DOI:
10.1039/D5DT01239B
(Paper)
Dalton Trans., 2025,
54, 10946-10955
Carboranyl diphosphenes: synthesis, structure and reactivity†
Received
27th May 2025
, Accepted 18th June 2025
First published on 18th June 2025
Abstract
Carboranyl-diphosphenes {1-P-2-[C(tBu) = N(Ar)]-1,2-C2B10H10}2 [Ar = Dmp (6) or Ph (7); Dmp = 2,6-Me2C6H3, Ph = C6H5] are prepared in good yields by the reduction of iminocarboranyldichlorophosphines 1-PCl2-2-[C(tBu) = N(Ar)]-1,2-C2B10H10 [where Ar = Dmp (4) or Ph (5)] with potassium graphite (KC8) in THF. Compounds 6 and 7 have similar dimeric structures in the solid state as confirmed by single-crystal X-ray analyses. Their solution structures are, however, different on the basis of 31P NMR studies, which could be ascribed to the steric effects of Dmp over Ph. As a result, 6 and 7 exhibit different coordination chemistry toward W(CO)5(THF). On the other hand, they show similar properties with oxidizing agents, giving a variety of phosphorus(III) compounds. The results indicate that 6 dissociates into carboranyl phosphinidene in THF.
Introduction
The chemistry of low-valent main group compounds has received much attention and is a rapidly developing research area of organometallic chemistry,1,2 which has not only promoted the understanding of the bonding interactions in main group compounds2,3 but also exhibited transition-metal-like behavior of these compounds to activate small molecules.4 Among these, low-valent phosphorus(I) compounds are among the most studied classes, including monomeric phosphinidenes (Lewis base5 or transition-metal6 stabilized) and diphosphenes7,8 (Fig. 1).
 |
| | Fig. 1 Presentation of phosphorus(I) species. | |
Diphosphenes (RP = PR), featuring small HOMO–LUMO energy gaps compared to their alkene analogues, have achieved remarkable advances in the last decades due to the development of diverse ligand sets with distinct stereoelectronic properties, enabling a comprehensive understanding of the structure, bonding, and reactivity within the diphosphene family.7,8 In 1981, the first isolation of diphosphene Mes*P = PMes* (Mes* = 2,4,6-tBu3C6H2) by Yoshifuji9 opened up the avenue for classical diphosphenes, which were kinetically stabilized by extremely bulky aryl or alkyl substituents.7,8 On the other hand, plenty of diphosphenes with diverse Lewis base ligands, including non-carbon (B, Ga, Si, Ge, N, P) donors, stable N-heterocyclic carbenes (NHCs) and other singlet carbenes, were realized in recent years.8 Recent selected examples include the works of Yamashita10a and Goicoechea10b on boryl-substituted diphosphene compounds, a chiral 1,1′-binaphthyl diphosphene reported by Tsurusaki and Kamikawa,11 vinyl-substituted diphosphenes12a,c and a di-1,2-dihydro-1,2-diphosphete-substituted diphosphene12b developed by Stephen and Ghadwal, a germyliumylidene-substituted diphosphene synthesized by Driess,13 a gallium carbenoid-substituted diphosphene prepared by Goicoechea,14 and the utilization of diphosphenes as ligands for gold(I) species by Protasiewicz15a and Jana15b and for a lead(0) complex by Mo.15c
In 2016, we described the preparation and reactivity of the imine-stabilized carboranyl phosphinidene 1,2-[C(tBu) = N(2′,6′-iPr2C6H3)P]-1,2-C2B10H101.16 We wondered if the substituents on the iminocarborane ligand would influence the structures of the resulting phosphorus(I) compounds. With this in mind, new iminocarborane ligands with different aryl groups (Ar = Dmp and Ph) (Dmp = 2,6-Me2C6H3, Ph = C6H5) were synthesized to study the steric effects of the aryl groups on carboranyl phosphorus(I) compounds. These results are reported in this article.
Results and discussion
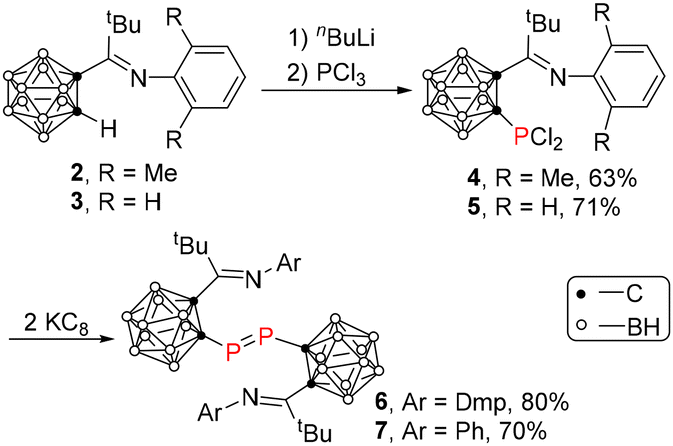
The iminocarboranes 2 and 3 were prepared in 77%–80% yields by a salt elimination reaction of 1-Li-1,2-C2B10H11 with ArN = CtBuCl (Ar = Dmp17 or Ph18) in Et2O. Treatment of 2 or 3 with 1 equiv. of n-BuLi in Et2O, followed by the addition of 1 equiv. of PCl3 afforded compounds 4 and 5 as yellow crystals in 63% and 70% yields, respectively. Reduction of 4 or 5 with 2 equiv. of potassium graphite (KC8) in THF at room temperature afforded the corresponding carboranyl-diphosphene 6 or 7 as orange-yellow crystals in 70%–80% yields (Scheme 1). The phosphorus chemical shift of 6 was observed at 214.1 ppm, which was comparable to that of 210.5 ppm in 1,16 suggesting the monomeric phosphinidene structure of 6 in solution. In contrast, it was shifted upfield significantly to −43.0 ppm for compound 7, suggesting a dimeric diphosphene structure in solution probably due to the less steric hindrance of the Ph group.
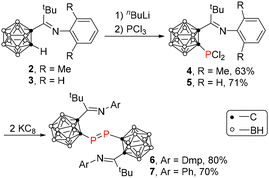 |
| | Scheme 1 Synthesis of carboranyl diphosphenes 6 and 7. | |
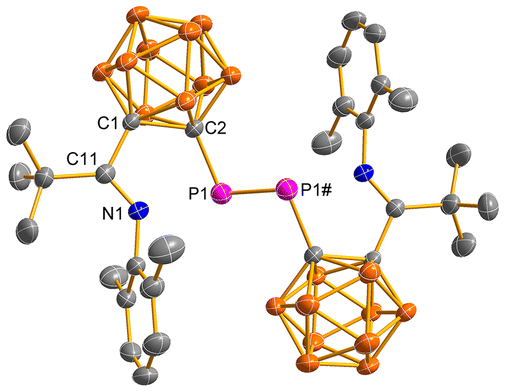
The solid-state structures of 6 and 7 were confirmed by single-crystal X-ray analyses and are shown in Fig. 2 and 3, respectively. They are very similar, displaying diphosphene structures. The phosphorus atom is bonded to one cage-carbon atom and another phosphorus atom in a bent geometry with a C(2)–P(1)–P(1)# angle of 99.4(6)° in 6 and 98.8(1)° in 7, respectively, which are within the range of the corresponding C–P–P angles of diphosphenes [93.2(1)–114.9(1)°].19 The P(1)–P(1)# bond distances of 2.037(1) Å in 6 and 2.037(2) Å in 7 fall in the range of previously reported diphosphenes [1.985(3)–2.051(2) Å].19a,20 The P(1)–C(2) bond distances of 1.871(2) Å in 6 and 1.876(2) Å in 7 are comparable to those of 1.883(2) Å in 4 and 5, respectively. The C(1)–C(2) distances of 1.730(2) Å in 6 and 1.729(3) Å in 7 are obviously elongated when compared to those of 1.702(3) Å in 4 and 1.699(3) Å in 5, respectively, suggesting a partial exo-π bonding nature of the cage C–P bond.
 |
| | Fig. 2 Molecular structure of 6. Selected bond lengths (Å) and angles (°): P(1)–P(1)# 2.037(1), C(1)–C(2) 1.730(2), P(1)–C(2) 1.871(2), C(11)–N(1) 1.262(2), C(11)–C(1) 1.545(2); C(2)–P(1)–P(1)# 99.4(1), N(1)–C(11)–C(1) 111.1(2), C(11)–C(1)–C(2) 113.8(2), C(1)–C(2)–P(1) 119.6(2). | |
 |
| | Fig. 3 Molecular structure of 7. Selected bond lengths (Å) and angles (°): P(1)–P(1)# 2.037(2), C(1)–C(2) 1.729(3), P(1)–C(2) 1.876(2), C(11)–N(1) 1.262(3), C(11)–C(1) 1.535(3); C(2)–P(1)–P(1)# 98.8(1), N(1)–C(11)–C(1) 110.9(2), C(11)–C(1)–C(2) 113.7(2), C(1)–C(2)–P(1) 120.0(2). | |
It is noteworthy that the P(1)–N(1) distances of 2.520 Å in 6 and 2.500 Å in 7 are shorter than the sum of the van der Waals radii of phosphorus and nitrogen (3.55 Å),21 implying the presence of certain interactions between the phosphorus and nitrogen atoms.
