
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modelling the binding of cytotoxic dinuclear nickel complexes to two neighboring phosphate esters of DNA using dicarboxylate ligands†
Thomas
Riediger
,
Maximilian
Böhm
,
Michael
Kapiza
,
Anja
Stammler
,
Jan
Oldengott
and
Thorsten
Glaser
 *
*
Lehrstuhl für Anorganische Chemie I, Fakultät für Chemie, Universität Bielefeld, Universitätsstr. 25, D-33615 Bielefeld, Germany. E-mail: thorsten.glaser@uni-bielefeld.de
First published on 3rd June 2025
Abstract
The cytotoxic complex [(Htom6-Me){NiII(OAc)}2](OAc) (H2tom6-Me = 2,7-bis(di(6-methylpyridine-2-yl-methyl)aminomethyl)-1,8-naphthalenediol) is supposed to bind in the aquated form [(Htom6-Me){NiII(OH2)2}2]3+ to two neighboring phosphate diesters of the DNA backbone. To further support this intended molecular mode of action, difunctional ligands in the form of the dicarboxylates succinate and glutarate are used here to mimic two neighboring phosphates of the DNA backbone. The complex [(Htom6-Me){NiII(OAc)}2](OAc) is treated with 3 equiv. HCl to protonate the acetates providing presumably [(Htom6-Me){NiII(OH2)2}2]3+, which is reacted with the dicarboxylates yielding the complexes [(Htom6-Me){NiII(μ-succ)NiII}]+ and [(Htom6-Me){NiII(μ-glut)NiII}]+ confirmed by single-crystal X-ray diffraction. The sterical constraints of the dicarboxylates enforces shorter Ni⋯Ni distances demonstrating the flexibility of the coordination compartments despite the rigid 1,8-naphthalenediol backbone. These steric constraints by the pull effect of the organic spacers affect the NiII–ligand bonds and are reflected in FTIR and UV-Vis-NIR spectroscopic but not magnetic signatures. The comparison to a related CuII complex indicates a severe impact of the 6-methyl groups of the pyridine donors on the relative orientation of the anticipated phosphate binding sites in these complexes. The consequences for a rational strengthening of the binding to DNA and hence increase of the cytotoxicity by possible ligand modifications are discussed.
Introduction
Cisplatin1 and related complexes are a potent class of anticancer drugs.2 It is supposed that a platinum-based complex fragment binds to the nucleobases of DNA that interferes with cellular processes leading finally to apoptosis. Although many new platinum-based complexes were synthesized and tested, several types of cancer cannot be treated with cisplatin-based drugs and there is a development of resistance of cancer cells towards cisplatin-based drugs.3 The serious toxicity of platinum-based anticancer drugs stimulated research for more biorelevant metal ions especially of the first row.4In order to allow a new and different molecular mode of action, we intended to coordinate not to the nucleobases but to the phosphate diesters of the DNA backbone. To increase the binding affinity to the oxygen atoms of the phosphate diesters, our ligand design was based on the multivalence-principle.5 The binding should occur to two neighboring phosphate diesters of the DNA backbone by molecular recognition with two phosphate binding sites held by a rigid backbone at the distance of two neighboring phosphate esters in the DNA backbone of 6–7 Å. Sterical demand around the phosphate binding sites should prevent the binding to the less exposed nucleobases. These design guidelines led to the new family of 2,7-disubstituted 1,8-naphthalenediol ligands with bulky pendant arms in the 2,7-position (Scheme 1).6 Indeed, the complexes [(Htom6-Me){CuII(OAc)}2](OAc) and [(Htom6-Me){NiII(OAc)}2](OAc) bind to DNA, interfere with DNA synthesis in PCR at lower concentrations than cisplatin, and kill human cancer cells more efficiently than human stem cells of the same proliferation rate.7 We have shown a preferential binding of phosphate diester models that exchanges the coordinated acetates.8,9 Infrared multiple photon dissociation (IRMPD) spectroscopy10 showed the binding to the phosphates in nucleobases and high resolution ultra-high vacuum atomic force microscopy (HR-UHV-AFM) provided strong evidence for the binding of the complexes to the phosphate diesters of the DNA backbone.11
 | ||
| Scheme 1 Intended molecular recognition of two neighboring phosphate diesters of the DNA backbone by dinuclear complexes with a 2,7-disubstituted 1,8-naphthalenediol ligand. | ||
Despite this evidence, a direct proof of the intended binding mode of the dinuclear complexes to two neighboring phosphate diesters of the DNA backbone is still missing. To further support this binding mode, we thought to use difunctional ligands, i.e. ligands that exhibit two binding sites to better model two neighboring phosphate diesters of the DNA backbone. In a first attempt, we used glutaric acid (pentanedioic acid, H2glut). However, the reaction of [(Htom6-Me){CuII(OAc)}2](OAc) with glutaric acid did not provide the intended dinuclear complex [(Htom6-Me){CuII(μ-glut)CuII}]+ with an intramolecular glutarate bridge but the tetranuclear macrocycle [{(Htom6-Me)CuII2}2(μ-glut)2]2+, where the two carboxylate donors of one glutarate do not bind to the CuII ions of the same dinuclear complex fragment [(Htom6-Me)CuII2]3+ but to two of these fragments.12 Here, we present the successful reaction of the difunctional ligands glutaric acid and succinic acid (butanedioic acid, H2succ) with [(Htom6-Me){NiII(OAc)}2](OAc) providing the dinuclear complexes [(Htom6-Me){NiII(μ-glut)NiII}]+ and [(Htom6-Me){NiII(μ-succ)NiII}]+, respectively, (Scheme 2) where the two intramolecularly bridging dicarboxylates mimic the molecular recognition of the complex fragment [(Htom6-Me)NiII2]3+ with two neighboring phosphate sites of DNA.
