
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The “pogo stick” complex [FeCp*{N(SiMe3)2}]: spin state properties, adduct formation with Lewis bases, and reactivity towards weakly Brønsted acidic protonated NHCs†
Julian
Zinke
a,
Clemens
Bruhn
a,
Serhiy
Demeshko
b and
Ulrich
Siemeling
 *a
*a
aInstitute of Chemistry, University of Kassel, Heinrich-Plett-Straße 40, 34132 Kassel, Germany. E-mail: siemeling@uni-kassel.de
bInstitut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstraße 4, 37077 Göttingen, Germany
First published on 3rd March 2025
Abstract
[FeCp*{N(SiMe3)2}] is a high-spin complex with four unpaired electrons according to SQUID magnetometry and Mößbauer spectroscopy. Its reactions with 4-dimethylaminopyridine (DMAP) and NnPr4X (X = Cl, Br) respectively afford [FeCp*{N(SiMe3)2}(DMAP)] (6), NnBu4[FeCp*Cl{N(SiMe3)2}] (7), and NnPr4[FeCp*Br{N(SiMe3)2}] (8). 7 and 8 react with formamidinium tetrafluoroborates fc[(NCH2R)2CH][BF4] (fc = 1,1′-ferrocenylene; R = Ph, Mes) at room temperature in THF to furnish [FeCp*X{fc[(NCH2R)2C]}] (4: X = Cl, R = Mes; 10: X = Br, R = Mes; 11: X = Cl, R = Ph; 12: X = Br, R = Ph), which contain a ferrocene-based NHC ligand. No formation of 10 from fc[(NCH2Mes)2CH][BF4] and 8 takes place under the same mild conditions in toluene, instead leading to the isolation of fc[(NCH2Mes)2CH][FeCp*Br{N(SiMe3)2}] (9). In contrast to its halide adducts 7 and 8, [FeCp*{N(SiMe3)2}] is inert towards formamidinium tetrafluoroborates. It reacts readily, however, with formamidinium halides, affording [FeCp*Cl(IPr)] (1, IPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene) with 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride and [FeCp*I(BzIMe)] (2, BzIMe = 1,3-dimethylbenzimidazolin-2-ylidene) or, depending on the crystallisation conditions, [FeCp*(BzIMe)2][FeCp*I2] (3) with 1,3-dimethylbenzimidazolium iodide, in accord with an equilibrium of the type 2 [FeCp*X(NHC)] ⇄ [FeCp*(NHC)2]+ + [FeCp*X2]−. The major product formed with fc[(NCH2Mes)2CH]Cl is fc[(NCH2Mes)2CH][FeCp*Cl2] (5) instead of the expected NHC complex [FeCp*Cl{fc[(NCH2Mes)2C]}] (4). Halide abstraction reactions attempted with [FeCp*X{fc[(NCH2R)2C]}] afforded only intractable material. [FeCp*{fc[(NCH2Ph)2C]}2][PF6] (13) was serendipitously obtained in this context in trace amounts from 12 and Tl[PF6]. Compounds 1–13 were structurally characterised by single-crystal X-ray diffraction. Metric parameters reveal different electronic configurations for 1 and its solvate 1·0.5 benzene (two vs. four unpaired electrons).
Introduction
The one-legged piano stool (“pogo stick”) structure of the half-sandwich iron(II) complex [FeCp*{N(SiMe3)2}] (Cp* = η5-pentamethylcyclopentadienyl) was unprecedented for open-shell organometallics when we reported this compound in 1998.1 Analogous structurally characterised complexes containing bulkier cyclopentadienyl ligands, viz. [FeCp′{N(SiMe3)R}] (Cp′ = η5-1,2,4-tri-tert-butylcyclopentadienyl; R = CMe3, C6H3-2,6-iPr2) and [Fe5Cp{N(SiMe3)2}] (5Cp = η5-pentaisopropylcyclopentadienyl),2,3 have subsequently been published by the groups of Walter and Sitzmann.4 As demonstrated by Ohki and Tatsumi, the Brønsted basic amido ligand present in [FeCp*{N(SiMe3)2}] allows specific reactions with suitable Brønsted acids under liberation of hexamethyldisilazane.5 The dinuclear complex [{FeCp*(μ-NMePh)}2] was obtained with N-methylaniline,5b and reactions with the N-heterocyclic carbene (NHC) precursors 1,3-dimesitylimidazolium chloride and 1,3-diisopropyl-4,5-dimethylmidazolium chloride afforded the NHC chlorido complexes [FeCp*Cl(IMes)] (IMes = 1,3-dimesitylimidazolin-2-ylidene) and [FeCp*Cl(MeIiPr)] (MeIiPr = 1,3-diisopropyl-4,5-dimethylimidazolin-2-ylidene).5a,6 The latter reactions leading to half-sandwich NHC complexes serve as the starting point for the work presented in the current paper. Our focus is on 1,1′-ferrocenylene-bridged, and hence redox-functionalised, NHCs of the type fc[(NR)2C:] (fc = 1,1′-ferrocenylene),7 which are special ring-expanded N-heterocyclic carbenes (reNHCs).8,9 The N–C–N angles of reNHCs are significantly larger than those of their five-membered ring counterparts (100–106°).10 As a consequence, reNHCs exhibit a higher steric impact together with a more pronounced ambiphilicity, and hence higher reactivity and lower thermal stability, than standard five-membered NHCs.8,9,11 Structurally characterised examples of the type fc[(NR)2C:] have particularly large carbene bond angles in the range from 119–122°,12 similar to the values determined for acyclic diaminocarbenes (ca. 121°).13 Thermal stability has been achieved for fc[(NR)2C:] only with rather bulky N-substituents. For example, among the congeners bearing primary alkyl substituents CH2R′, isolation was possible for R′ = tBu and Mes (Mes = mesityl),12a,c but not for R′ = H, iPr and Ph.7,12a,14 The steric demand of NHC ligands is reflected by their percent buried volume (%Vbur) value, which may be calculated from structural data of NHC metal complexes (usually [MX(NHC)] with M = Cu, Au and X = Cl, Br) using the SambVca 2.1 web application.15 The comparatively high steric demand of reNHCs is illustrated by %Vbur values determined from [MX(NHC)] for a series of N-benzyl substituted NHCs with different ring sizes, increasing from 31.5% for the five-membered imidazole-based NHC (IBn)16 to 33.6% for the six-membered and 36.1% for the seven-membered NHC.11b From a formal point of view, 1,1′-ferrocenylene-bridged NHCs fc[(NR)2C:] exhibit a six-membered FeC2N2C ring, whose Fe atom is much larger than the other atoms. Not surprisingly, therefore, the %Vbur value of 34.9% determined for fc[(NCH2Ph)2C:] is in between those of the six- and the seven-membered reNHCs; the thermally stable mesityl-containing congener fc[(NCH2Mes)2C:] has a significantly larger value of 38.0%.12aResults and discussion
Magnetic properties and electronic structure of [FeCp*{N(SiMe3)2}]
Before coming to our preparative work, we take the opportunity to address in detail the magnetic properties and electronic structure of [FeCp*{N(SiMe3)2}], which we had described as diamagnetic in C6D6 solution in our brief report published in 1998.1 This was soon noted to be unexpected17 and proved to be wrong. In our original publication, NMR signals due to diamagnetic impurities (silicon grease and decamethylferrocene) had erroneously been assigned to the product.18 Due to the paramagnetic nature of [FeCp*{N(SiMe3)2}], its NMR signals are broad and located far outside the region typical of diamagnetic compounds. The 1H NMR spectrum exhibits two signals at δ = 225 (ν½ 1180 Hz) and 44 ppm (ν½ 1440 Hz), which integrate for 15 and 18 protons, respectively, compatible with an assignment to Cp* and N(SiMe3)2, respectively (see Fig. S1 in the ESI†). For comparison, the 1H NMR signal due to the N(SiMe3)2 unit is observed at 39 and 45 ppm, respectively, for [Fe5Cp{N(SiMe3)2}] and [FeCp′{N(SiMe3)2}] in C6D6.2b,c Thanks to the excellent solubility of [FeCp*{N(SiMe3)2}] in benzene, a 13C{1H} NMR spectrum could be recorded, when a highly concentrated solution (ca. 400 mg mL−1) was used (see Fig. S2 in the ESI†). Two, instead of the expected three, signals were detected, which are positioned at δ = 1173 (ν½ 880 Hz) and −74 ppm (ν½ 310 Hz). We refrain from suggesting an assignment. Note that only 1H NMR data are available for [FeCp′{N(SiMe3)R}] and [Fe5Cp{N(SiMe3)2}] investigated by Walter and Sitzmann (vide supra). They demonstrated that these compounds are high-spin iron(II) complexes with four unpaired electrons (S = 2), suggesting that the same should hold true for [FeCp*{N(SiMe3)2}].2,3 This indeed turned out to be the case. We have studied the magnetic properties of our “pogo stick” complex in the solid state by SQUID magnetometry. The χMT value of 4.0 cm3 mol−1 K at 210 K unambiguously indicates an S = 2 high-spin state of iron(II) (expected spin-only value of 3.0 cm3 mol−1 K) with some orbital contribution. When the temperature was lowered, the χMT value decreased below ca. 80 K due to the pronounced zero-field splitting of −47.3 cm−1 (Fig. 1).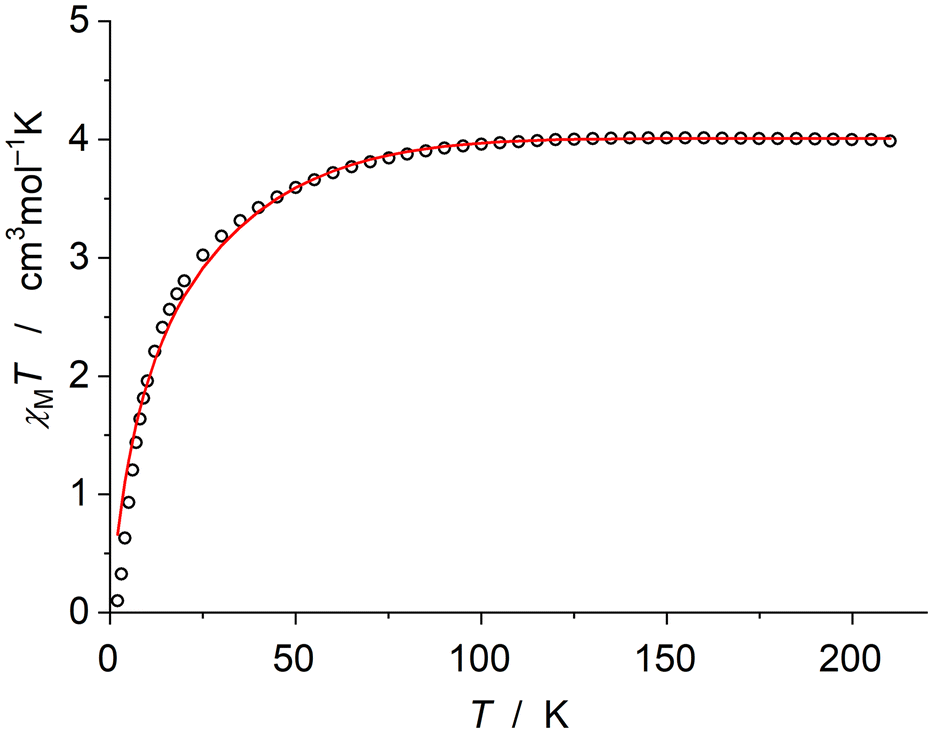 | ||
| Fig. 1 Variable-temperature χMT product for [FeCp*{N(SiMe3)2}] (open circles) and best fit curve (red line) with best fit parameters gx = gy = 1.45, gz = 2.90 and D = −47.3 cm−1 (where D is the axial zero-field splitting parameter). | ||
The large negative D parameter is an indicator of the possible slow relaxation of magnetisation between the ms = ±2 microstates separated by the energy barrier for spin reversal Ueff. The out-of-phase alternating current (ac) susceptibility (χ′′) was observed even in the zero direct current (dc) magnetic field, and analysis of the ac measurements revealed Ueff = 130 cm−1 (see the ESI† for more details), similar to that of the related [Fe5Cp{N(SiMe3)2}] with Ueff = 113 cm−1.2b
The results of Mößbauer experiments performed with [FeCp*{N(SiMe3)2}] are also in excellent agreement with a high-spin configuration of iron(II) and an S = 2 ground state, providing a typically high isomer shift δ = 0.89 mm s−1 and a quadrupole splitting ΔEQ = 0.51 mm s−1 at 7 K (Fig. 2).
