
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand-directed top-down synthesis of trivacant lacunary polyoxomolybdates from plenary Keggin-type [α-XMo12O40]3− (X = P, As, V) in organic media†
Atsuhiro
Jimbo
,
Chifeng
Li
,
Kentaro
Yonesato
 ,
Kazuya
Yamaguchi
and
Kosuke
Suzuki
*
,
Kazuya
Yamaguchi
and
Kosuke
Suzuki
*
Department of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ksuzuki@appchem.t.u-tokyo.ac.jp
First published on 26th February 2025
Abstract
Lacunary polyoxometalates (POMs), featuring highly reactive vacant sites, serve as valuable building blocks and precursors for the rational design of functional materials with widespread applications in catalysis, analytical chemistry, energy conversion and storage, medicine, and optical materials. While diverse Keggin-type polyoxotungstates, including both plenary and lacunary species, have been synthesized through dehydration condensation reactions and equilibrium displacement in aqueous solvents, the isolation and use of lacunary polyoxomolybdates remain challenging. To address this, the current study proposes a “ligand-directed top-down synthetic approach” for producing lacunary polyoxomolybdates from plenary Keggin-type species in organic solvents. By reacting plenary Keggin-type polyoxomolybdates [α-XMo12O40]3− (X = P, As, V) with 4-methoxypyridine (pyOMe) in acetonitrile, we successfully synthesized the corresponding trivacant lacunary polyoxomolybdates ([XMo9O31(pyOMe)3]3−), where the vacant sites are stabilized by three pyOMe ligands. Remarkably, this approach enables the synthesis of a lacunary vanadomolybdate, a species previously unattainable through equilibrium control in aqueous systems. Furthermore, the reversible coordination of pyOMe ligands to molybdenum atoms at the vacant sites makes these lacunary polyoxomolybdates highly versatile precursors for assembling POM–organic hybrids. Overall, this study introduces an innovative synthetic methodology for POMs, demonstrating notable potential for advancing the development of functional materials.
Introduction
Polyoxometalates (POMs), a class of precisely defined anionic metal oxide clusters (e.g., W6+, Mo6+, and V5+), have garnered considerable attention owing to their structural diversity and remarkable physicochemical properties.1,2 These clusters are highly versatile, with widespread applications in catalysis, analytical chemistry, energy conversion and storage, medicine, and optical materials. This versatility stems from their tunable properties, including redox potentials, acidity, and stability, which can be finely adjusted by modifying their structures, constituent elements, and oxidation states. Among the various types of POMs, Keggin-type POMs [XM12O40]n− stand out for their ability to exhibit diverse properties depending on their central heteroatoms (X; e.g., P5+, As5+, Si4+, Ge4+) and polyatoms (M; e.g., W6+, Mo6+). These compounds are typically synthesized through dehydration condensation and equilibrium displacement in aqueous solvents, with careful control over pH conditions, metal ion concentrations, and mixing ratios. To date, extensive research has been conducted on the equilibrium and speciation profiles of polyoxotungstates in aqueous solvents,3 enabling the isolation of fully occupied plenary [XW12O40]n− species, along with various lacunary species including monovacant [XW11O39]n−, divacant [XW10O36]n−, and trivacant [XW9O34]n− polyoxotungstates (e.g., X = P5+, As5+, Si4+, Ge4+) (Fig. 1a).4 These lacunary polyoxotungstates, with reactive vacant sites, serve as valuable building blocks and precursors for designing functional materials.4 For instance, our group synthesized various advanced materials, such as multinuclear metal-oxo nanoclusters,5 metal nanoclusters,6 and organic–POM hybrids, using lacunary POMs in organic solvents.7 | ||
