
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of titanium pyrazolates towards CO2, CS2 and N2O†
Felix
Kracht
,
Sophie
Mayer
,
Cäcilia
Maichle-Mössmer
and
Reiner
Anwander
 *
*
Institut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: reiner.anwander(at)uni-tuebingen.de
First published on 23rd June 2025
Abstract
The tetravalent titanium pyrazolate Ti(pzMe2)4 inserts two molecules of carbon dioxide instantly at ambient temperature and 1 bar heteroallene, forming the complex Ti(CO2·pzMe2)2(pzMe2)2 (pzMe2 = 3,5-dimethylpyrazolato). CO2 insertion is reversible as proven by thermogravimetric analysis (TGA). Solid Ti(CO2·pzMe2)2(pzMe2)2 exhibits an exceptionally high stability at ambient atmosphere but converts in solution over time via deoxygenation into the oxo-bridged species [Ti(CO2·pzMe2)2(pzMe2)]2(μ-O) and [Ti(μ-O)2(μ-pzMe2)4(Ti{CO2·pzMe2}{pzMe2})2]. Treatment of Ti(pzMe2)4 with CS2 results in a mono-insertion to Ti(CS2·pzMe2)(pzMe2)3, which is reversible at 110 °C. Applying similar conditions, trivalent Ti(pztBu2)3 inserts also two molecules of CO2 to afford Ti(CO2·pztBu2)2(pztBu2) displaying complete de-insertion at 70 °C. At elevated temperatures compound Ti(CO2·pztBu2)2(pztBu2) does neither display redox activity nor engages in a deoxygenation reaction. While tetravalent Ti(pzMe2)4 does not react with N2O under ambient conditions, trivalent Ti(pztBu2)3 converts into oxo-bridged [Ti(pztBu2)3]2(μ-O) via initial N2O insertion and subsequent fast release of N2.
Introduction
The rapidly rising greenhouse gas emissions and concomitant record-high concentrations in the Earth's atmosphere are the primary cause for the anthropogenic global climate change, with carbon dioxide (CO2) having the greatest impact, followed by methane (CH4) and nitrous oxide (N2O).1–13 Although the concentration of N2O (337.69 ppb, 2024) is relatively low when compared to CO2 (427 ppm, 2024; 50% increase since start of industrialization), its global warming potential is 300 times stronger, contributing 6% to the anthropogenic greenhouse effect.1,12,13 The activation of these small, mostly inert molecules leading to their capture, conversion, storage, and ultimately climate change mitigation, has become a major focus of research in the past decades. For the development of efficient capture materials, a fundamental understanding of the activation of such small molecules by metal centres is crucial. In particular, amine-functionalized and M–N(ligand) components/materials feature efficient systems for CO2 capture and valorization.14,15Titanium is attractive as a sustainable metal, not only because it is affordable and non-toxic, but also because of its high abundance in the Earth's crust. Already in the 1970s, CO2 insertion into the Ti–N bond of tetravalent Ti(NMe2)4 under formation of carbamate complex Ti(CO2·NMe2)4 was reported. However, such carbamate complexes were structurally elucidated only three decades later.16–18 Similar examples exist for trivalent titanium complexes.19–21 In addition, a twofold addition of CO2 across the Ti![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond of the tetravalent imide complex (C5Me5)Ti(NAr)(L) (Ar = 2,6-C6H3Me2; L = (NiPr2)2CR; R = Me, Ph) was achieved resulting in (C5Me5)Ti({CO2}2·NAr)(L).22,23 For sterically less demanding imido ligands, the mono-inserted congener (C5Me5)Ti(CO2·NtBu)(L) (L = (NR1)2CR2; R1 = SiMe3, iPr; R2 = Ph, Me) was shown to rapidly deoxygenate CO2 under formation of the oxo-bridged dimer [(C5Me5)Ti(L)(μ-O)]2 and elimination of the corresponding isocyanate (Scheme 1A).
N double bond of the tetravalent imide complex (C5Me5)Ti(NAr)(L) (Ar = 2,6-C6H3Me2; L = (NiPr2)2CR; R = Me, Ph) was achieved resulting in (C5Me5)Ti({CO2}2·NAr)(L).22,23 For sterically less demanding imido ligands, the mono-inserted congener (C5Me5)Ti(CO2·NtBu)(L) (L = (NR1)2CR2; R1 = SiMe3, iPr; R2 = Ph, Me) was shown to rapidly deoxygenate CO2 under formation of the oxo-bridged dimer [(C5Me5)Ti(L)(μ-O)]2 and elimination of the corresponding isocyanate (Scheme 1A).
