
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral CNN pincer Ir(III)–H complexes. Transient ligand and counterion influences in the asymmetric hydrogenation of imines and quinolines†
Yisong
Wen
 ab,
Albert
Cabré
a,
Jordi
Benet-Buchholz
c,
Antoni
Riera
*ab and
Xavier
Verdaguer
*ab
ab,
Albert
Cabré
a,
Jordi
Benet-Buchholz
c,
Antoni
Riera
*ab and
Xavier
Verdaguer
*ab
aInstitute for Research in Biomedicine (IRB Barcelona), The Barcelona Institute of Science and Technology, Baldiri Reixac 10, Barcelona E-08028, Spain. E-mail: xavier.verdaguer@irbbarcelona.org; antoni.riera@irbbarcelona.org
bDepartament de Química Inorgànica i Orgànica, Secció de Química Orgànica. Universitat de Barcelona, Martí i Franquès 1, Barcelona E-08028, Spain
cInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Av. Països Catalans 16, 43007 Tarragona, Spain
First published on 5th June 2025
Abstract
Building upon the success of Ir-IMMAX catalysts, we designed a novel cationic Ir(III)–H catalyst featuring a meridional CNN pincer ligand with a perpendicular monodentate phosphine, where chirality arises from both the chiral oxazoline moiety and the metal center itself. The catalyst is readily synthesized from commercially available precursors, obtained as a single diastereomer, and exhibits high activity in the asymmetric hydrogenation of imines and quinolines under mild conditions. One of the synthesized Ir(III)–H complexes efficiently reduced N-methylimines and 2-methylquinoline at room temperature under 3 bar of H2 pressure, positioning itself among the most active catalysts described to date. The incorporation of a weakly coordinating transient ligand, such as acetonitrile, significantly enhanced catalytic performance. Additionally, the counterion displayed an important influence on both enantioselectivity and activity, with the BArF complex providing higher selectivity and the triflate complex exhibiting superior reactivity.
Introduction
Asymmetric hydrogenation (AH) is an essential method in modern synthetic chemistry, providing an efficient approach to enantiomerically enriched pharmaceuticals, agrochemicals and fine chemicals.1 Iridium has been one of the most extensively studied transition metals for this application, since it provides highly active catalysts for the hydrogenation of challenging substrates such as non-functionalized alkenes, imines and nitrogen containing heteroaromatic compounds.2–7 In 2016, our group introduced a library of Ir-MaxPHOX catalysts featuring a P-stereogenic phosphino-oxazoline ligand (Fig. 1a), which have been successfully applied to the hydrogenation of both functionalized and non-functionalized alkenes.8–11 Building upon this foundation, we developed the Ir-IMMAX catalyst—an octahedral Ir(III) hydride complex featuring a MaxPHOX ligand, a cyclometallated imine, and a transient solvent ligand.12 The Ir-IMMAX catalyst has demonstrated outstanding efficiency and selectivity in the asymmetric hydrogenation of N-methyl, N-alkyl, and N-aryl imines, providing enantiomeric excesses (ee) of up to 99% under low hydrogen pressure (3 bar).13 | ||
| Fig. 1 Reported Ir-MaxPHOX and Ir-IMMAX catalysts, examples of iridium pincer catalysts for asymmetric transformations and the designed CNN pincer Ir(III)–H complex. | ||
Given the selectivity and efficiency achieved with the Ir-IMMAX catalyst in the hydrogenation of imines, we aimed to find alternative octahedral Ir(III) systems with comparable structural frameworks that allow straightforward synthesis, easier structural modification and larger reaction scope. This idea led us to explore the iridium pincer complexes, which have gained significant attention over the past decades due to their high modularity and exceptional stability.14 The first application of iridium pincer complexes was reported by Jensen, Kaska, and co-workers in 1996 for the transfer dehydrogenation of alkanes.15 Since then, many pincer complexes have been developed by fine-tuning various aspects of the ligand structure. However, their applications have predominantly focused on achiral transformations such as dehydrogenation, dehydroaromatization, and isomerization. In contrast, only a limited number of asymmetric transformations have been reported, with selected examples shown in Fig. 1b.16–20
With the idea to mimic the coordination environment of the Ir-IMMAX complexes with a pincer ligand, we designed complex A (Fig. 1c), a cationic Ir(III) species featuring a meridional phenyl-pyridine-oxazoline pincer ligand, and a monodentate phosphine positioned perpendicular to the pincer plane. We anticipated that the phosphine would be placed anti relative to the oxazoline's isopropyl group, and that the transient ligand (L) would occupy the position trans to the phosphine, as observed in Ir-IMMAX complexes.
Here, we report the highly stereoselective, one-step synthesis of complexes A (X = OTf and BArF) with different transient ligands from readily available metal precursors. The structure of A and its dihydride synthetic intermediate has been unambiguously confirmed by single-crystal X-ray diffraction. The resulting complexes have been tested in the asymmetric hydrogenation of imines and quinolines, with the results highlighting the strong influence of both the transient ligand and counterion on the performance of the catalysts.