| Fig. 1 (a) Typical synthesis of lacunary polyoxomolybdates via equilibrium control in aqueous solvents. (b) Proposed approach: ligand-directed top-down synthesis of trivacant lacunary polyoxomolybdates (IX, TBA3[XMo9O31(pyOMe)3], X = P, As, V) from plenary species [XMo12O40]3− in organic solvents and their application in the synthesis of polyoxomolybdate–organic hybrids (IIX). | ||
Polyoxomolybdates, another prominent class of POMs, are characterized by electrochemical, photochemical, and physicochemical properties that substantially differ from those of their tungsten-based counterparts.8 Similar to polyoxotungstates, the speciation profiles of phosphomolybdates (P/Mo),9a,b arsenomolybdates (As/Mo),9c,d and vanadomolybdates (V/Mo)9e in aqueous solvents have been extensively researched. Recently, computational studies have further explored the speciation profiles of phosphomolybdates10a and arsenomolybdates.10b However, achieving structural control of polyoxomolybdates through equilibrium displacement in aqueous solvents remains challenging, partly because most lacunary species exist as minor species in equilibrium mixtures (Fig. 1a). Although multivacant polyoxomolybdates are promising as building blocks for functional materials owing to the high reactivity of their vacant sites and their distinctive redox properties, the synthesis of these compounds using aqueous synthetic mixtures and their subsequent characterization remain challenging. To date, the trivacant lacunary Keggin-type phosphomolybdate [PMo9O34]9− is the only multivacant polyoxomolybdate that has been isolated from aqueous mixtures.11 The use of multivacant lacunary polyoxomolybdates is further limited by their inherent structural instability. For instance, [PMo9O34]9− is prone to structural transformations or decomposition during reactions with metal ions or organic ligands in both aqueous and organic solvents.12 To address these challenges, our group recently developed a method to stabilize [PMo9O34]9− by coordinating pyridine ligands to Mo atoms at the vacant sites in organic solvents.13 This reversible coordination afforded a pyridine-stabilized lacunary phosphomolybdate ([PMo9O31(py)3]3−; py = pyridine), serving as a versatile precursor for incorporating metal ions14 and constructing POM–organic hybrids.13,15 Using this method, we also achieved a ligand-directed structural transformation of [PMo9O34]9− into a novel divacant lacunary polyoxomolybdate, [PMo10O34(py)2]3−,15a which has not been previously observed in aqueous POM chemistry.3,9,10
Building on these insights, we hypothesized that equilibrium control using organic protecting ligands in organic solvents could mitigate undesired hydrolysis, condensation, and structural decomposition. This study aimed to develop a reproducible method for synthesizing and stabilizing new lacunary polyoxomolybdates while enabling their use in functional material assembly. Accordingly, in this study, we propose a “top-down synthetic approach” for preparing lacunary Keggin-type polyoxomolybdates from their plenary precursors through ligand-directed equilibrium control in organic solvents (Fig. 1b). By reacting tetra-n-butylammonium (TBA) salts of plenary Keggin-type polyoxomolybdates [α-XMo12O40]3− (X = P, As, V) with 4-methoxypyridine (pyOMe, C6H7NO) in acetonitrile, we selectively convert them into the corresponding trivacant lacunary polyoxomolybdates IX (TBA3[XMo9O31(pyOMe)3]). In these products, three pyOMe ligands stabilize the structure through coordination with Mo atoms. Notably, this method enables the synthesis of lacunary vanadomolybdate IV, a species previously unobserved under aqueous equilibrium conditions. Furthermore, the stabilized lacunary polyoxomolybdates IX serve as precursors for assembling POM–organic hybrids IIX. Overall, this study lays the foundation for the expanded exploration and application of lacunary polyoxomolybdates in the development of functional materials, offering promising new directions in advanced POM chemistry and the design of materials for diverse applications, such as catalysis, energy conversion, and energy storage.