In view of the solid-state structures and the 31P NMR chemical shifts of 1, 6 and 7, it was suggested that the size of the aryl group in the carboranyl imine moiety may play a role in the solution structures of the resulting compounds, monomeric carboranyl-phosphinidene vs. dimeric carboranyl-diphosphene. The experimental results indicated that the sterically hindered 2,6-diisopropylphenyl group led to a phosphinidene in both solution and solid state,16 whereas the phenyl group resulted in the formation of diphosphene 7 in both solution and solid-state as confirmed by single-crystal X-ray analysis and solution 31P NMR chemical shift. It was interesting to note that 6 with a moderate size substituent, 2,6-dimethylphenyl, existed as a dimeric carboranyl-diphosphene in the solid-state, but as a monomeric carboranyl-phosphinidene in solution. In this connection, we wondered if the addition of a Lewis acid could trap the monomeric phosphinidene.
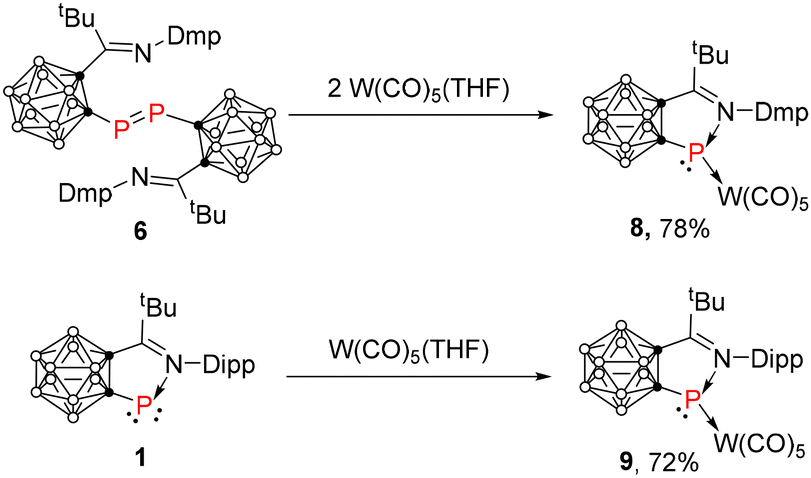
Treatment of 6 with in situ generated W(CO)5(THF), which was freshly prepared by UV irradiation of W(CO)6 in THF for 24 h at room temperature, gave the desired complex 8 as purple crystals in 78% yield (Scheme 2). Its 31P NMR chemical shift was observed at 233.4 ppm. For comparison, 1 also reacted with W(CO)5(THF) to afford the corresponding complex 9 as purple crystals in 72% yield with the phosphorus chemical shift being observed at 260.0 ppm (Scheme 2). However, no reaction was observed between 7 and W(CO)5(THF) under similar conditions as indicated by 31P NMR, implying the presence of only the dimeric diphosphene in solution for 7.
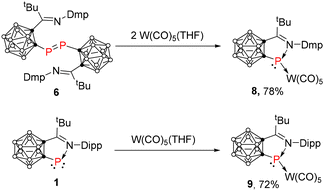 |
| | Scheme 2 Reaction of 1 or 6 with W(CO)5(THF). | |
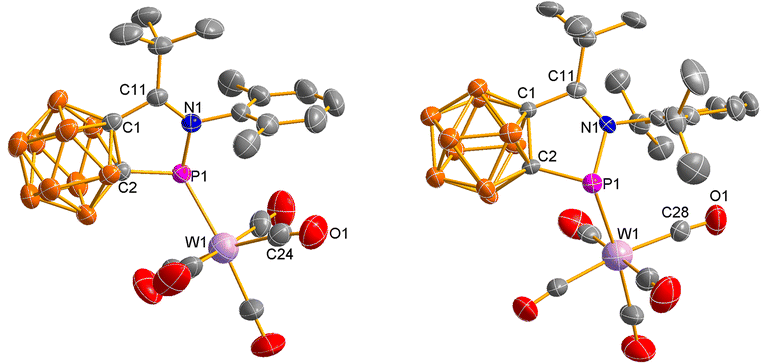
The solid-state structures of 8 and 9 were confirmed by single-crystal X-ray analyses and are shown in Fig. 4. The P(1) atoms in 8 and 9 are σ-bonded to one cage carbon atom, one imine nitrogen atom and one tungsten atom in a trigonal pyramidal geometry. The five atoms (C1, C2, P1, N1, C11) of the exo five-membered ring are nearly co-planar (the sum of internal pentagon angles = 538.9° for 8 and 537.9° for 9). The bond distances in the five-membered ring (C1, C2, P1, N1, and C11) in 8 and 9 are very close to each other, which are barely longer than the corresponding values observed in 1.16
 |
| | Fig. 4 Molecular structures of 8 (left) and 9 (right). | |
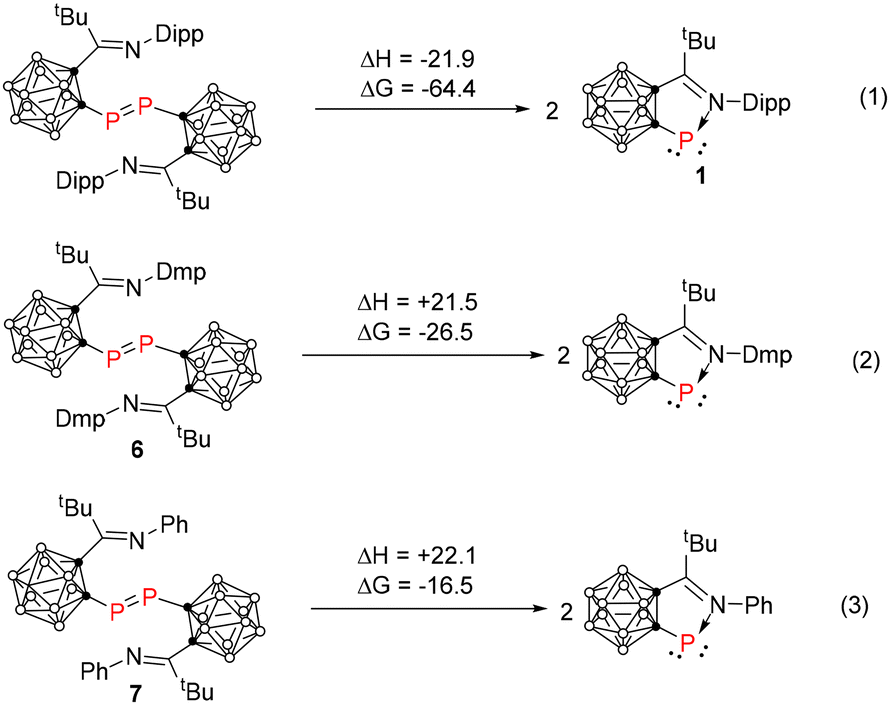
To better understand the dissociation of diphosphenes in solution, density functional theory (DFT) calculations at the B3LYP/6-31G(d,p) level of theory were performed. The optimized bond distances and angles of 1, 6 and 7 are in very good agreement with the experimental values. The structures of monomeric phosphinidenes with 2,6-dimethylphenyl or phenyl groups, as well as diphosphene with a 2,6-diisopropyl group, were also optimized. In addition, the dissociation enthalpies and Gibbs free energies of carboranyl-diphosphenes into the corresponding monomeric carboranyl-phosphinidenes were calculated at the B3LYP/6-31G(d,p) level of theory (Fig. 5). The three reactions presented the dissociation of three different aryl substituted carboranyl-diphosphenes to the corresponding monomeric carboranyl-phosphinidenes, respectively (aryl = Dipp, Dmp and Ph). Eqn (1) shows a negative enthalpy change, whereas it is positive for both eqn (2) and (3) in Fig. 5. Though ΔG is negative for all three reactions, the values are quite different. The largest negative value of Gibbs energy, as well as the negative value of enthalpy in eqn (1) implies that the sterically hindered 2,6-diisopropylphenyl group prohibits the formation of a dimer, leading to the isolation of the monomeric imine-stabilized phosphinidene 1. On the other hand, the positive values of enthalpy in eqn (2) and (3) suggest that the less sterically demanding 2,6-dimethylphenyl and phenyl groups favor the formation of diphosphenes with the tendency of phenyl > 2,6-dimethylphenyl. On the other hand, the solvation of the P center might contribute to the stabilization of the monomeric 2,6-dimethylphenyl phosphinidene in solution.
 |
| | Fig. 5 The calculated dissociation enthalpy and Gibbs energy of carboranyl-diphosphenes (in kcal mol−1) at the B3LYP/6-31G(d,p) level of theory. | |
We are interested in exploring the chemical properties of the resultant diphosphene compounds. Compound 6 reacted with 2 equiv. of Cu(OAc)2 in THF at room temperature to afford a redox product, phosphine 10, as colorless crystals in 60% yield (Scheme 3). Its 31P NMR spectrum exhibited a singlet at 95.5 ppm, which was similar to that of 105.8 ppm observed for a Dipp-substituted phosphine.16 Single-crystal X-ray analyses confirmed that the P(1) atom in 10 is σ-bonded to one cage carbon atom and two acetyl groups in a trigonal pyramidal geometry (Fig. 6). Treatment of 7 with 1 equiv. of sulfur (S) gave thiadiphosphirane 11 as yellow crystals in 80% yield (Scheme 3). This kind of reaction has been observed in aryl diphosphene compounds.22 Its 31P NMR chemical shift of −66.6 ppm was comparable to that of −82.4 ppm in the Dipp-substituted carboranyl thiadiphosphirane16 and those observed in aryl-thiadiphosphiranes.22 Single-crystal X-ray analyses show that the P(1) atom in 11 adopts a typical trigonal pyramidal geometry (Fig. 7). On the other hand, protonation of 7 with HCl in ether gave phosphine 12 as yellow crystals in 40% yield (Scheme 3). Its 31P NMR chemical shift was observed at 9.7 ppm as a doublet with JP–H = 256.0 Hz. The solid-state structure of 12 was further confirmed by single-crystal X-ray analysis (Fig. S1†).