 | ||
| Fig. 2 Zero-field 57Fe Mössbauer spectrum of [FeCp*{N(SiMe3)2}] at 7 K (open circles) and best fit curve (red line). | ||
Furthermore, the spectrum is additionally magnetically split by an internal magnetic hyperfine field Bhf = 95.2 T, in agreement with literature values in the range of 95 to 101 T found for similar [FeCp′{N(SiMe3)R}] complexes.2a
Synthetic work and crystal structures
In close analogy to the work by Ohki and Tatsumi mentioned above, [FeCp*{N(SiMe3)2}] was reacted in toluene with 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride (1 equivalent), which is the hydrochloride of the highly popular NHC IPr (Scheme 1).6 | ||
| Scheme 1 Synthesis of compounds 1–3 (Dipp = 2,6-diisopropylphenyl). | ||
The expected product [FeCp*Cl(IPr)] (1) was obtained as a brownish orange crystalline solid in 70% yield after work-up and crystallisation from diethyl ether or benzene. The 1H NMR spectrum of 1 (analytically pure according to CHN data; see Fig. S3 in the ESI†) shows broad signals at δ ≈ 158 (ν½ 270 Hz), 54 (ν½ 100 Hz), −13 (ν½ 120 Hz) and −14 ppm (ν½ 420 Hz), together with only slightly broadened signals at δ ≈ 1 (ν½ 16 Hz) and −12 ppm (ν½ 24 Hz). An assignment cannot be made with certainty. The eight IPr methyl groups are expected to give rise to two signals with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 integral ratio (12 H each). Consequently, the signal at 158 ppm, which exhibits the largest integral, is plausibly assigned to Cp* (15 H). However, the sum of the integrals of the remaining signals is less than half of the expected value (15 H vs. 36 H). If the signal at 158 ppm is assigned to the IPr methyl groups (assuming their accidental isochrony), the integrals of the other signals sum up to 23 H, which is reasonably close to the expected value of 27 H. For comparison, according to Ohki and Tatsumi, the 1H NMR signal of [FeCp*Cl(IMes)] due to the Cp* methyl groups is located at δ ≈ 169 ppm, while the methyl groups of the IMes ligand give rise to three signals at δ ≈ 76, −11 and −44 ppm; the mesityl CH units give rise to an extremely broad signal between −5 and −23 ppm, and the signal due to the CH units in the backbone of the IMes ligand is located at δ ≈ 39 ppm.5a In the same vein, the reaction with 1,3-dimethylbenzimidazolium iodide (1 equivalent) in THF furnished [FeCp*I(BzIMe)] (2, BzIMe = 1,3-dimethylbenzimidazolin-2-ylidene) in 63% yield, when diethyl ether was used for crystallisation (Scheme 1). The broad signals in the 1H NMR spectrum of 2 (analytically pure according to CHN data; see Fig. S4 in the ESI†) at δ ≈ 36 (ν½ 130 Hz) and −41 ppm (ν½ 160 Hz), respectively integrating for 15 and 6 protons, are assigned to the Cp* and BzIMe methyl groups, respectively, while the two only slightly broadened signals at δ ≈ 15 (ν½ 27 Hz) and 12 ppm (ν½ 21 Hz) are ascribed to the p-C6H4 backbone of the BzIMe ligand. Interestingly, crystallisation from a mixture of benzene and n-hexane furnished the isomeric, and ionic, product [FeCp*(BzIMe)2][FeCp*I2] (3) in a similarly high yield of 72% (Scheme 1). 2 and 3 are both brownish yellow and thus cannot be distinguished simply by visual inspection. However, XRD analysis of several crystals of a batch obtained from a diethyl ether solution invariably confirmed their identity as compound 2. In turn, when crystals of a batch obtained from a benzene solution layered with n-hexane were used, the same procedure confirmed their identity as compound 3. When crystalline 3 (analytically pure according to CHN data) was dissolved in C6D6, 1H NMR spectroscopic analysis revealed the presence of isomer 2 as the dominant species in the solution. In addition to the four signals due to 2, the spectrum also shows several slightly broadened signals between ca. 4 and 0 ppm (see Fig. S5 in the ESI†). It appears unlikely that these signals are due to the cation of 3, because a signal due to the anion of 3, which is expected to be located at ca. 200 ppm,19 was not observed. Our observations indicate that 2 and 3 are in equilibrium with one another in solution. The solubility of the ionic isomer 3 is expected to be substantially lower than that of 2 in solvents of low polarity. It is plausible, therefore, that it is 3 which crystallises from hydrocarbon solution, while the situation is inverse in the more polar solvent diethyl ether. Compounds 1–3 were structurally characterised by single-crystal X-ray diffraction (XRD). Their molecular structures are shown in Fig. 3–5. Pertinent metric parameters are collected in Table 1, which also contains data for closely related compounds for comparison. We note in this context that structurally characterised complexes of the type [FeCp*X(NHC)] (X = halogen) are known only for X = Cl,5a,20 and congeners containing other cyclopentadienyl ligands are limited to a single example each for X = Cl and Br, viz. [FeCp′Cl(IMes)]21 and [Fe{η5-C5(p-C6H4-Et)5}Br(MeIiPr)],22 and four examples for X = I, viz. [FeCp′I(NHC)] (NHC = IMes, IPr, MeIiPr and ItBu = 1,3-di-tert-butylimidazolin-2-ylidene).23
:1 integral ratio (12 H each). Consequently, the signal at 158 ppm, which exhibits the largest integral, is plausibly assigned to Cp* (15 H). However, the sum of the integrals of the remaining signals is less than half of the expected value (15 H vs. 36 H). If the signal at 158 ppm is assigned to the IPr methyl groups (assuming their accidental isochrony), the integrals of the other signals sum up to 23 H, which is reasonably close to the expected value of 27 H. For comparison, according to Ohki and Tatsumi, the 1H NMR signal of [FeCp*Cl(IMes)] due to the Cp* methyl groups is located at δ ≈ 169 ppm, while the methyl groups of the IMes ligand give rise to three signals at δ ≈ 76, −11 and −44 ppm; the mesityl CH units give rise to an extremely broad signal between −5 and −23 ppm, and the signal due to the CH units in the backbone of the IMes ligand is located at δ ≈ 39 ppm.5a In the same vein, the reaction with 1,3-dimethylbenzimidazolium iodide (1 equivalent) in THF furnished [FeCp*I(BzIMe)] (2, BzIMe = 1,3-dimethylbenzimidazolin-2-ylidene) in 63% yield, when diethyl ether was used for crystallisation (Scheme 1). The broad signals in the 1H NMR spectrum of 2 (analytically pure according to CHN data; see Fig. S4 in the ESI†) at δ ≈ 36 (ν½ 130 Hz) and −41 ppm (ν½ 160 Hz), respectively integrating for 15 and 6 protons, are assigned to the Cp* and BzIMe methyl groups, respectively, while the two only slightly broadened signals at δ ≈ 15 (ν½ 27 Hz) and 12 ppm (ν½ 21 Hz) are ascribed to the p-C6H4 backbone of the BzIMe ligand. Interestingly, crystallisation from a mixture of benzene and n-hexane furnished the isomeric, and ionic, product [FeCp*(BzIMe)2][FeCp*I2] (3) in a similarly high yield of 72% (Scheme 1). 2 and 3 are both brownish yellow and thus cannot be distinguished simply by visual inspection. However, XRD analysis of several crystals of a batch obtained from a diethyl ether solution invariably confirmed their identity as compound 2. In turn, when crystals of a batch obtained from a benzene solution layered with n-hexane were used, the same procedure confirmed their identity as compound 3. When crystalline 3 (analytically pure according to CHN data) was dissolved in C6D6, 1H NMR spectroscopic analysis revealed the presence of isomer 2 as the dominant species in the solution. In addition to the four signals due to 2, the spectrum also shows several slightly broadened signals between ca. 4 and 0 ppm (see Fig. S5 in the ESI†). It appears unlikely that these signals are due to the cation of 3, because a signal due to the anion of 3, which is expected to be located at ca. 200 ppm,19 was not observed. Our observations indicate that 2 and 3 are in equilibrium with one another in solution. The solubility of the ionic isomer 3 is expected to be substantially lower than that of 2 in solvents of low polarity. It is plausible, therefore, that it is 3 which crystallises from hydrocarbon solution, while the situation is inverse in the more polar solvent diethyl ether. Compounds 1–3 were structurally characterised by single-crystal X-ray diffraction (XRD). Their molecular structures are shown in Fig. 3–5. Pertinent metric parameters are collected in Table 1, which also contains data for closely related compounds for comparison. We note in this context that structurally characterised complexes of the type [FeCp*X(NHC)] (X = halogen) are known only for X = Cl,5a,20 and congeners containing other cyclopentadienyl ligands are limited to a single example each for X = Cl and Br, viz. [FeCp′Cl(IMes)]21 and [Fe{η5-C5(p-C6H4-Et)5}Br(MeIiPr)],22 and four examples for X = I, viz. [FeCp′I(NHC)] (NHC = IMes, IPr, MeIiPr and ItBu = 1,3-di-tert-butylimidazolin-2-ylidene).23