 | ||
| Scheme 1
A: addition of CO2 across a TiN bond and subsequent deoxygenation;22B: N2O insertion into titanium/zirconium complexes and subsequent N2 elimination;28C: reversible CO2 insertion into metal pyrazolates;32–34D: literature-known tetravalent and trivalent homoleptic titanium pyrazolate complexes.35,36 | ||
Light-induced deoxygenation of CO2 is feasible on a TiO2 surface in the presence of H2O under formation of CH4, CH3OH, H2 and O2,24,25 whereas CO2 deoxygenation reactions were also observed for divalent and trivalent titanium compounds.26,27
The activation of N2O with trivalent titanocene complex (Cp2TiCl)2 has been reported, proceeding via slow oxidation of the titanium centre under formation of oxo-bridged species [Cp2TiCl]2O and elimination of N2.28–31 Only a few examples of N2O-inserted compounds prior to N2 elimination have been reported,28,30 including azoxymetallacylopentene complex (C5Me5)2Ti(N{O}NCPhCPh) obtained from (C5Me5)2Ti(PhCCPh) (Scheme 1B).28
Recently, we found that homoleptic metal pyrazolates [M(pzR,R′)x] insert CO2 readily and reversibly, forming the corresponding carbamate complexes (see Scheme 1C).32 Structurally characterized examples comprise redox-active Ce(CO2·pzMe2)4, the tetranuclear cluster [Ce(CO2·pzMe2)3]4, and the light metal derivatives Mg(CO2·pztBu2)2(thf)2, Al(CO2·pztBu2)2(pztBu2) and [Sc(μ-CO2·pztBu2)(pztBu2)2]2.33,34 Using unsubstituted [Mg(pz)2]n, exceptionally high reversible CO2 capacities of up to 35.7 wt% have been achieved, highlighting its potential as an efficient CO2 capture material. Spurred by these findings, we envisaged the reactivity of pyrazolate complexes of the light metal titanium toward both carbon dioxide, CS2 and N2O. Homoleptic complexes, tetravalent Ti(pzMe2)4 (1) and trivalent Ti(pztBu2)3 (4), previously reported by Winter and Mösch-Zanetti, respectively, were chosen as discrete precursors (see Scheme 1D).35,36
Results and discussion
CO2 activation with tetravalent Ti(pzMe2)4
Exposure of tetravalent Ti(pzMe2)4 (1) to a 1 bar CO2 atmosphere in benzene or toluene gave the twofold inserted carbamate complex Ti(CO2·pzMe2)2(pzMe2)2 (1CO2) (Scheme 2). The insertion reaction occured instantly, as monitored by an immediate colour change of the solution from yellow to orange. The CO2 uptake of complex 1CO2 equals a mass fraction of 17.0 wt% CO2 or 3.8 mmol CO2 per gram. | ||
| Scheme 2 Reaction of Ti(pzMe2)4 (1) with an excess of CO2 and subsequent deoxygenation in solution affording oxo-bridged species. | ||
A single crystal X-ray diffraction (SCXRD) analysis of 1CO2 revealed two distinct molecules in the unit cell with slightly varying metrical parameters. The four ligands of 1CO2 arrange in a distorted square planar geometry, with the cis-positioned carbamato ligands coordinated in the κ2(N,O) mode (Fig. 1). The pyrazolato ligands are tilted out of the plane spanned by the titanium centre and the two coordinating nitrogen atoms, by 26.6° and 18.9°, respectively. The inserted CO2 exhibits localized C–O single (1.275(3)–1.277(3) Å) and double bonds (1.205(3)–1.207(3) Å) and OCO angles ranging from 128.5(2) to 129.0(3)°. The loss of aromaticity of the pyrazole ring of the carbamato ligand is indicated by distinct C–C and C–N double/single bonds. Additional CO2 insertion into 1CO2 is most likely impeded by the coordinative saturation of the small titanium centre, and would cause unfavourable tilting and hence significant steric interference of newly formed carbamato ligands. Fourfold inserted Ce(CO2·pzMe2)4 clearly supports the size-determining influence of the metal centre for CO2 insertion (effective ionic radii for 6-coordination: Ti4+ 0.605, Ce4+ 0.87 Å).37 Although an electronic effect caused by saturation of the titanium centre with two oxygen atoms might play a role as well, this is opposed by the comparatively higher oxophilicity of titanium (θ = 1.0) versus cerium (θ = 0.9).38
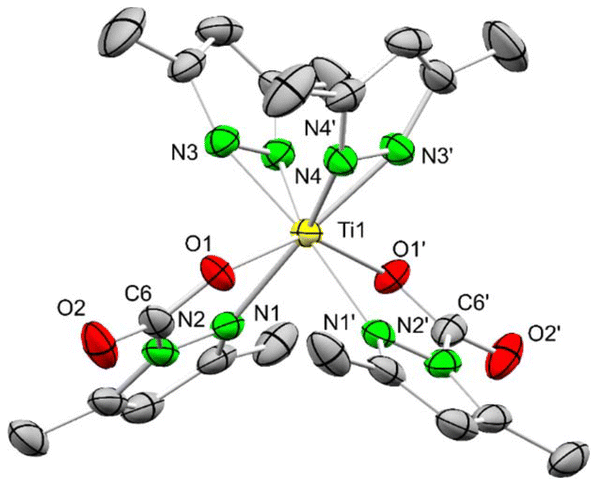 | ||
| Fig. 1 Crystal structure of Ti(CO2·pzMe2)2(pzMe2)2 (1CO2). Ellipsoids are set at the 50% probability level. Hydrogen, lattice toluene and a second molecule of 1CO2 are omitted for clarity. See ESI† for selected interatomic distances and angles. | ||
Formation of the twofold inserted complex 1CO2 was confirmed by solution NMR spectroscopy. The 1H NMR spectrum shows three methyl group signals at δ = 2.62, 2.21 and 2.17 ppm with an integral of 6, 6 and 12 protons, respectively. The two signals at lower field can be assigned to the distinct methyl groups of the two carbamato ligands, caused by CO2 insertion. The third signal is attributed to the magnetically equal methyl groups of the two unchanged pyrazolato ligands. Further, two signals for the ring protons at δ = 5.72 and 5.00 ppm were detected. The 13C NMR spectrum is consistent with the 1H NMR spectrum and a signal for the inserted CO2 could be resolved at δ = 150.5 ppm via experiments with labelled 13CO2 (see Fig. S4†). A diffuse reflectance infrared Fourier transform (DRIFT) measurement revealed a strong band at ṽ = 1740 cm−1 assigned to the CO stretching vibration. The CO2 uptake was corroborated by a thermogravimetric analysis (TGA) showing a CO2 releasing step of 17.5% between 90–150 °C which is in good agreement with the aforementioned calculated uptake of 17.0 wt%. When solid 1 is exposed to an atmosphere of 1 bar CO2 a colour change of the powder from yellow to orange was observed. The orange product was identified as 1CO2 by 1H NMR spectroscopy. This can be further interpreted as a fully reversible CO2 insertion process in the solid state of compound 1. Application of higher CO2 pressure of up to 10 bar did not lead to any additional insertion of CO2 but only the formation of 1CO2. The combination of the small titanium(IV) centre and the steric bulk of the pyrazolato ligand seem to suppress further insertion reactions.