Results and discussion
Starting from 6-bromopicolinaldehyde, the model pincer ligand 1 was readily synthesized following a two-step procedure from available literature in high yield (Scheme 1).21,22 With the ligand in hand, initial attempts to synthesize the designed complex A started from the Ir(cod)(PCy3)Cl (cod = 1,5-cyclooctadiene) precursor. We envisioned that removal of cod ligand by means of hydrogen and complexation with 1 would provide dihydride complex 2, which upon heating would lead to cyclometallated complex 3 (Scheme 2). Chloride complex 3 would be a convenient precursor of the desired final complex since halogen abstraction would allow the generation of the desired vacant coordination site, which could be stabilized with a transient solvent ligand (i.e. THF or ACN). To this end, ligand 1 was added to the Ir precursor and cod was removed via hydrogen bubbling. 1H NMR analysis of the reaction mixture revealed two doublets of doublets at −21.89 and −25.05 ppm suggesting that the corresponding dihydride 2 was obtained as major metal hydride species, along with substantial amounts (40–50%) of free starting ligand. Upon subsequent thermally induced cyclometallation (toluene, 100 °C), multiple hydride-containing products were observed from which the desired complex could not be isolated. More critically, we observed increased amounts of uncoordinated ligand post-cyclometallation, suggesting that the dihydride 2 was inherently unstable. This instability likely arises from the N,N-bidentate ligand adopting a trans arrangement relative to the two hydrides, making it prone to dissociation. An additional potential problem when using non-symmetric pincer ligands as 1 is the formation of different stereoisomers since the metal becomes a chiral center itself, as happens, for instance, in Huang's iridium complexes (Fig. 1b).19 Overall, we believed these factors were responsible for the inefficient synthesis of 3. | ||
| Scheme 1 Synthesis of CNN pincer ligand 1. | ||
 | ||
| Scheme 2 Initial attempts to synthesize the desired Ir(III) hydride CNN pincer complex. | ||
Recognizing the need for improved stability and selectivity, we sought an alternative approach using a cationic precursor instead of a neutral one. We reasoned that a cationic complex, being more electrophilic, should provide increased stability to the corresponding dihydride intermediates. With this idea in mind, we turned our attention to Crabtree's catalysts. Starting from the BArF analogue of Crabtree's catalyst, ligand 1 was successfully coordinated following cod removal under hydrogen in THF at room temperature, yielding the dihydride intermediate 4-BArF in significantly improved purity as a single diastereomer (Scheme 3). Notably, this intermediate exhibited sufficient stability to allow purification via column chromatography, affording a crystalline orange solid in 86% yield. In the 1H-NMR spectrum (Fig. 2), the two hydrides appear as doublet of doublets at −21.00 ppm and −24.13 ppm, with a J(H,P) of 23.4 Hz and a J(H,H) of 8.2 Hz, confirming that both hydrides are located cis to the phosphorous atom.
 | ||
| Scheme 3 Synthesis of complexes 4 and 5 from the corresponding Crabtree's catalyst. | ||
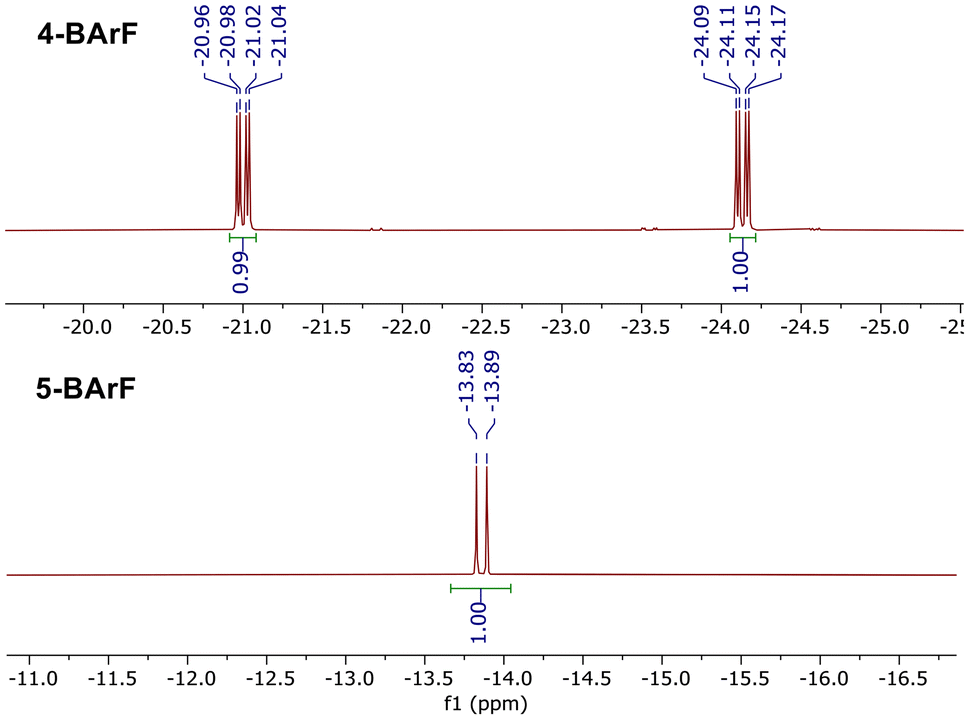 | ||
| Fig. 2 Hydride zone in the 1H-NMR spectra of 4-BArF and 5-BArF. | ||
Performing the cod ligand removal in toluene at room temperature and subsequently heating to 100 °C induced cyclometallation, producing the desired cyclometallated complex 5-BArF in high purity and selectivity (Scheme 3). Again, column chromatography provided complex 5-BArF as a crystalline orange solid in 77% yield. In the 1H-NMR spectrum, the hydride appears as a doublet at −13.86 ppm, with a J(H,P) of 26.2 Hz, characteristic of a hydride cis to phosphorous atom (Fig. 2). A similar strategy was employed to synthesize the triflate analogues of both complexes (4-OTf and 5-OTf, respectively), starting from the triflate analogue of Crabtree's catalyst.