 | ||
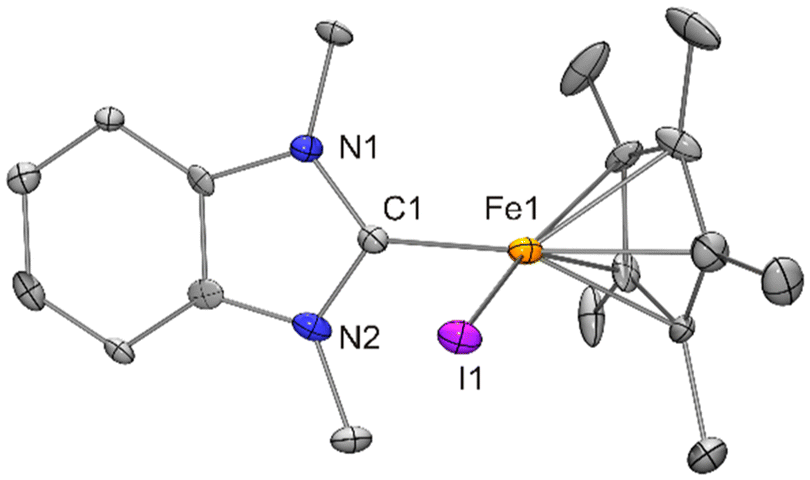
| Fig. 3 Molecular structures of [FeCp*Cl(IPr)]·0.5 benzene (1·0.5 benzene; top) and [FeCp*Cl(IPr)] (1; bottom) in the crystal (ORTEP with 30% probability ellipsoids, H atoms and solvent molecule not shown). | ||
 | ||
| Fig. 4 Molecular structure of [FeCp*I(BzIMe)] (2) in the crystal (ORTEP with 30% probability ellipsoids, H atoms not shown). | ||
 | ||
| Fig. 5 Molecular structure of [FeCp*(BzIMe)2][FeCp*I2]·1.5 benzene (3·1.5 benzene) in the crystal (ORTEP with 30% probability ellipsoids, H atoms and solvent molecules not shown). | ||
| Fe–Cp*/′centroid | Fe–X | Fe–Y | X–Fe–Y | Ref. | |
|---|---|---|---|---|---|
| a X = Cl. b Y = Ccarbene. c X = Br. d Five independent molecules with very similar bond parameters; data given for molecule 1. e X = I. f In the cation. g X = Y = Ccarbene. h In the anion. i X = Y = I. j X = Y = Cl. k Four independent molecules with very similar bond parameters; data given for molecule 1. l X = Namide. m Y = NDMAP. n Y = Cl. o Y = Br. p Two independent ion pairs with very similar bond parameters; data given for the anion of ion pair 1. | |||||
| [FeCp*Cl(IPr)]·0.5 benzene (1·0.5 benzene) | 2.00 | 2.2672(7)a | 2.147(2)b | 103.95(7) | This work |
| [FeCp*Cl(IPr)] (1) | 1.80 | 2.2352(6)a | 1.977(2)b | 92.04(6) | This work |
| [FeCp*Cl{fc[(NCH2Mes)2C]}] (4) | 1.80 | 2.2578(17)a | 1.972(6)b | 103.50(17) | This work |
| [FeCp*Cl{fc[(NCH2Ph)2C]}] (11) | 1.80 | 2.2474(13)a | 1.973(5)b | 99.60(13) | This work |
| [FeCp*Cl(IMes)]·toluene | 1.93 | 2.2715(7)a | 2.085(3)b | 98.14(7) | 5a |
| [FeCp*Cl(MeIiPr)] | 1.78 | 2.2434(8)a | 1.950(2)b | 95.46(6) | 5a |
| [FeCp′Cl(IMes)] | 2.02 | 2.3297(8)a | 2.168(3)b | 99.06(8) | 21 |
| [FeCp*Br{fc[(NCH2Mes)2C]}] (10) | 1.80 | 2.3968(9)c | 1.977(5)b | 103.54(13) | This work |
| [FeCp*Br{fc[(NCH2Ph)2C]}] (12) | 1.79 | 2.3899(4)c | 1.961(2)b | 99.40(7) | This work |
| [FeCp*I(BzIMe)] (2)d | 1.78 | 2.5796(15)e | 1.941(10)b | 94.6(3) | This work |
| [FeCp′I(IMes)] | 2.02 | 2.7128(6)e | 2.162(4)b | 99.07(9) | 23 |
| [FeCp*(BzIMe)2][FeCp*I2]·1.5 benzene (3·1.5 benzene) | 1.79f | 2.6193(9)e | 1.950(6)b | 95.6(2)g | This work |
| 1.98h | 2.6539(9)e | 1.944(6)b | 105.10(3)i | ||
| [FeCp*{fc[(NCH2Ph)2C]}2][PF6]·THF (13·THF) | 1.86 | 2.060(14)b | 108.8(5)g | This work | |
| 2.083(13)b | |||||
| [FeCp′(MeIMe)2]I·2 THF | 1.82 | 1.969(2)b | 93.64(8)g | 27 | |
| 1.979(2)b | |||||
| PPh4[FeCp*I2] | 1.96 | 2.6201(5)e | 106.56(3)i | 19 | |
| fc[(NCH2Mes)2CH][FeCp*Cl2]·2 toluene (5·2 toluene) | 1.98 | 2.2752(12)a | 106.90(5)j | This work | |
| 2.2732(12)a | |||||
| NnPr4[FeCp*Cl2] | 1.98 | 2.2953(8)a | 106.27(3)j | 19 | |
| 2.2814(8)a | |||||
| [FeCp*{N(SiMe3)2}(DMAP)] (6)k | 2.02 | 1.954(7)l | 2.134(7)m | 104.1(3) | This work |
| [FeCp′{N(SiMe3)2}(DMAP)] | 2.05 | 1.9827(10)l | 2.156(2)m | 92.92(7) | 33 |
| NnPr4[FeCp*Cl{N(SiMe3)2}] (7) | 2.04 | 1.983(2)l | 2.3360(8)n | 106.19(7) | This work |
| NnPr4[FeCp*Br{N(SiMe3)2}]·0.5 benzene (8·0.5 benzene) | 2.03 | 1.956(3)l | 2.4981(6)o | 104.44(8) | This work |
| fc[(NCH2Mes)2CH][FeCp*Br{N(SiMe3)2}] (9)p | 2.03 | 1.976(3)l | 2.4972(7)o | 103.59(10) | This work |
Crystallisation of [FeCp*Cl(IPr)] (1) from benzene afforded the solvate 1·0.5 benzene, whereas solvent-free crystals resulted from diethyl ether. The molecular structures differ substantially (Fig. 3 and Table 1). In comparison with unsolvated 1, the solvate 1·0.5 benzene has a much wider Cl–Fe–Ccarbene angle (104.0 vs. 92.0°) and much longer Fe–Ccarbene (2.15 vs. 1.98 Å) and Fe–Cp*centroid distances (2.00 vs. 1.80 Å). Previously reported compounds of the type [FeCp*Cl(NHC)] exhibit Fe–Cp*centroid distances between 1.78 and 1.99 Å.5a,20 However, this distance is usually only ca. 1.80 Å, as observed for solvent-free 1. Distances >1.90 Å have been found in two cases only, namely for NHC = IMes (1.93 Å)5a and 1-mesityl-3-[2-(N,N-dimethylamino)ethyl]imidazolin-2-ylidene (1.99 Å).20b In contrast to the Fe–Cl bond lengths of [FeCp*Cl(NHC)], which vary only slightly, the Fe–Ccarbene distances of [FeCp*Cl(NHC)] range from 1.92 to 2.13 Å.5a,20 Exceptionally long Fe–Cp*centroid and Fe–Ccarbene distances go hand in hand. The values determined for 1·0.5 benzene (2.00 and 2.15 Å) are larger than those of the previously reported Cp* containing congeners and similar to those of [FeCp′Cl(IMes)] (2.02 and 2.17 Å; Table 1). A comparison of [FeCp*Cl(IMes)]·toluene and [FeCp*Cl(MeIiPr)] reported by Ohki and Tatsumi in their seminal paper (Table 1)5a reveals that, although MeIiPr (%Vbur = 38.4%) has a higher steric impact than IMes (%Vbur = 36.5%),15 it is [FeCp*Cl(MeIiPr)] which exhibits the comparatively short Fe–Cp*centroid and Fe–Ccarbene distances typical of the majority of complexes of this type. It would be interesting to subject solvent-free [FeCp*Cl(IMes)] to a structural study by XRD. According to Ohki and Tatsumi, 16 VE iron(II) complexes [FeCp*Cl(NHC)] generally have two unpaired electrons (S = 1).5a However, experimental data in support of this claim are not available to date. The difference between the exceptionally long and the commonly observed (“ordinary”) Fe–Cp*centroid and Fe–Ccarbene distances is at least 0.15 Å, which is similar to the 0.17 Å difference of the ionic radii of hexacoordinate high-spin and low-spin iron(II)24 and clearly indicates that 1 and its solvate 1·0.5 benzene do not have the same electronic configuration at the temperature of the XRD experiments (T = 100 K). Note that the influence of lattice solvent on the spin state properties of FeII complexes is well documented.25
The iodido complex [FeCp*I(BzIMe)] (2) exhibits “ordinary” Fe–Cp*centroid and Fe–Ccarbene distances. Its Fe–I bond is ca. 0.3 Å longer than the Fe–Cl bonds of analogous chlorido complexes, which is in concert with the 0.29 Å difference of the tetrahedral covalent radii of I and Cl.26 In comparison with 2, the closely related complexes [FeCp′I(NHC)] reported by Walter exhibit exceptionally long distances (Table 1 exemplarily contains data for [FeCp′I(IMes)]) and have four unpaired electrons (S = 2) according to an in-depth experimental and computational study.23 [FeCp*Cl(IMes)] and [FeCp*Cl(MeIiPr)] were also included in the computational study by Walter, which revealed that a high-spin configuration with four unpaired electrons (S = 2) is preferred for the former, whereas the intermediate spin configuration with two unpaired electrons (S = 1) claimed by Ohki and Tatsumi5a is preferred for the latter.23 This lends further credence to the notion that 1 and its solvate 1·0.5 benzene have different electronic ground states (S = 1 for the former and S = 2 for the latter).