Results and discussion
In aqueous solutions, the equilibrium of Keggin-type POMs typically favors the formation of lacunary POMs from plenary species under high pH conditions (i.e., high hydroxide concentrations).3 Inspired by this, we explored the synthesis of lacunary polyoxomolybdates in organic solvents, beginning with the reaction of TBA3[α-PMo12O40] (PMo12) with TBAOH as a base in acetonitrile at room temperature (∼25 °C). Specifically, two different molar equivalents of TBAOH (1 or 10 equivalents relative to PMo12) were investigated. When treated with 1 equivalent of TBAOH, PMo12 retained its original structure, as confirmed by both phosphorus-31 nuclear magnetic resonance (31P NMR) spectroscopy and electrospray ionization mass (ESI-mass) spectrometry (Fig. 2a and e). However, upon adding 10 equivalents of TBAOH, the 31P NMR spectrum of the reaction mixture displayed peaks at 2.52 ppm (major species) and 5.48 ppm (minor species) (Fig. 2b). Correspondingly, the ESI-mass spectrum of the mixture revealed the formation of decomposed species, identified as [P2Mo5O23]6− (m/z = 1155.8, [TBAH4P2Mo5O23]−) and [PMo6O22]3− (m/z = 1443.8, [TBA2PMo6O22]−) (Fig. 2f). These species likely correspond to the 2.52 and 5.48 ppm signals observed in the 31P NMR spectrum.9a,16 Thus, the reaction between PMo12 and TBAOH did not yield the desired lacunary polyoxomolybdates but instead resulted in decomposition. To overcome this issue, we tested the addition of pyridine, intended to act as both a base and a stabilizing ligand for lacunary polyoxomolybdates. However, treating PMo12 with 250 equivalents of pyridine in acetonitrile for 1 h produced no observable structural changes in PMo12, as evidenced by its 31P NMR and ESI-mass spectra (Fig. 2c and g). | ||
| Fig. 2 (a–d) 31P NMR and (e–h) ESI-mass spectra of the reaction mixtures used for synthesizing lacunary POMs from PMo12 in acetonitrile under various conditions: (a and e) PMo12 after reacting with 1 equivalent of TBAOH for 1 h; (b and f) PMo12 after reacting with 10 equivalents of TBAOH for 1 h; (c and g) PMo12 after reacting with 250 equivalents of pyridine for 1 h; and (d and h) PMo12 after reacting with 250 equivalents of pyOMe for 15 min. (i) Crystal structure of the anionic component of IP. (j) ESI-mass spectrum of IP in acetonitrile. Inset: enlarged spectrum (top) and simulated pattern of [TBA2PMo9O31]− (m/z: 1875.5, bottom). Light green octahedra and the light purple tetrahedron represent [MoO6] and [PO4], respectively. Red, black, pink, and blue spheres denote O, C, H, and N atoms, respectively. | ||
Consequently, to facilitate the structural transformation, 4-methoxypyridine (pyOMe, C6H7NO; pKa = 14.23 in acetonitrile), a ligand with higher basicity than pyridine (pKa = 12.53 in acetonitrile), was used.17 When PMo12 was reacted with 250 equivalents of pyOMe at room temperature (∼25 °C) for 15 min, the peak corresponding to PMo12 disappeared from the 31P NMR spectrum, while a new peak appeared at 0.31 ppm, indicating the structural transformation of PMo12 into a new species (Fig. 2d). The ESI-mass spectrum confirmed the formation of a trivacant lacunary structure, with a prominent set of signals centered at m/z = 1875.5, attributed to [TBA2PMo9O31]− (theoretical m/z: 1875.5) (Fig. 2h). The ESI-mass spectrum displayed another set of signals centered at m/z = 1122.7, attributed to [TBAMo6O19]−, confirming the formation of a byproduct, [Mo6O19]2− (i.e., the Lindqvist-type cluster).