Single-crystal X-ray structures of complexes 4-BArF and 5-BArF are depicted in Fig. 3. As hypothesized, the introduction of a bulky phosphine ligand such as tricyclohexylphosphine resulted in the formation of a single diastereomer for both complexes, with the phosphine positioned trans to the directing isopropyl group of the chiral oxazoline and perpendicular to the ligand plane. The bidentate ligand is confirmed to be trans to two hydrides in 4-BArF, justifying the relatively low stability of this dihydride intermediate. In 5-BArF, cyclometallation occurred with the release of a hydrogen molecule, leaving the remaining hydride positioned trans to the pyridine moiety of ligand 1. The reactive site is occupied by the coordinated pyridine, situated trans to the monodentate phosphine.
 | ||
| Fig. 3 Single-crystal X-ray structure of 4-BArF (top) and 5-BArF (bottom). The Ortep diagram shows ellipsoids at 50% probability. The BArF counterions and solvent molecules are omitted for clarity. Only the hydrides attached to the iridium center are shown. | ||
With these complexes in hand, we next sought to replace the coordinated pyridine with acetonitrile, a weaker and more labile transient ligand, mirroring the ligand environment of the Ir-IMMAX catalyst, as the acetonitrile ligand would likely enhance catalyst activity. Initially, direct synthesis from the corresponding acetonitrile analogue of Crabtree's catalyst, [Ir(CH3CN)(cod)(PCy3)]BArF, was attempted following the successful synthesis of 5-BArF and 5-OTf. However, such a strategy proved unfeasible due to the lower stability of the acetonitrile dihydride intermediate, leading to intricate mixtures of iridium hydride species. Finally, we sought the direct pyridine-acetonitrile ligand exchange on complex 5-OTf (Scheme 4). Upon heating 5-OTf in acetonitrile, a new doublet appeared at −15.24 ppm corresponding to the acetonitrile ligand exchange product 6-OTf. Upon co-distillation of the outcoming pyridine with acetonitrile several times (×3), we could completely remove the pyridine to obtain the target 6-OTf. Purification via flash chromatography provided the new complex in 71% yield and 92% purity, as determined by 1H NMR.23 At this point we proceeded to test the newly obtained species 4, 5 and 6 as catalysts in AH.
 | ||
| Scheme 4 Displacement of pyridine with acetonitrile to form complex 6-OTf. | ||
To evaluate the catalytic performance of our synthesized complexes, we first investigated their activity and selectivity in the AH of imines (Table 1). As representative substrates, we selected 4-methoxy-substituted N-phenyl imine and 4-chloro-substituted N-methyl imine, owing to their enhanced stability and ease of handling as solids at room temperature. Hydrogenation was carried out at 1 mol% catalyst loading, using 50 bar and 3 bar of hydrogen pressure at room temperature in dichloromethane. Comparative analysis of conversions revealed that the dihydride complex 4-OTf was the least active catalyst, likely due to its inherent instability leading to rapid deactivation. In contrast, all the cyclometallated catalysts provided full conversion at 50 bar of hydrogen with both substrates (Table 1, entries 5, 6, 9, 10, 13 and 14). Surprisingly, at a lower hydrogen pressure of 3 bar, the acetonitrile complex 6-OTf was still able to provide full conversion for the hydrogenation of N-methyl imines (Table 1, entry 16). This is particularly notable, as reports of this transformation under such mild conditions are scarce, since N-methyl imines are challenging substrates due to the high nucleophilicity of the resulting amine product, which tends to coordinate to the catalyst causing deactivation.24 The superior reactivity of the acetonitrile-ligated complex supports our hypothesis that a weaker transient ligand enhances catalytic performance, whereas the corresponding pyridine-ligated complex exhibited lower conversion under identical conditions (Table 1, entry 12).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | R, R′ | H2 (bar) | Conv.a (%) | ee.b (%) |
| a Conversion rate was determined via1H-NMR. b Enantiomeric excesses were determined via chiral HPLC/GC. | |||||
| 1 | 4-OTf | Ph, OMe | 50 | 68 | 13 (S) |
| 2 | 4-OTf | Me, Cl | 50 | 30 | — |
| 3 | 4-OTf | Ph, OMe | 3 | 0 | — |
| 4 | 4-OTf | Me, Cl | 3 | 0 | — |
| 5 | 5-BArF | Ph, OMe | 50 | 100 | 48 (R) |
| 6 | 5-BArF | Me, Cl | 50 | 100 | 89 (R) |
| 7 | 5-BArF | Ph, OMe | 3 | 60 | 48 (R) |
| 8 | 5-BArF | Me, Cl | 3 | 16 | — |
| 9 | 5-OTf | Ph, OMe | 50 | 100 | 16 (S) |
| 10 | 5-OTf | Me, Cl | 50 | 100 | 48 (R) |
