
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and ion conducting properties of mixed-alkali Na3–xLixInCl6 solid electrolytes†
Dat
Le Thanh
 abc,
Long Hoang Bao
Nguyen
bd,
Emmanuelle
Suard
a and
Romain
Berthelot
*bd
abc,
Long Hoang Bao
Nguyen
bd,
Emmanuelle
Suard
a and
Romain
Berthelot
*bd
aInstitut Laue-Langevin (ILL), 38042, Grenoble, France
bICGM, Univ. Montpellier, CNRS, ENSCM, 34095, Montpellier, France. E-mail: romain.berthelot@umontpellier.fr
cAlistore-European Research Institute, CNRS FR 3104, 80039, Amiens, France
dRéseau sur le Stockage électrochimique de l’énergie (RS2E), CNRS, 80039, Amiens, France
First published on 2nd May 2025
Abstract
Li substitution and its impact on the structures and ion transport of Na3−xLixInCl6 polymorphs, synthesized via ball-milling and further annealing, were investigated. Despite a solubility limit of x = 0.6, Li incorporation seems to form a connected Li+ network to enhance the ionic conductivity of the resulting materials.
All-solid-state batteries (ASSBs) have attracted considerable interest in recent years thanks to their enhanced energy density, improved safety, and extended lifetime. The development of ASSBs has stimulated the discovery of new inorganic materials possessing high ionic conductivity, even at ambient temperature.1 Depending on the nature of the anion, inorganic solid-state electrolytes (SSEs) are usually classified into oxides, sulphides, or halides, with the latter being well recognized thanks to their high oxidation stability and excellent mechanical flexibility required for material processing and cell assembly.2–4 The development of halide SSEs has advanced rapidly since the pioneering work of Asano et al., reporting Li3YCl6 and Li3YBr6 that possess room-temperature Li+ conductivities of up to 0.5 and 1.7 mS cm−1, respectively.5 Since then, different families of halide-based Li+ SSEs have been thoroughly investigated, including aliovalent substitution solid solutions, such as Li3−xIn1−xZrxCl6, which exhibits a high ionic conductivity of up to 1.25 mS cm−1 for x = 0.4.6
Besides halide-based Li+ SSEs, the number of studies on Na+ counterparts has surged in the last few years. The most commonly investigated halide-based Na+ SSEs are Na3MX6 where M = Sc, Y, In, and Sm–Lu and X = Cl, Br, or I; nonetheless, most of them exhibit relatively low ionic conductivity at ambient temperature.7–10 Different strategies, including compositional and structural modifications, have been carried out to improve their conducting properties. One of the most widely adopted approaches is aliovalent substitution at the transition metal site, which can enhance Na+ conductivity by about three or four orders of magnitude compared with undoped compositions. For instance, Na2ZrCl6 exhibits an Na+ conductivity of 6.9 × 10−5 mS cm−1, compared to 6.6 × 10−2 and 3.5 × 10−2 mS cm−1 of Na2.25Zr0.75Y0.25Cl6 and Na2.4Zr0.6Er0.4Cl6, respectively.11–13 The other strategy involves anionic mixing to modify bond strength between the alkali and halogen atoms, thereby facilitating ion conduction.14,15 This approach was reported for halide-based Na+ SSEs, including Na3GdBrxI6−x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 and Na3InCl6−xBrx,10 though little improvement in the ionic conductivity was observed.
16 and Na3InCl6−xBrx,10 though little improvement in the ionic conductivity was observed.
The structures of halide-based Li+/Na+ SSEs, AxMXn, have several similarities. Indeed, they can be described as a close packed lattice of X− anions, while the cations, Li+/Na+ and Mn+, occupy the available interstitial sites. Both hexagonal and cubic close packed anion lattices have been observed; nonetheless, subtle structural distortions can occur depending on the amount of alkali vacancies and the charge and ionic radii of alkali, metal, and halide ions, leading to a structural diversity of the resulting compound. The difference in the energy of hexagonal and cubic lattices is subtle and thermal treatment can lead to a structural conversion between the two forms. It was suggested that cubic packing offers more favorable interstitial connectivity and diffusion pathways than hexagonal packing, potentially resulting in higher ionic conductivity.17 Alkali vacancies and aliovalent and isovalent substitution on the metal/anion site are the three degrees of freedom that are commonly used to monitor the structure and ionic conductivity of halide-based Li+/Na+ SSEs. Isovalent substitution on the alkali site also represents a degree of freedom that can directly affect the structure and conduction properties but is rarely reported in the literature.