Crystallizing the reaction mixture by adding diethyl ether yielded single crystals suitable for X-ray diffraction analysis. This analysis revealed that the anionic component of the product (IP) was a trivacant lacunary A-α-Keggin-type phosphomolybdate, [A-α-PMo9O31(pyOMe)3]3−, featuring three pyOMe molecules coordinated to Mo atoms at the vacant sites (Fig. 2i and Table S1†). Notably, the ESI-mass spectrum of the IP crystals in acetonitrile displayed a set of signals centered at m/z = 1875.5, attributed to [TBA2PMo9O31]−; however, no signals corresponding to [Mo6O19]2− were apparent (Fig. 2j). The ESI-mass spectrum also suggested the dissociation of pyOMe ligands from the Mo atoms during the ionization process, which occurred during the ESI-mass spectrometry analysis. Further, elemental analysis confirmed that [Mo6O19]2− did not crystallize, resulting in the high-purity isolation of IP. Comprehensive characterization using various techniques—including X-ray crystallography, ESI-mass spectrometry, thermogravimetric (TG) analysis (Fig. S1†), and elemental analysis—validated the molecular formula of IP as TBA3[A-α-PMo9O31(pyOMe)3]·3(H2O). These findings indicate that IP was synthesized via a top-down transformation of plenary PMo12 in the presence of a base and protective pyOMe ligands, yielding [Mo6O19]2− as a byproduct. This transformation is represented by the following reaction equation: [α-PMo12O40]3− + OH− + 3pyOMe → [A-α-PMo9O31(pyOMe)3]3− + 0.5[Mo6O19]2− + 0.5H2O. Density functional theory (DFT) calculations further supported the thermodynamic feasibility of this reaction, with a standard Gibbs energy change of the reaction (ΔrG°) of −51.02 kJ mol−1 (equilibrium constant, K = 8.77 × 108 at 298 K).
The reaction of PMo12 with 250 equivalents of 4-cyanopyridine (pyCN, C6H4N2; pKa = 8.50 in acetonitrile), a ligand with lower basicity than pyridine (pKa = 12.53 in acetonitrile),17,18 in acetonitrile did not induce any structural transformation in PMo12 (Fig. S2†). However, when IP was reacted with 250 equivalents of pyCN in acetonitrile for 1 h, followed by crystallization using diethyl ether, single crystals of PMo9-pyCN were obtained. X-ray crystallographic analysis revealed that PMo9-pyCN was structurally analogous to IP, featuring three pyCN molecules coordinated to the Mo atoms at the vacant sites of the trivacant A-α-Keggin-type {PMo9} structure (Fig. S3 and Table S2†). Based on the results of X-ray crystallography, ESI-mass spectrometry (Fig. S4†), TG analysis (Fig. S5†), and elemental analysis, the molecular formula for PMo9-pyCN was determined to be TBA3[A-α-PMo9O31(pyCN)3]. Notably, PMo9-pyCN could not be synthesized directly from PMo12via the proposed direct top-down approach using pyCN, likely owing to the ligand's low basicity and weak coordination ability. Instead, it was successfully afforded through a ligand exchange reaction with IP, as indicated by the following equation: [A-α-PMo9O31(pyOMe)3]3− + 3pyCN → [A-α-PMo9O31(pyCN)3]3− + 3pyOMe. These results highlight the critical role of both ligand basicity and coordination ability in the ligand-directed synthesis of lacunary species derived from PMo12. To further examine this dependency, a combined approach employing pyridine and an additional base was explored. When PMo12 was reacted with 250 equivalents of pyridine and 2 equivalents of TBAOH in acetonitrile at room temperature (∼25 °C) for 1 h, the 31P NMR and ESI-mass spectra confirmed the conversion of PMo12 into IP. These observations showed that the combination of a ligand and a base effectively facilitates the structural transformation of PMo12 (Fig. S6†).