| 11 | 5-OTf | Ph, OMe | 3 | 100 | 19 (S) |
| 12 | 5-OTf | Me, Cl | 3 | 34 | — |
| 13 | 6-OTf | Ph, OMe | 50 | 100 | 16 (S) |
| 14 | 6-OTf | Me, Cl | 50 | 100 | 45 (R) |
| 15 | 6-OTf | Ph, OMe | 3 | 100 | 17 (S) |
| 16 | 6-OTf | Me, Cl | 3 | 100 | 47 (R) |

Interestingly, we observed a pronounced effect of the counterion on both selectivity and activity. Specifically, the cyclometallated complex 5-BArF provided significantly higher enantioselectivity towards the (R)-enantiomer with up to 89% ee in the reduction of N-methyl imine (Table 1, entry 6); compared to 5-OTf which afforded only 48% ee (Table 1, entry 10). In contrast, the triflate analogue 5-OTf exhibited increased catalytic activity (Table 1, entries 7 vs. 11). This discrepancy could be rationalized by considering the potential involvement of the triflate anion in the transition state through hydrogen bonding interactions, as reported by Fan, Yu and co-workers,25 which may stabilize a lower-energy transition state and thus enhance conversion. However, this interaction appears to stabilize the transition state leading to the (S)-enantiomer, as evidenced by the lower enantioselectivity observed for N-methyl imine (Table 1, entries 6 vs. 10) and the shift in selectivity toward the (S)-enantiomer in the case of N-phenyl imine (Table 1, entries 5 vs. 9). Finally, it is worth noting that enantioselectivity was nearly identical between the pyridine (5-OTf) and acetonitrile (6-OTf) variants (Table 1, entries 9–11 vs. 13–15). This fact aligns with the expectation that the dissociation of the transient ligand occurs before the catalytic cycle begins.
It has been proposed that the AH of imines catalyzed by cationic octahedral Ir(III) complexes follows an outer-sphere mechanism,26 and a plausible catalytic cycle for the reduction of imines with catalysts 5–6 is depicted in Fig. 4. The transient ligand of the pre-catalyst is exchanged with a dihydrogen molecule leading to the active catalyst I. Subsequent activation of the imine substrate through protonation by the acidic Ir(III) dihydrogen complex I leads to the neutral dihydride II and the corresponding iminium ion. The stereodetermining step is the transfer of the hydride trans to the phosphine in II to the protonated substrate. For complexes 5-OTf and 6-OTf, we believe the activated iminium ion is H-bonded to the triflate counterion, which impacts both activity and selectivity of the system.
 | ||
| Fig. 4 Hydrogenation mechanism with Ir(III) octahedral catalysts. | ||
The catalytic activity of the synthesized complexes was then tested with 2-methylquinoline and 2-phenylquinoline (Table 2). The AH of heteroaromatic substrates is less explored than that of prochiral imines, primarily due to the increased stability imparted by the aromaticity of these compounds, which may require harsher reaction conditions.3,4 The AH of quinolines is thought to follow an outer-sphere mechanism as in the case of imines, although it proceeds through a cascade reaction pathway.25 The AH of quinolines was carried out under identical conditions to those used for the imine substrates at 1 mol% catalyst loading in dichloromethane. Overall, the conversion data in Table 2 confirms that quinolines are more difficult to hydrogenate, with the 2-methylquinoline being easier to reduce than the phenyl substituted counterpart. With respect to the activity of catalysts 4–6, a similar trend is observed, with dihydride 4-OTf providing very low conversions (Table 2, entries 1–4). Among the cyclometallated pyridine complexes, 5-OTf demonstrated higher activity than the corresponding 5-BArF counterpart (Table 2, entries 9–11 vs. 5–7). A major reactivity difference was observed when changing the transient ligand from pyridine to acetonitrile. Full conversions were obtained for both quinolines at 50 bar of hydrogen pressure using 6-OTf (Table 2, entries 13 and 14). Most notably, at 3 bar of hydrogen, 2-methyl and 2-phenylquinoline were reduced with 100% and 77% conversion respectively (Table 2, entries 15 and 16). Catalyst 6-OTf is more active than the ruthenium catalyst described by Fan, Yu and co-workers,25 and compares favorably with the achiral diphosphine-carbene iridium complex described by Crabtree and co-workers, which hydrogenates quinolines at 1 bar of H2 pressure.27 Hence, 6-OTf is among the most active catalysts described in the literature for the reduction of quinolines. Unfortunately, the selectivity is very low and only 19% ee is obtained for the reduction of 2-methylquinoline (Table 2, entry 13). In this regard, we believe the selectivity could be improved by introducing secondary interactions (i.e., H-bonding) between the substrate and the ligand structure like we recently demonstrated with the IMMAX catalyst system in the hydrogenation of imines.13