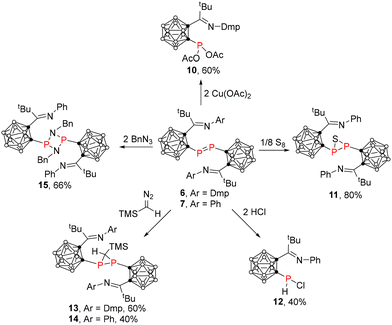 |
| | Scheme 3 Reactivity of 6 and 7. | |
 |
| | Fig. 6 Molecular structure of 10. Selected bond lengths (Å) and angles (°): C(1)–C(2) 1.699(3), P(1)–C(2) 1.883(2), C(11)–N(1) 1.266(3), C(11)–C(1) 1.545(3), P(1)–O(1) 1.673(2), P(1)–O(3) 1.692(2); N(1)–C(11)–C(1) 109.9(2), C(11)–C(1)–C(2) 113.0(2), C(1)–C(2)–P(1) 118.0(2), C(2)–P(1)–O(1) 97.0(1), C(2)–P(1)–O(3) 93.7(2), O(1)–P(1)–O(3) 92.7(2). | |
 |
| | Fig. 7 Molecular structure of 11. Selected bond lengths (Å) and angles (°): C(1)–C(2) 1.725(4), P(1)–C(2) 1.900(3), C(11)–N(1) 1.271(3), C(11)–C(1) 1.538(4), P(1)–P(2) 2.224(1), P(1)–S(1) 2.145(1), P(2)–S(1) 2.077(1); N(1)–C(11)–C(1) 110.6(2), C(11)–C(1)–C(2) 115.1(2), C(1)–C(2)–P(1) 116.8(2), C(2)–P(1)–P(2) 99.3(1), C(2)–P(1)–S(1) 99.7(1), S(1)–P(1)–P(2) 56.7(1), P(1)–S(1)–P(2) 63.6(1), S(1)–P(2)–P(1) 56.8(1). | |
Furthermore, carboranyl-diphosphenes could undergo different cycloaddition reactions. Treatment of 6 or 7 with 1 equiv. of trimethylsilyldiazomethane afforded carboranyl-diphosphiranes 13 or 14 as yellow crystals in moderate yields via a [2 + 1] cycloaddition reaction (Scheme 3). The solid-state structures were further confirmed by single-crystal X-ray analyses. The representative structure of 14 was shown in Fig. 8. The [2 + 1 + 1] cycloaddition reaction of 7 with 2 equiv. of benzyl azide yielded carboranyl-cyclodiphosphazane 15 as pale yellow crystals in 66% yield (Scheme 3). This reaction may proceed via coupling of 7 with a benzyl nitrene (generated from benzyl azide) to form an azadiphosphiridine intermediate,23 subsequently followed by coupling with another equiv. of benzyl nitrene to produce 15. Its 31P NMR chemical shift was observed as a singlet at 195.2 ppm. Single-crystal X-ray analyses confirmed the formation of a four-membered co-planar heterocyclic ring (Fig. 9).
 |
| | Fig. 8 Molecular structure of 14. Selected bond lengths (Å) and angles (°): C(1)–C(2) 1.745(3), P(1)–C(2) 1.881(2), C(11)–N(1) 1.269(3), C(11)–C(1) 1.543(4), P(1)–P(2) 2.192(2), P(1)–C(22) 1.837(3); N(1)–C(11)–C(1) 110.4 (2), C(11)–C(1)–C(2) 116.2(2), C(1)–C(2)–P(1) 117.1(2), C(2)–P(1)–P(2) 98.8(1), P(2)–P(1)–C(22) 53.4(1), P(1)–C(22)–P(2) 73.3(2). | |
 |
| | Fig. 9 Molecular structure of 15. Selected bond lengths (Å) and angles (°): C(1)–C(2) 1.703(3), P(1)–C(2) 1.943(2), C(11)–N(1) 1.268(3), C(11)–C(1) 1.540(3), P(1)–N(2) 1.712(2), P(1)–N(2)# 1.723(23); N(1)–C(11)–C(1) 110.8 (2), C(11)–C(1)–C(2) 113.8(2), C(1)–C(2)–P(1) 117.1 (2), C(2)–P(1)–N(2) 102.8(1), C(2)–P(1)–N(2)# 102.2(1), N(2)–P(1)–N(2)# 80.6 (1), P(1)–N(2)–P(1)# 99.4 (1). | |
Conclusions
In summary, the structural diversity of the carboranyl phosphorus(I) compounds correlates with the steric properties of the aryl group and the strength of the N → P intramolecular interaction. The computational studies suggest that the aryl group in the carboranyl moiety can affect the N → P intramolecular interaction in compounds 1, 6 and 7. These observations are further evidenced by the 31P NMR chemical shifts of the carboranyl phosphorus(I) compounds, as well as the isolation of complex 8via the reaction of 6 with W(CO)5(THF). Furthermore, carboranyl-diphosphenes can undergo diverse reactions with Cu(OAc)2, S8, HCl, (TMS)CHN2 and BnN3 to give phosphines 10 to 15. It is noteworthy that the reaction of diphosphenes with BnN3 provides an alternative synthetic method to afford cyclodiphosphazanes, which have found wide applications in coordination chemistry as versatile ligands and in supramolecular chemistry as scaffolds. In addition, the resulting carborane-phosphorus compounds might serve as valuable synthons for the development of 3D boron cluster-based heterocyclic systems, which exhibit unique photophysical properties and great potential in functional materials.24
Experimental
General remarks
All procedures were carried out under a dry argon atmosphere using standard Schlenk and glovebox techniques. 1H, 13C, 11B and 31P NMR spectra were recorded on a Bruker DPX 400 spectrometer at 400, 100, 128 and 162 MHz, respectively. All chemical shifts were reported in δ units with references to the residual solvent resonances of the deuterated solvents for proton and carbon chemical shifts and to external BF3·OEt2 (0.00 ppm) for boron chemical shifts. NMR multiplicities were abbreviated as follows: s = singlet, d = doublet, t = triplet, m = multiplet, and br = broad signal. Mass spectra were obtained on a Thermo Finnigan MAT 95 XL spectrometer. All organic solvents were freshly distilled from sodium benzophenone ketyl immediately prior to use. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 and ArN = CtBuCl (Ar = Dmp17 or Ph18) were prepared according to literature procedures. All other chemicals were purchased from Aldrich, J&K or Acros Chemical Co. and used as received unless otherwise specified.
16 and ArN = CtBuCl (Ar = Dmp17 or Ph18) were prepared according to literature procedures. All other chemicals were purchased from Aldrich, J&K or Acros Chemical Co. and used as received unless otherwise specified.
X-ray structure determination.
Single crystals were immersed in Paratone-N oil and sealed under argon in thin-walled glass capillaries. All data were collected at 296 K for 2–6 and 8–15 or 297 K for 7 on a Bruker D8 Venture diffractometer or a Bruker Kappa ApexII Duo diffractometer using Mo-Kα radiation. An empirical absorption correction was applied using the SADABS program.25 All structures were solved by direct methods and subsequent Fourier difference techniques and were refined anisotropically for all non-hydrogen atoms by full-matrix least-squares calculations on F2 using the SHELXTL program package.26 All hydrogen atoms were geometrically fixed using the riding model. Crystal data and details of data collection and refinement are given in Table S1.†
Preparation of 2.
To an ether (50 mL) solution of o-C2B10H12 (2.30 g, 16.0 mmol), n-BuLi (1.6 M, 10.0 mL, 16.0 mmol) was slowly added via a syringe at 0 °C under stirring. The mixture was allowed to slowly warm to room temperature and stirred at room temperature for 2 h, to which (2,6-Me2C6H3)N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(tBu)Cl (3.60 g, 16.0 mmol) was slowly added. The reaction mixture was stirred at room temperature overnight. After the removal of the solvent, column chromatographic separation (silica gel, 230–400 mesh) using hexane/AcOEt (40:1) as the eluent afforded 2 as a pale yellow solid (4.20 g, 80%). 1H NMR (400 MHz, CDCl3): δ 1.21 (s, 9H; CMe3), 1.99 (s, 6H; Me) 4.61 (br s, 1H; cage CH), 6.85 (t, J = 7.2 Hz, 1H; aromatic CH), 6.96 (d, J = 7.2 Hz, 2H; aromatic CH). 13C{1H} NMR (100 MHz, CDCl3): δ 18.6 (Me), 29.4 (CMe3), 45.5 (s, CMe3), 63.7 (cage C), 78.2 (cage C), 121.6, 122.5, 127.8, 146.6 (aromatic C), 162.0 (CN). 11B{1H} NMR (128 MHz, CDCl3): δ −13.7 (2B), −11.3 (2B), −9.0 (4B), −3.1 (1B), −2.2 (1B). HRMS: m/z calcd for C15H2910B211B8N+: 331.3305. Found: 331.3308.