To extend this ligand-directed synthesis methodology to arsenomolybdate, we examined AsMo12 (TBA3[α-AsMo12O40]), a compound structurally analogous to PMo12 with heteroatom substitution. While previous studies examining the speciation profiles of arsenomolybdates in aqueous solvents have suggested the potential formation of a trivacant lacunary species,9c,d,10b successful isolation of this species is yet to be reported. When AsMo12 was reacted with 250 equivalents of pyOMe in acetonitrile at room temperature (∼25 °C) for 1 h, the ESI-mass spectrum of the reaction solution displayed a set of signals centered at m/z = 1919.5, attributed to [TBA2AsMo9O31]−, indicating the formation of a trivacant lacunary species (Fig. S7†). This ESI-mass spectrum also revealed the formation of [Mo6O19]2− (m/z = 1122.6) as a byproduct. Further, crystallization of the reaction mixture using p-xylene yielded single crystals of IAs. X-ray crystallographic analysis confirmed that the anionic structure of IAs closely resembled that of IP, with a trivacant lacunary configuration stabilized by three pyOMe ligands (Fig. 3a and Table S3†). The ESI-mass spectrum of IAs crystals in acetonitrile displayed a set of signals centered at m/z = 1919.5, attributed to [TBA2AsMo9O31]−; however, no signals corresponding to [Mo6O19]2− were apparent (Fig. 3b). Combined with the elemental analysis outcomes, these results demonstrate the exclusion of the byproduct ([Mo6O19]2−) during crystallization, ensuring the high purity of IAs. Comprehensive characterization through several techniques, including X-ray crystallography, ESI-mass spectrometry, TG analysis (Fig. S8†), and elemental analysis, confirmed the molecular formula of IAs as TBA3[A-α-AsMo9O31(pyOMe)3]·2(CH3CN). The formation of IAs likely followed a reaction mechanism similar to that of IP: [α-AsMo12O40]3− + OH− + 3pyOMe → [A-α-AsMo9O31(pyOMe)3]3− + 0.5[Mo6O19]2− + 0.5H2O. DFT calculations again confirmed the thermodynamic feasibility of this reaction, yielding a ΔrG° value of −76.78 kJ mol−1 (K = 2.87 × 1013 at 298 K).
 | ||
| Fig. 3 (a) Crystal structure of the anionic component of IAs. (b) ESI-mass spectrum of IAs in acetonitrile. Inset: enlarged spectrum (top) and simulated pattern of [TBA2AsMo9O31]− (m/z: 1919.5, bottom). Light green octahedra and the light blue tetrahedron represent [MoO6] and [AsO4], respectively. Red, black, pink, and blue spheres denote O, C, H, and N atoms, respectively. | ||
Building on these findings, we sought to synthesize previously unreported lacunary structures, focusing on vanadomolybdates containing a pentavalent heteroatom (V5+), analogous to phosphomolybdates (P5+) and arsenomolybdates (As5+). Notably, lacunary vanadomolybdate species are yet to be identified in speciation studies using aqueous solvents.9e Previous reports in this context have been limited to the synthesis of a lacunary vanadoxomolybdate capped by a triol at Mo atoms lying opposite the vacant site.19 To investigate the formation of lacunary vanadomolybdates, TBA3[α-VMo12O40] (VMo12) was reacted with 250 equivalents of pyOMe in acetonitrile at room temperature (∼25 °C) for 1 h. The ESI-mass spectrum of the reaction mixture displayed a set of signals centered at m/z = 1895.5, attributed to [TBA2VMo9O31]−, confirming the successful formation of a trivacant species. The spectrum also indicated the formation of [Mo6O19]2− (m/z = 1122.6) as a byproduct (Fig. S9†). Crystallization using mesitylene as a precipitant yielded single crystals of IV. X-ray crystallographic analysis revealed that IV adopted a trivacant lacunary Keggin-type vanadomolybdate structure stabilized by three pyOMe ligands coordinated to Mo atoms at the vacant sites (Fig. 4 and Table S4†). Intriguingly, some {VMo9} units transformed from an α-type structure to a β-type structure via an approximately 60° rotation of the [Mo3O13] unit, yielding an α/β ratio of 0.75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.25, as confirmed by X-ray crystallography and 51V NMR spectroscopy (Fig. S10†). The ESI-mass spectrum of IV crystals in acetonitrile displayed a set of signals centered at m/z = 1895.6, attributed to [TBA2VMo9O31]−, with no detectable presence of [Mo6O19]2−. This confirmed the effective exclusion of the byproduct from crystallization (Fig. 4b). Combining the results of X-ray crystallography, ESI-mass spectrometry, TG analysis (Fig. S11†), and elemental analysis, the molecular formula of IV was determined to be TBA3[VMo9O31(pyOMe)3]·3(H2O)·0.5(CH3CN). DFT calculations further revealed ΔrG° values of −94.43 and −89.70 kJ mol−1 for the transformation of TBA3[α-VMo12O40] into the α- and β-type structures of IV, respectively, demonstrating that the α-type structure is thermodynamically more favorable.