Strikingly, different to our previous studies on magnesium, aluminium and rare-earth-metal pyrazolates, solid 1CO2 remained stable under ambient atmosphere. After one month only small amounts of the hydrolysis product HOOCpzMe2 were observed in the 1H NMR spectrum and the solid kept its orange colour (Fig. S6†). In stark contrast, decomposition of 1CO2 readily occurred in solution, both under an argon or CO2 atmosphere (Fig. S7†). Overnight a new signal set appeared in the 1H NMR spectrum which accumulated over time and after one week only a minor amount of 1CO2 was left. The observed decomposition is reproducible at ambient temperature. The obtained solution provided access to crystals suitable for a SCXRD analysis, which revealed the formation of the oxo-bridged bimetallic complex [Ti(CO2·pzMe2)2(pzMe2)]2(μ-O) (2) (Scheme 2 and Fig. S35†). Each titanium centre of 2 is coordinated by two terminal carbamato and one terminal pyrazolato ligand. However, NMR experiments showed the presence of more than one species ruling out a clear signal assignment to 2. Further, after three months morphologically different crystals formed in the same solution and could be identified by a SCXRD measurement as the oxo-bridged trimetallic complex [Ti(μ-O)2(μ-pzMe2)4(Ti{CO2·pzMe2}{pzMe2})2] (3, Scheme 2 and Fig. S36†). The central titanium of 3 is bridged by one oxo and two pyrazolato ligands in the μ-N,N mode to each of the outer titanium atoms. The coordination sphere of the outer titanium centres is completed by one terminal pyrazolato ligand in the κ2(N,N) mode and one carbamato ligand in the κ2(N,O) mode. The Ti–O(oxo) interatomic distances of 2 and 3 range from 1.790(2) to 1.8199(12) Å with one exception for the inner titanium centre of 3 which is slightly elongated (1.8465(12) Å). The latter is comparable with the reported bis-oxo [(C5Me5)Ti((NSiMe3)2CPh)(μ-O)]2 (1.8429(13)–1.8604(12) Å) and mono-oxo [(C5H4Me)2TiCl]2O (1.837(2) Å).23,39 Similarly, the Ti–O(carbamato) interatomic distances of oxo-bridged 2 and 3 are slightly shorter (1.9930(18)–2.0286(19) Å) than in oxo-free 1CO2 (2.0445(18)–2.0484(16) Å), with again one exception detected for 3 (2.0437(13) Å).
Decomposition of 1CO2 was not observed when the solution is kept at 0 °C. Deoxygenation of inserted CO2 or hydrolysis and a subsequent dissociation of pyrazole/carbamic acid are likely decomposition pathways of 1CO2 (Fig. S42†), which would result in oxo complexes like 2 and 3. Note that single-crystalline 1CO2 was obtained after three years at −40 °C, while the corresponding 1H NMR spectrum revealed that it remained stable during this time span. This makes a hydrolysis pathway highly unlikely. However, reactions of titanium compounds with CO2 involving a deoxygenation process and subsequent formation of oxo bridges have been reported previously.22 Unfortunately, conclusive organic co-products of a putative deoxygenation reaction involving 1CO2 could not be identified by 13C NMR spectroscopy, even when labelled 13CO2 was used. The formation of CO was not observed. Mass spectrometry experiments revealed a [M + H]+-fragment of 219.16 g mol−1 which fits well to the urea derivative OC(pzMe2)2 (219.12 g mol−1). This is in good agreement with the displacement of two pzMe2-moieties upon the formation of 2. Experiments in amber glass NMR tubes showed the same decomposition pattern, excluding a light-induced mechanism. The elongation of the titanium-oxo chain indicates that this decomposition pathway might ultimately lead to the formation of titania, TiO2. However, it has remained unclear whether trimetallic 3 is formed via bimetallic 2.
CS2 activation with tetravalent Ti(pzMe2)4
To examine the feasibility of a sulfido-bridged compound similar to complexes 2 or 3,401 was reacted with an excess of CS2. 1H NMR monitoring at ambient temperature indicated a rather slow conversion which was not complete even after several weeks. Therefore, the reaction was conducted at 60 °C affording full conversion to the mono-inserted complex Ti(CS2·pzMe2)(pzMe2)3 (1CS2) after five days (Scheme 3). The thiocarbamato ligand coordinates to the metal centre in the κ2(N,S) mode similar to the carbamato ligand in complex 1CO2 (Fig. 2). The C–S interatomic distances of the inserted CS2 moiety reveal localized double and single bonds of 1.6543(19) Å and 1.7019(19) Å, respectively, forming an angle of 121.64(12)°. | ||
| Scheme 3 Reaction of Ti(pzMe2)4 (1) with an excess of CS2. | ||
 | ||
| Fig. 2 Crystal structure of Ti(CS2·pzMe2)(pzMe2)3 (1CS2). Ellipsoids are set at the 50% probability level. Hydrogen atoms and one toluene lattice are omitted for clarity. See ESI† for selected interatomic distances and angles. | ||
Accordingly, the degree of electron delocalization is comparatively larger in 1CS2 (ΔC–S 0.0476 Å) than in 1CO2 (ΔC–O 0.068–0.072 Å). An additional insertion of CS2 seems disfavoured for steric reasons considering the larger size of the sulphur atoms. Moreover, the HSAB mismatch according to Pearson, the low thiophilicity of titanium (S = 0.0),38 and changed electrophilicity of the heteroallene will counteract a second insertion.
At ambient temperature, the 1H NMR spectrum of 1CS2 shows the thiocarbamato ligand as sharp signals, with the aromatic backbone proton resonating at δ = 5.05 ppm and distinct methyl groups at δ = 2.38 and 2.36 ppm. In the aromatic region two broad signals appeared at δ = 5.75 and 5.61 ppm in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 integral ratio, while the remaining methyl groups gave one broad signal. The integral ratios of the aromatic protons indicate that the pyrazolato ligands positioned trans and cis to the thiocarbamato ligand are magnetically unequal. Correspondingly, a variable temperature (VT) 1H NMR experiment at −60 °C revealed a sharpening of the broadened resonances resulting in three distinct signals sets with an integral ratio of 1:1:2, assigned to the thiocarbamato ligand, the trans- and the two cis-positioned pyrazolato ligands (Fig. S12†). The 13C signal of the inserted CS2 is resolved at δ = 209.2 ppm being consistent with the literature.41 In the DRIFT spectrum of 1CS2 vibrations characteristic of C–S double and single bonds are detected in the fingerprint region at ṽ = 1071 and 895 cm−1, respectively.42,43 A TGA experiment revealed a CS2 releasing step of 14.8% between 110–180 °C, which is close to the theoretical value of 15.1%.