C(tBu)Cl (3.60 g, 16.0 mmol) was slowly added. The reaction mixture was stirred at room temperature overnight. After the removal of the solvent, column chromatographic separation (silica gel, 230–400 mesh) using hexane/AcOEt (40:1) as the eluent afforded 2 as a pale yellow solid (4.20 g, 80%). 1H NMR (400 MHz, CDCl3): δ 1.21 (s, 9H; CMe3), 1.99 (s, 6H; Me) 4.61 (br s, 1H; cage CH), 6.85 (t, J = 7.2 Hz, 1H; aromatic CH), 6.96 (d, J = 7.2 Hz, 2H; aromatic CH). 13C{1H} NMR (100 MHz, CDCl3): δ 18.6 (Me), 29.4 (CMe3), 45.5 (s, CMe3), 63.7 (cage C), 78.2 (cage C), 121.6, 122.5, 127.8, 146.6 (aromatic C), 162.0 (CN). 11B{1H} NMR (128 MHz, CDCl3): δ −13.7 (2B), −11.3 (2B), −9.0 (4B), −3.1 (1B), −2.2 (1B). HRMS: m/z calcd for C15H2910B211B8N+: 331.3305. Found: 331.3308.
Preparation of 3.
Compound 3 was prepared in a similar manner to that of 2 using o-C2B10H12 (2.30 g, 16.0 mmol), n-BuLi (1.6 M, 10.0 mL, 16.0 mmol) and PhNC(tBu)Cl (3.1 g, 16.0 mmol) as a pale yellow solid (3.90 g, 80%). 1H NMR (400 MHz, CDCl3): δ 1.25 (s, 9H; CMe3), 4.68 (br s, 1H; cage CH), 6.51 (d, J = 7.6 Hz, 2H; aromatic CH), 6.99 (t, J = 7.6 Hz, 1H; aromatic CH), 7.27 (t J = 8.0 Hz, 2H; aromatic CH). 13C{1H} NMR (100 MHz, CDCl3): δ 31.4 (CMe3), 45.4 (s, CMe3), 63.5 (cage C), 78.3 (cage C), 115.6, 122.7, 128.8, 148.8 (aromatic C), 163.0 (CN). 11B{1H} NMR (128 MHz, CDCl3): δ −14.1 (2B), −11.7 (2B); −9.2 (4B), −3.4 (1B), −2.6 (1B). HRMS: m/z calcd for C13H2510B211B8N+: 303.2991. Found: 303.2994.
Preparation of 4.
To an ether solution (20 mL) of 2 (0.79 g, 2.4 mmol), a hexane solution of n-BuLi (1.6 M, 1.5 mL, 2.4 mmol) was slowly added via a syringe at 0 °C under stirring. The reaction mixture was allowed to warm to room temperature and stirred overnight. PCl3 (0.25 mL, 2.9 mmol) was then added via a syringe at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 4 h. After the removal of the precipitate by filtration, the filtrate was concentrated to about 5 mL, to which n-hexane (2 mL) was added. Compound 4 was isolated as orange crystals after the solution stood at room temperature overnight (0.65 g, 63%). 1H NMR (400 MHz, C6D6): δ 0.79 (s, 9H; CMe3), 1.80 (s, 6H; Me), 6.72 (d, J = 6.8 Hz, 2H; aromatic CH), 6.78 (d, J = 6.4 Hz, 1H; aromatic CH). 13C{1H} NMR (100 MHz, C6D6): δ 19.3, 19.4 (Me), 29.4 (CMe3), 44.6 (CMe3), 85.9 (d, J = 12.2 Hz, cage C), 87.1 (cage C), 125.7, 126.5, 128.8, 142.0 (aromatic C), 168.8 (CN). 11B{1H} NMR (128 MHz, C6D6): δ −13.6 (2B), −6.9 (6B), −0.1 (2B). 31P NMR (162 MHz, C6D6): 89.2. Anal. calcd for C15H2810B211B8Cl2NP: C 41.67, H 6.53, N 3.24. Found: C 41.94, H 6.83, N 3.17.
Preparation of 5.
5 was prepared as pale yellow crystals (0.69 g, 71%) in a similar manner to that of 4 using 3 (0.73 g, 2.4 mmol), n-BuLi (1.6 M, 1.5 mL, 2.4 mmol) and PCl3 (0.25 mL, 2.9 mmol). 1H NMR (400 MHz, C6D6): δ 0.73 (s, 9H; CMe3), 6.23 (d, J = 7.2 Hz, 2H; aromatic CH), 6.84 (t, J = 7.2 Hz, 1H; aromatic CH), 6.91 (t, J = 7.2 Hz, 2H; aromatic CH). 13C{1H} NMR (100 MHz, C6D6): δ 29.9 (CMe3), 44.5 (CMe3), 85.0 (d, J = 12.2 Hz, cage C) 87.0 (d, J = 131.8 Hz, cage C), 120.6, 126.0, 128.4, 142.4 (aromatic C), 169.5 (CN). 11B{1H} NMR (128 MHz, C6D6): δ −10.1 (4B), −8.1 (4B), −1.8 (1B), −0.1 (1B). 31P NMR (162 MHz, C6D6): 109.3. Anal. calcd for C13H2410B211B8Cl2NP: C 38.62, H 5.98, N 3.46. Found: C 39.07, H 6.07, N 2.93.
Preparation of 6.
A THF solution (20 mL) of 4 (518 mg, 1.2 mmol) was slowly added to a mixture of KC8 (324 mg, 2.4 mmol) in THF at room temperature, and the reaction mixture was stirred overnight at room temperature. The color of the solution was changed to orange yellow. After filtration, the orange yellow filtrate was concentrated to about 5 mL. Compound 6 was isolated as orange yellow crystals after the solution stood at room temperature overnight (347 mg, 80%). 1H NMR (400 MHz, C6D6): δ 1.23 (s, 18H; CMe3), 2.26 (s, 12 H; Me2), 7.08 (d, J = 8.8 Hz, 4H; aromatic CH), 7.24 (t, J = 7.2 Hz, 2H; aromatic CH). 13C{1H} NMR (100 MHz, C6D6): δ 18.5 (Me), 30.4 (CMe3), 45.2 (CMe3), 86.5 (d, 1JPC = 85.0 Hz, cage C) 87.6 (cage C), 121.8, 122.7, 128.8, 146.8 (aromatic C), 161.9 (CN). 11B{1H} NMR (128 MHz, C6D6): δ −11.9 (4B), −9.9 (6B), −8.4 (6B), −6.7 (4B). 31P NMR (162 MHz, C6D6): 214.1. Anal. calcd for C30H5610B411B16N2P2: C 49.84, H 7.81, N 3.88. Found: C 50.46, H 7.69, N 3.37.
Preparation of 7.
7 was prepared as orange yellow crystals (280 mg, 70%) in a similar manner to that of 6 using 5 (485 mg, 1.2 mmol) and KC8 (324 mg, 2.4 mmol) in THF. 1H NMR (400 MHz, C6D6): δ 0.87 (s, 18H; CMe3), 6.59 (m, J = 6.8 Hz, 4H; aromatic CH), 6.82 (m, 6H; aromatic CH). 13C{1H} NMR (100 MHz, C6D6): δ 31.3 (CMe3), 45.2 (CMe3), 85.9 (d, J = 62.0 Hz, cage C), 86.9 (cage C), 115.8, 122.8, 128.8, 149.1 (aromatic C), 163.0 (CN). 11B{1H} NMR (128 MHz, C6D6): δ −9.5 (8B), −8.2 (10B), −4.9 (2B). 31P NMR (162 MHz, C6D6): −43.0. Anal. calcd for C26H4810B411B16N2P2: C 46.83, H 7.26, N 4.20. Found: C 46.89, H 7.40, N 4.29.
Preparation of 8.
