
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceσ-BH activation of NHC → BH3 fragments in dinuclear Cu complexes†
Aurèle
Camy
a,
David
Martins-Bessa
a,
Laure
Vendier
a,
Israel
Fernández
 *b and
Sébastien
Bontemps
*a
*b and
Sébastien
Bontemps
*a
aLCC-CNRS, Université de Toulouse, CNRS, 205 route de Narbonne, 31077 Toulouse Cedex 04, France. E-mail: sebastien.bontemps@lcc-toulouse.fr
bDepartamento de Química Orgánica I and Centro de Innovación en Química Avanzada (ORFEO—CINQA), Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: israel@quim.ucm.es
First published on 1st April 2025
Abstract
We explored herein the coordination properties of the bis(phosphine)NHC → BH3 ligand toward cationic and neutral Cu centres. The synthesis of three dinuclear Cu complexes featuring σ-BH interactions is reported. Noticeably, in each Cu complex, a BH moiety bridges two Cu centres via two σ-BH interactions in a μ2–η1η1 BH coordination mode. NMR, IR, deuterium incorporation and X-ray diffraction analyses, supported by DFT investigation, enabled us to finely measure the level of activation of the B–H bond depending on the coordination environment. The study shows a gradual increase of activation whether the BH fragment is involved in none, one or two σ-BH interactions with Cu centres.
Introduction
The activation of B–H bonds at a metal centre is a key elementary step in hydroboration and dehydrogenative borylation reactions, often consisting of the formation of a σ-BH interaction with a transition metal centre (TM).1 Such interaction has been investigated with a variety of anionic boron compounds featuring a BH fragment such as (i) the simple borohydride [BH4]−,2 (ii) scorpionate ligands and related systems3 and (iii) anionic carboranes.4 Notably, σ-BH interactions have been reported with anionic BH fragments in mononuclear and dinuclear Cu complexes. In dinuclear Cu systems, three types of coordination modes were characterised (Chart 1a): (i) [BH4]− was shown to bridge two Cu centres via two η2 σ-BH interactions (Chart 1a, I),5 (ii) diborate systems were shown to bridge two Cu centres via two η1 σ-BH interactions of two BH fragments (Chart 1a, II),4,6 and (iii) finally, a BH fragment can bridge two Cu centres via two σ-BH interactions of the same B–H bond in a μ2–η1η1 BH coordination mode (Chart 1a, III).4a,6b | ||
| Chart 1 (a–c) Relevant literature precedents of σ-BH interactions with metal centres, (d) present work. | ||
Complementarily, the coordination of Lewis base-stabilised borane compounds (LB → BH3)7 enabled us to characterize σ-BH interactions with neutral systems.1,8 In the case of Cu, examples are limited to mononuclear Cu adducts of two different nitrogen-based LB (R3N → BH3) (Chart 1b).9 There is however no example of NHC → BH3 coordination to Cu. In fact, despite the high versatility of N-heterocyclic carbene (NHC), σ-BH interactions with NHC → BH3 compounds were characterised only in Mn, Cr, Mo and W complexes (Chart 1c).10 In these compounds, the σ-BH bond features a weak end-on η1 coordination of the NHC → BH3 ligand with a wide B–H–TM angle and negligible π-back bonding from the metal to the σ*(B–H) orbital. In the solid state, the B–H bond is negligibly elongated compared to the other B–H bonds, and in solution the three hydrides are in fast exchange, even at low temperature. As a result of the weak interaction, the NHC → BH3 fragment was shown to be easily displaced in the Mn complex.10 The labile nature of the coordination of NHC → BH3 could explain the scarcity of studies on this topic.
Recently, we synthesised a new bis(phosphine) ligand platform aiming at stabilizing two metal centres with a bridging NHC → BH3 central moiety. We reported the coordination chemistry of this ligand toward the Au(I) precursor, leading to the complete breaking of a B–H bond and the formation of an original Au4 cluster after reductive elimination of dihydrogen.11 Pursuing the exploration of coinage metal, we report herein the coordination of this ligand platform toward neutral and cationic Cu(I) precursors and show that (i) σ-BH interactions of NHC → BH3 fragments can be observed with Cu and (ii) the ligand platform supports dinuclear Cu systems (Chart 1d). In this context, various σ-BH interactions were characterised. X-ray diffraction, IR and DFT analyses were used to finely assess the different levels of B–H activation in the three dinuclear copper complexes synthesised.
