Wang, M. F. Rettig and A. H. Maki, J. Am. Chem. Soc., 1974, 96, 7669–7674 CrossRef CAS.
- J. L. Robbins, N. M. Edelstein, S. R. Cooper and J. C. Smart, J. Am. Chem. Soc., 1979, 101, 3853–3857 CrossRef CAS.
- M. D. Walter, C. D. Sofield, C. H. Booth and R. A. Andersen, Organometallics, 2009, 28, 2005–2019 CrossRef CAS.
- A. M. May, M. Deegbey, K. Edme, K. J. Lee, R. N. Perutz, E. Jakubikova and J. L. Dempsey, Inorg. Chem., 2024, 63, 1858–1866 CrossRef CAS PubMed.
- A. E. Ashley, A. R. Cowley and D. O'Hare, Eur. J. Org. Chem., 2007, 2239–2242 CrossRef CAS.
- A. E. Ashley, R. T. Cooper, G. G. Wildgoose, J. C. Green and D. O'Hare, J. Am. Chem. Soc., 2008, 130, 15662–15677 CrossRef CAS PubMed.
- G. Balazs, F. G. N. Cloke, L. Gagliardi, J. C. Green, A. Harrison, P. B. Hitchcock, A. R. M. Shahi and O. T. Summerscales, Organometallics, 2008, 27, 2013–2020 CrossRef CAS.
- S. A. Westcott, A. K. Kakkar, G. Stringer, N. J. Taylor and T. B. Marder, J. Organomet. Chem., 1990, 394, 777–794 CrossRef CAS.
- M. J. Calhorda, V. Félix and L. s. F. Veiros, Coord. Chem. Rev., 2002, 230, 49–64 CrossRef CAS.
- D. P. Freyberg, J. L. Robbins, K. N. Raymond and J. C. Smart, J. Am. Chem. Soc., 1979, 101, 892–897 CrossRef CAS.
- A. H. Maki and T. E. Berry, J. Am. Chem. Soc., 1965, 87, 4437–4441 CrossRef CAS.
- S. Mugiraneza and A. M. Hallas, Commun. Phys., 2022, 5, 95 CrossRef.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- M. B. Meredith, J. A. Crisp, E. D. Brady, T. P. Hanusa, G. T. Yee, M. Pink, W. W. Brennessel and V. G. Young Jr., Organometallics, 2008, 27, 5464–5473 CrossRef CAS.
- J. Cirera, M. Via-Nadal and E. Ruiz, Inorg. Chem., 2018, 57, 14097–14105 CrossRef CAS PubMed.
- S. Gómez-Coca and E. Ruiz, Dalton Trans., 2024, 53, 11895–11902 RSC.
- C. G. Collins Rice, L. J. Morris, J.-C. Buffet, Z. R. Turner and D. O'Hare, Chem. Commun., 2023, 59, 12128–12131 RSC.
- P. Ransom, A. E. Ashley, N. D. Brown, A. L. Thompson and D. O'Hare, Organometallics, 2011, 30, 800–814 CrossRef CAS.
- C. G. Collins Rice, A. Evans, Z. R. Turner, J. Wattoom and D. O'Hare, Ind. Chem. Mater., 2025, 3, 178–190 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2421029 and 2421030. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt01345c |
| This journal is © The Royal Society of Chemistry 2025 |