Compound 6 (72.2 mg, 0.1 mmol), W(CO)6 (70.4 mg, 0.2 mmol) and THF (1 mL) were mixed in a closable Schlenk tube. The resulting mixture was irradiated with Xe light (300 W, 290–1500 nm) under argon for 24 h at room temperature. The color was changed from orange yellow to reddish purple. The 31P NMR spectrum indicated a complete conversion of 6. After the removal of the solvent, the residue was recrystallized from hexane (3 mL) at room temperature to give 8 as reddish purple crystals (106.9 mg, 78%). 1H NMR (400 MHz, C6D6): δ 0.73 (s, 9H; CMe3), 1.89 (s, 6H; CHMe2), 6.60 (d, J = 7.6 Hz, 2H; aromatic CH), 6.81 (t, J = 7.6 Hz, 1H; aromatic CH). 13C{1H} NMR (100 MHz, C6D6): δ 18.8 (d, J = 5.5 Hz, Me), 30.2 (d, J = 6.4 Hz, CMe3), 44.7 (d, J = 10.4 Hz, CMe3), 83.7 (d, J = 65.2 Hz, cage C), 85.4 (d, J = 13.0 Hz, cage C), 122.7, 130.2, 133.7, 137.8 (aromatic C), 164.6 (d, J = 31.0 Hz, CN), 191.1, 196.2 (CO), 200.6 (d, J = 21.9 Hz, CO). 11B{1H} NMR (128 MHz, C6D6): δ −10.4 (5B), −8.3 (4B), −4.5 (1B). 31P NMR (162 MHz, C6D6): δ 233.4. HRMS: m/z calcd for C20H2810B211B8NO5PW+: 685.2190. Found: 685.2190.
Preparation of 9.
This compound was prepared as reddish purple crystals (53.4 mg, 72%) in a similar manner to that of 8 from 1 (41.7 g, 0.1 mmol) and W(CO)6 (35.2 mg, 0.1 mmol). 1H NMR (400 MHz, C6D6): δ 0.85 (s, 9H; CMe3), 0.96 (d, J = 6.8 Hz, 6H; CHMe2), 1.27 (d, J = 6.8 Hz, 6H; CHMe2), 2.74 (m, 2H; CHMe2), 6.81 (d, J = 8.0 Hz, 2H; aromatic CH), 7.00 (t, J = 8.0 Hz, 1H; aromatic CH). 13C{1H} NMR (100 MHz, C6D6): δ 24.6, 25.6, 28.6 (CMe3, CHMe2), 30.2 (CHMe2), 45.9 (d, J = 10.5 Hz, CMe3), 83.5 (d, J = 61.7 Hz, cage C), 85.2 (d, J = 12.5 Hz, cage C), 123.1, 123.8, 125.8, 131.5 (aromatic C), 135.2 (d, J = 9.4 Hz, aromatic C), 143.9 (aromatic C), 165.1 (d, J = 35.4 Hz, CN), 195.9 (CO), 199.7 (d, J = 25.2 Hz, CO). 11B{1H} NMR (128 MHz, C6D6): δ −10.0 (4B), −8.3 (4B), −4.2 (2B). 31P NMR (162 MHz, C6D6): δ 260.0. HRMS: m/z calcd for C24H3610B211B8NO5PW+: 741.2817. Found: 741.2814.
Preparation of 10.
To a THF solution (15 mL) of 6 (72.2 mg, 0.1 mmol) was added Cu(OAc)2 (36.4 mg, 0.2 mmol) at room temperature, and the mixture was stirred at room temperature overnight. The color of the solution was changed from orange yellow to deep brown. The 31P NMR spectrum indicated a complete conversion of 6. After the removal of the solvent, the resulting mixture was dissolved in n-hexane (20 mL). After the removal of the precipitate, the hexane solution was concentrated to about 5 mL. Compound 10 was isolated as colorless crystals after slow evaporation of the solvent at room temperature (57.5 mg, 60%). 1H NMR (400 MHz, CD2Cl2): δ 1.25 (s, 9H; CMe3), 1.95 (s, 6H; Me), 2.12 (s, 6H; OCMe), 6.99 (m, 3H; aromatic CH). 13C{1H} (100 MHz, CD2Cl2): δ 19.7 (d, J = 2.8 Hz, OCMe), 21.9 (Me), 29.6 (CMe3), 45.1 (CMe3), 84.0 (d, J = 85.8 Hz, cage C), 85.4 (d, J = 9.7 Hz, cage C), 124.8, 125.0, 128.6, 143.4 (aromatic C), 165.9 (CN), 170.0 (d, J = 10.8 Hz, CO). 11B{1H} NMR (128 MHz, CD2Cl2): δ −11.0 (4B), −8.7 (4B), −2.0 (2B). 31P NMR (162 MHz, CD2Cl2): δ 95.5. HRMS: m/z calcd for C19H3410B211B8NO4P+: 479.3233. Found: 479.3237.
Preparation of 11.
To a THF solution (15 mL) of 7 (66.6 mg, 0.1 mmol) was added S8 (3.2 mg, 0.0125 mmol) at room temperature, and the reaction mixture was stirred at room temperature overnight. The 31P NMR spectrum indicated a complete conversion of 7. After the removal of the solvent, the resulting yellow powder was dissolved in toluene (5 mL). Compound 11 was isolated as yellow crystals after slow evaporation of the solvent at room temperature (55.8 mg, 80%). 1H NMR (400 MHz, C6D6): δ 0.90 (s, 9H; CMe3), 1.09 (s, 9H; CMe3), 6.93 (m, 6H; aromatic CH), 7.09 (m, 4H; aromatic CH). 13C{1H} (100 MHz, C6D6): δ 30.8, 31.1 (CMe3), 46.1 (CMe3), 87.4 (cage C), 115.1, 115.8, 122.8, 123.3, 128.8, 148.3 (aromatic C), 159.6 (CN); another cage C was not observed. 11B{1H} NMR (128 MHz, C6D6): δ −7.5 (8B), −5.8 (8B), −0.4 (4B). 31P NMR (162 MHz, C6D6): δ −66.6. Anal. calcd for C26H4810B411B16N2P2S-0.5C7H8: C 47.56, H 7.04, N 3.76. Found: C 47.54, H 7.26, N 3.54.
Preparation of 12.
To a THF solution (20 mL) of 7 (66.6 mg, 0.1 mmol), an ether solution of HCl (1.0 M, 0.2 mL, 0.2 mmol) was slowly added via a syringe at 0 °C under stirring. The reaction mixture was allowed to warm to room temperature and stirred overnight. The 31P NMR spectrum indicated a complete conversion of 7. After the removal of the solvent, the resulting yellow powder was dissolved in toluene (5 mL). Compound 12 was isolated as light yellow crystals after slow evaporation of the solvent at room temperature (29.6 mg, 40%). 1H NMR (400 MHz, C6D6): δ 0.71 (s, 9H; CMe3), 6.01 (d, J = 7.2 Hz, 1H; aromatic CH), 6.25 (d, J = 7.6 Hz, 1H; aromatic CH), 6.34 (d, JHP = 254.0 Hz, 1H; PH), 6.84 (m, 3H; aromatic CH). 13C{1H} (400 MHz, C6D6): δ 30.7 (CMe3), 44.1 (CMe3), 79.5 (d, J = 81.0 Hz, cage C), 83.2 (d, J = 9.0 Hz, cage C), 120.0, 121.1, 126.5, 128.9, 142.0 (aromatic C), 171.3 (CN). 11B{1H} NMR (128 MHz, C6D6): δ −9.8 (4B), −7.7 (3B), −4.7 (1B), −2.7 (1B), 0.3 (1B). 31P NMR (162 MHz, C6D6): δ 9.7 (d, JPH = 256.0 Hz). HRMS: m/z calcd for C13H2510B211B8ClNP+: 369.2411. Found: 369.2413.
Preparation of 13.
To a THF solution (20 mL) of 6 (144.4 mg, 0.2 mmol), a hexane solution of trimethylsilyldiazomethane (2.0 M, 0.1 mL, 0.2 mmol) was slowly added at room temperature, and the mixture was stirred at room temperature overnight. The 31P NMR spectrum indicated a complete conversion of 6. After the removal of the solvent, the resulting yellow powder was dissolved in toluene (5 mL). Compound 13 was isolated as yellow crystals after slow evaporation of the solvent at room temperature (97.1 mg, 60%). 1H NMR (400 MHz, C6D6): δ −0.20 (s, 9H; SiMe3), 0.29 (s, 1H; CHTMS), 0.97 (s, 18H; CMe3), 1.88 (s, 3H; Me), 1.89 (s, 3H; Me), 2.27 (s, 3H; Me), 2.35 (s, 3H; Me), 6.83 (m, 6H; aromatic CH). 13C{1H} (100 MHz, C6D6): δ −0.66, 0.44 (SiMe3), 19.4 (CHTMS), 28.6 (CMe3), 45.6, 45.4 (CMe3), 121.5, 122.0, 122.1, 122.3, 122.4, 122.5, 127.6, 127.9, 128.0, 128.3, 145.8, 146.3 (aromatic CH), 161.2, 162.1 (CN); the signals of cage C were not observed. 11B{1H} NMR (128 MHz, C6D6): δ −7.7 (12B), −1.0 (8B). 31P NMR (162 MHz, C6D6): δ −120.3 (d, J = 171.7 Hz; 1P), −90.3 (d, J = 171.7 Hz; 1P). HRMS: m/z calcd for (C34H6610B411B16N2P2Si + Na)+: 831.6369. Found: 831.6384.
Preparation of 14.