:1 integral ratio, while the remaining methyl groups gave one broad signal. The integral ratios of the aromatic protons indicate that the pyrazolato ligands positioned trans and cis to the thiocarbamato ligand are magnetically unequal. Correspondingly, a variable temperature (VT) 1H NMR experiment at −60 °C revealed a sharpening of the broadened resonances resulting in three distinct signals sets with an integral ratio of 1:1:2, assigned to the thiocarbamato ligand, the trans- and the two cis-positioned pyrazolato ligands (Fig. S12†). The 13C signal of the inserted CS2 is resolved at δ = 209.2 ppm being consistent with the literature.41 In the DRIFT spectrum of 1CS2 vibrations characteristic of C–S double and single bonds are detected in the fingerprint region at ṽ = 1071 and 895 cm−1, respectively.42,43 A TGA experiment revealed a CS2 releasing step of 14.8% between 110–180 °C, which is close to the theoretical value of 15.1%.
Any decomposition of 1CS2 could not be observed at ambient temperature, while heating to 110 °C led to de-insertion of CS2 under full recovery of 1. Again, this is most likely caused by the soft Lewis base character of sulphur, which also disfavours the formation of sulphido bridges. Crucially, complex 1 features a hard titanium(IV) centre featuring a significantly higher oxophilicity (θ = 1.0) than thiophilicity (S = 0.0).38
CO2 and N2O activation with trivalent Ti(η2-pztBu2)3
Exposure of trivalent Ti(pztBu2)3 (4) to a 1 bar CO2 atmosphere in toluene gave an immediate colour change of the solution from blue over green and yellow to finally red. An SCXRD analysis revealed the formation of the twofold-inserted titanium(III) complex Ti(CO2·pztBu2)2(pztBu2) (4CO2) (Scheme 4). The CO2 uptake of complex 4CO2 amounts to a mass fraction of 13.1 wt% CO2 or 3.0 mmol CO2 per gram. | ||
| Scheme 4 Reactions of Ti(pztBu2)3 (4) with an excess of CO2 and N2O. | ||
Complex 4CO2 is isostructural to the aluminium complex Al(CO2·pztBu2)2(pztBu2) reported recently (Fig. 3).34 Accordingly, two carbamato ligands in the κ2(N,O) mode and one pyrazolato in the κ2(N,N) mode adopt a propeller-like geometry with both stereoisomers (Λ,Δ) present in the unit cell. Another CO2 insertion into the third pyrazolato ligand is impeded by the bulky tBu moieties, which prevent the pyrazolato ligand from the necessary tilting (cf., effective ionic radii for 6-coordination: Ti3+ 0.670, Al3+ 0.535 Å).37 The unit cell holds three distinct molecules of 4CO2 and six lattice toluene, which form a superstructure with alternating layers of complex 4CO2 and toluene (Fig. S39†).
 | ||
| Fig. 3 Connectivity of Ti(CO2·pztBu2)2(pztBu2) (4CO2). Ellipsoids are set at the 50% probability level and tBu moieties are displayed in wireframe projection for clarity. Hydrogen atoms, two additional molecules of 4CO2 and six lattice toluene are omitted for clarity. | ||
The DRIFT spectrum of 4CO2 shows two characteristic bands for CO stretching vibration at ṽ = 1765 and 1753 cm−1 similar to the aluminium congener. Due to the paramagnetic character of 4CO2 the signals in the 1H NMR spectrum are significantly broadened and only two signals are resolved. One very broad signal between δ = 7–3 ppm can most likely be assigned to aromatic protons in the pyrazolato/carbamato backbones while the other broadened signal at δ = 1.55 ppm can be ascribed to tBu moieties. Despite the broadened signals, the CO2 insertion reaction can be easily monitored by 1H NMR spectroscopy (Fig. S16†). At the start of the reaction signals for 4 and 4CO2 in addition to another signal at δ = 1.67 ppm could be observed. The additional signal likely arises from a mono-inserted product, which was also found for the aluminium congener. The EPR spectrum of 4CO2 is shown in Fig. S41.†
After one hour the conversion of 4 to 4CO2 is complete, which is also indicated by the red colour of the solution. A VT 1H NMR study revealed a complete CO2 release already at 70 °C suggesting that the overall CO2 insertion process into 4 is reversible (Fig. S18†). This is again also indicated by the colour of the solution which turns blue after the experiment and again red after several hours at ambient temperature. Shining UV light on compound 4CO2 led to oxidation of the titanium centre, however, the formation of a significant amount of oxalate was not observed by 13C NMR spectroscopy. A TGA experiment revealed an initial toluene solvent release of 13.9% between 77–110 °C, which is in line with the high lattice toluene content in the crystalline material. Subsequently, a CO2 releasing step of 12.1% between 110–150 °C was observed. Both releasing steps fit to a toluene/4CO2 ratio of 1:1 (toluene: 12.0%; CO2: 11.5%). Interestingly, different to tetravalent 1CO2 no deoxygenation reaction was observed for 4CO2 over time in solution, however, a fast decomposition of neat 4CO2 occurred at ambient atmosphere to unidentified products.
Treatment of tetravalent 1 with an excess of N2O led to no visible reaction as suggested by 1H NMR spectroscopy and the unchanged yellow colour of the solution. In contrast, exposure of trivalent 4 to N2O resulted in an immediate colour change from blue to green and after 10 minutes to yellow. A 1H NMR spectrum revealed the presence of a paramagnetic (not 4) and a diamagnetic species. Overnight, orange crystals formed in a toluene solution and a SCXRD measurement revealed the formation of the oxo-bridged species [Ti(pztBu2)3]2(μ-O) (5) (Scheme 4). Both titanium centres of 5 underwent N2O-promoted +III → +IV oxidation under elimination of N2. The 1H NMR spectrum of the supernatant solution revealed a full conversion of 4 to 5, while the signals of the new paramagnetic species disappeared completely. The latter paramagnetic species is most likely the elusive N2O insertion complex in which the titanium centre is still in the formal oxidation state +III. So far, attempts to isolate this paramagnetic species were not successful.