 | ||
| Scheme 2 Synthesis of complexes. | ||
Experimental section
Solvents and starting materials were of the highest commercially available purity and used as received. The complex [(Htom6-Me){NiII(OAc)}2](OAc) was synthesized according to the procedure reported previously.7[(Htom6-Me){NiII(μ-succ)NiII}](BPh4)·2CH3CN
A solution of hydrochloric acid (557 μmol, 3.0 equiv.) in MeOH (6 mL) and H2O (2 mL) was added to a dark green solution of [(Htom6-Me){NiII(OAc)}2](OAc)·9H2O (206 mg, 188 μmol, 1 equiv.) in MeOH (10 mL) at 40 °C, which resulted in a light green solution. After stirring for 30 minutes at 40 °C, the solution was added to a solution of NaBPh4 (391 mg, 1.14 mmol, 6.1 equiv.) in MeOH (4.5 mL) and H2O (0.5 mL) at room temperature. The resulting precipitate was isolated and complete precipitation was enforced by adding H2O to the filtrate. The combined solids were washed with H2O and redissolved in CH3CN (13.3 mL) and H2O (0.7 mL). A solution of NEt3 (54.0 μL, 390 μmol, 2.1 equiv.) and succinic acid (23 mg, 195 μmol, 1.0 equiv.) in CH3CN (13.3 mL) and H2O (0.7 mL) was added. The solution was stirred at ambient temperatures for 15 minutes and filtered. Diffusion of Et2O into the filtrate provided dark green crystals, which were suitable for single-crystal X-ray diffraction. The crystals were filtered off and washed three times with Et2O. Yield: 228 mg (179 μmol, 95%). ESI-MS (pos. mode, CH3CN): m/z = 869.1 [(Htom6-Me){NiII(μ-succ)NiII}]+. IR (KBr): v∼/cm−1 = 3055 m, 3033 m, 2999 w, 2984 w, 2923 w, 2846 w, 2247 w, 1951 w, 1885 w, 1827 w, 1770 w, 1608 s, 1579 s, 1541 s, 1468 s, 1450 s, 1400 m, 1370 m, 1335 m, 1308 w, 1299 w, 1273 w, 1244 w, 1166 m, 1138 w, 1121 w, 1099 w, 1085 w, 1056 m, 1032 w, 1016 w, 1000 w, 970 w, 892 m, 828 m, 791 m, 750 m, 734 s, 707 s, 612 m, 512 w, 452 w. UV-Vis-NIR (2,2,2-trifluoroethanol): v∼/cm−1 (ε/M−1 cm−1) = 42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 (47400), 37700 (18700), 36600 (14400), 30900 (5100), 29300 (8000), 28200 (10500), 22300 (42), 21200 (37), 20900 (38), 20000 (27), 19500 (28), 16800 (24), 12600 (11), 10300 (27). Anal. calcd for [(Htom6-Me){NiII(μ-succ)NiII}](BPh4)·2CH3CN (C72H71BN8Ni2O6): C 67.95, H 5.62, N 8.81%. Found: C 68.06, H 5.81, N 8.60%.
800 (47400), 37700 (18700), 36600 (14400), 30900 (5100), 29300 (8000), 28200 (10500), 22300 (42), 21200 (37), 20900 (38), 20000 (27), 19500 (28), 16800 (24), 12600 (11), 10300 (27). Anal. calcd for [(Htom6-Me){NiII(μ-succ)NiII}](BPh4)·2CH3CN (C72H71BN8Ni2O6): C 67.95, H 5.62, N 8.81%. Found: C 68.06, H 5.81, N 8.60%.