Results and discussion
Bis(phosphine)NHC → BH3 ligand A reacted cleanly with two equivalents of the cationic copper complex [Cu(CH3CN)4][BF4] in dichloromethane (CH2Cl2) to afford complex 1, isolated in 73% yield (Scheme 1). The in situ31P{1H} NMR monitoring of the reaction indicated the quasi-complete replacement of the ligand signal at 17.6 ppm by a singlet at 30.7 ppm within 3 h. The BH3 fragment was characterised by a quartet at −33.1 ppm (1JB–H = 85 Hz) in 11B NMR analysis and by a broad signal between 3.4 and 2.4 ppm in 1H NMR analysis. Upon selective 11B decoupling, the BH3 fragment appears as a triplet in 1H{11B} NMR analysis due to a 2JH–P coupling of 11.4 Hz (cf. the ESI† for multinuclei selective and broad-band decoupling analyses). While solid-state analyses showed that the B–H bonds are involved in three different types of coordination toward the dinuclear copper core (vide infra), low temperature did not enable the freezing of the exchange process between the three H atoms of the BH3 moiety in NMR analyses (Fig. S15–17†). Lowering the temperature led to the replacement of the broad quartet by a broad singlet at 183 K, presumably due to quadrupole-induced thermal decoupling between H and B, similar to what was shown with NHC → BH3 coordinated to the W centre (Chart 1c)10 and in a L2Cu(BH4) complex.12 The chemical shift of the carbenic carbon (deduced from the coupling with methylene protons in the 2D HMBC 13C–1H NMR spectrum) is towards high fields compared to the ligand (δC 167.6 in 1, 183.3 in A). | ||
| Scheme 1 Synthesis of compound 1. | ||
Colourless crystals suitable for X-ray diffraction (XRD) analysis were grown from the slow diffusion of pentane into a concentrated CH2Cl2 solution. This analysis showed that compound 1 is a dicationic dinuclear complex stabilised by one ligand A and three acetonitrile molecules (Fig. 1a). Each Cu centre is linked to a phosphine moiety, while the central NHC → BH3 fragment bridges both Cu centres, each hydride being involved in a different coordination mode. More precisely, while H100 remains too far from the metal centres for any interaction, H101 bridges Cu1 and Cu2 in a μ2–η1η1BH coordination mode and H102 interacts with Cu2 in an η1BH coordination mode. Acetonitrile molecules complete the tetra-coordinated environment of the Cu centres.
 | ||
| Fig. 1 (a) Structure of complex 1. Thermal ellipsoids are drawn at 30% probability (co-crystallised CH2Cl2, two BF4 anions and hydrogen atoms are omitted for clarity except for the BH3 fragment). H100, H101 and H102 were located in the difference Fourier map and refined freely without constraint; (b) selected distances and angles in complex 1 from XRD and DFT analyses. | ||
With complex 1 featuring BH fragments in three different coordination environments in hand, we aimed at evaluating the level of activation of the B–H bonds depending on the bonding situation. While (i) the crystal structure is of acceptable quality and (ii) the hydrogen atoms of the BH3 fragment were located on the Fourier map and refined freely without constraint, the localization of hydrogen atoms is not good enough in the X-ray diffraction method for an accurate comparison of the B–H bond distances. Theoretical investigations were thus used in support of the XRD analysis to discuss the B–H distances.13 Geometry optimization was performed on complex 1, starting from the crystal structure, with the BP86/def2-TZVP level of theory using the D3 dispersion correction and the resolution-of-identity (RI) approximation (RI-BP86-D3/def2-TZVP). Vibrational analysis was performed to ensure that the found stationary point corresponds to an energy minimum, at the same RI-BP86-D3/def2-TZVP level. The main structural parameters obtained in the XRD and DFT analyses are shown in Fig. 1b. As expected, the major differences involve the hydrogen atoms. The DFT provided B–H bond distances of 1.205, 1.246 and 1.298 Å for H100, H102 and H101, respectively. This indicates a continuum of activation of the B–H bond whether it is involved in none (H100), one (H102) or two (H101) σ bonds with the metal centres. The B–H–Cu angle of 123.2° with Cu1 is comparable to the 127.09° of the related Mn complex,10 while the angles are more acute with Cu2 (91.6° and 89.7°).