:0.25, as confirmed by X-ray crystallography and 51V NMR spectroscopy (Fig. S10†). The ESI-mass spectrum of IV crystals in acetonitrile displayed a set of signals centered at m/z = 1895.6, attributed to [TBA2VMo9O31]−, with no detectable presence of [Mo6O19]2−. This confirmed the effective exclusion of the byproduct from crystallization (Fig. 4b). Combining the results of X-ray crystallography, ESI-mass spectrometry, TG analysis (Fig. S11†), and elemental analysis, the molecular formula of IV was determined to be TBA3[VMo9O31(pyOMe)3]·3(H2O)·0.5(CH3CN). DFT calculations further revealed ΔrG° values of −94.43 and −89.70 kJ mol−1 for the transformation of TBA3[α-VMo12O40] into the α- and β-type structures of IV, respectively, demonstrating that the α-type structure is thermodynamically more favorable.
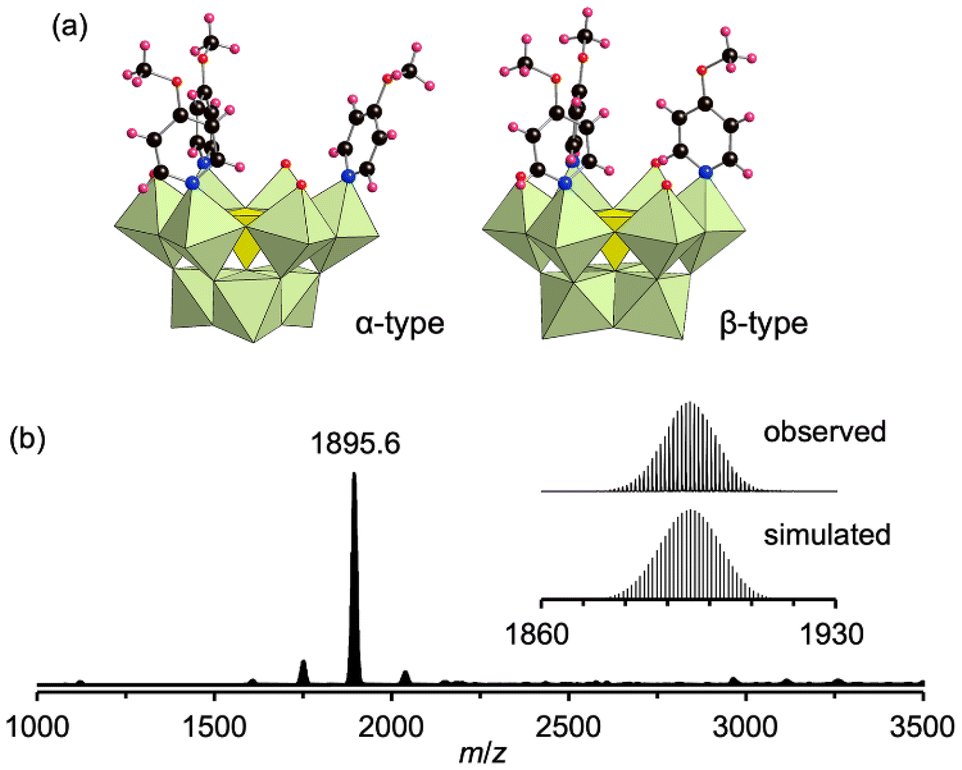 | ||
| Fig. 4 (a) Crystal structure of the anionic component of IV: α-type (left) and β-type (right). (b) ESI-mass spectrum of IV in acetonitrile. Inset: enlarged spectrum (top) and simulated pattern of [TBA2VMo9O31]− (m/z: 1895.5, bottom). Light green octahedra and the yellow tetrahedron represent [MoO6] and [VO4], respectively. Red, black, pink, and blue spheres denote O, C, H, and N atoms, respectively. | ||
Finally, we explored the potential of the IX species as precursors for the synthesis of functional materials. Leveraging the reversible coordination of pyOMe ligands with the Mo atoms at the vacant sites, we investigated their reactivity with imidazole ligands, which effectively coordinate with lacunary polyoxomolybdates, as demonstrated in our recent study.15c The reaction of IV with 2 equivalents of 1,3,5-tris[(1H-imidazol-1-yl)methyl]benzene (L, C18H18N6, Fig. 1b) in acetonitrile at 80 °C for 30 min, followed by the addition of mesitylene, afforded single crystals of IIV. X-ray crystallographic analysis revealed that the anionic structure of IIV adopted a capped monomeric configuration, comprising a {VMo9} unit and a single L ligand coordinated to three Mo atoms at the vacant site (Fig. 5a and Table S5†). Interestingly, X-ray crystallography analysis revealed that IIV contained a mixture of α- and β-type {VMo9} units, with an α/β ratio of 0.20:0.80. This indicates that some α-type {VMo9} units from IV (α/β ratio of 0.75:0.25) transformed into β-type units. The ESI-mass spectrum of IIV in acetonitrile displayed a set of signals centered at m/z = 2457.0 and 2698.2, attributed to [TBA3HVMo9O31L]+ and [TBA4VMo9O31L]+, respectively, confirming that the POM–organic hybrid structure was retained in the solution phase (Fig. 5b). Similarly, the reaction of IAs with ligand L afforded IIAs, a structural analogue of IIV, comprising α- and β-type {AsMo9} units with an α/β ratio of 0.08:0.92 (Fig. S12a and Table S5†). Further, the ESI-mass spectrum of IIAs in acetonitrile presented a set of signals centered at m/z = 2480.9 and 2723.2, attributed to [TBA3HAsMo9O31L]+ and [TBA4AsMo9O31L]+, respectively (Fig. S12b†). These results confirm the first successful synthesis of POM–organic hybrids using trivacant lacunary vanadomolybdate and arsenomolybdate as precursors.
 | ||
| Fig. 5 (a) Crystal structure of the anionic component of IIV: α-type (left) and β-type (right). (b) ESI-mass spectrum of IIV in acetonitrile. Inset: enlarged spectrum (top) and simulated pattern of [TBA3HVMo9O31L]+ (m/z: 2457.0, bottom) and [TBA4VMo9O31L]+ (m/z: 2698.2, bottom) (L = C18H18N6). Light green octahedra and the yellow tetrahedron represent [MoO6] and [VO4], respectively. Red, black, pink, and blue spheres denote O, C, H, and N atoms, respectively. | ||
Conclusions
In conclusion, we successfully developed a novel ligand-directed top-down synthetic approach for producing trivacant lacunary Keggin-type polyoxomolybdates IX (TBA3[XMo9O31(pyOMe)3], X = P, As, V) from their plenary counterparts TBA3[XMo12O40] using 4-methoxypyridine (pyOMe) as a protecting ligand in organic solvents. Our investigations using alternative pyridine ligands highlighted the critical roles of ligand basicity and coordination ability in driving this synthesis. Notably, the proposed approach enabled the synthesis of a new trivacant lacunary vanadomolybdate, a species not previously observed in aqueous systems. Furthermore, pyOMe-protected IX species were successfully transformed into POM–organic hybrids IIX through reactions with a multidentate imidazole-based ligand (L), demonstrating their versatility as precursors. The proposed ligand-directed synthetic strategy provides access to previously unexplored lacunary POMs and opens new pathways for the development of diverse functional materials leveraging these lacunary POMs as building blocks.Data availability
The data supporting this manuscript is available in the ESI† of and available on request. Crystallographic data for IP, PMo9-pyCN, IAs, IV, IIAs, and IIV have been deposited at the CCDC (deposition numbers 2408026–2408031).†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported in part by JST FOREST (JPMJFR213M), JSPS KAKENHI (24K01448, 22H04971), and the JSPS Core-to-Core program. A part of computations was performed using Research Center for Computational Science, Okazaki, Japan (Project: 23-IMS-C106, 24-IMS-C101).References
- (a) M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983 CrossRef; (b) C. L. Hill, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier Pergamon, Amsterdam, 2004, vol. 4, p. 6792 Search PubMed.