[(Htom6-Me){NiII(μ-glut)NiII}](BPh4)·2CH3CN
A solution of hydrochloric acid (296 μmol, 3.0 equiv.) in H2O (6 mL) was added to a dark green solution of [(Htom6-Me){NiII(OAc)}2](OAc)·5H2O·0.1ACN (102 mg, 99.4 μmol, 1 equiv.) in H2O (10 mL) at 40 °C. The resulting light green solution was stirred for 10 minutes at 40 °C, cooled to ambient temperature, and added to a solution of NaBPh4 (204 mg, 596 mmol, 6.0 equiv.) in H2O (5 mL). The precipitate was filtered off, washed with H2O, redissolved in CH3CN (15 mL), and treated with a solution of NEt3 (29.0 μL, 209 μmol, 2.1 equiv.) and glutaric acid (13.8 mg, 105 μmol, 1.1 equiv.) in CH3CN (5 mL). After addition of H2O (1 mL) the mixture was filtered. Slow diffusion of methyl tert-butyl ether into the filtrate provided dark green crystals, which were suitable for single-crystal X-ray diffraction. The crystals were filtered off and washed three times with Et2O. Yield: 68 mg (53 μmol, 53%). ESI-MS (pos. mode, CH3CN): m/z = 883.1 [(Htom6-Me){NiII(μ-glut)NiII}]+, 442.1 {[(Htom6-Me){NiII(μ-glut)NiII}]+ + H+}2+. IR (KBr): v∼/cm−1 = 3158 w, 3114 w, 3054 m, 3030 m, 2998 w, 2983 w, 2928 w, 2907 w, 2845 w, 2248 w, 2004 w, 1951 w, 1885 w, 1826 w, 1769 w, 1608 s, 1579 s, 1530 s, 1468 s, 1449 w, 1437 w, 1399 s, 1370 m, 1335 m, 1304 m, 1286 w, 1274 w, 1244 w, 1225 w, 1203 w, 1185 w, 1165 m, 1139 m, 1121 w, 1098 w, 1085 w, 1056 m, 1032 w, 1017 m, 1000 w, 968 m, 937 w, 922 w, 885 w, 860 w, 832 m, 791 m, 749 w, 734 s, 708 s, 669 w, 660 w, 644 w, 624 w, 611 m, 561 w, 512 m, 489 w, 453 w, 438 w. UV-Vis-NIR (2,2,2-trifluoroethanol): v∼/cm−1 (ε/M−1 cm−1) = 42900 (46000), 38400 (17700), 37700 (17600), 36500 (13200), 30900 (4990), 29300 (7880), 28200 (10270), 22300 (30), 21300 (27), 20900 (28), 20000 (19), 19600 (21), 16400 (21), 12590 (11), 10180 (30). Anal. calcd for [(Htom6-Me){NiII(μ-glut)NiII}](BPh4)·2CH3CN (C73H73N8O6BNi2): C 68.15, H 5.72, N 8.71%. Found: C 67.84, H 5.76, N 8.57%.
Crystal structure determination
Single-crystals were removed from the mother liquor, coated with oil, and measured at 100(2) K on a Bruker KAPPA APEX II four-circle diffractometer equipped with 4K CCD detector. MoKα radiation with a focusing graphite monochromator was used to measure the crystals. Absorption was corrected numerically for [(Htom6-Me){NiII(μ-glut)NiII}](BPh4)·2CH3CN and using equivalent reflections for [(Htom6-Me){NiII(μ-succ)NiII}](BPh4)·2CH3CN. Corrections were performed with the program SADABS-2016/2.13 The structures were solved and refined vs. F2 with the programs SHELXT/L14 using OLEX2.15The hydrogen atom between O1 and O3 was found and refined, all other hydrogen positions were generated for all structures. [(Htom6-Me){NiII(μ-succ)NiII}](BPh4)·2CH3CN shows pseudo-symmetry along a C2 axis through C5 and C6, while [(Htom6-Me){NiII(μ-glut)NiII}](BPh4)·2CH3CN shows pseudo C-centration.
CCDC numbers 2445426–2445427† contain the supplementary crystallographic data for this paper.
Other physical measurements
Infrared spectra (400–4000 cm−1) of solid samples were recorded on a Bruker Vertex 70 as KBr disks. ESI mass spectra were recorded on a Bruker Esquire 3000 ion trap mass spectrometer equipped with a standard ESI source. Elemental analyses were carried out on a HEKAtech Euro EA analyzer. UV-Vis-NIR absorption spectra were measured on a JASCO V770 spectrophotometer at 20 °C. Magnetic susceptibility data were measured on powdered samples in the temperature range 2–300 K by using a SQUID magnetometer (Quantum Design MPMS XL-7 EC) with a field of 1.0 T. Variable-temperature variable-field (VTVH) measurements were performed in various static fields (1–7 T) in the range 2–10 K with the magnetization equidistantly sampled on a 1/T temperature scale. For calculations of the molar magnetic susceptibilities, χm, the measured susceptibilities were corrected for the underlying diamagnetism of the sample holder and the sample by using tabulated Pascal's constants. The susceptibility data were analyzed using the program package JulX written by Eckhard Bill for spin-Hamiltonian simulations and fittings of the data by a full-matrix diagonalization approach. Magnetic moments were obtained from numerically generated derivatives of the eigenvalues of eqn (1), and summed up over 16 field orientations along a 16-point Lebedev grid to account for the powder distribution of the sample.Results and discussion
Synthesis
We have recently shown that the coordinated acetates in the cytotoxic complex [(Htom6-Me){CuII(OAc)}2](OAc) can be de-coordinated by protonation delivering HOAc resulting in the complex [(Htom6-Me){CuII(OH2)}2]3+.16 Moreover, we have shown that the reaction of the dicopper complex [(Htom6-Me){CuII(OH2)}2]3+ in CH3CN with a mixture of glutaric acid and NEt3 provided the tetranuclear macrocycle [{(Htom6-Me)CuII2}2(μ-glut)2]2+.12 In order to evaluate the influence of the metal ion on the coordination mode, we thought to use the also cytotoxic dinickel complex [(Htom6-Me){NiII(OAc)}2](OAc) to obtain upon protonation the corresponding aqua complex [(Htom6-Me){NiII(OH2)2}2]3+ for the reaction with dicarboxylic acids.The reaction of [(Htom6-Me){NiII(OAc)}2](OAc) with 3 equiv. of HCl in a mixture of MeOH/H2O provides after addition of NaBPh4 a precipitate. Based on our previous results, this procedure should deliberate the coordinated acetates and protonate the free acetates to acetic acid providing presumably [(Htom6-Me){NiII(OH2)2}2](BPh4)3. This precipitate was redissolved in CH3CN with a small amount of H2O. Reaction with 2.1 equiv. NEt3 and either 1 equiv. succinic acid or glutaric acid provides the complexes [(Htom6-Me){NiII(μ-succ)NiII}](BPh4) and [(Htom6-Me){NiII(μ-glut)NiII}](BPh4), respectively (Scheme 2).