The evaluation of the B–H bond activation was also performed by IR analysis. In order to help in the determination of the ν(B–H) stretching frequencies, the corresponding 1–BD3 complex was synthesised and theoretical IR spectra of complexes 1 and 1-BD3 were simulated (see ESI_IR_ppt†). The 1-BD3 complex was prepared on a small scale (0.055 mmol) and analysed by IR in situ, from the reaction of the ligand A-BD3 with [Cu(CH3CN)4][BF4]. A-BD3 was synthesised with 82% deuterium enrichment from the reaction between the bis(phosphine) carbene compound and Me3N → BD3. This BD3 source was prepared by deuteriation of Me3N → BH3 with D2 catalysed by the ruthenium polyhydride catalyst Ru(H)2(H2)2(PCy3)2 (see the ESI†), which was used previously for the deuteriation of HBpin.14
The experimental and theoretical frequencies of the B–H and B–D stretching modes are reported in Fig. 2. The ν(B–H/D) frequencies were calculated to be 2495, 2213 and 1901 cm−1, attributed to B-H100, B-H102 and B-H101, respectively, for complex 1 and were 1841, 1625 and 1383 cm−1 for the corresponding B–D bonds in 1-BD3.
 | ||
Fig. 2 Experimental and theoretical stretching frequencies (cm−1) of B–H, B–D and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds in 1 and 1-BD3. N bonds in 1 and 1-BD3. | ||
In the experimental IR spectra, the ν(B–H and B–D) of the non-interacting BH/BD and of the σ-BH/BD interacting with one Cu centre could be assigned. The latter appears close to the stretching bands of the acetonitrile (Fig. 2). However, the ν(B–H and B–D) of the fragment bridging two Cu centres could not be detected. Since the area in which the ν(B–H) of the bridging fragment is relatively clear of other signals, we believe that the signal must be too weak or too broad for detection. In the case of the ν(B–D), the theoretical value indicates that this band could be overlapping with other vibrational bending modes. The low frequency calculated for ν(B–H101) of 1901 cm−1 is noticeable since it is lower than the 1933 cm−1 measured for a diphosphite ((OEt)3P)2Cu–BH4 complex,15 the lowest ν(B–H) involved in σ-BH with Cu that was found in the literature.16
The nature of the σ-BH bonding was then explored by means of QTAIM (Quantum Theory of Atoms in Molecules) and NBO (Natural Bond Orbital) calculations. The QTAIM calculations (Fig. 3a) located three different Cu–H bonds according to the occurrence of three bond critical points (BCPs) along with the corresponding bond paths running between Cu1, Cu2 and H102 and H101 atoms. Cu2 is linked to H102 and H101, resulting in a BH2Cu four-membered ring which is characterised by the presence of the corresponding ring critical point (RCP, red sphere in Fig. 3a), while Cu1 is solely attached to H101.