The connectivity structure of 5 indicates a highly symmetrical structure with only one symmetrically independent pyrazolato ligand, the titanium centre and bridging oxygen in the asymmetric unit (Fig. 4).
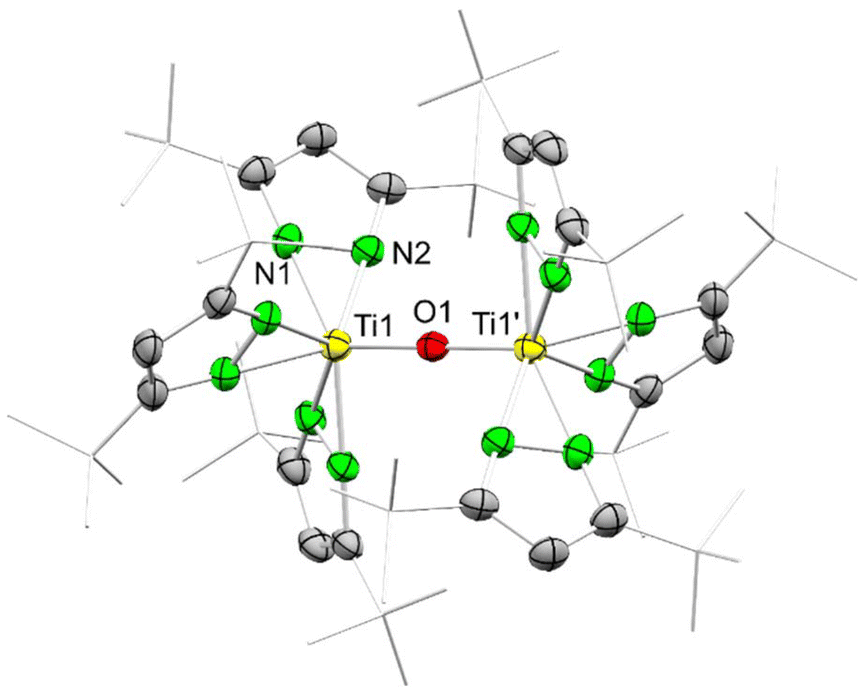 | ||
| Fig. 4 Connectivity of [Ti(pztBu2)3]2(μ-O) (5). Ellipsoids are set at the 50% probability level. Hydrogen atoms are omitted and tBu moieties are displayed in the wireframe projection for clarity. | ||
Each titanium centre is coordinated by three terminal pyrazolato ligands in the κ2(N,N) mode adopting a propeller-like geometry. NMR studies corroborate the highly symmetric character of 5 showing only one signal set for all six pyrazolato ligands. The 1H NMR spectrum displays a sharp signal at δ = 6.36 ppm with an integral of six protons assignable to the aromatic ring protons and a slightly broadened signal at δ = 1.01 ppm with an integral of 108 protons for the tBu moieties. Similarly, the 13C NMR spectrum revealed one signal set, however, the quaternary carbon ring atom could only be resolved in a 1H–13C HMBC NMR experiment at δ = 157.5 ppm.
The core structure of 5 is reminiscent of 2 except the inserted CO2. Therefore, we wondered about whether 5 can be transformed into putative {[Ti(CO2·pztBu2)2(pztBu2)]2(μ-O)} or {[Ti(CO2·pztBu2)(pztBu2)2]2(μ-O)} when exposed to an excess of CO2. Due to the insolubility of 5 in non-donating solvents these experiments were conducted in THF, which however did not result in any CO2 insertion at ambient conditions as indicated by 1H NMR experiments. This behaviour can be ascribed to the enhanced steric bulk of the tBu moieties blocking the titanium centres, in addition to the low solubility of 5 in THF. Attempts to synthesize the sterically less demanding Ti(pzMe2)3 as a precursor in order to access putative [Ti(pzMe2)3]2(μ-O) were not successful.