The FTIR spectra of [(Htom6-Me){NiII(μ-succ)NiII}](BPh4) and [(Htom6-Me){NiII(μ-glut)NiII}](BPh4) and for comparison of [(Htom6-Me){Ni(OAc)}2](BPh4)7 (Fig. 1 and S1†) are almost superimposable and show the characteristic bands of the coordinated pyridines at 1608 and 1579 cm−1. Slight but significant differences were observed for the stretching modes νas(CO2−) and νs(CO2−) in the ranges 1530–1540 and 1450–1470 cm−1, respectively (Fig. 1). The differences Δ(νas–νs) of 78 cm−1 for OAc−, 62 cm−1 for glut2−, and 91 cm−1 for succ2− indicate a bidentate binding mode of the carboxylates but with slight structural variations.17 [(Htom6-Me){NiII(μ-succ)NiII}](BPh4) shows one extra band at 892 cm−1 that might be characteristic for the succinate ligand.
 | ||
| Fig. 1 FTIR spectra of [(Htom6-Me){NiII(μ-succ)NiII}](BPh4), [(Htom6-Me){NiII(μ-glut)NiII}](BPh4), and [(Htom6-Me){NiII(OAc)}2](BPh4)7 for comparison. | ||
Structural characterization
The complexes [(Htom6-Me){NiII(μ-succ)NiII}](BPh4)·2CH3CN and [(Htom6-Me){NiII(μ-glut)NiII}](BPh4)·2CH3CN crystallize with one complex in the asymmetric unit, i.e. without crystallographically imposed symmetry although the two complexes are close to C2 symmetry (Fig. 2). The overall molecular structures are very similar with the N3 binding pockets coordinating facially to the NiII ions. The octahedral coordination environments are completed by bidentate carboxylates and the naphthalenediolato donors. | ||
| Fig. 2 Molecular structures of (a) [(Htom6-Me){NiII(μ-succ)NiII}]+ in single-crystals of [(Htom6-Me){NiII(μ-succ)NiII}](BPh4)·2CH3CN and (b) [(Htom6-Me){NiII(μ-glut)NiII}]+ in single-crystals of [(Htom6-Me){NiII(μ-glut)NiII}](BPh4)·2CH3CN. Hydrogen atoms except those bound at oxygen atoms have been omitted for clarity. | ||
The coordination around the NiII ions differs slightly for the two complexes. To evaluate the influence of the organic spacer, these variations are analyzed using the molecular structure of [(Htom6-Me){NiII(OAc)}2](BPh4)7 with monodentate acetates as reference. Fig. 3 shows on the left side truncated structures including selected bond lengths and on the right side an orientation viewing from the top of the acetates with the naphthalene unit almost perpendicular to the projection plane. Differences in bond lengths are emphasized only if they are outside the ± 3σ range (Table 1).
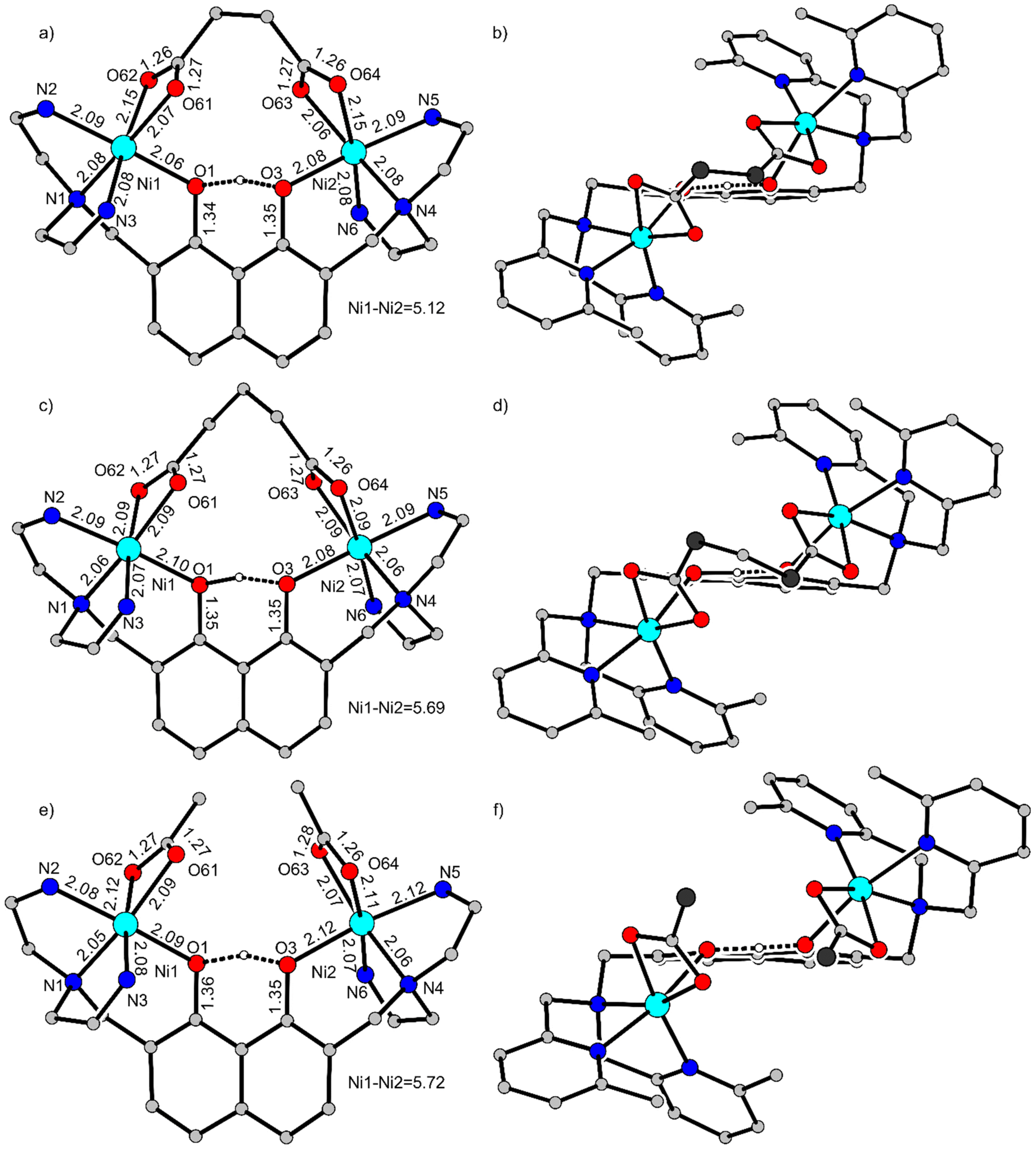 | ||
| Fig. 3 Comparison of the molecular structures of (a) + (b) [(Htom6-Me){NiII(μ-succ)NiII}]+ in single-crystals of [(Htom6-Me){NiII(μ-succ)NiII}](BPh4)·2CH3CN, (c) + (d) [(Htom6-Me){NiII(μ-glut)NiII}]+ in single-crystals of [(Htom6-Me){NiII(μ-glut)NiII}](BPh4)·2CH3CN, and (e) + (f) [(Htom6-Me){NiII(OAc)}2]+ in single-crystals of [(Htom6-Me){Ni(OAc)}2](BPh4)·CH2Cl2.7 Left side: Molecular structures with selected bond lengths (Å) and with the pyridine donors truncated for clarity. Right side: View from the top of the carboxylate ligands with the naphthalene units oriented almost perpendicular to the projection plane. The methyl groups of the acetates in (f) and the corresponding carbon atoms in (b) and (d) are highlighted with larger spheres and dark grey filling. | ||