In the present work, the impact of isovalent alkali substitution on the structure and ion transport properties of halide-based SSEs will be investigated using Na3−xLixInCl6. The parental compound Li3InCl6 adopts a trigonal structure with a hexagonal packing lattice of Cl− while Li+ and In3+ occupy octahedral sites.6,18 On the other hand, Na3InCl6 can adopt either the same trigonal structure or a monoclinic one, which is a distorted cubic packing lattice of Cl− with part of the Na+ occupying the prismatic site.10,19 The existence of two different polymorphs for Na3InCl6 allows an insightful study on the solubility limit and impacts of Li+ substitution in each structure. Several Na3−xLixInCl6 compounds with different Li contents are synthesized using mechanochemistry, which might favor the formation of metastable phases, followed by a subsequent thermal treatment to obtain thermodynamically stable products. A combination of powder X-ray diffraction (PXRD), neutron diffraction (ND), and electrochemical impedance spectroscopy (EIS) is employed to investigate the solubility limit, structural modifications and conduction properties of the resulting compounds.
A series of Na3−xLixInCl6 (0 ≤ x ≤ 1.5) compositions was prepared using mechanochemistry. Up to x = 0.6, the PXRD patterns of the as-obtained samples can be indexed using the monoclinic structure (P21/n space group), in which the Cl− lattice forms a distorted cubic close packing structure while In3+ occupies octahedral sites; Na+ and Li+ are distributed over octahedral and prismatic sites (Fig. 1a and c). The Na/LiCl6 trigonal prisms and octahedra are inter-connected via corner- and edge-sharing linkages, creating a tri-dimensional diffusion pathway for alkali ions. The cell parameters obtained from Rietveld refinement (Fig. 2a and c) show that the variation in lattice parameters upon Li+ substitution is subtle; nonetheless, a linear contraction of the unit cell is observed up to x = 0.6, suggesting a lithium incorporation process. From x = 0.9 up to x = 1.5, Bragg peaks of the desired phase become progressively broader and less intense (Fig. S2†), indicating a decrease in crystallinity. Furthermore, new peaks corresponding to impurities are observed, suggesting that a solid solution might not have been formed in this compositional range.
 | ||