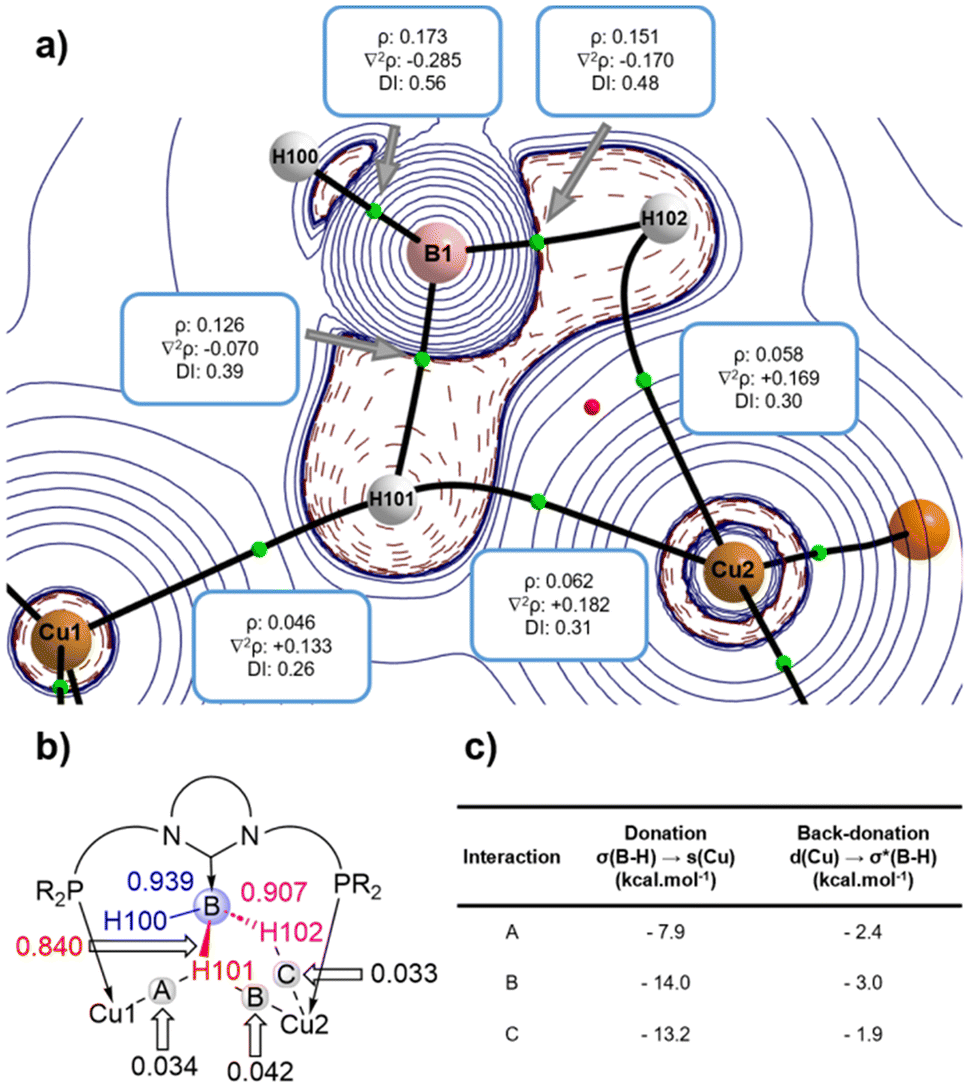 | ||
| Fig. 3 (a) Contour map of the Laplacian function (∇2ρ) of compound 1 in the B–H101–Cu2–Cu1 molecular plane (green and red spheres represent BCPs and RCPs, respectively) together with relevant QTAIM values, (b) relevant Wiberg Bond Indexes (WBI) obtained by NBO analysis and (c) strength and decomposition of B–H⋯Cu interactions determined by the SOPT-NBO analysis. | ||
The electron density ρ, the delocalization index (DI) and the electron density Laplacian ∇2ρ values enabled a more quantitative description of the B–H and Cu–H interactions (Fig. 3a). According to the sign of ∇2ρ values, the B–H bonds are covalent (i.e., electron-sharing) while the Cu–H bonds are dative. The electron density around the B–H101 bond is shared between both copper centres, which results in a smaller ∇2ρ value computed for BCP(B-H101) than for BCP(B-H102), thus indicating a weaker bond. This feature is further supported by the corresponding DI values (Fig. 3a) and the NBO-WBIs (Wiberg Bond Index) (Fig. 3b), which follow the same trend (DI: 0.39 vs. 0.48; WBI: 0.840 vs. 0.907). Interestingly, although H101 interacts with both Cu centres, the interaction with Cu2 is stronger than that with Cu1, as suggested by both the WBI (0.042 vs. 0.034) and DI values (0.31 vs. 0.26).
Further quantitative insight into the bonding can be gained with the Second Order Perturbation Theory (SOPT) of the NBO method. This SOPT-NBO approach indicates that the σ-BH–Cu interactions can be viewed as dative bonds consisting of the donation of electron density from the doubly occupied σ(B–H) molecular orbital, for H102 and H101, to the empty 4s(Cu) atomic orbital coupled with a backdonation from doubly occupied 3d(Cu) atomic orbitals towards the vacant σ*(B–H). As expected, the associated SOPT-stabilization energies (ΔE(2), Fig. 3c) indicated that the donations are stronger (−7.9 to −14.0 kcal mol−1) than the backdonations (−2.4 to −3.0 kcal mol−1).17 In agreement with the relative strength of the bonds based on the WBI and DI values commented above, the SOPT-NBO values confirm that the Cu2–H101 interaction is stronger than the Cu1–H101 interaction.