Catalytic formation of cyclic carbonates
Titanium complexes are widely used in the copolymerization of CO2 and epoxides.44–48 Since metal pyrazolates effectively promote the catalytic cycloaddition of CO2 and epoxides, a similar behaviour of the titanium derivatives could be anticipated.32–34 For better comparability, the conditions of the catalytic transformations were adapted to our previous studies (Scheme 5). | ||
| Scheme 5 Catalytic conversion of CO2 and epoxide into cyclic carbonates by applying titanium pyrazolate 1 as catalyst. aReaction conditions: 1 bar CO2, 0.5 mol% catalyst (1 and 2), 1 mol% tetra-n-butylammonium bromide (TBAB) as cocatalyst at ambient temperature for 24 h in neat epoxide; bConversion determined by comparison of the integral protons at the α-position of the epoxide and the corresponding cyclic carbonate (exception: 2-tert-butyloxirane/3,3-dimethyl-1,2-butene carbonate where the integral of the tBu moieties was used). | ||
Tetravalent Ti(pzMe2)4 (1) exhibits moderate catalytic activity for the conversion of propylene oxide (59%), ranging between magnesium (42–59%) and rare-earth-metal pyrazolates (84–99%) and close to Al(pztBu2)3 (65%).32–34 This is in line with the carboxopilicity criterion (as derived from the CO2-release temperature),33 and reflects both the enhanced oxophilicity and Lewis acidity of titanium(IV).41 As expected, the conversion of sterically more demanding epoxides was considerably lower, being lowest for 2-tert-butyloxirane (3%) and highest for epoxy hexane (11%). Trivalent Ti(pztBu2)3 (4) showed a significantly lower conversion of 27% for propylene oxide compared to tetravalent 1. This can be ascribed to side reactions, as indicated by an immediate colour change upon the addition of epoxide. Trivalent titanium compounds are known to catalytically convert epoxides to alcohols under ring opening and oxidation of the titanium center.49 Examples of titanium-based catalysts in this cycloaddition reaction are rather rare.50–55 The highest catalytic activity was observed for the tetrazole containing (C5Me5)TiCl2(O,N-L) (L = 5-(2-hydroxyphenolate)-1H-tetrazole) featuring a maximum TOF of 422 for propylene oxide at 75 °C and 22 bar CO2.52 Recently, the Ti31 cluster (C2H8N)8[Ti31O52(OiPr)4(PhCO2)24] was reported to reach a quantitative conversion for propylene oxide when a catalyst load of 0.83 mol% is used at 80 °C and 1 bar CO2 over five days, according to a photocatalytic mechanism.55
Conclusions
Tetravalent Ti(pzMe2)4 undergoes a twofold CO2 insertion to Ti(CO2·pzMe2)2(pzMe2)2 when exposed to an excess of CO2 as proven by SCXRD, NMR and DRIFT experiments. A thermogravimetric analysis showed an exhaustive CO2 release between 90–150 °C pointing to the reversibility of the CO2 insertion. In solid form, Ti(CO2·pzMe2)2(pzMe2)2 showed an exceptionally high stability, when compared to derivatives of other light metals. Even after a month at ambient atmosphere only minor hydrolysis products were observed. Strikingly, in solution under a 1 bar CO2 atmosphere Ti(CO2·pzMe2)2(pzMe2)2 converted into oxo-bridged species [Ti(CO2·pzMe2)2(pzMe2)]2(μ-O) and [Ti(μ-O)2(μ-pzMe2)4(Ti{CO2·pzMe2}{pzMe2})2], en route to titania. This transformation reflects the highly polarizing nature of titanium(IV) and is accelerated at elevated temperatures. Heteroallene CS2 undergoes a fully reversible mono-insertion into Ti(pzMe2)4via formation of Ti(CS2·pzMe2)(pzMe2)3. The thiocarbamate complex does not engage in the formation of sulfido-bridged species, even under thermal treatment. Trivalent Ti(pztBu2)3 also undergoes a twofold CO2 insertion and the obtained bis(carbamato) complex Ti(CO2·pztBu2)2(pztBu2) is stable in solution against oxidation and deoxygenation. Ti(CO2·pztBu2)2(pztBu2) de-inserts CO2 exhaustively already at 70 °C without a decomposition side reaction. Distinct reactivity of tetravalent Ti(pzMe2)4 and trivalent Ti(pztBu2)3 is also found when treated with nitrous oxide. The tetravalent pyrazolate displayed inert behaviour toward N2O, whereas Ti(pztBu2)3 gets oxidized to afford the oxo-bridged complex [Ti(pztBu2)3]2(μ-O) via fast release of N2.Conflicts of interest
There are no conflicts of interest.Data availability
The data that support the findings of this study are available in the ESI† of this article.Acknowledgements
We are grateful to the VECTOR foundation for generous support. We thank Adrian Jenner for performing the EPR measurement, Dr Markus Ströbele for running the TGA experiments and Dr Peter Haiss for running the mass spectrometry experiments.References
- H. Lee and J. Romero, IPCC, 2023: Climate Change 2023: Synthesis Report. Contribution of Working Groups I, II and III to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Geneva, Switzerland, 2023, pp. 35–115 Search PubMed.
- P. Falkowski, R. J. Scholes, E. Boyle, J. Canadell, D. Canfield, J. Elser, N. Gruber, K. Hibbard, P. Högberg, S. Linder, F. T. Mackenzie, B. Moore III, T. Pedersen, Y. Rosenthal, S. Seitzinger, V. Smetacek and W. Steffen, The Global Carbon Cycle: A Test of Our Knowledge of the Earth as a System, Science, 2000, 290, 291–296 CrossRef CAS PubMed.
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Irreversible climate change due to carbon dioxide emissions, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS PubMed.
- R. S. Haszeldine, Carbon Capture and Storage: How Green Can Black Be?, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- D. W. Keith, Why Capture CO2 from the Atmosphere, Science, 2009, 325, 1654–1655 CrossRef CAS PubMed.
- G. Centi and S. Perathoner, Opportunities and prospects in the chemical recycling of carbon dioxide to fuels, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- N. von der Assen, P. Voll, M. Peters and A. Bardow, Life cycle assessment of CO2 capture and utilization: a tutorial review, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- M. Aresta, A. Dibenedetto and A. Angelini, Catalysis for the Valorization of Exhaust Carbon: from CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Using carbon dioxide as a building block in organic synthesis, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- R. E. Siegel, S. Pattanayak and L. A. Berben, Reactive Capture of CO2: Opportunities and Challenges, ACS Catal., 2023, 13, 766–784 CrossRef CAS.
- M. J. Prather, Time Scales in Atmospheric Chemistry: Coupled Perturbations to N2O, NOy, and O3, Science, 1998, 279, 1339–1341 CrossRef CAS PubMed.
- T. S. Ledley, E. T. Sundquist, S. E. Schwartz, D. K. Hall, J. D. Fellows and T. L. Killeen, Climate Change and Greenhouse Gases, EOS., Trans. AGU, 1999, 80, 453–458 CrossRef.
- A. C. Forse and P. J. Milner, New chemistry for enhanced carbon capture: beyond ammonium carbamates, Chem. Sci., 2021, 12, 508–516 RSC.
- R. Ayyappan, I. Abdalghani, R. C. D. Costa and G. R. Owen, Recent developments on the transformation of CO2 utilising ligand cooperation and related strategies, Dalton Trans., 2022, 51, 11582–11611 RSC.
- C. Forte, M. Hayatifar, G. Pampaloni, A. M. R. Galletti, F. Renili and S. Zacchini, Ethylene Polymerization Using Novel Titanium Catalytic Precursors Bearing N,N-Dialkylcarbamato Ligands, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3338–3345 CrossRef CAS.