| Fig. 1 PXRD patterns and crystal structures of the Na3−xLixInCl6 (0 ≤ x ≤ 0.9) series synthesized by mechanochemistry (a and c) followed by annealing at 200 °C (b and d). The low-intensity peaks, marked with * in one representative pattern, may indicate the presence of impurities, although they cannot be attributed to any specific compound. | ||
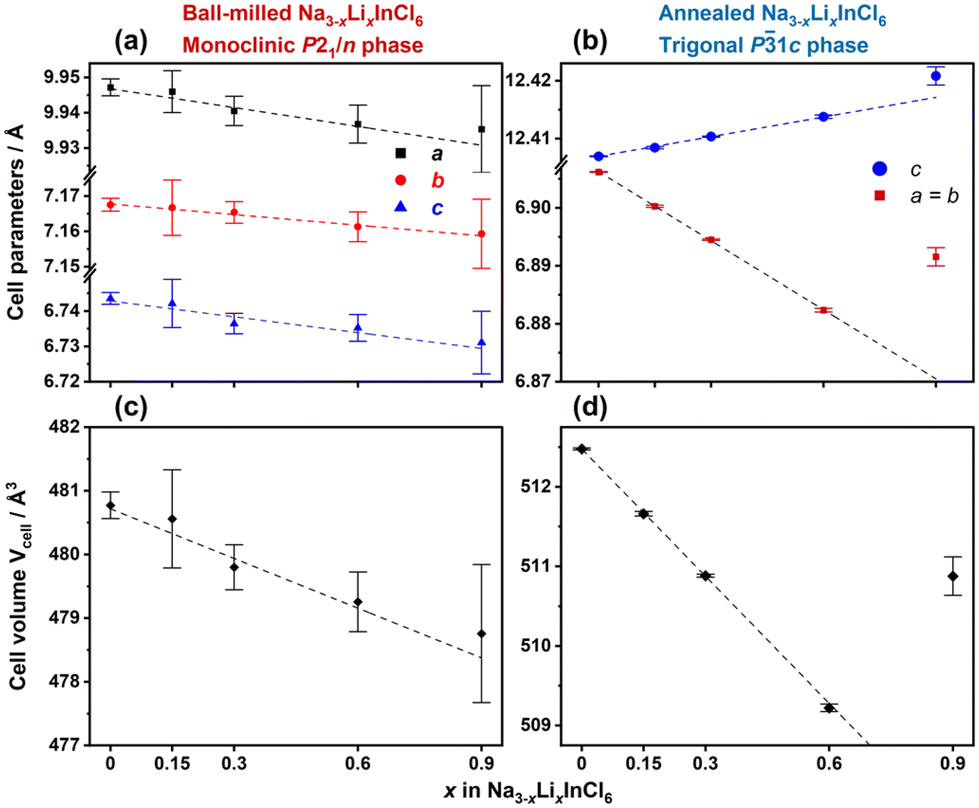 | ||
| Fig. 2 Evolution of cell parameters and cell volume for the Na3−xLixInCl6 (0 ≤ x ≤ 0.9) series determined by Rietveld refinement of the PXRD patterns. Samples were synthesized by ball-milling (a and c) followed by thermal treatment at 200 °C (b and d). | ||
As thermal treatment can induce a significant impact on the structures and transport properties of mechanochemically prepared chloride SSEs,10,19 an annealing process at 200 °C under vacuum was carried out on the as-obtained ball-milled Na3−xLixInCl6 samples (0 ≤ x ≤ 1.5). The PXRD patterns of the annealed samples show an evident phase transformation with an enhanced crystallinity (Fig. 1b). Most diffraction peaks can be indexed using the trigonal polymorph (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 1c space group) of Na3InCl6. This observation agrees with previous studies, showing that monoclinic Na3InCl6 would convert to a trigonal polymorph upon thermal treatment.10,19 Unlike the monoclinic P21/n space group, all metal sites in the trigonal P1c phase group are octahedrally coordinated by chloride ions. The structure consists of two distinct alternating layers stacked along the c-axis: one contains only edge-sharing Na/Li4fCl6 octahedra and the other contains Na/Li2aCl6 and In2dCl6 octahedra alternately linked by edge-sharing to form honeycomb layers (Fig. 1d). The Na/LiCl6 octahedra between the two layers are connected via corner-sharing, creating here also a tri-dimensional diffusion pathway for alkali ions.
1c space group) of Na3InCl6. This observation agrees with previous studies, showing that monoclinic Na3InCl6 would convert to a trigonal polymorph upon thermal treatment.10,19 Unlike the monoclinic P21/n space group, all metal sites in the trigonal P1c phase group are octahedrally coordinated by chloride ions. The structure consists of two distinct alternating layers stacked along the c-axis: one contains only edge-sharing Na/Li4fCl6 octahedra and the other contains Na/Li2aCl6 and In2dCl6 octahedra alternately linked by edge-sharing to form honeycomb layers (Fig. 1d). The Na/LiCl6 octahedra between the two layers are connected via corner-sharing, creating here also a tri-dimensional diffusion pathway for alkali ions.