The coordination of compound A toward neutral copper chloride (CuCl) was then investigated. The in situ monitoring of the reaction by 31P{1H} NMR suggested dynamic processes at play in the coordination toward one, two and three equivalents of CuCl (Fig. 4). After the addition of one equivalent of CuCl on the ligand A, the sharp signal of compound A at 17.6 ppm (Fig. 4a) was replaced by two broad signals at 17.6 and 21.7 ppm after 90 min (Fig. 4b). While this feature suggested the expected coordination of one phosphine arm, the other phosphine moiety remaining pendent, and the situation further evolved at room temperature since, after 96 h, the signal at 17.6 ppm disappeared and a very large signal appeared at around 25.2 ppm, while the signal at 21.7 ppm remained (Fig. 4c). Upon the addition of a second equivalent of CuCl, the signal at 21.7 ppm disappeared and the signal at 25.2 ppm broadened significantly (Fig. 4d). The system kept evolving with time since after 24 h, a new signal at around 30.3 ppm appeared along with the signal at 25.2 ppm (Fig. 4e). It is only with the addition of a third equivalent of CuCl that a single signal is observed at 30.3 ppm which did not evolve with time (Fig. 4f).
 | ||
| Fig. 4 Stacked 31P{1H} NMR spectra in CD2Cl2 at room temperature of the reaction between compound A and one, two and three equivalents of CuCl. | ||
Following this in situ monitoring, we first aimed at synthesizing the species featuring the 31P{1H} NMR signal at around 30.3 ppm. Under the optimised stoichiometric conditions, the addition of 2.5 equivalents of CuCl to the ligand A in CH2Cl2 led to the clean formation of complex 2 in 2.5 h (Fig. 5). Complex 2 was isolated in only 40% yield, because several CH2Cl2 washings were necessary to remove the excess of CuCl. Elemental analysis showed nonetheless that a small amount of CuCl remained. Complex 2 was characterised in solution by broad signals at 30.1 and −33.9 ppm in 31P{1H} and 11B NMR spectra, respectively. The BH3 fragment was further characterised in 1H NMR analysis by a broad signal at 2.59 ppm, which sharpened upon 11B decoupling (cf. the ESI†). Monocrystals suitable for X-ray diffraction (XRD) analysis were grown from a saturated CH2Cl2/diethyl ether solution. Complex 2 features a cluster of four copper centres bridged by three chloride atoms (Cu4Cl3) (Fig. 5). Halogen-bridged Cu complexes are commonly encountered with copper halide precursors.18 Upon complexation, a chloride has been formally abstracted from a presumed Cu4Cl4 core by the additional equivalent of CuCl resulting in a cationic Cu4Cl3 cluster and an anionic CuCl2 counterion. The chloride abstraction appears easily accessible since it is already observed for a sub-stoichiometric equivalent of CuCl (eqn (2), Fig. 4e). The cluster is stabilised by two ligands A: one phosphine moiety for each Cu centre and a bridging BH moiety in a μ2–η1η1BH coordination mode, like in complex 1, for each ligand part. The two other BH fragments do not interact with the Cu centres, but exhibit hydrogen bonding with the protic hydrogen atom of the methylene arm of the ligand with H⋯H distances of 2.27 and 2.31 Å, associated with C–H–H angles of 112° and 111°, and B–H–H angles of 113° and 114°. This feature was observed in the free ligand A11 but not in complex 1. Compared to 1, NBO calculations indicated that the bridging μ2–η1η1BH coordination mode consists of the same donation/backdonation interaction having rather similar stabilization energies: −12.9 and −13.5 kcal mol−1 for the σ(B–H) → s(Cu) donation and −2.6 and −2.7 kcal mol−1 for the d(Cu) → σ*(B–H) backdonation. WBI indicates a slightly weaker B–H bond (0.789 for 2vs. 0.840 for 1) and stronger Cu–H interactions (0.055 and 0.058 for 2vs. 0.034 and 0.042 for 1).
 | ||
| Fig. 5 (a) Synthesis of complexes 2 and 3; (b and c) crystal structures of complexes 2 and 3, respectively. Thermal ellipsoids are drawn at 30% probability, hydrogen atoms are omitted for clarity except for the BH3 fragment, and they were located in the difference Fourier map and refined freely without any constraint. | ||
The shortest Cu–Cu bond length of 2.9174(6) Å for 2 is beyond the sum of copper van der Waals radii (2.80 Å).19 Cuprophilic interactions are nonetheless characterised within this range.20 The analysis of these interactions using the NBO method calculations indicates that this interaction is rather weak (ΔE(2) < 0.5 kcal mol−1) and can be considered as negligible between Cu centres. In the case of 1, the Cu–Cu bond length is larger (>3.5 Å) and no interaction is detected according to QTAIM (no BCP (Cu–Cu) was located) or NBO analysis.