In the context of the ionic isomer of 2, [FeCp*(BzIMe)2][FeCp*I2] (3), we note that cations of the type [Fe(η5-C5R5)(NHC)2]+ are extremely scarce. We are aware of only two structurally characterised examples, viz. [FeCp′(MeIMe)2]I (MeIMe = 1,3,4,5-tetramethylimidazolin-2-ylidene) reported by Walter27 and the zwitterionic chelate [FeCp*{Ph2B(cyclo-NCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHNtBuC)2}] containing a borate-bridged dicarbene ligand described very recently by Prokopchuk.28 Essentially the same holds true for anions of the type [Fe(η5-C5R5)X2]− (X = halogen), whose range was expanded only recently by our work introducing Cp* congeners.19 The structural features of the cation present in 3·1.5 benzene strongly resemble those of the cation of [FeCp′(MeIMe)2]I (Table 1). Due to its chelate nature, the Ccarbene–Fe–Ccarbene bond angle of ca. 86° determined for the zwitterionic complex [FeCp*{Ph2B(cyclo-NCHCHNtBuC)2}] is substantially more acute (by 10°) than that of 3·1.5 benzene. While the Fe–Ccarbene bond lengths of this chelate (1.97 Å) are almost indistinguishable from those of 3·1.5 benzene, the Fe–Cp*centroid distance is significantly longer (1.85 vs. 1.78 Å for the cation of 3) but still lies in the “ordinary” range. The structural features of the [FeCp*I2]− anion present in 3·1.5 benzene are very similar to those determined for PPh4[FeCp*I2],19 which is the only structurally characterised compound known to date containing this anion.
CHNtBuC)2}] containing a borate-bridged dicarbene ligand described very recently by Prokopchuk.28 Essentially the same holds true for anions of the type [Fe(η5-C5R5)X2]− (X = halogen), whose range was expanded only recently by our work introducing Cp* congeners.19 The structural features of the cation present in 3·1.5 benzene strongly resemble those of the cation of [FeCp′(MeIMe)2]I (Table 1). Due to its chelate nature, the Ccarbene–Fe–Ccarbene bond angle of ca. 86° determined for the zwitterionic complex [FeCp*{Ph2B(cyclo-NCHCHNtBuC)2}] is substantially more acute (by 10°) than that of 3·1.5 benzene. While the Fe–Ccarbene bond lengths of this chelate (1.97 Å) are almost indistinguishable from those of 3·1.5 benzene, the Fe–Cp*centroid distance is significantly longer (1.85 vs. 1.78 Å for the cation of 3) but still lies in the “ordinary” range. The structural features of the [FeCp*I2]− anion present in 3·1.5 benzene are very similar to those determined for PPh4[FeCp*I2],19 which is the only structurally characterised compound known to date containing this anion.
We next turned our attention to 1,1′-ferrocenylene-bridged NHCs. Their precursors are formamidinium tetrafluoroborates fc[(NR)2CH][BF4],7,12,14,29 which are accessible by the formylative cyclisation of diaminoferrocenes fc(NHR)2 with a trialkyl orthoformate in the presence of NH4[BF4]. This method is based on seminal work by Saba and Kaloustian30 and provides the most popular route to NHC precursors.31 We were inspired by the 15 VE iron(I) complex [Fe{η5-C5(p-C6H4-Et)5}(MeIiPr)] containing a rather bulky cyclopentadienyl ligand in combination with a moderately bulky NHC, which was recently reported by Wolf.22 We speculated that the reaction of [FeCp*{N(SiMe3)2}] with a formamidinium tetrafluoroborate fc[(NR)2CH][BF4] could lead to [FeCp*{fc[(NR)2C]}][BF4], a compound containing a cationic 14 valence electron (VE) complex, which might be sufficiently stable for experimental observation thanks to steric protection by the moderately bulky Cp* ligand in combination with an intrinsically rather bulky ferrocene-based reNHC ligand. In addition, electronic stabilisation by the formation of a redox isomer analogous to Wolf's 15 VE iron(I) complex seemed feasible thanks to the redox-active nature of the ferrocene-based reNHC ligand. However, [FeCp*{N(SiMe3)2}] proved to be inert towards fc[(NR)2CH][BF4] (Scheme 2), indicating that the reactions observed for [FeCp*{N(SiMe3)2}] with (benz-)imidazolium halides (vide supra) rely on the presence of coordinating halide anions.
 | ||
| Scheme 2 Reactivity of [FeCp*{N(SiMe3)2}] towards fc[(NR)2CH][BF4] and fc[(NCH2Mes)2CH]Cl. A tentative stoichiometrically balanced equation for the formation of 5 is shown in grey (bottom). | ||
Indeed, the previously reported formamidinium chloride fc[(NCH2Mes)2CH]Cl (obtained from the corresponding stable NHC with triethylammonium chloride)12a showed a swift reaction with [FeCp*{N(SiMe3)2}] (used in a 1:1 stoichiometric ratio) under mild conditions. The crude product was taken up in diethyl ether. Slow evaporation of the solvent afforded green crystals together with a small amount of orange crystals. The 1H NMR spectrum (see Fig. S7 in the ESI†) is very complex and does not allow a meaningful interpretation. The most downfield-shifted signal at δ = 209 ppm is located in the region typical of anions of the type [FeCp*X2]−.19 XRD studies revealed that the expected product [FeCp*Cl{fc[(NCH2Mes)2C]}] (4) had been isolated only as a very minor component (orange crystals), while the major component was identified as fc[(NCH2Mes)2CH][FeCp*Cl2] (5; Scheme 2), consistent with the conspicuous signal at δ = 209 ppm. In view of the presence of small amounts of 4 in the crystalline material obtained, the isolated yield of 5 cannot be determined accurately and is estimated to be 60% with respect to fc[(NCH2Mes)2CH]Cl (in view of the two Cl atoms present in 5, two equivalents of this starting material are consumed). As indicated in Scheme 2, the reaction of [FeCp*{N(SiMe3)2}] with two equivalents of fc[(NCH2Mes)2CH]Cl to furnish 5 is expected to afford the stable NHC fc[(NCH2Mes)2C]12a and hexamethyldisilazane as additional products. However, this has not been further investigated by us because it was compound 4 that was the focus of our interest (vide infra). The molecular structures of 4 and 5·2 toluene are shown in Fig. 6 and 7.
 | ||
| Fig. 6 Molecular structure of [FeCp*Cl{fc[(NCH2Mes)2C]}] (4) in the crystal (ORTEP with 30% probability ellipsoids, H atoms not shown). | ||
 | ||
| Fig. 7 Molecular structure of fc[(NCH2Mes)2CH][FeCp*Cl2]·2 toluene (5·2 toluene) in the crystal (ORTEP with 30% probability ellipsoids, H atoms except H1 and solvent molecules not shown). A short N2CH⋯Carene contact (2.39 Å, C1–H1–C23 ca. 107°) is indicated by a dashed line. | ||
[FeCp*Cl{fc[(NCH2Mes)2C]}] (4) exhibits “ordinary” Fe–Cp*centroid and Fe–Ccarbene distances essentially identical to those of 1 and indicative of two unpaired electrons (S = 1, vide supra), but has a much wider Cl–Fe–Ccarbene angle (103.50(17) vs. 92.04(6)°), which is indistinguishable within experimental error from that of the solvate 1·0.5 benzene (103.95(7)°). The structures of the ions present in 5, viz. fc[(NCH2Mes)2CH]+ and [FeCp*Cl2]−, are unexceptional in comparison with fc[(NCH2Mes)2CH][BF4]·THF12a on the one hand and NnPr4[FeCp*Cl2] (Table 1) on the other hand.19
Unlike in the rational synthesis of NnPr4[FeCp*Cl2],19 the formation of [FeCp*Cl2]− in the reaction of [FeCp*{N(SiMe3)2}] leading to 5 is unexpected. NnPr4[FeCp*Cl2] was obtained as green crystals in 60% yield by addition of NnPr4Cl (1 equivalent) to [FeCp*Cl] (generated in situ from LiCp* and FeCl2 in THF at low temperatures). Despite being aggregated in solution,32 [FeCp*Cl] cannot be isolated and undergoes decomposition already well below 0 °C, which is in contrast to the thermally stable, monomeric “pogo stick” complex [FeCp*{N(SiMe3)2}]. Nevertheless, we expected [FeCp*{N(SiMe3)2}], being a low-coordinate 14 VE complex, to be capable of ligand association. The only previously reported example in this context was published by Walter, demonstrating that [FeCp′{N(SiMe3)2}] reacts with 4-dimethylaminopyridine (DMAP) to afford the adduct [FeCp′{N(SiMe3)2}(DMAP)], which contains a high-spin iron(II) centre with four unpaired electrons (S = 2) according to solid-state magnetic studies.33 Not surprisingly, the analogous synthesis of [FeCp*{N(SiMe3)2}(DMAP)] (6) proved to be straightforward (Scheme 3).
 | ||
| Scheme 3 Synthesis of compounds 6–8. | ||
Analytically pure 6 was obtained as a green solid. The rather low yield of 24% is due to fact that the crude product contained decamethylferrocene, which was not easy to remove due to its solubility being quite similar to that of 6. The 1H NMR spectrum of 6 (analytically pure according to CHN data; see Fig. S8 in the ESI†) exhibits two broad signals at δ ≈ 185 (ν½ 540 Hz) and 4 ppm (ν½ 420 Hz), which are assigned to the Cp* and the N(SiMe3)2 ligand, respectively. Both signals are high-field shifted by ca. 40 ppm with respect to [FeCp*{N(SiMe3)2}]. In addition, a broad signal integrating for 10 protons is observed at δ ≈ 3 ppm (ν½ 100 Hz), which is ascribed to the DMAP protons. The structure of 6 was determined by XRD (Fig. 8) and closely resembles that of [FeCp′{N(SiMe3)2}(DMAP)],33 indicating that both DMAP adducts have the same electronic structure with four unpaired electrons. The Fe bonds of [FeCp′{N(SiMe3)2}(DMAP)] are slightly elongated in comparison with 6 due to the bulky Cp′ ligand, which also causes a substantially smaller N–Fe–N bond angle of 92.92(7) vs. 104.1(3)° for 6. In comparison with the parent “pogo stick” complex [FeCp*{N(SiMe3)2}], its DMAP adduct 6 has a substantially larger Fe–Cp*centroid distance (2.02 vs. 1.90 Å), whereas the increase in coordination number causes only a slight increase of the Fe–Namide bond length (1.954(7) vs. 1.900(2) Å).1 NHC complex 1 and DMAP complex 6 are both complexes of the general type [FeCp*X(L)]. The Fe–Cp*centroid distances of 6 and the solvate 1·0.5 benzene, for which a high-spin configuration with four unpaired electrons (S = 2) seems most likely (vide supra), are very similar (2.02 and 2.00 Å, respectively), and the same holds true for the Fe–NDMAP distance of 6 and the Fe–Ccarbene distance of 1·0.5 benzene (2.134(7) and 2.147(2) Å, respectively). Due to the much larger radius of Cl in comparison with N, the Fe–Cl bond of 1·0.5 benzene is much longer than the Fe–Namide bond of 6 (2.2672(7) vs. 1.954(7) Å). However, the L–Fe–X bond angles of 1·0.5 benzene and 6 are essentially identical (104°).