This compound was prepared in a similar manner to that of 13 as yellow crystals (60.2 mg, 40%) from 7 (133.2 mg, 0.2 mmol) and trimethylsilyldiazomethane (2.0 M, 0.1 mL, 0.2 mmol). 1H NMR (400 MHz, C6D6): δ −0.17 (s, 9H; SiMe3), 0.29 (s, 1H; CHTMS), 0.96 (s, 18H; CMe3), 6.77 (m, 3H; aromatic CH), 6.98 (m, 7H; aromatic CH). 13C{1H} (100 MHz, C6D6): δ 2.12 (SiMe3), 21.4 (CHTMS), 31.2, 31.3 (CMe3), 46.3, 46.4 (CMe3), 85.7 (d, J = 114 Hz, cage C), 87.8 (d, J = 13.0 Hz, cage C), 115.8, 117.7, 123.3, 123.5, 125.7, 129.3, 147.3, 147.4 (aromatic CH), 162.0, 162.4 (CN). 11B{1H} NMR (128 MHz, C6D6): δ −8.9 (12B), −2.1 (8B). 31P NMR (162 MHz, C6D6): δ −123.2 (d, J = 178.2 Hz; 1P), −88.3 (d, J = 178.2 Hz; 1P). HRMS: m/z calcd for (C30H5810B411B16N2P2Si + Na)+: 775.5743. Found: 775.5762.
Preparation of 15.
To a THF solution of 7 (66.6 mg, 0.1 mmol) (20 mL), benzyl azide (26.6 mg, 0.2 mmol) was slowly added at room temperature, and the mixture was stirred at room temperature overnight. The 31P NMR spectrum indicated a complete conversion of 7. After the removal of the solvent, the resulting yellow powder was dissolved in toluene (5 mL) to give 15 as yellow crystals after slow evaporation of the solvent at room temperature (57.9 mg, 66%). 1H NMR (400 MHz, CD2Cl2): δ 1.01 (s, 18H; CMe3), 2.34 (s, 4H; CH2Ph), 6.39 (d, J = 7.6 Hz, 4H; aromatic CH), 7.26 (m, 12H; aromatic CH), 7.34 (d, J = 7.6 Hz, 4H; aromatic CH). 13C{1H} (100 MHz, CD2Cl2): δ 31.2 (CMe3), 45.7 (CMe3), 49.7 (CH2Ph), 66.0 (cage C), 84.6 (cage C), 118.6, 123.8, 125.6, 127.5, 128.3, 128.4, 128.5, 129.3, 136.6, 146.0 (aromatic CH), 162.0 (CN). 11B{1H} NMR (128 MHz, CD2Cl2): δ −8.9 (12B), −2.1 (8B). 31P NMR (162 MHz, CD2Cl2): δ 195.2. Anal. calcd for C40H6210B411B16N4P2: C 54.78, H 7.13, N 6.39. Found: C 54.35, H 7.35, N 6.64.
Author contributions
J. Z. and Z. X. directed and conceived this project. T. L. C. conducted the experiments. All authors discussed the results and wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers 2446505–2446518 for 2–15.† Experimental details and spectral data are available in the ESI.†
Acknowledgements
This work was supported by grants from the Research Grants Council of the Hong Kong Special Administrative Region (Project No. 14307421 to J. Z.), the National Natural Science Foundation of China (Project No. 22331005 to Z. X.), and the Shenzhen Science and Technology Program (Project No. KQTD20221101093558015 to Z. X.).
References
- For reviews, see:
(a) Y. Wang and G. H. Robinson, Carbene Stabilization of Highly Reactive Main-Group Molecules, Inorg. Chem., 2011, 50, 12326–12337 CrossRef CAS PubMed;
(b) Y. Wang and G. H. Robinson, N-Heterocyclic Carbene Main-Group Chemistry: A Rapidly Evolving Field, Inorg. Chem., 2014, 53, 11815–11832 CrossRef CAS PubMed.
- For selected reviews, see:
(a) P. P. Power, π-Bonding and the Lone Pair Effect in Multiple Bonds between Heavier Main Group Elements, Chem. Rev., 1999, 99, 3463–3504 CrossRef CAS PubMed;
(b) R. C. Fischer and P. P. Power, π-Bonding and the Lone Pair Effect in Multiple Bonds Involving Heavier Main Group Elements: Developments in the New Millennium, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed;
(c) A. Stasch and C. Jones, Stable dimeric magnesium(I) compounds: from chemical landmarks to versatile reagents, Dalton Trans., 2011, 40, 5659–5672 RSC;
(d) M. Asay, C. Jones and M. Driess,
N-Heterocyclic Carbene Analogues with Low-Valent Group 13 and Group 14 Elements: Syntheses, Structures, and Reactivities of a New Generation of Multitalented Ligands, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- For selected examples, see:
(a) K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, B. Niepotter, H. Wolf, R. Herbst-Irmer and D. Stalke, A Stable Singlet Biradicaloid Siladicarbene:(L:)2Si, Angew. Chem., Int. Ed., 2013, 52, 2963–2967 CrossRef CAS PubMed;
(b) K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, I. Tkach, H. Wolf, R. Kratzer, R. Herbst-Irmer, B. Niepotter and D. Stalke, Conversion of a Singlet Silylene to a stable Biradical, Angew. Chem., Int. Ed., 2013, 52, 1801–1805 CrossRef CAS PubMed;
(c) J. Flock, A. Suljanovic, A. Torvisco, W. Schoefberger, B. Gerke, R. Pöttgen, R. C. Fischer and M. Flock, The Role of 2,6-Diaminopyridine Ligands in the Isolation of an Unprecedented, Low-Valent Tin Complex, Chem. – Eur. J., 2013, 19, 15504–15517 CrossRef CAS PubMed;
(d) T. Chu, L. Belding, E. van der Art, T. Dudding, I. Korobkov and G. I. Nikonov, A Coordination Compound of Ge0 Stabilized by a Diiminopyridine Ligand, Angew. Chem., Int. Ed., 2014, 53, 2711–2715 CrossRef CAS PubMed;
(e) G. Frenking, R. Tonner, N. Klein, N. Takagi, T. Shimizu, A. Krapp, K. K. Pandey and P. Parameswaran, New bonding modes of carbon and heavier group 14 atoms Si–Pb, Chem. Soc. Rev., 2014, 43, 5106–5139 RSC;
(f) M. Lein, G. Krapp and G. Frenking, Why Do the Heavy-Atom Analogues of Acetylene E2H2 (E = Si-Pb) Exhibit Unusual Structures?, J. Am. Chem. Soc., 2005, 127, 6290–6299 CrossRef CAS PubMed.
- For selected reviews and examples, see:
(a) P. P. Power, Main-group elements as transition metals, Nature, 2010, 463, 171–177 CrossRef CAS PubMed;
(b) P. P. Power, Interaction of Multiple Bonded and Unsaturated Heavier Main Group Compounds with Hydrogen, Ammonia, Olefins, and Related Molecules, Acc. Chem. Res., 2011, 44, 627–637 CrossRef CAS PubMed;
(c) S. K. Mandal and H. W. Roesky, Group 14 Hydrides with Low Valent Elements for Activation of Small Molecules, Acc. Chem. Res., 2012, 45, 298–307 CrossRef CAS PubMed;
(d) H. W. Roesky and S. S. Kumar, Chemistry of aluminium(I), Chem. Commun., 2005, 4027–4028 RSC;
(e) P. P. Power, Reactions of heavier main-group compounds with hydrogen, ammonia, ethylene and related small molecules, Chem. Rec., 2012, 12, 238–255 CrossRef CAS PubMed;
(f) C. E. Kefalidis, A. Stasch, C. Jones and L. Maron, On the mechanism of the reaction of a magnesium(I) complex with CO2: a concerted type of pathway, Chem. Commun., 2014, 50, 12318–12321 RSC;
(g) T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, Low Coordinate Germanium(II) and Tin(II) Hydride Complexes: Efficient Catalysts for the Hydroboration of Carbonyl
Compounds, J. Am. Chem. Soc., 2014, 136, 3028–3031 CrossRef CAS PubMed;
(h) O. T. Summerscales, C. A. Caputo, C. E. Knapp, J. C. Fettinger and P. P. Power, The Role of Group 14 Element Hydrides in the Activation of C–H Bonds in Cyclic Olefins, J. Am. Chem. Soc., 2012, 134, 14595–14603 CrossRef CAS PubMed;
(i) C. A. Caputo, J. D. Guo, S. Nagase, J. C. Fettinger and P. P. Power, Reversible and Irreversible Higher-Order Cycloaddition Reactions of Polyolefins with a Multiple-Bonded Heavier Group 13 Alkene Analogue: Contrasting the Behavior of Systems with π–π, π–π*, and π–n+ Frontier Molecular Orbital Symmetry, J. Am. Chem. Soc., 2012, 134, 7155–7164 CrossRef CAS PubMed;
(j) A. G. Baradzenka, M. Pilkington, A. Dmitrienko and G. I. Nikonov, Small Molecule Activation on a Base-stabilized Phosphinidene, Chem. – Eur. J., 2023, 29, e202300523 CrossRef CAS PubMed.