Different reactivities of complex 2 were then investigated. We first probed if H/D exchange could occur with D2. However, in the presence or not of a catalytic amount of Ru(H)2(H2)2(PCy3)2, used for the deuteriation of Me3N → BH3 (see the ESI†), no H/D exchange could be detected, with complex 2 remaining unchanged. In contrast, the reaction with borohydride reactants NaBH4 or NaHBEt3 led to the rapid decomposition of complex 2. Interestingly, the reaction of potassium graphite (KC8) with complex 2 led to the clean formation of the neutral complex 3 (Fig. 5). Complex 3 is characterised in solution by broad signals at 27.1 (s) and −33.1 ppm (quart, 1JB–H = 88 Hz) in 31P{1H} and 11B NMR spectra, respectively, close to the signal of complex 2. The XRD analysis indicated that complex 3 is composed of two ligands A, four Cu and four Cl atoms (Fig. 5c). In this reaction, KC8 formally removes one equivalent of CuCl from 2 to generate 3, although the exact role of KC8 remains unclear to us. The retention of the dimeric form of complexes 2 and 3 in solution was confirmed by a DOSY experiment showing lower diffusion coefficients (D = 6.3 and 6.0 × 10−10 m2 s−1, respectively) than that of ligand A (D = 9.3 × 10−10 m2 s−1). Moreover, while the phosphorus and boron moieties of complex 3 appear equivalent in solution on the NMR time scale, highlighting a fluxional behaviour already observed in its in situ generation (Fig. 4), it is not the case in the solid state. There exists a Cu3Cl3 core stabilised by (i) one ligand A, tridentated via the two phosphine moieties and a BH fragment being involved in the bridging of two Cu centres via two σ-BH interactions and (ii) only one phosphorus atom of the second ligand. The other phosphine moiety and the BH3 fragment of this second ligand stabilize a Cu–Cl counterpart via a σ-BH interaction. In this complex, there are three different BH bonding situations, similar to complex 1: a non-bonding BH, a BH bond bridging two Cu centres via two σ-BH interactions and a BH bond involved in a single σ-BH interaction with a fourth Cu centre. Overall, complexes 2 and 3 show that η1σ-BH and μ2–η1η1σ-BH coordination are also accessible in neutral Cu complexes.
Conclusions
In conclusion, we showed that σ-BH interactions could be observed with Cu in the case of NHC → BH3 fragments. The use of bis(phosphine)NHC → BH3 was key to the characterisation of these relatively weak σ-BH–Cu interactions, extending the family of coordinated NHC → BH3 to Cu centres (Chart 1c).10 In addition, the present results show that the ligand platform stabilises dinuclear systems in the case of Cu, in addition to the Au complexes reported earlier.11Three different structures of cationic and also neutral Cu complexes were synthesised. The σ-BH interactions are composed of the σ(B–H) bond donation to the 4s orbital of the Cu centres coupled with a comparatively weak backdonation from the 3d orbitals of Cu to the σ*(B–H) orbital. More precisely, σ-BH interactions could be involved in either η1BH or μ2–η1η1 BH coordination mode. The coordination proved to be dynamic in solution in the case of the neutral CuCl precursor, leading to the isolation of complexes 2 and 3. In complex 1, the presence of three different coordination environments for the same BH3 fragment enabled us to finely assess the level of activation of these B–H bonds thanks to in-depth investigations by XRD, NMR and IR supported by DFT. It shows a gradual increase of activation whether the BH fragment is involved in none, one or two σ-BH interactions with Cu centres.