 | ||
| Fig. 8 Molecular structure of [FeCp*{N(SiMe3)2}(DMAP)] (6) in the crystal (ORTEP with 30% probability ellipsoids, H atoms not shown). | ||
We next addressed ligand association reactions with halide anions. In analogy to our recently published preparation of NnPr4[FeCp*X2], NnPr4X (X = Cl, Br) served as the halide source.19 Gratifyingly, NnPr4[FeCp*Cl{N(SiMe3)2}] (7) and NnPr4[FeCp*Br{N(SiMe3)2}] (8) were obtained in yields of 75% and 83%, respectively, as green crystalline solids from [FeCp*{N(SiMe3)2}] and NnPr4X in toluene at room temperature after work-up (Scheme 3). An NMR spectroscopic investigation of 7 and 8 (analytically pure according to CHN data) revealed that each paramagnetic anion causes two broad 1H NMR signals (ν½ ≈ 660–780 Hz) located at δ ≈ 165 and −5 ppm, which may be assigned to the Cp* and the N(SiMe3)2 ligand, respectively (see Fig. S9 and S10 in the ESI†). Although substantially broadened (ν½ ≈ 220–350 Hz), the signals due to the diamagnetic tetra-n-propylammonium cations of 7 and 8 do not experience pronounced paramagnetic shifts (δ ≈ 5–2 ppm). Both compounds were structurally characterised by XRD (Fig. 9 and 10).
 | ||
| Fig. 9 Molecular structure of NnPr4[FeCp*Cl{N(SiMe3)2}] (7) in the crystal (ORTEP with 30% probability ellipsoids, H atoms not shown). | ||
 | ||
| Fig. 10 Molecular structure of NnPr4[FeCp*Br{N(SiMe3)2}]·0.5 benzene (8·0.5 benzene) in the crystal (ORTEP with 30% probability ellipsoids, H atoms and solvent molecule not shown). | ||
The Fe–Cp*centroid and Fe–Namide distances of 7 and 8·0.5 benzene are very similar to one another and also resemble those of [FeCp*{N(SiMe3)2}(DMAP)] (6, vide supra). The Fe–Br bond of 8·0.5 benzene is ca. 0.16 Å longer than the Fe–Cl bond of 7, which is in fair agreement with the difference of 0.12 Å of the tetrahedral covalent radii of Cl and Br.26
With compound 7 in hand, we studied its suitability for the synthesis of [FeCp*Cl{fc[(NCH2Mes)2C]}] (4). An equimolar mixture of 7 and fc[(NCH2Mes)2CH][BF4] was stirred in THF for 2 h, and colourless NnPr4[BF4] was filtered off after changing the solvent to diethyl ether. Slow evaporation of the filtrate afforded 4 in 17% yield (Scheme 4). 4 shows a low solubility in benzene and was therefore subjected to 1H NMR spectroscopic analysis in THF-d8. The complexity of the spectrum (obtained with an analytically pure sample according to CHN data; see Fig. S6 in the ESI†) indicates the presence of more than one chemical species and unfortunately does not allow a meaningful interpretation. The prominent signal at δ = 202 ppm lies in the region typical of anions of the type [FeCp*X2]−,19 suggesting that an equilibrium 2 [FeCp*Cl{fc[(NCH2Mes)2C]}] ⇄ [FeCp*{fc[(NCH2Mes)2C]}2]+ + [FeCp*Cl2]− is operative under these conditions, with ion formation being favourable in the polar solvent THF. It seems plausible that the other two species, viz. [FeCp*Cl{fc[(NCH2Mes)2C]}] and [FeCp*{fc[(NCH2Mes)2C]}2]+, give rise to the remaining, and partially overlapping, signals between 19 and 1 ppm. Performing the reaction in toluene resulted in a slightly lower yield of 12%. Interestingly, when the bromido homologue 8 was combined with fc[(NCH2Mes)2CH][BF4] in toluene, analogous work-up afforded the ionic compound fc[(NCH2Mes)2CH][FeCp*Br{N(SiMe3)2}] (9) in 49% yield due to mutual inertness of fc[(NCH2Mes)2CH]+ and [FeCp*Br{N(SiMe3)2}]− under these conditions (Scheme 4). However, a swift reaction furnishing [FeCp*Br{fc[(NCH2Mes)2C]}] (10) took place upon dissolving 9 in THF. This reaction also occurs in toluene or benzene, but only at elevated temperatures. When an NMR sample of 9 in C6D6 was heated to 60 °C for 45 min and subsequently allowed to cool to ambient temperature, 10 crystallised out in 59% yield, corresponding to 29% with respect to 8 over two steps (Scheme 4). 10 is obtained straightforwardly in 41% yield in a single step by combining 8 with fc[(NCH2Mes)2CH][BF4] in THF (Scheme 4). 10 exhibits a substantially higher solubility in benzene than the chlorido homologue 4. Its 1H NMR spectrum in C6D6 (obtained with an analytically pure sample according to CHN data; see Fig. S12 in the ESI†) exhibits 13 signals between δ = 48 ppm and −17 ppm, disregarding a few very minor signals in the range of δ = 7–0 ppm. If the signal with the largest integral at δ = 45 ppm is assigned to Cp* (15 H), the sum of the integrals of the other signals corresponds to ca. 39 H, which is reasonably close to the expected value of 34 H. However, a meaningful interpretation has not been possible. Compounds 9 and 10 were both obtained as orange crystals suitable for structural characterisation by XRD (Fig. 11 and 12).
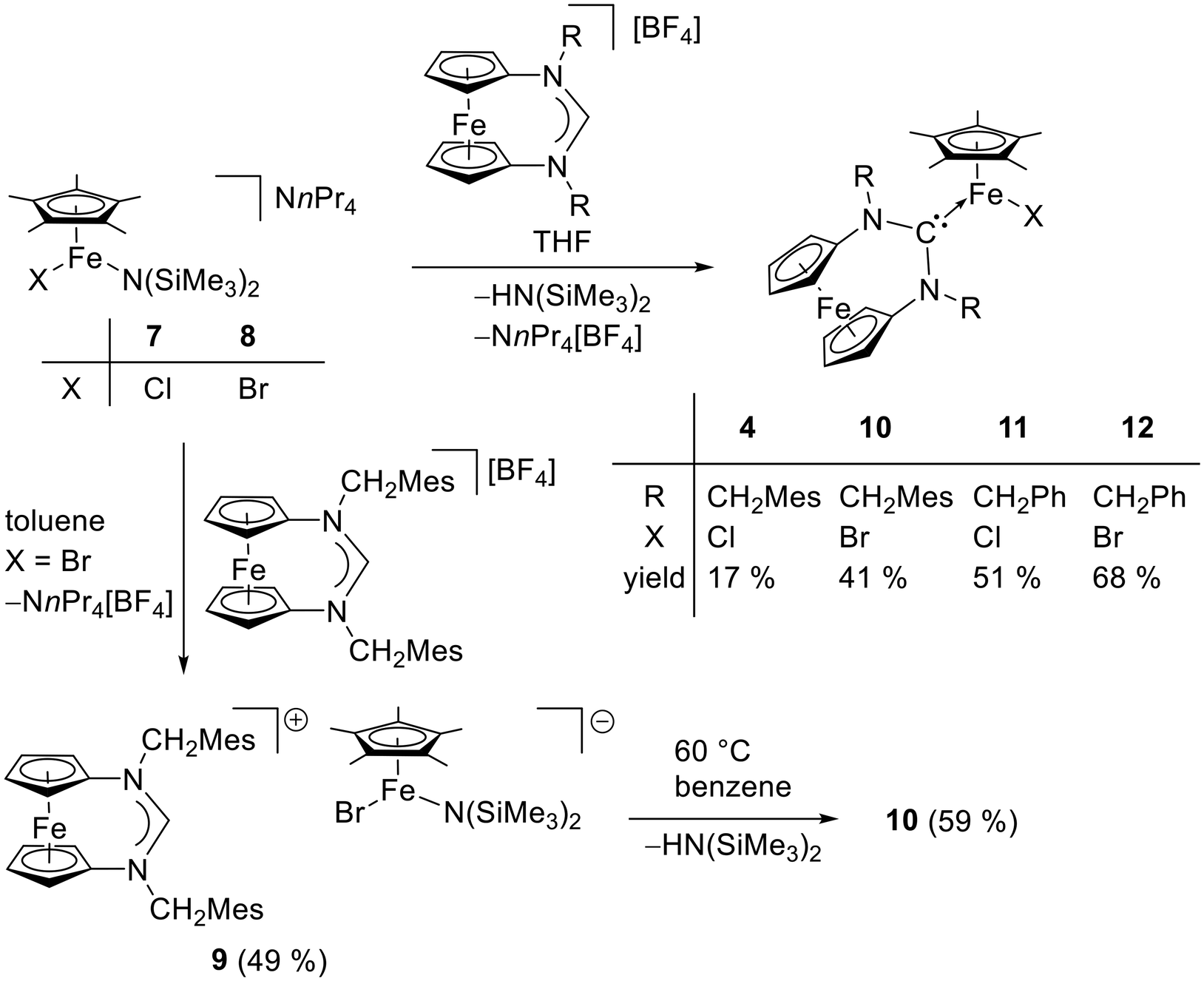 | ||
| Scheme 4 Synthesis of compounds 4 and 9–12. | ||
 | ||
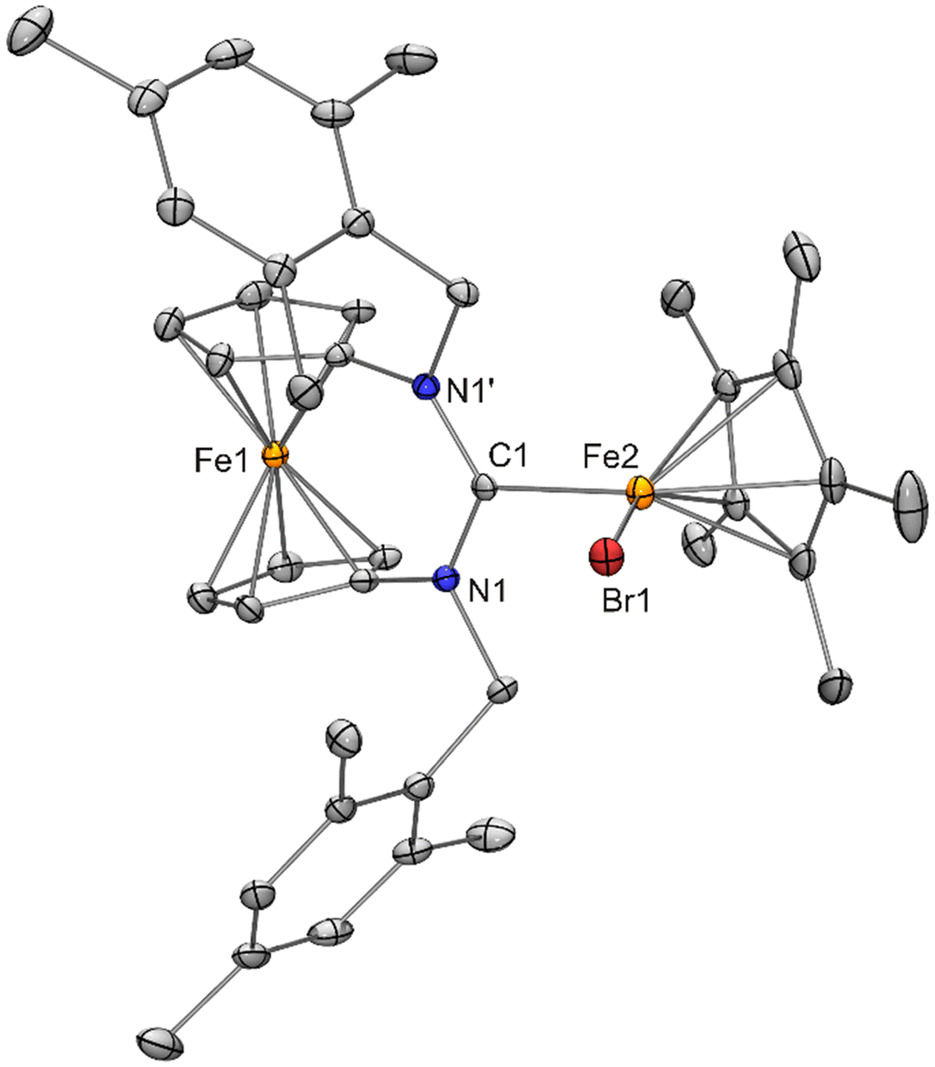
| Fig. 11 Molecular structure of fc[(NCH2Mes)2CH][FeCp*Br{N(SiMe3)2}] (9) in the crystal (ORTEP with 30% probability ellipsoids, H atoms except H1 not shown). Short N2CH⋯Carene contacts (H1–C13 2.36 Å, C1–H1–C13 108°; H1–C23 2.47 Å, C1–H1–C23 105°) are indicated by dashed lines. | ||
 | ||