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | R | H2 (bar) | Conv.a (%) | ee.b (%) |
| a Conversion rate was determined via1H-NMR. b Enantiomeric excesses were determined via chiral HPLC. | |||||
| 1 | 4-OTf | Me | 50 | 23 | — |
| 2 | 4-OTf | Ph | 50 | 10 | — |
| 3 | 4-OTf | Me | 3 | 0 | — |
| 4 | 4-OTf | Ph | 3 | 0 | — |
| 5 | 5-BArF | Me | 50 | 43 | — |
| 6 | 5-BArF | Ph | 50 | <5 | — |
| 7 | 5-BArF | Me | 3 | <5 | — |
| 8 | 5-BArF | Ph | 3 | 0 | — |
| 9 | 5-OTf | Me | 50 | 100 | 18 |
| 10 | 5-OTf | Ph | 50 | 81 | 15 |
| 11 | 5-OTf | Me | 3 | 25 | — |
| 12 | 5-OTf | Ph | 3 | <5 | — |
| 13 | 6-OTf | Me | 50 | 100 | 19 |
| 14 | 6-OTf | Ph | 50 | 100 | 12 |
| 15 | 6-OTf | Me | 3 | 100 | 0 |
| 16 | 6-OTf | Ph | 3 | 77 | 0 |

Conclusions
In summary, we have developed a novel octahedral Ir(III)–H catalyst featuring a meridional CNN pincer ligand with a monodentate phosphine positioned perpendicular to the pincer plane. The catalyst was readily synthesized as a single diastereomer with its structure confirmed by single-crystal X-ray diffraction, revealing its unique chiral environment derived from both the chiral oxazoline moiety and the metal center itself.Evaluation of the catalysts in the AH of imines and quinolines demonstrated high activity under mild conditions. Catalyst 6-OTf was able to completely reduce N-methylamine and 2-methylquinoline at room temperature under 3 bar of H2 pressure, positioning itself among the most active catalysts described to date. The presence of a weakly coordinating transient ligand, such as acetonitrile, significantly enhanced catalytic performance compared to the pyridine-ligated analogue. Additionally, counterions played a crucial role in both enantioselectivity and activity, with the triflate complex exhibiting superior reactivity, but lower enantioselectivity.
These results show that the novel iridium pincer–phosphine system can provide highly active catalysts, providing valuable insights for the rational design of next-generation iridium catalysts for asymmetric hydrogenation.
Experimental section
General methods
All reactions were carried out under nitrogen atmosphere in dried solvents. THF and CH2Cl2 were dried with PureSolv purification system from Innovative Technology, Inc. Other commercially available anhydrous solvents were used with no further purification. Silica gel chromatography was performed by using 35–70 mm silica and flash chromatography was performed on automated Combiflash® system from Teledyne Isco. NMR spectra were recorded at 23 °C on Varian Mercury 400 apparatus, with spectra referenced to the residual solvent peak. IR spectra were recorded with Thermo Nicolet Nexus FT-IR apparatus. HRMS were recorded in LTQ-FT Ultra (Thermo Scientific) using Nanoelectrospray technique. HPLC chromatography was performed on Agilent Technologies Series 1100 chromatograph with UV detector. GC chromatography was performed on Agilent Technologies 6890N with FID detector.Imines substrates were prepared according to the previously described procedure.13 Commercial 2-methylquinoline was purified by Kugelrohr distillation. Commercial 2-phenylquinoline was purified via flash chromatography (silica gel, hexanes/EtOAc).
Synthesis of CNN pincer ligand
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4), providing 9.51 g of the desired intermediate as yellowish oil (97% yield). 1H NMR (400 MHz, CDCl3): δ 10.18 (d, J = 0.6 Hz, 1H), 8.12–8.07 (m, 2H), 7.99–7.88 (m, 3H), 7.56–7.44 (m, 3H) ppm.
:15), providing 11.77 g of the desired ligand as white solid (85% yield). 1H NMR (400 MHz, CDCl3): δ 8.01–8.07 (m, 3H), 7.87–7.79 (m, 2H), 7.51–7.38 (m, 3H), 4.55 (dd, J = 9.6, 8.2 Hz, 1H), 4.26 (t, J = 8.3 Hz, 1H), 4.22–4.14 (m, 1H), 1.99–1.85 (m, 1H), 1.08 (d, J = 6.7 Hz, 3H), 0.96 (d, J = 6.8 Hz, 3H) ppm.
:4), providing 9.51 g of the desired intermediate as yellowish oil (97% yield). 1H NMR (400 MHz, CDCl3): δ 10.18 (d, J = 0.6 Hz, 1H), 8.12–8.07 (m, 2H), 7.99–7.88 (m, 3H), 7.56–7.44 (m, 3H) ppm.
:15), providing 11.77 g of the desired ligand as white solid (85% yield). 1H NMR (400 MHz, CDCl3): δ 8.01–8.07 (m, 3H), 7.87–7.79 (m, 2H), 7.51–7.38 (m, 3H), 4.55 (dd, J = 9.6, 8.2 Hz, 1H), 4.26 (t, J = 8.3 Hz, 1H), 4.22–4.14 (m, 1H), 1.99–1.85 (m, 1H), 1.08 (d, J = 6.7 Hz, 3H), 0.96 (d, J = 6.8 Hz, 3H) ppm.