| [(Htom6-Me){NiII(μ-succ)NiII}]+ | [(Htom6-Me){NiII(μ-glut)NiII}]+ | |||
|---|---|---|---|---|
| Ni1 | Ni2a | Ni1 | Ni2a | |
| a The numbering scheme of the Ni2 side or molecule has been adapted according to the Ni1 side. | ||||
| Ni1–O1 | 2.0585(9) | 2.0787(9) | 2.1027(9) | 2.0780(9) |
| Ni1–N1 | 2.0779(11) | 2.0784(11) | 2.0608(11) | 2.0623(11) |
| Ni1–N2 | 2.0873(11) | 2.0868(11) | 2.0924(11) | 2.0882(11) |
| Ni1–N3 | 2.0819(11) | 2.0775(11) | 2.0681(10) | 2.0726(10) |
| Ni1–O61 | 2.0648(9) | 2.0636(9) | 2.0898(9) | 2.0870(9) |
| Ni1–O62 | 2.1465(10) | 2.1499(10) | 2.0932(9) | 2.0940(9) |
| Ni1⋯Ni2 | 5.5239(4) | 5.6904(4) | ||
| O1–Ni1–N1 | 89.90(4) | 89.25(4) | 90.72(4) | 91.61(4) |
| O1–Ni1–N2 | 165.63(4) | 164.85(4) | 167.50(4) | 167.81(4) |
| O1–Ni1–N3 | 92.33(4) | 92.41(4) | 89.86(4) | 90.59(4) |
| O1–Ni1–O61 | 85.96(4) | 86.02(4) | 86.05(4) | 87.55(4) |
| O1–Ni1–O62 | 85.77(4) | 85.36(4) | 85.74(4) | 85.90(4) |
| N1–Ni1–N2 | 79.91(4) | 79.56(4) | 79.97(4) | 80.02(4) |
| N1–Ni1–N3 | 82.49(4) | 83.50(4) | 83.84(4) | 82.67(4) |
| N1–Ni1–O61 | 166.50(4) | 166.63(4) | 162.68(4) | 162.87(4) |
| N1–Ni1–O62 | 104.16(4) | 104.44(4) | 99.68(4) | 99.67(4) |
| N2–Ni1–N3 | 96.31(4) | 96.39(4) | 97.34(4) | 97.06(4) |
| N2–Ni1–O61 | 101.63(4) | 102.59(4) | 100.36(4) | 97.82(4) |
| N2–Ni1–O62 | 86.93(4) | 87.56(4) | 87.62(4) | 86.78(4) |
| N3–Ni1–O61 | 110.49(4) | 109.16(4) | 113.12(4) | 114.45(4) |
| N3–Ni1–O62 | 173.06(4) | 171.70(4) | 174.39(4) | 175.82(4) |
| O61–Ni1–O62 | 62.75(4) | 62.75(4) | 63.14(4) | 63.20(4) |
The coordination of the carboxylates is the same for all three complexes (central coordination site A as defined in ref. 9). The Ni1–Ni2 distance is 5.72 Å in [(Htom6-Me){NiII(OAc)}2]+ (Fig. 3e), that decreases only slightly in [(Htom6-Me){NiII(μ-glut)NiII}]+ to 5.69 Å (Fig. 3c) but strongly to 5.12 Å in [(Htom6-Me){NiII(μ-succ)NiII}]+ (Fig. 3a) indicating that the shorter ethylene spacer of succinate exerts a strong pull effect on the two coordinated carboxylates. This pull effect is also nicely reflected by the distance between the carbon atoms of the methyl groups of the acetates of 3.78 Å in [(Htom6-Me){NiII(OAc)}2]+ (Fig. 3f, carbon atoms highlighted in dark grey). This distance decreases for the corresponding carbon atoms by insertion of a bridging methylene unit in [(Htom6-Me){NiII(μ-glut)NiII}]+ to 2.63 Å (Fig. 3d) and more extremely by connecting those two methyl groups to one ethylene bridge in [(Htom6-Me){NiII(μ-succ)NiII}]+ to 1.53 Å. This increased pull effect of the organic spacers