Rietveld refinements of PXRD data from thermally treated Na3−xLixInCl6 compositions show an expansion along the c-axis and a contraction in the (ab) plane upon Li+ substitution. A linear evolution in the cell parameters consistent with Vegard's law is observed only until the composition x = 0.6. In this low Li+ incorporation range, the mixed-alkali Na3−xLixInCl6 is thermodynamically stable as no impurity phase emerges upon heating. These results indicate the formation of solid solutions up to 20% (x = 0.6) in both monoclinic and trigonal Na3−xLixInCl6 polymorphs, despite the difference in the ionic radius of Li+ and Na+ (0.76 vs. 1.02 Å).20
In order to understand the impact of Li+ substitution on the structure and ion conducting properties, insightful information on Li+ and Na+ site occupation is essential. The high crystallinity of trigonal Na3−xLixInCl6 compositions enables the use of the diffraction technique for Na/Li distribution analysis. Since 7Li possesses a negative coherent scattering length, in contrast to the positive values for Na, Cl, and In,21 ND is suitable for probing the Li position and investigating site preference for both alkali ions between two crystallographic sites (2a and 4f). An indirect approach, including ND pattern simulations for different scenarios of alkali ion distribution and comparison with experimental patterns, is used. Simulated patterns for Na3−xLixInCl6 (x = 0.15, 0.3, and 0.6) are generated according to three scenarios: (1) all lithium at the 2a site, (2) all lithium at the 4f site, and (3) lithium evenly distributed between both sites.
For each composition, most diffraction peaks overlap significantly (Fig. S5†), except in the low-Q region where the (002) and (100) reflections exhibit notable intensity differences (Fig. 3). These differences become more pronounced as the Li+ content increases. Comparison with the experimental ND data indicates that the extreme case, where Li+ occupies only one site exclusively, can qualitatively be ruled out (Fig. 3 and S5†). Due to significant peak overlap in the simulated patterns and weak intensities in the region of interest, refining exact site occupation factors was challenging. However, the indirect simulation comparison approach was sufficiently reliable to suggest that both alkali ions likely occupy both sites, though their proportions remain undetermined.
 | ||
| Fig. 3 Simulated (a) and experimental (b) ND patterns for the three compositions of Na3−xInxCl6 in the low-Q region. For each composition, three simulated ND patterns correspond to different lithium distributions between the 2a and 4f sites. | ||
The occupation of Li+ in both 2a and 4f sites may help explain the limited solubility of Li+ in an Na3InCl6 trigonal structure. At a low degree of substitution (x ≤ 0.6), the replacement of Na+ by Li+ would lead to a contraction of cell parameters; however, the interstitial site would be big enough to accommodate both Na+ and Li+. For more Li-rich compositions, the contraction of the cell parameters and the size of the interstitial sites would be so great that Na+ could not be fit in the structure.
To further understand the dual-site occupation of Li+ and its effect on the conduction properties within the solid solution Na3−xLixInCl6 (x ≤ 0.6), EIS was used. As electronic conductivity is considerably lower than ionic conductivity (Fig. S7, Tables S7 and S8†), only ionic contributions are considered in impedance analysis. The impedance spectra of all the samples show a suppressed semicircle, corresponding to the ionic conduction, followed by the capacitive blocking behavior of the stainless-steel electrodes (Fig. 4a, b and S6†). In this case, bulk and grain boundary contributions to the impedance cannot be deconvoluted; therefore, the values of reported conductivities herein represent the total ionic conductivities (Table S7†). All ball-milled and annealed phases at each composition exhibit Arrhenius behavior from 30 °C to 70 °C, though a deviation is observed at 80 °C. The ionic conductivity values of both Na3InCl6 polymorphs at 30 °C are of the same order of magnitude as those reported in other studies, both around 1.4 × 10−5 mS cm−1, indicating that Na3InCl6 is an ionic insulator. Upon Li+ substitution, the ionic conductivity gradually increases. For instance, a 10% Li+ substitution (x = 0.3) results in small improvements, with ionic conductivities of 8.6 × 10−5 and 4.9 × 10−5 mS cm−1 at 30 °C for the monoclinic and trigonal phases, respectively. At Na+-rich compositions (x ≤ 0.3), the ionic conductivities of both polymorphs are of similar magnitude across the whole temperature range (30–80 °C), indicating that the packing sequence of Cl− has little impact on the ionic conductivity (Fig. 4c and d).