Synthesis of octahedral Ir(III) complexes
:5) collecting the bright orange band, providing 95 mg of the desired complex as orange solid (86% yield). 1H NMR (400 MHz, CDCl3): δ 8.33–8.26 (m, 2H), 8.00 (t, J = 7.8 Hz, 1H), 7.91 (dd, J = 7.7, 1.5 Hz, 1H), 7.79–7.74 (m, 2H), 7.71 (s, 8H), 7.51 (s, 4H), 7.46 (t, J = 7.5 Hz, 1H), 7.32 (t, J = 7.9 Hz, 2H), 7.20–7.10 (m, 4H), 4.71 (t, J = 10.4 Hz, 1H), 4.50 (t, J = 9.3 Hz, 1H), 4.21–4.12 (m, 1H), 2.21–2.08 (m, 1H), 1.86–0.66 (m, 33H), 0.84 (d, J = 7.1 Hz, 3H), 0.10 (d, J = 6.7 Hz, 3H), −21.00 (dd, J = 23.1, 8.2 Hz, 1H), −24.13 (dd, J = 23.7, 8.2 Hz, 1H) ppm. 13C NMR (101 MHz, CDCl3): δ 171.2, 165.1, 161.8 (q, J = 49.8 Hz), 154.6, 146.0, 139.8, 138.9, 137.6, 134.9, 130.9, 130.1, 129.9, 129.0 (qq, J = 31.3, 3.0 Hz), 128.0, 126.4 (d, J = 2.5 Hz), 125.7, 124.7 (q, J = 272.7 Hz), 117.6, 72.1, 70.6, 35.8 (d, J = 31.3 Hz), 30.8, 29.9, 28.6 (d, J = 3.9 Hz), 27.6 (d, J = 11.1 Hz), 27.3 (d, J = 9.5 Hz), 27.1, 26.3, 19.8, 14.1 ppm. 31P NMR (162 MHz, CDCl3): δ 14.79 (dd, J = 19.1, 4.1 Hz) ppm. 19F NMR (376 MHz, CDCl3): δ −62.4 ppm. HRMS (ESI+, direct infusion): m/z [M–BArF]+ calculated for [C40H58IrN3OP]+: 820.3941, found 820.3953. IR-ATR (cm−1): 2928, 2853, 1351, 1273, 1124, 885.
:1) collecting the bright orange band, providing 116 mg of the desired complex as orange solid (65% yield). 1H NMR (400 MHz, CDCl3): δ 8.44–8.36 (m, 3H), 8.29 (dd, J = 7.8, 1.4 Hz, 1H), 7.89–7.93 (m, 2H), 7.39–7.45 (m, 1H), 7.37–7.32 (m, 2H), 7.30 (dd, J = 8.3, 7.2 Hz, 2H), 7.15–7.09 (m, 2H), 4.83 (dd, J = 10.4, 9.2 Hz, 1H), 4.69 (t, J = 9.1 Hz, 1H), 4.15 (ddd, J = 10.3, 8.8, 3.7 Hz, 1H), 2.16 (m, 1H), 1.80–0.67 (m, 33H), 0.87 (d, J = 7.1 Hz, 3H), 0.21 (d, J = 6.7 Hz, 3H), −21.16 (dd, J = 23.2, 8.2 Hz, 1H), −24.03 (dd, J = 23.6, 8.2 Hz, 1H) ppm. 13C NMR (101 MHz, CDCl3): δ 171.9, 164.5, 155.0, 146.0, 140.4, 140.0, 137.7, 131.0, 130.3, 129.5, 127.7, 127.0, 126.6 (d, J = 2.5 Hz), 121.1 (q, J = 320.9 Hz), 72.1, 70.9, 35.8 (d, J = 30.9 Hz), 30.8, 30.5, 28.7 (d, J = 3.9 Hz), 27.7 (d, J = 11.1 Hz), 27.4 (d, J = 9.4 Hz), 26.4, 20.1, 14.8 ppm. 31P NMR (162 MHz, CDCl3): δ 14.54 (dd, J = 20.1, 5.9 Hz) ppm. 19F NMR (376 MHz, CDCl3): δ –78.1 ppm. HRMS (ESI+, direct infusion): m/z [M–OTf]+ calculated for [C40H58IrN3OP]+: 820.3941, found 820.3953. IR-ATR (cm−1): 2922, 2848, 2225, 1588, 1444, 1426, 1262, 1220, 1146, 1031.
:5) collecting the bright orange band, providing 84 mg of the desired complex as orange solid (77% yield). 1H NMR (400 MHz, CDCl3): δ 8.07–8.01 (m, 2H), 7.96 (d, J = 8.2 Hz, 1H), 7.82 (t, J = 8.2 Hz, 1H), 7.72 (s, 8H), 7.62 (d, J = 7.3 Hz, 1H), 7.57 (tt, J = 7.7, 1.5 Hz, 1H), 7.52 (s, 4H), 7.51–7.49 (m, 1H), 7.39–7.34 (m, 1H), 7.03–6.95 (m, 4H), 4.70 (t, J = 9.9 Hz, 1H), 4.52 (dd, J = 9.5, 6.8 Hz, 1H), 4.6 (m, 1H), 2.11–0.81 (m, 34H), 0.79 (d, J = 7.0 Hz, 3H), 0.01 (d, J = 6.8 Hz, 3H), −13.86 (d, J = 26.2 Hz, 1H) ppm. 13C NMR (101 MHz, CDCl3): δ 172.8, 167.4, 161.9 (q, J = 49.8 Hz), 153.1, 144.6, 143.4, 141.7 (d, J = 7.6 Hz), 140.6, 139.3, 137.9, 134.9, 132.7, 129.0 (qq, J = 31.3, 2.9 Hz), 128.7, 126.3 (d, J = 2.5 Hz), 125.5, 124.7 (q, J = 273.7 Hz), 122.8, 121.1 (d, J = 4.9 Hz), 117.6, 71.2, 70.8, 35.7, 30.9, 29.7, 27.6, 27.1, 26.6, 19.3, 14.2 ppm. 31P NMR (162 MHz, CDCl3): δ 1.86 ppm. 19F NMR (376 MHz, CDCl3): δ −62.4 ppm. HRMS (ESI+, direct infusion): m/z [M–BArF]+ calculated for [C40H56IrN3OP]+: 818.3785, found 818.3776; IR-ATR (cm−1): 2932, 2853, 1353, 1273, 1116, 885.