also affects slightly the mean Ni–Onaph bond length that decreases from 2.11 Å in [(Htom6-Me){NiII(OAc)}2]+ to 2.09 Å in [(Htom6-Me){NiII(μ-glut)NiII}]+ and to 2.07 Å in [(Htom6-Me){NiII(μ-succ)NiII}]+. The mean Ni–Ocarb bond lengths are slightly longer for the carboxylate oxygen atoms coordinated trans to a pyridine. This asymmetry in the mean Ni–Ocarb distances (0.03 Å in [(Htom6-Me){NiII(OAc)}2]+) almost vanishes in [(Htom6-Me){NiII(μ-glut)NiII}]+ but increases significantly to 0.08 Å in [(Htom6-Me){NiII(μ-succ)NiII}]+. It is interesting to note, that these asymmetries correlate with the difference of the vibrational modes Δ(νas–νs) of the carboxylates, which is the largest for succinate and the smallest for glutarate. Thus, the carboxylate stretching modes reflect the pull effect of the organic spacers.
Magnetic properties
The magnetic data of both complexes are shown in Fig. 4. The effective magnetic moment, μeff, is almost temperature independent but decreases slightly below 30 K. This behavior can be attributed to a weak antiferromagnetic interaction or to zero-field-splitting or to a combination of both but cannot be differentiated from the simulation of μeffvs. T alone. However, the variable field–variable temperature (VTVH) magnetization data show a nesting behavior of the isofield lines indicating the presence of significant zero-field splitting.18 The simultaneous simulation of the μeffvs. T and the VTVH data allows to analyze these contributions. Therefore, we have simulated and fitted the magnetic data using the spin-Hamiltonian (eqn (1)).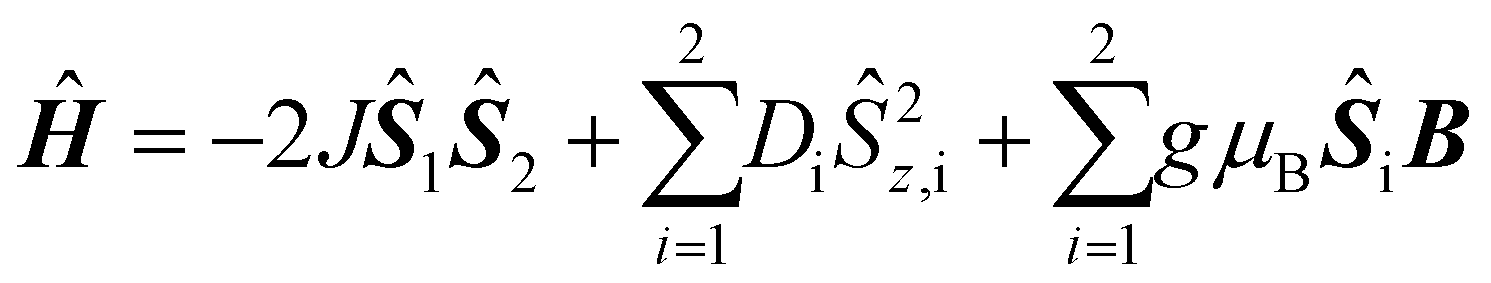 | (1) |
 | ||
| Fig. 4 Temperature-dependence of the effective magnetic moment, μeff, of (a) [(Htom6-Me){NiII(μ-succ)NiII}](BPh4)·2CH3CN and (b) [(Htom6-Me){NiII(μ-glut)NiII}](BPh4)·2CH3CN: and variable-field variable-temperature (VTVH) magnetization data in the insets. The solid lines correspond to spin-Hamiltonian fits with the following parameter sets: (a) J12 = −0.075 cm−1, gi = 2.26, Di = 3.50 cm−1, χTIP = 50 × 10−6 cm3 mol−1, (b) J12 = −0.20 cm−1, gi = 2.235, Di = 4.00 cm−1, χTip = 150 × 10−6 cm3 mol−1. Contributions from χTIP were subtracted from experimental and simulated data. | ||
The first term is the isotropic HDvV exchange Hamiltonian, the second term the local axial zero-field splitting, and the last term the local Zeeman Hamiltonian. The simultaneous simulation and fitting provided the parameter sets given in Fig. 4. Please note that the signs of Di correspond only to the fits provided but are not determined by these powder measurements. Both complexes exhibit very small antiferromagnetic exchange and a moderate zero-field splitting parameter. There is no correlation between the exchange coupling constant J and the slight variation of the Ni–Onaph bond lengths, which should mainly influence the exchange pathway.