- M. H. Chisholm and M. W. Extine, Reactions of Transition Metal-Nitrogen σ Bonds. 3. Early Transition Metal N,N-Dimethylcarbamates. Preparation, Properties, and Carbon Dioxide Exchange Reactions, J. Am. Chem. Soc., 1977, 99, 782–792 CrossRef CAS.
- M. H. Chisholm and M. W. Extine, Reactions of Transition Metal-Nitrogen σ Bonds. 4. Mechanistic Studies of the Carbon Dioxide Insertion and Carbon Dioxide Exchange Reactions Involving Early Transition Metal Dimethylamido and N,N-Dimethylcarbamato Compounds, J. Am. Chem. Soc., 1977, 99, 792–802 CrossRef CAS.
- A. Mendiratta, C. C. Cummins, F. A. Cotton, S. A. Ibragimov, C. A. Murillo and D. Villagrán, A Diamagnetic Ditatanium(III) Paddlewheel Complex with No Direct Metal–Metal Bond, Inorg. Chem., 2006, 45, 4328–4330 CrossRef CAS PubMed.
- D. B. Dell’Amico, F. Calderazzo, U. Girulani and G. Pelizzi, N,N-Dialkylcarbamato complexes of titanium(III) and vanadium(III), Gazz. Chim. Ital., 1986, 116, 609–611 Search PubMed.
- D. B. Dell′Amico, F. Calderazzo, U. Giurlani and G. Pelizzi, Synthesis and Reactivity of N,N-Dialkylcarbamato Complexes of Titanium(III) and Vanadium(III). Crystal and Molecular Structure of an Anionic Dimeric Titanium(III) Derivative, Chem. Ber., 1987, 120, 955–964 CrossRef.
- A. E. Guiducci, C. L. Boyd, E. Clot and P. Mountford, Reactions of cyclopentadienyl-amidinate titanium imido compounds with CO2: cycloaddition-extrusion vs. cycloaddition-insertion, Dalton Trans., 2009, 5960–5979 RSC.
- P. J. Tiong, A. Nova, L. R. Groom, A. D. Schwarz, J. D. Selby, A. D. Schofield, E. Clot and P. Mountford, Reactions of Cyclopentadienyl-Amidinate Titanium Hydrazides with CO2, CS2, and Isocyanates: Ti=Nα Cycloaddition-NNR2 Group Transfer Reactions, Organometallics, 2011, 30, 1182–1201 CrossRef CAS.
- M. Anpo and K. Chiba, Photocatalytic reduction of CO2 on anchored titanium oxide catalysts, J. Mol. Catal., 1992, 74, 207–212 CrossRef CAS.
- M. Anpo, H. Yamashita, Y. Ichihashi and S. Ehara, Photocatalytic reduction of CO2 with H2O on various titanium oxide catalysts, J. Electroanal. Chem., 1995, 396, 21–26 CrossRef.
- A. F. R. Kilpatrick and F. G. N. Cloke, Reductive deoxygenation of CO2 by a bimetallic titanium bis(pentalene) complex, Chem. Commun., 2014, 50, 2769–2771 RSC.
- G. Fachinetti, C. Floriani, A. Chiesi-Villa and C. Guastini, Carbon Dioxide Activation. Deoxygenation and Disproportionation of Carbon Dioxide Promoted by Bis(cyclopentadienyl)titanium and -zirconium Derivatives. A Novel Bonding Mode of the Carbonato and a Trimer of the Zirconyl Unit, J. Am. Chem. Soc., 1979, 101, 1767–1775 CrossRef CAS.
- G. A. Vaughan, C. D. Sofield, G. L. Hillhouse and A. L. Rheingold, Oxygen-Atom Transfer from Nitrous Oxide. Identification of Intermediates in the Oxidation of Diphenylacetylene at Group 4 Metal Centers and the Structural Characterization of (η-C5Me5)2Ti{N(O)NCPh=CPH}·1/2 C7H8, J. Am. Chem. Soc., 1989, 111, 5491–5493 CrossRef CAS.
- D. J. Mindiola, L. A. Watson, K. Meyer and G. L. Hillhouse, Functionalization of Complexed N2O in Bis(pentamethylcyclopentadienyl) Systems of Zirconium and Titanium, Organometallics, 2014, 33, 2760–2769 CrossRef CAS PubMed.
- Y. Liang, I. Efremenko, Y. Diskin-Posner, L. Avram and D. Milstein, Calcium-Ligand Cooperation Promoted Activation of N2O, Amine, and H2 as well as Catalytic Hydrogenation of Imines, Quinoline, and Alkenes, Angew. Chem., Int. Ed., 2024, 63, e202401702 CrossRef CAS PubMed.
- F. Bottomley, I. J. B. Lin and M. Mukaida, Reactions of Dinitrogen Oxide (Nitrous Oxide) with Dicyclopentadienyltitanium Complexes Including a Reaction in Which Carbon Monoxide Is Oxidized, J. Am. Chem. Soc., 1980, 102, 5238–5242 CrossRef CAS.
- U. Bayer, D. Werner, C. Maichle-Mössmer and R. Anwander, Effective and Reversible Carbon Dioxide Insertion into Cerium Pyrazolates, Angew. Chem., Int. Ed., 2020, 59, 5830–5836 CrossRef CAS PubMed.
- F. Kracht, P. Rolser, K. Eichele, C. Maichle-Mössmer and R. Anwander, Carbon dioxide affinity (“carboxophilicity”) of trivalent light metal pyrazolates, Inorg. Chem. Front., 2024, 11, 6948–6959 RSC.
- F. Kracht, P. Rolser, P. Preisenberger, C. Maichle-Mössmer and R. Anwander, Organomagnesia: Reversibly High Carbon Dioxide Uptake by Magnesium Pyrazolates, Adv. Sci., 2024, 11, 2403295 CrossRef CAS PubMed.
- N. C. Mösch-Zanetti, R. Krätzner, C. Lehmann, T. R. Schneider and I. Usón, Titanium(III) Compounds with η2-Pyrazolato Ligands, Eur. J. Inorg. Chem., 2000, 13–16 CrossRef.