:1) collecting the bright orange band, providing 130 mg of the desired complex as orange solid (62% yield). 1H NMR (400 MHz, CDCl3): δ 8.20–8.12 (m, 2H), 8.08–8.00 (m, 2H), 7.64 (tt, J = 7.6, 1.5 Hz, 1H), 7.56–7.51 (m, 1H), 7.41–7.36 (m, 1H), 7.14 (t, J = 6.7 Hz, 1H), 7.02–6.93 (m, 2H), 4.86 (dd, J = 10.2, 9.4 Hz, 1H), 4.76 (dd, J = 9.4, 7.2 Hz, 1H), 4.04 (ddd, J = 10.1, 7.2, 4.4 Hz, 1H), 2.08–0.86 (m, 34H), 0.84 (d, J = 7.0 Hz, 3H), 0.18 (d, J = 6.7 Hz, 3H), −13.96 (d, J = 26.2 Hz, 1H) ppm. 13C NMR (101 MHz, CDCl3): δ 173.4, 166.8, 153.3, 144.7, 144.1, 141.8 (d, J = 7.6 Hz), 140.4, 140.2, 137.9, 132.2, 129.1, 127.0, 126.5 (d, J = 2.6 Hz), 125.3, 122.6, 122.2 (q, J = 323.2 Hz), 121.5, 71.8, 70.8, 35.4, 31.3, 29.2, 27.7, 26.7, 19.5, 15.2 ppm. 31P NMR (162 MHz, CDCl3): δ 1.47 ppm. 19F NMR (376 MHz, CDCl3): δ −78.1 ppm. HRMS (ESI+, direct infusion): m/z [M–OTf]+ calculated for [C40H56IrN3OP]+: 818.3785, found 818.3800. IR-ATR (cm−1): 2926, 2850, 2238, 2136, 1571, 1446, 1258, 1148, 1029, 915.
:1) collecting the bright orange band, providing 111 mg of the desired complex as orange solid (92% purity, 71% yield). 1H NMR (400 MHz, CD3CN): δ 8.14 (d, J = 8.3 Hz, 1H), 8.05 (t, J = 7.9 Hz, 1H), 7.79 (d, J = 7.5 Hz, 1H), 7.76–7.66 (m, 1H), 7.40–7.32 (m, 1H), 7.11–7.01 (m, 1H), 4.92 (t, J = 9.8 Hz, 1H), 4.82 (dd, J = 9.4, 5.8 Hz, 1H), 4.13 (ddd, J = 9.6, 5.8, 3.2 Hz, 1H), 1.85–0.90 (m, 34H), 1.03 (d, J = 7.0 Hz, 3H), 0.86 (d, J = 6.7 Hz, 3H), −15.24 (d, J = 25.4 Hz, 1H) ppm. 13C NMR (101 MHz, CD3CN): δ 173.6, 167.7, 146.2, 145.6, 142.3, 140.6, 139.1 (d, J = 8.1 Hz), 132.4, 126.0, 123.4, 122.2 (d, J = 14.5 Hz) 72.5, 70.8, 35.3, 31.7, 29.5, 28.0, 27.3 (d, J = 1.4 Hz), 19.3, 15.6 ppm. 31P NMR (162 MHz, CD3CN): δ 2.92 (d, J = 6.8 Hz) ppm. 19F NMR (376 MHz, CD3CN): δ −79.3 ppm. HRMS (ESI+, direct infusion): m/z [M–OTf–CH3CN]+ calculated for [C35H51IrN2OP]+: 739.3363, found 739.3325; m/z [M–OTf–CH3CN–2H]+ calculated for [C35H49IrN2OP]+: 737.3206, found 737.3230. IR-ATR (cm−1): 2924, 2850, 2125, 1575, 1148, 1258, 1146, 1029.
Asymmetric hydrogenation of imines and quinolines
2.0 mg of catalyst (0.01 equiv.), substrate (1 equiv.) and 1 mL of anhydrous CH2Cl2 were placed in a glass tube inside a hydrogenation reactor. After purging with vacuum-nitrogen–hydrogen cycles, the system was adjusted to 3 bar of hydrogen. After stirring at room temperature for 14–20 h, the pressure was released and the solvent was removed under reduced pressure. Conversions were determined via1H-NMR of the reaction crude and enantiomeric excesses were determined via chiral HPLC/GC analysis.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for complexes 4-BArF and 5-BArF have been deposited at the Cambridge Crystallographic Data Centre under CCDC 2430455 and 2430460,† respectively.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by a grant to A. R. and X. V. from Ministerio de Ciencia, Innovación y Universidades (PID2023-147298NB-I00 funded by MCIU/AEI/10.13039/501100011033/FEDER, UE). IRB Barcelona is the recipient of institutional funding from MICINN through the Centres of Excellence Severo Ochoa Award and from the CERCA Program of the Catalan Government. We thank Generalitat de Catalunya for grant 2021 SGR 00866. Y. W. thanks MICINN for a predoctoral fellowship.References
-
G. Q. Lin, J. G. Zhang and J. F. Cheng, in Chiral Drugs: Chemistry and Biological Action, 2011, pp. 3–28 Search PubMed
.