UV-Vis-NIR spectroscopy
The UV-Vis-NIR spectra of [(Htom6-Me){NiII(μ-succ)NiII}](BPh4) and [(Htom6-Me){NiII(μ-glut)NiII}](BPh4) are shown with [(Htom6-Me){NiII(OAc)}2](OAc) as reference in Fig. 5. The spectra exhibit above 25000 cm−1 the typical π → π* transitions and below 25000 cm−1 the typical d–d transitions of NiII. Although the differences are not tremendous, the 3A2g(F) → 3T2g(F) and 3A2g(F) → 3T1g(F)19 are slightly shifted to higher energies for [(Htom6-Me){NiII(μ-succ)NiII}]+ (10280 and 16770 cm−1) than in [(Htom6-Me){NiII(μ-glut)NiII}]+ (10180 and 16400 cm−1), and [(Htom6-Me){NiII(OAc)}2]+ (10060 and 16100 cm−1). This higher ligand field in [(Htom6-Me){NiII(μ-succ)NiII}]+ correlates with the shorter Ni–Onaph bond lengths and the stronger Ni–Ocarb asymmetry (one shorter Ni–Ocarb bond) induced by the shorter ethylene spacer between the two coordinated carboxylates. Extraction of the Racah parameter B from these transitions20 provides 850 cm−1 for both [(Htom6-Me){NiII(μ-glut)NiII}]+ and [(Htom6-Me){NiII(OAc)}2]+ but 940 cm−1 for [(Htom6-Me){NiII(μ-succ)NiII}]+. The significantly higher value for [(Htom6-Me){NiII(μ-succ)NiII}]+ indicates an overall lower covalence of the NiII–ligand bonds, i.e. the higher covalence of the shorter Ni–Onaph is overcompensated by the other bonds leading an overall lower covalence of the NiII ion.
 | ||
| Fig. 5 Electronic absorption spectra of [(Htom6-Me){NiII(μ-succ)NiII}](BPh4) and [(Htom6-Me){NiII(μ-glut)NiII}](BPh4) in 2,2,2-trifluoroethanol and of [(Htom6-Me){NiII(OAc)}2](OAc)7 in CH3CN for comparison. 2,2,2-Trifluorethanol was used as solvent as (i) those two complexes were not soluble enough in CH3CN for concentrated solutions required for measuring the d–d transitions and (ii) we have had a good experience previously16 with 2,2,2-trifluoroethanol as a slightly polar solvent without strong coordinating properties. | ||
Conclusion
It is assumed that the cytotoxicity of the dinuclear complex [(Htom6-Me){NiII(OAc)}2](OAc) is based on the substitution of the acetates by H2O ligands in aqueous media forming the complex [(Htom6-Me){NiII(OH2)2}2]3+ and that after electrostatic attraction by the polyanionic DNA the complex fragment [(Htom6-Me)NiII2]3+ binds to two neighboring phosphate diesters of the DNA backbone. This molecular mode of action has been mimicked by protonation of the acetates forming presumably [(Htom6-Me){NiII(OH2)2}2]3+ that reacts with the difunctional donors succinate and glutarate providing the anticipated complexes [(Htom6-Me){NiII(μ-succ)NiII}]+ and [(Htom6-Me){NiII(μ-glut)NiII}]+, respectively. The comparison of the molecular structures of these two complexes with that of [(Htom6-Me){NiII(OAc)}2]+ as reference demonstrates a pull effect of the organic spacer that is stronger the shorter the spacer is. FTIR and UV-Vis-NIR spectroscopies reflect the induced changes on NiII coordination environment indicating the potential to serve as spectroscopic probes. The coordination compartments show enough flexibility to compensate for this pull effect demonstrating the flexibility despite the rigid 1,8-naphthalenediol backbone.It seems to be worthwile to speculate on the origin of the different complexes obtained from [(Htom6-Me)MII2]3+ with glutarate for NiII (dinuclear complex with intramolecularly bridging glutarate) and CuII (tetranuclear complex with intermolecularly bridging glutarates).12 NiII ions with this donor set prefer six-coordination enforcing the carboxylates to bind in a bidentate fashion, while the Jahn–Teller active CuII ions prefer five-coordination with monodentate carboxylates. The 6-methyl groups of the pyridine donors exhibit sterical strain on ligands coordinated cis to the pyridine with the 6-methyl group pointing to these ligands.21 As a consequence, the N3 binding pockets prefer a facial binding to the NiII ions with bidentate carboxylates but a meridional binding to the CuII ions with monodentate carboxylates (Fig. 6).7 This results in an orientation of the acetates in the NiII complexes turned towards each other (distance between the two carboxylate carbon atoms of 4.16 Å), while those in the CuII complexes are turned away from each other (distance between the two carboxylate carbon atoms of 9.46 Å). The latter seems not only to be unfavorable for the intramolecular bridging mode of a dicarboxylate model but also for the intended coordination to two neighboring phosphate diesters of the DNA backbone. Thus, this study shows a severe impact of the 6-methyl groups of the pyridines on the coordination mode of the metal ions that could enforce sterical constraints for the binding to two neighboring phosphates of the DNA backbone and hence weaken the binding affinity. To prevent these sterical constraint from the beginning, the dinuclear CuII2 and NiII2 complexes without 6-methyl groups (i.e. [(Htom)MII2]3+) could enhance the binding affinity to the DNA backbone enhancing the cytotoxicity. Corresponding studies have been started in our lab.