Further work is ongoing in our laboratories toward the variation of the boron fragment in such bis(phosphine)NHC → BR3 platforms.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2429977–2429979 for 1–3 and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Conflicts of interest
There are no conflicts to declareAcknowledgements
AC, DM-B and SB thank the CNRS and the French Ministry for financial support. This work has benefited from French State aid managed by the Agence Nationale de la recherche under France 2030 plan (ANR-22-PESP-0010 Projet ciblé “POWERCO2”) within the PEPR project SPLEEN. Dr Aswin Chandran is acknowledged for the synthesis of RuH2(H2)2P(Cy3)2. IF is thankful for the financial support from the Spanish MCIN/AEI/10.13039/501100011033 (PID2022-139318NB-I00 and RED2022-134287-T). For the purpose of Open Access, a CC-BY public copyright license has been applied by the authors to the present document and will be applied to all subsequent versions up to the Author Accepted Manuscript arising from this submission.References
-
(a)
I. M. Riddlestone, J. A. B. Abdalla and S. Aldridge, in Adv. Organomet. Chem., ed. P. J. Pérez, Academic Press, 2015, vol. 63, pp. 1–38 Search PubMed
; (b) K. Saha, D. K. Roy, R. D. Dewhurst, S. Ghosh and H. Braunschweig, Acc. Chem. Res., 2021, 54, 1260–1273 CrossRef CAS
-
(a) Z. Xu and Z. Lin, Coord. Chem. Rev., 1996, 156, 139–162 CrossRef CAS
-
(a) M. Jackson, S. D. Thomas, G. J. Tizzard, S. J. Coles and G. R. Owen, Molecules, 2023, 28, 4825 CrossRef CAS PubMed
-
(a) A. E. Dziova, V. V. Avdeeva, I. N. Polyakova, L. V. Goeva, E. A. Malinina and N. T. Kuznetsov, Russ. Chem. Bull., 2011, 60, 1608–1611 CrossRef CAS
-
(a) V. Ferraro, J. Castro, E. Trave and M. Bortoluzzi, J. Organomet. Chem., 2022, 957, 122171 CrossRef CAS
-
(a) M. Hata, J. A. Kautz, X. L. Lu, T. D. McGrath and F. G. A. Stone, Organometallics, 2004, 23, 3590–3602 CrossRef CAS
- It is noteworthy that the use of neutral diborane stabilized by phosphine or an amine Lewis base was shown to give rise to dinuclear Cu complexes featuring two σ-BH interactions following the coordination mode II (Chart 1a), see:
(a) A. Wagner, E. Kaifer and H.-J. Himmel, Chem. – Eur. J., 2013, 19, 7395–7409 CrossRef PubMed
- P. Ríos, A. Rodríguez and S. Conejero, Chem. Sci., 2022, 13, 7392–7418 RSC
-
(a) A. Hicken, A. J. P. White and M. R. Crimmin, Inorg. Chem., 2017, 56, 8669–8682 CrossRef CAS PubMed
- P. Bissinger, H. Braunschweig, T. Kupfer and K. Radacki, Organometallics, 2010, 29, 3987–3990 CrossRef CAS
- A. Camy, L. Vendier, C. Bijani, I. Fernández and S. Bontemps, Inorg. Chem., 2023, 62, 9035–9043 CrossRef CAS PubMed
-
(a) J. C. Bommer and K. W. Morse, Inorg. Chem., 1978, 17, 3708–3710 CrossRef CAS
- DFT was shown to accurately locate hydride in σ bonding, notably toward Ru, see:
(a) M. Grellier, S. A. Mason, A. Albinati, S. C. Capelli, S. Rizzato, C. Bijani, Y. Coppel and S. Sabo-Etienne, Inorg. Chem., 2012, 52, 7329–7337 CrossRef
-
(a) S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS
- V. D. Makhaev, A. P. Borisov, É. B. Lobkovskii, V. B. Polyakova and K. N. Semenenko, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1985, 34, 1731–1736 CrossRef
- N. V. Belkova, I. E. Golub, E. I. Gutsul, K. A. Lyssenko, A. S. Peregudov, V. D. Makhaev, O. A. Filippov, L. M. Epstein, A. Rossin, M. Peruzzini and E. S. Shubina, Crystals, 2017, 7, 318 CrossRef
- Although values below 5 kcal mol−1 given by the SOPT-NBO method should be taken with caution, the method clearly indicates that the backdonations are weaker than the corresponding donations.
- R. Peng, M. Li and D. Li, Coord. Chem. Rev., 2010, 254, 1–18 CrossRef CAS
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS
-
(a) M. Kakavand, M. Cheraghi, A. Mahdavi, A. Neshat, A. Kozakiewicz-Piekarz, P. Bazargani and Y. Balmohammadi, Inorg. Chem., 2025, 64, 1272–1286 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2429977–2429979. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00586h |
| This journal is © The Royal Society of Chemistry 2025 |