- A. Baeza and A. Pfaltz, Chem. – Eur. J., 2010, 16, 4003–4009 CrossRef CAS PubMed
- A. Cabré, X. Verdaguer and A. Riera, Chem. Rev., 2022, 122, 269–339 CrossRef PubMed
- A. N. Kim and B. M. Stoltz, ACS Catal., 2020, 10, 13834–13851 CrossRef CAS PubMed
- J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130–2169 CrossRef CAS PubMed
- S. F. Zhu and Q. L. Zhou, Acc. Chem. Res., 2017, 50, 988–1001 CrossRef CAS PubMed
- C. Margarita and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 1346–1356 CrossRef CAS PubMed
- E. Salomó, S. Orgué, A. Riera and X. Verdaguer, Angew. Chem., Int. Ed., 2016, 55, 7988–7992 CrossRef PubMed
- A. Cabré, E. Romagnoli, P. Martínez-Balart, X. Verdaguer and A. Riera, Org. Lett., 2019, 21, 9709–9713 CrossRef PubMed
- M. Biosca, P. de la Cruz-Sánchez, J. Faiges, J. Margalef, E. Salomó, A. Riera, X. Verdaguer, J. Ferré, F. Maseras, M. Besora, O. Pàmies and M. Diéguez, ACS Catal., 2023, 13, 3020–3035 CrossRef CAS PubMed
- M. Biosca, E. Salomó, P. De La Cruz-Sánchez, A. Riera, X. Verdaguer, O. Pàmies and M. Diéguez, Org. Lett., 2019, 21, 807–811 CrossRef CAS PubMed
- E. Salomó, A. Gallen, G. Sciortino, G. Ujaque, A. Grabulosa, A. Lledós, A. Riera and X. Verdaguer, J. Am. Chem. Soc., 2018, 140, 16967–16970 CrossRef PubMed
- Y. Wen, M. Fernández-Sabaté, A. Lledós, G. Sciortino, J. Eills, I. Marco-Rius, A. Riera and X. Verdaguer, Angew. Chem., Int. Ed., 2024, 63, e202404955 CrossRef CAS PubMed
- J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed
- M. Gupta, C. Hagen, R. J. Flesher, W. C. Kaska and C. M. Jensen, Chem. Commun., 1996, 36, 2083–2084 RSC
- P. Paredes, J. Díez and M. P. Gamasa, Organometallics, 2008, 27, 2597–2607 CrossRef CAS
- C. P. Owens, A. Varela-Álvarez, V. Boyarskikh, D. G. Musaev, H. M. L. Davies and S. B. Blakey, Chem. Sci., 2013, 4, 2590–2596 RSC
- L. Qian, X. Tang, Z. Huang, Y. Wang, G. Liu and Z. Huang, Org. Lett., 2021, 23, 8978–8983 CrossRef CAS PubMed
- L. Qian, X. Tang, Y. Wang, G. Liu and Z. Huang, Chin. J. Chem., 2022, 40, 1131–1136 CrossRef CAS
- L. Qian, C. Yu, L. Gan, X. Tang, Y. Wang, G. Liu, X. Leng, Z. Sun, Y. Guo, X. S. Xue and Z. Huang, J. Am. Chem. Soc., 2024, 146, 3427–3437 CrossRef CAS PubMed
- M. P. Jensen, S. J. Lange, M. P. Mehn, E. L. Que and L. Que, J. Am. Chem. Soc., 2003, 125, 2113–2128 CrossRef CAS PubMed
- T. Wang, X. Q. Hao, J. J. Huang, K. Wang, J. F. Gong and M. P. Song, Organometallics, 2014, 33, 194–205 CrossRef CAS
- We believe the remaining 8% corresponds to an isomer of 6-OTf with a hydride peak at −23.46 ppm (d, J = 20.6 Hz). The mixture provides a single peak by HRMS.
- For steric and electronic reasons, we believe that hydrogen is a better ligand at the iridium reactive site and thus no major product inhibition was observed.
- T. Wang, L. G. Zhuo, Z. Li, F. Chen, Z. Ding, Y. He, Q. H. Fan, J. Xiang, Z. X. Yu and A. S. C. Chan, J. Am. Chem. Soc., 2011, 133, 9878–9891 CrossRef CAS PubMed
- B. Tutkowski, S. Kerdphon, E. Limé, P. Helquist, P. G. Andersson, O. Wiest and P. O. Norrby, ACS Catal., 2018, 8, 615–623 CrossRef CAS
- G. E. Dobereiner, A. Nova, N. D. Schley, N. Hazari, S. J. Miller, O. Eisenstein and R. H. Crabtree, J. Am. Chem. Soc., 2011, 133, 7547–7562 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: HPLC/GC methods, chromatograms, NMR spectra and X-ray crystallographic data. CCDC 2430455 and 2430460. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00667h |
| This journal is © The Royal Society of Chemistry 2025 |