| Fig. 12 Molecular structure of [FeCp*Br{fc[(NCH2Mes)2C]}] (10) in the crystal (ORTEP with 30% probability ellipsoids, H atoms not shown). | ||
The structure of the fc[(NCH2Mes)2CH]+ cation of 9 is very similar to that previously determined for fc[(NCH2Mes)2CH][BF4]·THF.12a The formamidinium proton is involved in a short N2CH⋯Carene contact (2.36 Å, C1–H1–C13 ca. 108°; 2.47 Å, C1–H1–C33 ca. 105°) to each of the mesityl rings, compatible with weak CH⋯π(arene) interactions.34 As already indicated by their essentially identical metric parameters given in Table 1, the structures of the [FeCp*Br{N(SiMe3)2}]− anions of 9 and 8 (discussed above) strongly resemble one another. The structure of [FeCp*Br{fc[(NCH2Mes)2C]}] (10) may be compared with that of the chlorido homologue 4. Both exhibit molecular Cs symmetry, with the mirror plane containing the two Fe atoms, the halogen atom and the Ccarbene atom. Pertinent metric parameters given in Table 1 are essentially identical for 10 and 4, except, of course, for the iron–halogen bond lengths (2.3968(9) vs. 2.2578(17) Å), whose difference of 0.14 Å is in good agreement with the 0.12 Å difference of the tetrahedral covalent radii of Cl and Br.26
The mesityl-containing compounds 4, 5, 9 and 10 did not give rise to 1H NMR spectra for which a meaningful interpretation seemed possible. We therefore turned our attention to corresponding analogues containing Ph, instead of Mes, groups. The reaction of 7 (prepared in situ from [FeCp*{N(SiMe3)2}] and NnPr4Cl) with fc[(NCH2Ph)2CH][BF4] in THF furnished [FeCp*Cl{fc[(NCH2Ph)2C]}] (11) in 51% yield; the bromido homologue [FeCp*Br{fc[(NCH2Ph)2C]}] (12) was obtained analogously in 68% yield from 8 and fc[(NCH2Ph)2CH][BF4] in THF (Scheme 4). A swift reaction was also observed in toluene at ambient temperature, which furnished 12 in a yield of 51% (not shown in Scheme 4). This is in contrast to the inertness of the anion of 8 towards the cation of fc[(NCH2Mes)2CH][BF4] under the same mild conditions in toluene. The different behaviour of fc[(NCH2Mes)2CH]+vs. fc[(NCH2Ph)2CH]+ is plausibly ascribed to the higher steric demand of the CH2Mes vs. the CH2Ph N-substituents (as reflected by the %Vbur values of 38.0 vs. 34.9% determined for the corresponding carbenes; vide supra), causing a significantly increased kinetic barrier. 11 and 12 were both isolated as orange, crystalline solids. Their 1H NMR spectra (obtained with analytically pure samples according to CHN data) are very similar (see Fig. S13 and S14 in the ESI†). 11 exhibits nine signals in the spectral range from 39 to −14 ppm. If the broad signal at δ = 39 ppm (ν½ 100 Hz), which exhibits the largest integral in the spectrum, is assigned to the Cp* ligand and thus represents 15 protons, the eight signals at δ = 31 (ν½ 140 Hz), 18 (ν½ 29 Hz), 15 (ν½ 24 Hz), 8 (ν½ 22 Hz), 5 (ν½ 16 Hz), 2 (ν½ 13 Hz), −1 (ν½ 15 Hz) and −14 ppm (ν½ 24 Hz) integrate for ca. 2, 2, 4, 2, 2, 2, 2 and 2 protons, respectively, representing a total of ca. 18 protons. In view of the uncertainties due to signal broadening, this appears to be more or less compatible with the total number of 22 protons of the phenyl (10 H), ferrocenylene (8 H) and methylene (4 H) groups. In addition to the signals assigned to 11, the 1H NMR spectrum exhibits several rather small signals between ca. 4 and 1 ppm and a broad down-field shifted signal at δ = 213 ppm (ν½ 320 Hz), which is compatible with the anion [FeCp*Cl2]−.19 It is tempting to surmise that [FeCp*Cl{fc[(NCH2Ph)2C]}] (11) is in equilibrium with the ionic compound [FeCp*{fc[(NCH2Ph)2C]}2][FeCp*Cl2] in C6D6 solution, with 11 as the dominant component (ca. 85%). In the case of the bromido homologue [FeCp*Br{fc[(NCH2Ph)2C]}] (12), no such down-field shifted signal close to 200 ppm, which would be compatible with [FeCp*Br2]−,19 is present in the 1H NMR spectrum. If the broad signal at δ = 32 ppm (ν½ 94 Hz) is assigned to the Cp* ligand (in analogy to 11), the sum of the integrals of the remaining eight signals corresponds to ca. 16 protons, which, in comparison with 11, deviates more from the expected value of 22 protons, indicating that our suggested assignment can only be tentative and has to be treated with caution in both cases. 11 and 12 have been structurally characterised by XRD (Fig. 13 and 14).
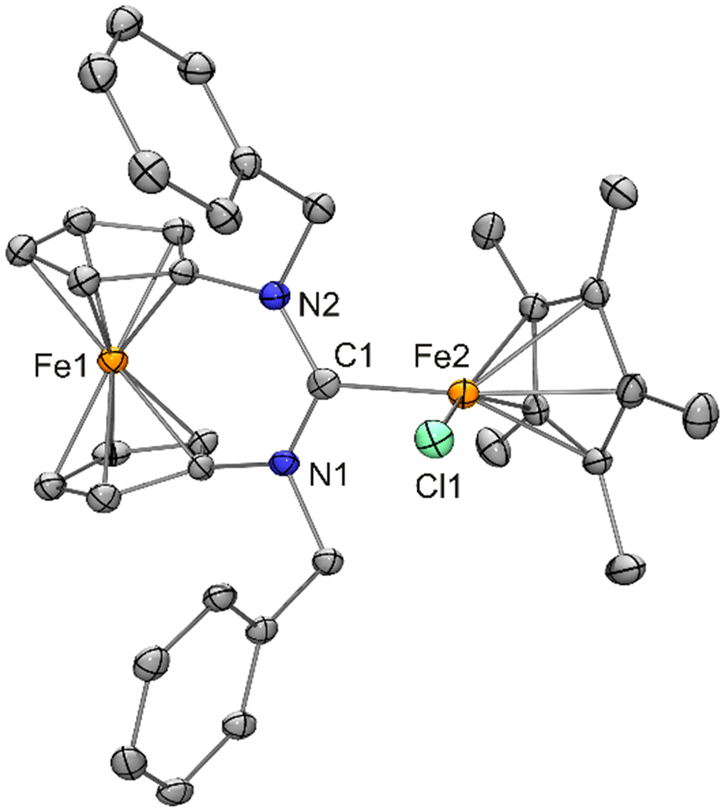 | ||
| Fig. 13 Molecular structure of [FeCp*Cl{fc[(NCH2Ph)2C]}] (11) in the crystal (ORTEP with 30% probability ellipsoids, H atoms not shown). | ||
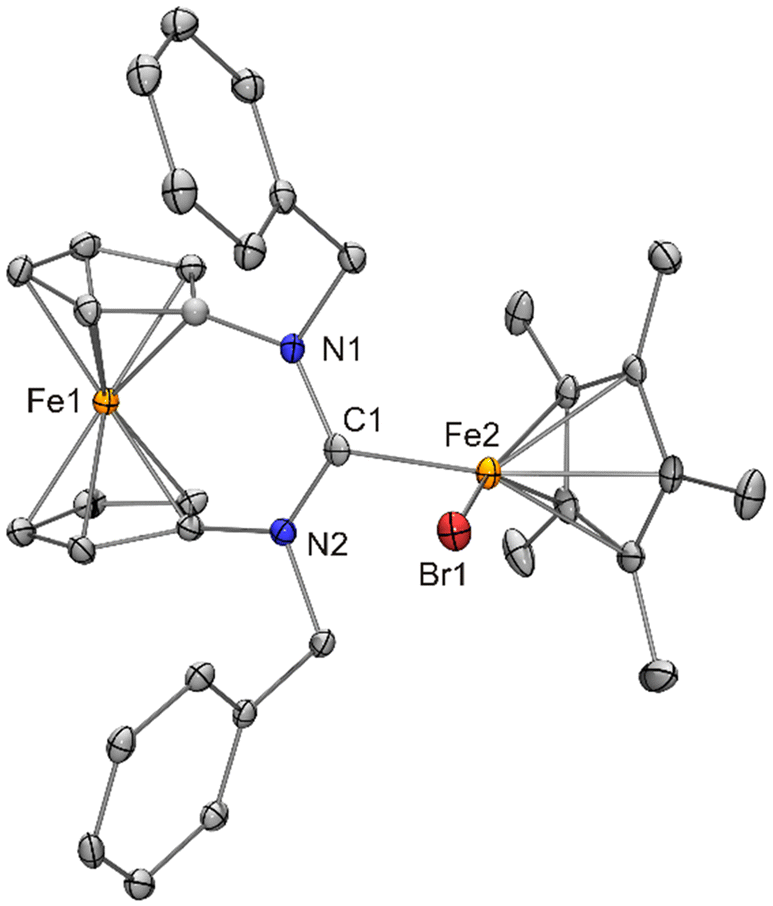 | ||
| Fig. 14 Molecular structure of [FeCp*Br{fc[(NCH2Ph)2C]}] (12) in the crystal (ORTEP with 30% probability ellipsoids, H atoms not shown). | ||
While their mesityl-containing analogues 4 and 10 both exhibit crystallographically imposed molecular Cs symmetry, 11 and 12 are only approximately Cs symmetric. The distances given in Table 1 for the bromido complex pair 10 and 12 are very similar and are even essentially identical for the chlorido complex pair 4 and 11. The halogen–Fe–Ccarbene bond angles of 4 and 10 are slightly wider (by 4°) than those of 11 and 12, reflecting the higher steric demand of fc[(NCH2Mes)2C] vs. fc[(NCH2Ph)2C] (vide supra).