 | ||
| Fig. 4 Representative Nyquist plots at 50 °C (a and b) and Arrhenius plots from the temperature-dependent impedance (c and d), corresponding to the ball-milled and annealed samples in the Na3−xLixInCl6 series. Comparison of ionic conductivities at 30 °C (e) and activation energies for ionic conduction (f) of these samples. | ||
Remarkably, higher lithium substitution of 20% (x = 0.6) leads to a conductivity enhancement by one order of magnitude, reaching 5.1 × 10−4 and 6.0 × 10−4 mS cm−1 at 30 °C for the monoclinic and trigonal phases, respectively. This significant gain in ionic conductivity could be contributed by the enhanced motion of both Na+ and Li+ ions; nonetheless, the contribution from Li+ is expected to be more significant than that from Na+. As illustrated in Fig. 4e, the ionic conductivity of Na3−xLixInCl6 (x ≤ 0.6) does not increase linearly with the amount of Li+ present in the structure, but its evolution follows an exponential trend. This behaviour suggests that Na+ in Na3−xLixInCl6 participates negligibly in ion conduction, but the ionic conductivity predominantly arises from Li+ motion. At a low substitution amount, each Li+ site is surrounded by In3+ or Na+, and thus little improvement in the ionic conductivity is observed. As the substitution degree increases, due to the occupation of Li+ at both sites, Li+ might form a 3D network, creating a diffusion pathway for Li+ and leading to a significant conductivity enhancement. Additionally, both phases of Na2.4Li0.6InCl6 possess the lowest activation energy, indicating that this composition might contain sufficient Li+ to create a connected Li+ network to facilitate Li+ diffusion. The ionic conductivity of the annealed sample is slightly higher than the one obtained from ball-milling, implying that the hexagonal packing of Cl− might facilitate the diffusion in this case. This is an interesting example showing that Na+ in these mixed-alkali compositions would be immobile, but Li+ would be mobile if the threshold amount of Li+ was reached. Li/Na mixed compositions are also observed in some layered oxides,22 and the resulting compounds can be used as electrode materials for Li- or Na-ion batteries. The flexibility in the structural properties of A3InCl6 and layered oxides could be due to the similar stacking sequence of the InCl6/MO6 structural units, and the size of the interstitial sites can vary to accommodate the guest ions.
Conclusions
Several mixed-alkali solid solutions of Na3–xLixInCl6 were prepared via ball-milling and subsequent annealing, showing a phase transition from monoclinic to trigonal upon annealing. Lattice parameters evolve linearly up to a Li+ solubility limit at x = 0.6, beyond which interstitial contraction and ion size mismatch hinder further incorporation. ND revealed that the newly introduced lithium occupies both alkali sites. EIS analysis shows an exponential increase of ionic conductivity with higher Li+ content, suggesting that the smaller lithium ions become progressively more connected and mobile, thus dominating the transport behaviour. These findings demonstrate the potential of alkali substitution to tailor the structure and ionic transport properties of halide-based solid-state electrolytes.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
As a part of the DESTINY PhD program, this publication acknowledges funding from the European Union's Horizon2020 Research and Innovation Program under the Marie Skłodowska-Curie Actions COFUND (Grant Agreement #945357). The authors acknowledge MSPD staff at ALBA synchrotron for help and data collection on the 2023097745 proposal beamtime.References
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- H. Kwak, S. Wang, J. Park, Y. Liu, K. T. Kim, Y. Choi, Y. Mo and Y. S. Jung, ACS Energy Lett., 2022, 7, 1776–1805 CrossRef CAS.
- L. Huang, L. Zhang, J. Bi, T. Liu, Y. Zhang, C. Liu, J. Cui, Y. Su, B. Wu and F. Wu, Energy Mater. Adv., 2024, 5, 0092 CrossRef CAS.