- For selected examples, see:
(a) A. J. Arduengo III, J. C. Calabrese, A. H. Cowley, H. V. R. Dias, J. R. Goerlich, W. J. Marshall and B. Riegel, Carbene–Pnictinidene Adducts, Inorg. Chem., 1997, 36, 2151–2158 CrossRef PubMed;
(b) A. J. Arduengo III, C. J. Carmalt, J. A. C. Clyburne, A. H. Cowley and R. Pyati, Nature of the bonding in a carbene–phosphinidene: a main group analogue of a Fischer carbene complex? Isolation and characterisation of a bis(borane) adduct, Chem. Commun., 1997, 981–982 RSC;
(c) B. D. Ellis, C. A. Dyker, A. Decken and C. L. B. Macdonald, The synthesis, characterisation and electronic structure of N-heterocyclic carbene adducts of PI cations, Chem. Commun., 2005, 1965–1967 RSC;
(d) Y. Wang, Y. Xie, M. Y. Abraham, R. J. Gilliard Jr, P. Wei, H. F. Schaefer III, P. V. R. Schleyer and G. H. Robinson, Carbene-Stabilized Parent Phosphinidene, Organometallics, 2010, 29, 4778–4780 CrossRef CAS;
(e) K. Schwedtmann, M. H. Holthausen, K.-O. Feldmann and J. J. Weigand, NHC-Mediated Synthesis of an Asymmetric, Cationic Phosphoranide, a Phosphanide, and Coinage-Metal Phosphanido Complexes, Angew. Chem., Int. Ed., 2013, 52, 14204–14208 CrossRef CAS PubMed;
(f) J. F. Binder, A. Swidan, M. Tang, J. H. Nguyen and C. L. B. Macdonald, Remarkably stable chelating bis-N-heterocyclic carbene adducts of phosphorus(I) cations, Chem. Commun., 2015, 51, 7741–7744 RSC;
(g) H. Cui, J. Zhang and C. Cui, 2-Hydro-2-aminophosphasilene with N–Si–P π Conjugation, Organometallics, 2013, 32, 1–4 CrossRef CAS;
(h) K. Hansen, T. Szilvási, B. Blom, S. Inoue, J. Epping and M. Driess, A Fragile Zwitterionic Phosphasilene as a Transfer Agent of the Elusive Parent Phosphinidene (:PH), J. Am. Chem. Soc., 2013, 135, 11795–11798 CrossRef CAS PubMed;
(i) K. Hansen, T. Szilvási, B. Blom and M. Driess, Transition Metal Complexes of a “Half-Parent” Phosphasilene Adduct Representing Silylene→Phosphinidene→Metal Complexes, Organometallics, 2015, 34, 5703–5708 CrossRef CAS;
(j) K. Hansen, T. Szilvási, B. Blom and M. Driess, A Persistent 1,2-Dihydrophosphasilene Adduct, Angew. Chem., Int. Ed., 2015, 54, 15060–15063 CrossRef CAS PubMed;
(k) S. Roy, P. Stollberg, R. Herbst-Irmer, D. Stalke, D. M. Andrada, G. Frenking and H. W. Roesky, Carbene-Dichlorosilylene Stabilized Phosphinidenes Exhibiting Strong Intramolecular Charge Transfer Transition, J. Am. Chem. Soc., 2015, 137, 150–153 CrossRef CAS PubMed;
(l) B. D. Ellis and C. L. B. Macdonald, Stable compounds containing heavier group 15 elements in the +1 oxidation state, Coord. Chem. Rev., 2007, 251, 936–973 CrossRef CAS;
(m) J. D. Protasiewicz, Coordination-Like Chemistry of Phosphinidenes by Phosphanes, Eur. J. Inorg. Chem., 2012, 4539–4549 CrossRef CAS;
(n) B. A. Surgenor, M. Bühl, A. M. Z. Slawin, J. D. Woollins and P. Kilian, Isolable Phosphanylidene Phosphorane with a Sterically Accessible Two-Coordinate Phosphorus Atom, Angew. Chem., Int. Ed., 2012, 51, 10150–10153 CrossRef CAS PubMed;
(o) J. W. Dube, C. L. B. Macdonald and P. I. Ragogna, Accessing the Coordination Chemistry of Phosphorus(I) Zwitterions, Angew. Chem., Int. Ed., 2012, 51, 13026–13030 CrossRef CAS PubMed;
(p) C. D. Martin and P. J. Ragogna, Substitution matters: isolating phosphorus diiminopyridine complexes, Dalton Trans., 2011, 40, 11976–11980 RSC;
(q) L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, A Singlet Phosphinidene Stable at Room Temperature, Chem, 2016, 1, 147–153 CrossRef CAS;
(r) Q. Luo, T. Liu, L. Huang, C. Yang and W. Lu, Aromative Dephosphinidenation of a Bisphosphirane-Fused Anthracene toward E−H (E=H, Si, N and P) Bond Activation, Angew. Chem., Int. Ed., 2024, 63, e202405122 CrossRef CAS PubMed;
(s) Q. Luo, T. Liu, L. Huang, Q. Li and W. Lu, On the Reactivity of Bisphosphirane-Fused Anthracene towards Dichalcogenide Bonds, IPr Carbene, 2,6-Diisopropylphenyl Isocyanide and Trimethylsilyl Azide, Eur. J. Org. Chem., 2025, e202500071 CrossRef CAS;
(t) For selected reviews, see: L. Dostál, Quest for stable or masked pnictinidenes: Emerging and exciting class of group 15 compounds, Coord. Chem. Rev., 2017, 353, 142–158 CrossRef;
(u) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, NHCs in Main Group Chemistry, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed;
(v) T. Krachko and J. C. Slootweg, N-Heterocyclic Carbene–Phosphinidene Adducts: Synthesis, Properties, and Applications, Eur. J. Inorg. Chem., 2018, 2734–2754 CrossRef CAS;
(w) M. Soleilhavoup and G. Bertrand, Stable Carbenes, Nitrenes, Phosphinidenes, and Borylenes: Past and Future, Chem, 2020, 6, 1275–1282 CrossRef CAS.
- For selected examples, see:
(a) F. Mathey, Developing the chemistry of monovalent phosphorus, Dalton Trans., 2007, 1861–1868 RSC;
(b) L. Weber, Phospha- and Arsaalkenes RE=C(NMe2)2 (E = P, As) as Novel Phosphinidene- and Arsinidene-Transfer Reagents, Eur. J. Inorg. Chem., 2007, 4095–4117 CrossRef CAS;
(c) D. W. Stephan, Zirconium-Phosphorus Chemistry: Strategies in Syntheses, Reactivity, Catalysis, and Utility, Angew. Chem., Int. Ed., 2000, 39, 314–329 CrossRef;
(d) F. Mathey, N. H. T. Huy and A. Marinetti, Electrophilic Terminal-Phosphinidene Complexes: Versatile Phosphorus Analogues of Singlet Carbenes, Helv. Chim. Acta, 2001, 84, 2938–2957 CrossRef CAS;
(e) H. Aktaş, J. C. Slootweg and K. Lammertsma, Nucleophilic Phosphinidene Complexes: Access and Applicability, Angew. Chem., Int. Ed., 2010, 49, 2102–2113 CrossRef PubMed;
(f) R. Waterman and T. D. Tilley, Terminal hafnium phosphinidene complexes and phosphinidene ligand exchange, Chem. Sci., 2011, 2, 1320–1325 RSC;
(g) B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Triamidoamine–Uranium(IV)-Stabilized Terminal Parent Phosphide and Phosphinidene Complexes, Angew. Chem., Int. Ed., 2014, 53, 4484–4488 CrossRef CAS PubMed;
(h) H. Braunschweig, J. O. C. Jimenez-Halla, K. Radacki and R. Shang, Direct Conversion from Terminal Borylene into Terminal Phosphinidene, Angew. Chem., Int. Ed., 2016, 55, 12673–12677 CrossRef CAS PubMed;
(i) H. Stafford, T. M. Rookes, E. P. Wildman, G. Balázs, A. J. Wooles, M. Scheer and S. T. Liddle, Terminal Parent Phosphanide and Phosphinidene Complexes of Zirconium(IV), Angew. Chem., Int. Ed., 2017, 56, 7669–7673 CrossRef CAS PubMed;
(j) C. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, A Base-Free Terminal Actinide Phosphinidene Metallocene: Synthesis, Structure, Reactivity, and Computational Studies, J. Am. Chem. Soc., 2018, 140, 14511–14525 CrossRef CAS PubMed;
(k) A. Doddi, D. Bockfeld, T. Bannenberg and M. Tamm, N-Heterocyclic Carbene Analogues of Nucleophilic Phosphinidene Transition Metal Complexes, Chem. – Eur. J., 2020, 26, 14878–14887 CrossRef CAS PubMed;
(l) T. E. Rieser, P. Wetzel, C. Maichle-Mossmer, P. Sirsch and R. Anwander, A Terminal Yttrium Phosphinidene, J. Am. Chem. Soc., 2023, 145, 17720–17733 CrossRef CAS PubMed;
(m) Q. Wen, B. Feng and Y. Chen, Rare-Earth Metal Phosphinidene Complexes: A Trip from Bridging One to Terminal One, Acc. Chem. Res., 2023, 56, 3343–3357 CrossRef CAS PubMed;
(n) T. G. Saint-Denis, T. A. Wheeler, Q. Chen, G. Balázs, N. S. Settineri, M. Scheer and T. D. Tilley, A Ruthenophosphanorcaradiene as a Synthon for an Ambiphilic Metallophosphinidene, J. Am. Chem. Soc., 2024, 146, 4369–4374 CrossRef CAS PubMed;
(o) M. C. Neben, N. Wegerich, T. A. A. Said, R. R. Thompson, S. Demeshko, K. Dollberg, I. Tkach, G. P. V. Trieste III, H. Verplancke, C. von Hänisch, M. C. Holthausen, D. C. Powers, A. Schnegg and S. Schneider, Transient Triplet Metallopnictinidenes M–Pn (M = PdII, PtII; Pn = P, As, Sb): Characterization and Dimerization, J. Am. Chem. Soc., 2025, 147, 5330–5339 CrossRef CAS PubMed.