Despite many attempts, our efforts to convert 4 or 10–12 into the corresponding 14 VE cations [FeCp*{fc[(NR)2C]}]+ by halide abstraction with reagents like Na[BPh4], Ag[BF4] or Tl[PF6] were unsuccessful, usually affording only intractable material. However, in one instance, it has been possible to identify [FeCp*{fc[(NCH2Ph)2C]}2][PF6] (13) as a product by XRD. The crystalline solvate 13·THF was isolated from the reaction of [FeCp*Br{fc[(NCH2Ph)2C]}] (12) with Tl[PF6] in THF, albeit in trace amounts only (Scheme 5). The structure is shown in Fig. 15.
 | ||
| Scheme 5 Serendipitous formation of 13. | ||
 | ||
| Fig. 15 Molecular structure of [FeCp*{fc[(NCH2Ph)2C]}2][PF6]·THF (13·THF) in the crystal (ORTEP with 30% probability ellipsoids, H atoms, anion and solvent molecule not shown). | ||
The [FeCp*{fc[(NCH2Ph)2C]}2]+ cation of 13·THF has a Ccarbene–Fe–Ccarbene bond angle of 108.8(5)°, which is much wider than the corresponding angles determined for [FeCp*(BzIMe)2][FeCp*I2]·1.5 benzene (3·1.5 benzene) and [FeCp′(MeIMe)2]I·2 THF, viz. 95.6(2) and 93.64(8)°, respectively. This may be ascribed to the much higher steric demand of the ferrocene-based NHC fc[(NCH2Ph)2C] vs. BzIMe and MeIMe, as reflected by their respective %Vbur values of 34.9%,12a 26.5%35 and 26.1%.15c Notably, the Fe–Ccarbene distances of 13 (average value 2.07 Å) are much longer (by at least 0.1 Å) than those of 3·1.5 benzene and [FeCp′(MeIMe)2]I·2 THF (average values: 1.95 and 1.97 Å, respectively). The difference is less pronounced for the Fe–Cp*centroid distances, which are 1.79 and 1.82 Å for 3 and [FeCp′(MeIMe)2]I·2 THF, respectively, and 1.86 Å for 13. The trend, however, is clearly the same. Taken together, these data seem to suggest a higher number of unpaired electrons in 13 in comparison with 3·1.5 benzene and [FeCp′(MeIMe)2]I·2 THF. However, we refrain from speculation.
Conclusion
We have investigated the “pogo stick” complex [FeCp*{N(SiMe3)2}] in terms of its spin state and chemical behaviour. The results obtained with crystalline samples by SQUID magnetometry and Mößbauer experiments show that [FeCp*{N(SiMe3)2}] is a high-spin FeII complex containing four unpaired electrons and an S = 2 ground state, which is in concert with the closely related complexes [FeCp′{N(SiMe3)R}] (R = SiMe3, CMe3, C6H3-2,6-iPr2) and [Fe5Cp{N(SiMe3)2}] studied by Walter and Sitzmann.2 [FeCp*{N(SiMe3)2}] readily forms adducts with DMAP (compound 6) and with chloride and bromide anions (compounds 7 and 8; Scheme 3). The presence of halide anions plays a crucial role in the reaction of [FeCp*{N(SiMe3)2}] with formamidinium salts, whose cations may be viewed as protonated NHCs. No reaction of [FeCp*{N(SiMe3)2}] was observed with formamidinium salts containing the essentially non-coordinating tetrafluoroborate anion. In contrast, facile formation of complexes of the type [FeCp*X(NHC)] (X = Cl, Br, I; compounds 1, 2, 4, 10, 11, 12) was possible by reacting [FeCp*{N(SiMe3)2}] with formamidinium halides (Schemes 1 and 2) or by reacting formamidinium tetrafluoroborates with [FeCp*X{N(SiMe3)2}]− (Scheme 4). This behaviour strongly suggests that the reactivity of the Brønsted basic amido ligand of [FeCp*{N(SiMe3)2}] is enhanced by association with a halide anion to a degree that the resulting adduct [FeCp*X{N(SiMe3)2}]− reacts readily with weakly Brønsted acidic formamidinium cations, whereas DMAP adduct 6 is inert. Structural data obtained by XRD at 100 K for 1 and for the benzene solvate 1·0.5 benzene reveal a crucial influence of the lattice solvent on the electronic ground state of the FeII complex. It can be reasonably assumed that the solvate 1·0.5 benzene has a high-spin configuration with four unpaired electrons (S = 2), whereas unsolvated 1 has only two unpaired electrons (S = 1). Experimental data point to an equilibrium of the type 2 [FeCp*X(NHC)] ⇄ [FeCp*(NHC)2]+ + [FeCp*X2]− in several cases, with particularly convincing evidence coming from the solvent-dependent isolation of either 2 or 3 (Scheme 1).Experimental section
General methods and instrumentation
All reactions involving air-sensitive compounds were performed under an inert atmosphere (argon or dinitrogen) using standard Schlenk techniques or a conventional glovebox. Starting materials were procured from standard commercial sources and used as received. Iron(II) chloride,36 pentamethylcyclopentadiene (Cp*H),37 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride,38 fc[(NCH2Ph)2CH][BF4], fc[(NCH2Mes)2CH][BF4], and fc[(NCH2Mes)2CH]Cl were prepared using adapted versions of the published procedures.12a NMR spectra were recorded at ambient temperature using Varian NMRS-500 and MR-400 spectrometers operating at 500 and 400 MHz, respectively, for 1H. Elemental analyses were carried out using a HEKAtech Euro EA-CHNS elemental analyser at the Institute of Chemistry.Synthesis and characterisation of Fe complexes
X-ray crystallography
For each data collection, a single crystal was mounted on a micro-mount and all geometric and intensity data were taken from this sample by ω-scans at 100(2) K. Data collections were carried out using either a Stoe StadiVari diffractometer equipped with a 4-circle goniometer and a DECTRIS Pilatus 200K detector or a Stoe IPDS2 diffractometer equipped with a 2-circle goniometer and an area detector. The data sets were corrected for absorption (by multi scans), Lorentz and polarisation effects. The structures were solved by direct methods (SHELXT) and refined using alternating cycles of least-squares refinements against F2 (SHELXL-2014/7).40 H atoms were included in the models at calculated positions with an isotropic displacement parameter 1.2 times that of their bonding partner. Experimental details for each diffraction experiment are given in Table S1 (see the ESI†).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Prof. Franc Meyer (Georg-August-Universität Göttingen) for granting access to his SQUID magnetometer and Mößbauer spectrometer and Prof. Eva Rentschler (Johannes-Gutenberg-Universität Mainz) for helpful discussions.References
- U. Siemeling, U. Vorfeld, B. Neumann and H.-G. Stammler, Organometallics, 1998, 17, 483–484 Search PubMed.
- (a) K. Münster, D. Baabe, B. Kintzel, M. Böhne, W. Plass, J. Raeder and M. D. Walter, Inorg. Chem., 2022, 61, 18883–18898 CrossRef PubMed; (b) O. A. Groß, S. Lauk, C. Müller, W. Gidt, Y. Sun, S. Demeshko, F. Meyer and H. Sitzmann, Eur. J. Inorg. Chem., 2017, 3635–3643 CrossRef; (c) M. D. Walter and P. S. White, Inorg. Chem., 2012, 51, 11860–11872 CrossRef PubMed.
- [FeCp′{N(SiMe3)2}] and [FeCp′(NHC6H2-2,4,6-tBu3)] have also been described, but were not structurally characterised; see ref. 2c.
- For a recent review on half-sandwich iron complexes, see: K. Münster and M. D. Walter, in Comprehensive Organometallic Chemistry, ed. G. Parkin, K. Meyer and D. O'Hare, Elsevier, Kidlington, 4th edn, 2022, vol. 7, pp. 46–184 Search PubMed.
- (a) Y. Ohki, T. Hatanaka and K. Tatsumi, J. Am. Chem. Soc., 2008, 130, 17174–17186 CrossRef; (b) Y. Ohki, Y. Takikawa, T. Hatanaka and K. Tatsumi, Organometallics, 2006, 25, 3111–3113 CrossRef CAS.
- For commonly used abbreviations of popular NHCs, see: S. Díez-González, in N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Díez-González, Royal Society of Chemistry, Cambridge, 2nd edn, 2017, p. x Search PubMed.
- Seminal paper: D. M. Khramov, E. L. Rosen, V. M. Lynch and C. W. Bielawski, Angew. Chem., Int. Ed., 2008, 47, 2267–2270 CrossRef CAS PubMed.
- Seminal papers: (a) N. Matsumura, J.-i. Kawano, N. Fukunishi and H. Inoue, J. Am. Chem. Soc., 1995, 117, 3523–3624 CrossRef; (b) R. W. Alder, M. E. Blake, C. Bortolotti, S. Bufali, C. P. Butts, E. Linehan, J. M. Oliva, A. G. Orpen and M. J. Quayle, Chem. Commun., 1999, 241–242 RSC.
- Recent reviews: (a) T. Zhou, G. Utecht-Jarzyńska and M. Szostak, Coord. Chem. Rev., 2024, 512, 215867 CrossRef CAS; (b) J. Li, W.-X. Shen and X.-R. Li, Curr. Org. Chem., 2012, 16, 2879–2891 CrossRef.