- X. Nie, J. Hu and C. Li, Interdiscip. Mater., 2023, 2, 365–389 Search PubMed.
- T. Asano, A. Sakai, S. Ouchi, M. Sakaida, A. Miyazaki and S. Hasegawa, Adv. Mater., 2018, 30, 1803075 CrossRef.
- B. Helm, R. Schlem, B. Wankmiller, A. Banik, A. Gautam, J. Ruhl, C. Li, M. R. Hansen and W. G. Zeier, Chem. Mater., 2021, 33, 4773–4782 CrossRef CAS.
- G. Meyer, S. Peter Ax, T. Schleid and M. Irmler, Z. Anorg. Allg. Chem., 1987, 554, 25–33 CrossRef CAS.
- M. S. Wickleder and G. Meyer, Z. Anorg. Allg. Chem., 1995, 621, 457–463 CrossRef CAS.
- A. Bohnsack and G. Meyer, Z. Anorg. Allg. Chem., 1996, 622, 173–178 CrossRef CAS.
- T. Zhao, M. A. Kraft and W. G. Zeier, Inorg. Chem., 2023, 62, 11737–11745 CrossRef CAS PubMed.
- H. Kwak, J. Lyoo, J. Park, Y. Han, R. Asakura, A. Remhof, C. Battaglia, H. Kim, S.-T. Hong and Y. S. Jung, Energy Storage Mater., 2021, 37, 47–54 CrossRef.
- E. A. Wu, S. Banerjee, H. Tang, P. M. Richardson, J.-M. Doux, J. Qi, Z. Zhu, A. Grenier, Y. Li, E. Zhao, G. Deysher, E. Sebti, H. Nguyen, R. Stephens, G. Verbist, K. W. Chapman, R. J. Clément, A. Banerjee, Y. S. Meng and S. P. Ong, Nat. Commun., 2021, 12, 1256 CrossRef CAS PubMed.
- R. Schlem, A. Banik, M. Eckardt, M. Zobel and W. G. Zeier, ACS Appl. Energy Mater., 2020, 3, 10164–10173 CrossRef CAS.
- P. Till, M. T. Agne, M. A. Kraft, M. Courty, T. Famprikis, M. Ghidiu, T. Krauskopf, C. Masquelier and W. G. Zeier, Chem. Mater., 2022, 34, 2410–2421 CrossRef CAS.
- X. Zhang, K. C. Phuah and S. Adams, Chem. Mater., 2021, 33, 9184–9193 CrossRef CAS.
- A. Bohnsack and G. Meyer, Z. Anorg. Allg. Chem., 1997, 623, 837–843 CrossRef CAS.
- X. Li, J. T. Kim, J. Luo, C. Zhao, Y. Xu, T. Mei, R. Li, J. Liang and X. Sun, Nat. Commun., 2024, 15, 53 CrossRef CAS PubMed.
- X. Li, J. Liang, J. Luo, M. Norouzi Banis, C. Wang, W. Li, S. Deng, C. Yu, F. Zhao, Y. Hu, T.-K. Sham, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li, K. R. Adair and X. Sun, Energy Environ. Sci., 2019, 12, 2665–2671 RSC.
- T. Zhao, A. N. Sobolev, R. Schlem, B. Helm, M. A. Kraft and W. G. Zeier, ACS Appl. Energy Mater., 2023, 6, 4334–4341 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- Neutron data booklet, ed. A. J. Dianoux and Institut Laue-Langevin, Old City, Philadelphia, PA, 2nd edn, 2003 Search PubMed.
- C. Vallée, M. Saubanère, P. Sanz-Camacho, Y. Biecher, B. Fraisse, E. Suard, G. Rousse, D. Carlier and R. Berthelot, Inorg. Chem., 2019, 58, 11546–11552 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, powder X-ray and neutron diffraction patterns, and electrochemical impedance spectra. See DOI: https://doi.org/10.1039/d5dt00817d |
| This journal is © The Royal Society of Chemistry 2025 |