-
(a) L. Weber, The chemistry of diphosphenes and their heavy congeners: synthesis, structure, and reactivity, Chem. Rev., 1992, 92, 1839–1906 CrossRef CAS;
(b) M. Yoshifuji, Sterically Protected Diphosphenes, Eur. J. Inorg. Chem., 2016, 607–615 CrossRef CAS;
(c) T. Sasamori and N. Tokitoh, Doubly bonded systems between heavier Group 15 elements, Dalton Trans., 2008, 1395–1408 RSC;
(d) M. C. Simpson and J. D. Protasiewicz, Phosphorus as a carbon copy and as aphotocopy: New conjugated materials featuring multiply bonded phosphorus, Pure Appl. Chem., 2013, 85, 801–815 CrossRef CAS.
- For recent reviews, see:
(a) L. Weber, F. Ebeler and R. S. Ghadwal, Advances and recent trends in dipnictenes chemistry, Coord. Chem. Rev., 2022, 461, 214499 CrossRef CAS;
(b)
T. Sasamori, V. Lee, N. Nagahora and S. Morisako, Low-coordinate compounds of heavier group 14–16 elements, in Comprehensive Inorganic Chemistry III, Elsevier, 3rd edn, 2023, pp. 118–164 Search PubMed.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, Synthesis and structure of bis(2,4,6-tri-tert-butylphenyl)diphosphene: isolation of a true phosphobenzene, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
-
(a) S. Asami, M. Okamoto, K. Suzuki and M. Yamashita, A Boryl-Substituted Diphosphene: Synthesis, Structure, and Reactionwith n-Butyllithium To Form a Stabilized Adduct by pπ-pπ Interaction, Angew. Chem., Int. Ed., 2016, 55, 12827–12831 CrossRef CAS PubMed;
(b) D. W. N. Wilson, M. P. Franco, W. K. Myers, J. E. McGrady and J. M. Goicoechea, Base Induced Isomerisation of a Phosphaethynolato-Borane: Mechanistic Insights into Boryl Migration and Decarbonylation to Afford a Triplet Phosphinidene, Chem. Sci., 2020, 11, 862–869 RSC.
-
(a) A. Tsurusaki, R. Ura and K. Kamikawa, 1,1′-Binaphthyl-substituted diphosphene: synthesis, structures, and chiral optical properties, Dalton Trans., 2018, 47, 4437–4441 RSC;
(b) A. Tsurusaki, R. Ura and K. Kamikawa, A Gold(I) Complex with a 1,1′-Binaphthyl-Substituted Diphosphene: Synthesis, Structure, and Catalytic Application to Intramolecular Hydroarylation Reactions, Organometallics, 2020, 39, 87–92 CrossRef CAS;
(c) A. Tsurusaki, S. Takechi and K. Kamikawa, Diphosphene with a phosphineborane tether and its rhodium complex, Dalton Trans., 2024, 53, 2929–2936 RSC.
-
(a) L. L. Liu, L. L. Cao, J. Zhou and D. W. Stephan, Facile Cleavage of the P=P Double Bond in Vinyl-Substituted Diphosphenes, Angew. Chem., Int. Ed., 2019, 58, 273–277 CrossRef CAS PubMed;
(b) L. L. Liu, J. Zhou, Y. Kim, L. L. Cao and D. W. Stephan, Oligomerization of Phosphaalkynes Mediated by Bulky N-Heterocyclic Carbenes: Avenues to Novel Phosphorus Frameworks, Dalton Trans., 2019, 48, 14242–14245 RSC;
(c) D. Rottschäfer, M. K. Sharma, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, A Modular Access to Divinyldiphosphenes with a Strikingly Small HOMO–LUMO Energy Gap, Chem. – Eur. J., 2019, 25, 8127–8134 CrossRef PubMed.
- Y. Xiong, S. Yao, T. Szilvási, E. Ballestero-Martínez, H. Grützmacher and M. Driess, Unexpected Photodegradation of a Phosphaketenyl-Substituted Germyliumylidene Borate Complex, Angew. Chem., Int. Ed., 2017, 56, 4333–4336 CrossRef CAS PubMed.
-
(a) D. W. N. Wilson, W. K. Myers and J. M. Goicoechea, Synthesis and Decarbonylation Chemistry of Gallium Phosphaketenes, Dalton Trans., 2020, 49, 15249–15255 RSC;
(b) M. K. Sharma, S. Chabbra, C. Wölper, H. M. Weinert, E. J. Reijerse, A. Schnegg and S. Schulz, Modulating the frontier orbitals of L(X)Ga-substituted diphosphenes [L(X)GaP]2 (X = Cl, Br) and their facile oxidation to radical cations, Chem. Sci., 2022, 13, 12643–12650 RSC.
-
(a) D. V. Partyka, M. P. Washington, T. G. Gray, J. B. Updegraff III, J. F. Turner II and J. D. Protasiewicz, Unusual Phosphorus-Phosphorus Double Bond Contraction upon Mono- and Di-auration of a Diphosphene, J. Am. Chem. Soc., 2009, 131, 10041–10048 CrossRef CAS PubMed;
(b) D. Dhara, S. Das, S. K. Pati, D. Scheschkewitz, V. Chandrasekhar and A. Jana, NHC-Coordinated Diphosphene-Stabilized Gold(I) Hydride and Its Reversible Conversion to Gold(I) Formate with CO2, Angew. Chem., Int. Ed., 2019, 58, 15367–11537 CrossRef CAS PubMed;
(c) M. Chen, Z. Zhang, J. Liu, G. Li, L. Zhao and Z. Mo, Isolation and Reactivity of Homoleptic Diphosphene Lead Complexes, Angew. Chem., Int. Ed., 2023, 62, e202312837 CrossRef CAS PubMed.
- T. L. Chan and Z. Xie, The synthesis, structure and reactivity of an imine-stabilized carboranylphosphorus(I) compound, Chem. Commun., 2016, 52, 7280–7283 RSC.
- J. E. Steves, M. D. Kennedy, K. P. Chiang, W. S. Kassel, W. G. Dougherty, T. J. Dudley and D. L. Zubris, Synthesis of a bulky bis(imino)pyridine compound: a methodology for systematic variation of steric bulk and energetic implications for metalation, Dalton Trans., 2009, 1214–1222 RSC.
- X. Liu, P. Wu, J. Li and C. Cui, Synthesis of 1,2-Borazaronaphthalenes from Imines by Base-Promoted Borylation of C–H bond, J. Org. Chem., 2015, 80, 3737–3744 CrossRef CAS PubMed.
-
(a) T. Sasamori, N. Takeda and N. Tokitoh, Synthesis and reactions of new diphosphenes bearing extremely bulky substituents, J. Phys. Org. Chem., 2003, 16, 450–462 CrossRef CAS;
(b) M. Sakagami, T. Sasamori, H. Sakai, Y. Furukawa and N. Tokitoh, 1,2-Bis(ferrocenyl)dipnictenes: Bimetallic Systems with a Pn=Pn Heavy π-Spacer (Pn: P, Sb, and Bi), Bull. Chem. Soc. Jpn., 2013, 86, 1132–1143 CrossRef CAS.
- E. Urnéžius and J. D. Protasiewicz, Synthesis and Structural Characterization of New Hindered Aryl Phosphorus Centers (Aryl = 2,6-Dimesitylphenyl), Main Group Chem., 1996, 1, 369–372 CrossRef.
- S. S. Batsanov, van der Waals Radii of Elements, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- M. Yoshifuji, K. Ando, K. Shibayama, N. Inamoto, K. Hitotsu and T. Higuchi, The First X-Ray Structure Analysis of a Thiadiphosphirane, Angew. Chem., Int. Ed. Engl., 1983, 22, 418–419 CrossRef.
- K. Schwedtmann, F. Hennersdorf, A. Bauzá, A. Frontera, R. Fischer and J. J. Weigand, Isolation of Azadiphosphiridines and Diphosphenimines by Cycloaddition of Azides and a Cationic Diphosphene, Angew. Chem., Int. Ed., 2017, 56, 6218–6222 CrossRef CAS PubMed.
- Z. Sun, J. Zong, H. Ren, C. Lu, D. Tu, J. Poater, M. Solà, Z. Shi and H. Yan, Couple-close construction of non-classical boron cluster-phosphonium conjugates, Nat. Commun., 2024, 15, 7934 CrossRef CAS PubMed.
-
G. M. Sheldrick, SADABS: Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1996 Search PubMed.
-
G. M. Sheldrick, SHELXTL 5.10 for Windows NT: Structure Determination Software Programs, Bruker Analytical X-ray systems, Inc., Madison, Wisconsin, USA, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectroscopic, and crystallographic details. CCDC 2446505–2446518. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt01239b |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jie
Zhang
a,
Jie
Zhang