- I. A. Guzei, A. G. Baboul, G. P. A. Yap, A. L. Rheingold, H. B. Schlegel and C. H. Winter, Surprising Titanium Comnplexes Bearing η2-Pyrazolato Ligands: Synthesis, Structure, and Molecular Orbital Studies, J. Am. Chem. Soc., 1997, 119, 3387–3388 CrossRef CAS.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides, Acta Cryst. A, 1976, 32, 751–767 CrossRef.
- K. P. Kepp, A Quantitative Scale of Oxophilicity and Thiophilicity, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- Y. Le Page, J. D. McCowan, B. K. Hunter and R. D. Heyding, The crystal and molecular structure of bis(biscyclopentadienylchlorotitanium)oxide, J. Organomet. Chem., 1980, 193, 201–207 CrossRef CAS.
- A. F. R. Kilpatrick, J. C. Green and F. G. N. Cloke, The Reductive Activation of CO2 Across a Ti=Ti Double Bond: Synthetic, Structural, and Mechanistic Studies, Organometallics, 2015, 34, 4816–4829 CrossRef CAS PubMed.
- H. L. M. Van Gaal, J. W. Diesveld, F. W. Pijpers and J. G. M. Van der Linden, 13C NMR Spectra of Dithiocarbamates. Chemical Shifts, Carbon–Nitrogen Stretching Vibration Frequencies, and π Bonding in the NCS2 Fragment, Inorg. Chem., 1979, 18, 3251–3260 CrossRef CAS.
- D. M. Wiles, B. A. Gingras and T. Suprunchuk, The C=S stretching vibration in the infrared spectra of some thiosemicarbazones, Can. J. Chem., 1967, 45, 469–473 CrossRef CAS.
- S. Jaiswal, S. K. Pandey, J. Prajapati, S. Chandra, M. K. Gond, M. K. Bharty, I. Tiwari and R. J. Butcher, Cd(II) complexes derived from thiazoline, hydrazide and carbodithioate ligands: synthesis, crystal structures and electrochemical sensing of uric acid, Appl. Organomet. Chem., 2023, 37, e7085 CrossRef CAS.
- K. Nakano, K. Kobayashi and K. Nozaki, Tetravalent Metal Complexes as a New Family of Catalysts for Copolymerization of Epoxides with Carbon Dioxide, J. Am. Chem. Soc., 2011, 133, 10720–10723 CrossRef CAS PubMed.
- C. C. Quadri and E. Le Roux, Copolymerization of cyclohexene oxide with CO2 catalyzed by tridentate N-heterocyclic carbene titanium(IV) complexes, Dalton Trans., 2014, 43, 4242–4246 RSC.
- S. K. Raman, A. C. Deacy, L. Pena Carrodeguas, N. V. Reis, R. W. F. Kerr, A. Phanopoulos, S. Morton, M. G. Davidson and C. K. Williams, Ti(IV)–Tris(phenolate) Catalyst Systems for the Ring-Opening Copolymerization of Cyclohexene Oxide and Carbon Dioxide, Organometallics, 2020, 39, 1619–1627 CrossRef CAS PubMed.
- I. Sancho, M. Navarro, M. Montilla, P. Salvador, C. Santamaría, J. M. Luis and A. Hernán-Gómez, Ti(III) Catalysts for CO2/Epoxide Copolymerization at Unusual Ambient Pressure Conditions, Inorg. Chem., 2023, 62, 14873–14887 CrossRef CAS PubMed.
- L. Suresh, R. Lalrempuia, T. Fjermestad, K. W. Törnroos, J. Bour, G. Frache, A. Nova and E. Le Roux, Trapping of Key “Ate” Intermediates of NHC-Group IV Relevant to Catalyzing Copolymerization of Cyclohexene Oxide with CO2, Organometallics, 2025, 44, 68–81 CrossRef CAS.
- C. Yao, T. Dahmen, A. Gansäuer and J. Norton, Anti-Markovnikov alcohols via epoxide hydrogenation through cooperative catalysis, Science, 2019, 364, 764–767 CrossRef CAS PubMed.
- D. Bai, H. Jing, Q. Liu, Q. Zhu and X. Zhao, Titanocene dichloride-Lewis base: An efficient catalytic system for coupling of epoxides and carbon dioxide, Catal. Commun., 2009, 11, 155–157 CrossRef CAS.
- D. Bai, G. Nian, G. Wang and Z. Wang, Titanocene dichloride/KI: an efficient catalytic system for synthesis of cyclic carbonates from epoxides and CO2, Appl. Organomet. Chem., 2013, 27, 184–187 CrossRef CAS.
- M. J. Go, K. M. Lee, C. H. Oh, Y. Y. Kang, S. H. Kim, H. R. Park, Y. Kim and J. Lee, New Titanium Catalysts Containing Tetrazole for Cycloaddition of CO2 to Epoxides, Organometallics, 2013, 32, 4452–4455 CrossRef CAS.
- Y. Wang, Y. Qin, X. Wang and F. Wang, Coupling reaction between CO2 and cyclohexene oxide: selective control from cyclic carbonate to polycarbonate by ligand design of salen/salalen titanium complexes, Catal. Sci. Technol., 2014, 4, 3964–3972 RSC.
- F. Al-Qaisi, E. Streng, A. Tsarev, M. Nieger and T. Repo, Titanium Alkoxide Complexes as Catalysts for the Synthesis of Cyclic Carbonates from Carbon Dioxide and Epoxides, Eur. J. Inorg. Chem., 2015, 5363–5367 CrossRef CAS.
- C. Liu, Y. Liu, G. Zhang, D. Wang, G. Chen, F. Gao, L. Liu, C.-H. Tung and Y. Wang, Seed-Mediated Growth of a High-nuclearity Titanium-oxide Cluster with Enhanced Photocatalytic Activities in CO2/Epoxide Cycloaddition, Eur. J. Inorg. Chem., 2024, 27, e202400189 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures, detailed crystallographic data, spectroscopic data (NMR, IR), analytical details. CCDC 2423931–2423936. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00957j |
| This journal is © The Royal Society of Chemistry 2025 |