- See, for example: (a) M. C. Jahnke and F. E. Hahn, in N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Díez-González, Royal Society of Chemistry, Cambridge, 2nd edn, 2017, pp. 1–45 Search PubMed; (b) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef PubMed; (c) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef PubMed.
- See, for example: (a) F. Vermersch, V. T. Wang, M. Abdellaoui, R. Jazzar and G. Bertrand, Chem. Sci., 2024, 15, 3707–3710 RSC; (b) A. Kumar, D. Yuan and H. V. Huynh, Inorg. Chem., 2019, 58, 7545–7553 CrossRef; (c) M. Iglesias, D. J. Beetstra, J. C. Knight, L.-L. Ooi, A. Stasch, S. Coles, L. Male, M. B. Hursthouse, K. J. Cavell, A. Dervisi and I. A. Fallis, Organometallics, 2008, 27, 3279–3289 CrossRef.
- (a) J. Zinke, C. Bruhn and U. Siemeling, Z. Anorg. Allg. Chem., 2023, 649, e202200334 CrossRef; (b) B. A. Correia Bicho, R. Guthardt, C. Bruhn, D. Großhennig, T. Orth, F. Pfeiffer and U. Siemeling, Eur. J. Inorg. Chem., 2022, e202101014 CrossRef; (c) U. Siemeling, C. Färber, M. Leibold, C. Bruhn, P. Mücke, R. F. Winter, B. Sarkar, M. von Hopffgarten and G. Frenking, Eur. J. Inorg. Chem., 2009, 4607–4612 CrossRef; (d) U. Siemeling, C. Färber and C. Bruhn, Chem. Commun., 2009, 98–100 RSC.
- (a) L. Wallbaum, D. Weismann, D. Löber, C. Bruhn, P. Prochnow, J. E. Bandow and U. Siemeling, Chem. – Eur. J., 2019, 25, 1488–1497 CrossRef PubMed; (b) T. Schulz, D. Weismann, L. Wallbaum, R. Guthardt, C. Thie, M. Leibold, C. Bruhn and U. Siemeling, Chem. – Eur. J., 2015, 21, 14107–14121 Search PubMed; (c) M. S. Collins, E. L. Rosen, V. M. Lynch and C. W. Bielawski, Organometallics, 2010, 29, 3047–3053 CrossRef; (d) R. W. Alder, P. R. Allen, M. Murray and A. G. Orpen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1121–1123 Search PubMed.
- C. D. Varnado Jr., E. L. Rosen, M. S. Collins, V. M. Lynch and C. W. Bielawski, Dalton Trans., 2013, 42, 13251–13264 Search PubMed.
- Reviews: (a) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed; (b) A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 Search PubMed; (c) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- Determined from the structure of [AuCl(IBn)]; see: S. Patil, A. Deally, F. Hackenberg, L. Kaps, H. Müller-Bunz, R. Schobert and M. Tacke, Helv. Chim. Acta, 2011, 94, 1551–1562 CrossRef CAS.
- See, for example: V. N. Sapunov, R. Schmid, K. Kirchner and H. Nagashima, Coord. Chem. Rev., 2003, 238–239, 363–382 CrossRef CAS.
- The wrong assignment was due to a lack of experience and was facilitated by the fact that the paramagnetic nature of the sample had apparently not been noticed or considered by the NMR operator, who provided a print-out of the 1H NMR spectrum, which showed the routine spectral range and contained, apart from the solvent signal, only two singlet signals, whose integrals happened to be compatible with the ratio of protons expected for the product.
- J. Zinke, C. Bruhn and U. Siemeling, Inorganics, 2023, 11, 437 CrossRef CAS.
- (a) Q. Liang, K. Hayashi, K. Rabeda, J. L. Jimenez-Santiago and D. Song, Organometallics, 2020, 39, 2320–2326 CrossRef CAS; (b) Q. Liang, K. Sheng, A. Salmon, V. Y. Zhou and D. Song, ACS Catal., 2019, 9, 810–818 CrossRef CAS.
- P. G. Jones, M. Freytag, M. Reiners and M. D. Walter, Private Communication to the Cambridge Structural Data Base, 2022, CCDC 2165746 Search PubMed.
- U. Chakraborty, S. Demeshko, F. Meyer, C. Rebreyend, B. de Bruin, M. Atanasov, F. Neese, B. Mühldorf and R. Wolf, Angew. Chem., Int. Ed., 2017, 56, 7995–7999 CrossRef CAS.
- M. Reiners, D. Baabe, K. Harms, M. Maekawa, C. G. Daniliuc, M. Freytag, P. G. Jones and M. D. Walter, Inorg. Chem. Front., 2016, 3, 250–262 RSC.
- (a) R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef. For a typical example for the crystallographic resolution of the high-spin and low-spin isomers of a hexacoordinate iron(II) complex, see: (b) B. A. Katz and C. E. Strouse, J. Am. Chem. Soc., 1979, 101, 6214–6221 CrossRef CAS.
- See, for example: (a) S. Sundaresam, J. Eppelsheimer, E. Gera, L. Wiener, L. M. Carella, K. R. Vignesh and E. Rentschler, Dalton Trans., 2024, 53, 10303–10317 RSC; (b) L. J. K. Cook, R. Kulmaczewski, O. Cespedes and M. A. Halcrow, Chem. – Eur. J., 2016, 22, 1789–1799 Search PubMed; (c) R. G. Miller and S. Brooker, Chem. Sci., 2016, 7, 2501–2505 RSC; (d) Y. Garcia, P. J. von Koningsbruggen, R. Lapouyade, L. Fournès, L. Rabardel, O. Kahn, V. Ksenofontov, G. Levchenko and P. Gütlich, Chem. Mater., 1998, 10, 2426–2433 CrossRef.
- Pauling's covalent tetrahedral radii reflect a convolution of ionic and covalent bonding; see: L. Pauling and M. L. Huggins, Z. Kristallogr., 1934, 87, 205–238 Search PubMed.
- P. G. Jones, M. Freytag, M. Maekawa and M. D. Walter, Private Communication to the Cambridge Structural Data Base, 2021, CCDC 2055400 Search PubMed.
- D. S. Tresp and D. E. Prokopchuk, Polyhedron, 2024, 248, 116745 CrossRef.
- (a) C. Thie, C. Bruhn and U. Siemeling, Eur. J. Inorg. Chem., 2015, 5457–5466 CrossRef; (b) S. Rittinghaus, C. Färber, C. Bruhn and U. Siemeling, Dalton Trans., 2014, 43, 3508–3520 Search PubMed.
- S. Saba, A. Brescia and M. K. Kaloustian, Tetrahedron Lett., 1991, 32, 5031–5034 CrossRef.
- Review: L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. Cécar, Chem. Rev., 2011, 111, 2705–2733 CrossRef PubMed.
- [FeCp*Cl]n has been formulated to be dimeric, tetrameric or oligomeric; selected examples for n = 2: (a) M. Tamizmani, J. R. Tidwell, E. W. Reinheimer, B. M. Lindley and C. D. Martin, Inorg. Chem., 2023, 62, 7150–7154 CrossRef PubMed; (b) H. Gao, J. Jia, C.-H. Tung and W. Wang, Organometallics, 2023, 42, 944–951 CrossRef; (c) Y. Li, Y. Li, B. Wang, Y. Luo, D. Yang, P. Tong, J. Zhao, L. Luo, Y. Zhou, S. Chen, F. Cheng and J. Qu, Nat. Chem., 2013, 5, 320–326 CrossRef PubMed; (d) M. Stephan, P. Müller, U. Zenneck, H. Pritzkow, W. Siebert and R. N. Grimes, Inorg. Chem., 1995, 34, 2058–2067 CrossRef. For n = 4, see: (e) R. Hettrich, M. Kaschke, H. Wadepohl, W. Weinmann, M. Stephan, H. Pritzkow, W. Siebert, I. Hyla-Kryspin and R. Gleiter, Chem. – Eur. J., 1996, 2, 482–494 CrossRef PubMed. Selected examples for aggregates with unknown n: (f) S. Takemoto, S.-i. Ogura, H. Yo, Y. Hosokoshi, K. Kamikawa and H. Matsuzaka, Inorg. Chem., 2006, 45, 4871–4873 CrossRef PubMed; (g) R. Shintani and G. C. Fu, Org. Lett., 2002, 4, 3699–3702 CrossRef PubMed.
- M. D. Walter and P. S. White, Dalton Trans., 2012, 41, 8506–8508 RSC.
- For selected reviews, see: (a) M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC; (b) M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757–1788 RSC; (c) M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC; (d) O. Takahashi, Y. Kohno, S. Iwasaki, K. Saito, M. Iwaoka, S. Tomoda, Y. Umezawa, S. Tsuboyama and M. Nishio, Bull. Chem. Soc. Jpn., 2001, 74, 2421–2430 CrossRef CAS; (e) For an example of an imidazolium N2CH⋯π(phenyl) interaction, see: J. Dupont, P. A. Z. Suarez, R. F. De Souza, R. A. Burrow and J.-P. Kintzinger, Chem. – Eur. J., 2000, 6, 2377–2381 CrossRef CAS PubMed.
- Determined from the structure of [AuBr(BzIMe)]; see: M. C. Jahnke, T. Pape and F. E. Hahn, Z. Anorg. Allg. Chem., 2010, 636, 2309–2314 CrossRef CAS.
- G. Winter, D. W. Thompson and R. J. Loehe, Inorg. Synth., 1973, 14, 101–104 CrossRef CAS.
- (a) C. M. Fendrick, L. D. Schertz, E. A. Mintz, T. J. Marks, T. E. Bitterwolf, P. A. Horine, T. L. Hubler, J. A. Sheldon and D. D. Belin, Inorg. Synth., 1992, 29, 193–198 CrossRef CAS; (b) F. X. Kohl and P. Jutzi, J. Organomet. Chem., 1983, 243, 119–121 CrossRef CAS.
- L. Jafarpour, E. D. Stevens and S. P. Nolan, J. Organomet. Chem., 2000, 606, 49–54 CrossRef CAS.
- See, for example: (a) A. Logallo, L. C. H. Maddock, M. Mu, L. Gravogel, N. Jin, M. N. Peñas-Defrutos, K. Meyer, M. García-Melchor and E. Hevia, Angew. Chem., Int. Ed., 2024, 63, e202402907 CrossRef CAS; (b) J. M. Smith, R. J. Lachicotte and P. L. Holland, Chem. Commun., 2001, 1542–1543 RSC; (c) U. Siemeling, U. Vorfeld, B. Neumann and H.-G. Stammler, Inorg. Chem., 2000, 39, 5159–5160 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of single-crystal X-ray diffraction analyses, magnetic measurements and Mößbauer spectroscopy, and plots of NMR spectra. CCDC 2421670–2421683. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00300h |
| This journal is © The Royal Society of Chemistry 2025 |