 | ||
| Fig. 6 Molecular structures of (a) [(Htom6-Me){NiII(OAc)}2]+ and (b) [(Htom6-Me){CuII(OAc)}2]+ to demonstrate the influence of the 6-methyl groups of the pyridines on the coordination mode of the N3 ligand compartments and hence on the relative orientation of the acetates to each other. | ||
Data availability
The supplementary crystallographic data for this study have been deposited at the Cambridge Crystallographic Data Centre under accession codes 2445426–2445427.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Financial support from Bielefeld University is gratefully acknowledged.References
- B. Rosenberg and L. van Camp, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- (a) S. Dilruba and G. V. Kalayda, Cancer Chemother. Pharmacol., 2016, 77, 1103–1124 CrossRef CAS PubMed; (b) S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS PubMed; (c) S. Rottenberg, C. Disler and P. Perego, Nat. Rev. Cancer, 2021, 21, 37–50 CrossRef CAS PubMed; (d) C. Zhang, C. Xu, X. Gao and Q. Yao, Theranostics, 2022, 12, 2115–2132 CrossRef CAS PubMed; (e) T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 Search PubMed; (f) Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 Search PubMed.
- (a) R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 Search PubMed; (b) L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- (a) S. Sen, M. Won, M. S. Levine, Y. Noh, A. C. Sedgwick, J. S. Kim, J. L. Sessler and J. F. Arambula, Chem. Soc. Rev., 2022, 51, 1212–1233 RSC; (b) D. Denoyer, S. Masaldan, S. La Fontaine and M. A. Cater, Metallomics, 2015, 7, 1459–1476 CrossRef CAS PubMed; (c) C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755–2794 CrossRef CAS.
- T. Jany, A. Moreth, C. Gruschka, A. Sischka, A. Spiering, M. Dieding, Y. Wang, S. H. Samo, A. Stammler, H. Bögge, G. Fischer von Mollard, D. Anselmetti and T. Glaser, Inorg. Chem., 2015, 54, 2679–2690 CrossRef CAS PubMed.
- S. Schwarzbich, C. Horstmann née Gruschka, J. Simon, L. Siebe, A. Moreth, C. Wiegand, A. Lavrentieva, T. Scheper, A. Stammler, H. Bögge, G. Fischer von Mollard and T. Glaser, Inorg. Chem., 2020, 59, 14464–14477 CrossRef CAS PubMed.
- J. Simon, A. Stammler, J. Oldengott, H. Bögge and T. Glaser, Inorg. Chem., 2020, 59, 14615–14619 CrossRef CAS PubMed.
- J. Simon, C. Horstmann née Gruschka, A. Mix, A. Stammler, J. Oldengott, H. Bögge and T. Glaser, Dalton Trans., 2022, 51, 2863–2875 RSC.
- M. Giampà, D. Corinti, A. Maccelli, S. Fornarini, G. Berden, J. Oomens, S. Schwarzbich, T. Glaser and M. E. Crestoni, Inorg. Chem., 2023, 62, 1341–1353 CrossRef PubMed.
- N. Biere, D. Kreft, V. Walhorn, S. Schwarzbich, T. Glaser and D. Anselmetti, J. Nanobiotechnol., 2023, 21, 26 CrossRef CAS PubMed.
- T. Riediger, Z. Aydin, A. Stammler, J. Oldengott and T. Glaser, Z. Anorg. Allg. Chem., 2024, e202300231 CrossRef CAS.
- SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- T. Riediger, A. Stammler, J. Oldengott, S. Walleck and T. Glaser, Inorg. Chem., 2025, 64, 9035–9043 CrossRef CAS PubMed.
- G. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- J.-J. Girerd and Y. Journaux, in Physical Methods in Bioinorganic Chemistry, ed. L. Que Jr., University Science Books, Sausalito, 2000, pp. 321–374 Search PubMed.
- (a) T. R. Holman, M. P. Hendrich and L. Que Jr., Inorg. Chem., 1992, 31, 937–939 CrossRef CAS; (b) L. C. Nathan, J. H. Nelson, G. L. Rich and R. O. Ragsdale, Inorg. Chem., 1969, 8, 1494–1497 CrossRef CAS; (c) R. Whyman, W. E. Hatfield and J. S. Paschal, Inorg. Chim. Acta, 1967, 1, 113 CrossRef CAS.
- A. B. P. Lever, J. Chem. Educ., 1968, 45, 711–712 CrossRef CAS.
- T. P. Zimmermann, S. Dammers, A. Stammler, H. Bögge and T. Glaser, Eur. J. Inorg. Chem., 2018, 48, 5229–5237 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2445426 and 2445427. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00941c |
| This journal is © The Royal Society of Chemistry 2025 |