- (a) M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219 CrossRef CAS PubMed; (b) A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed., 2020, 59, 596 CrossRef CAS PubMed; (c) U. Kortz, A. Müller, J. Van Slageren, J. Schnack, N. S. Dalal and M. Dressel, Coord. Chem. Rev., 2009, 253, 2315 CrossRef CAS; (d) H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572 RSC; (e) H. N. Miras, J. Yan, D.-L. Long and L. Cronin, Chem. Soc. Rev., 2012, 41, 7403 Search PubMed; (f) M. Nyman and P. C. Burns, Chem. Soc. Rev., 2012, 41, 7354 Search PubMed; (g) I. A. Weinstock, R. E. Schreiber and R. Neumann, Chem. Rev., 2018, 118, 2680 CrossRef CAS PubMed; (h) N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112 CrossRef CAS; (i) M. Lechner, R. Güttel and C. Streb, Dalton Trans., 2016, 45, 16716 RSC; (j) K. Suzuki, N. Mizuno and K. Yamaguchi, ACS Catal., 2018, 8, 10809 CrossRef CAS; (k) S. Uchida, Chem. Sci., 2019, 10, 7670 RSC.
- (a) N. I. Gumerova and A. Rompel, Chem. Soc. Rev., 2020, 49, 7568 RSC; (b) N. I. Gumerova and A. Rompel, Sci. Adv., 2023, 9, eadi0814 CrossRef CAS PubMed; (c) E. Petrus, D. Garay-Ruiz, M. Reiher and C. Bo, J. Am. Chem. Soc., 2023, 145, 18920 CrossRef CAS PubMed.
- (a) O. Oms, A. Dolbecq and P. Mialane, Chem. Soc. Rev., 2012, 41, 7497 RSC; (b) S.-T. Zheng and G.-Y. Yang, Chem. Soc. Rev., 2012, 41, 7623 RSC; (c) J. M. Cameron, G. Guillemot, T. Galambos, S. S. Amin, E. Hampson, K. M. Haidaraly, G. N. Newton and G. Izzet, Chem. Soc. Rev., 2022, 51, 293 RSC; (d) L.-L. Liu, L. Wang, X.-Y. Xiao, P. Yang, J. Zhao and U. Kortz, Coord. Chem. Rev., 2024, 506, 215687 CrossRef CAS.
- (a) K. Suzuki, N. Mizuno and K. Yamaguchi, J. Jpn. Pet. Inst., 2020, 63, 258 CrossRef CAS; (b) K. Suzuki, F. Tang, Y. Kikukawa, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 5356 CrossRef CAS PubMed; (c) T. Minato, D. Salley, N. Mizuno, K. Yamaguchi, L. Cronin and K. Suzuki, J. Am. Chem. Soc., 2021, 143, 12809 CrossRef CAS PubMed; (d) K. Sato, K. Yonesato, T. Yatabe, K. Yamaguchi and K. Suzuki, Chem. – Eur. J., 2022, 28, e202104051 CrossRef CAS PubMed.
- (a) K. Yonesato, H. Ito, H. Itakura, D. Yokogawa, T. Kikuchi, N. Mizuno, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2019, 141, 19550 CrossRef CAS PubMed; (b) K. Yonesato, D. Yanai, S. Yamazoe, D. Yokogawa, T. Kikuchi, K. Yamaguchi and K. Suzuki, Nat. Chem., 2023, 15, 940 CrossRef CAS PubMed; (c) Y. Koizumi, K. Yonesato, S. Kikkawa, S. Yamazoe, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2024, 146, 14610 CrossRef CAS PubMed.
- M. Yamgauchi, K. Shioya, C. Li, K. Yonesato, K. Murata, K. Ishii, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2024, 146, 4549 CrossRef PubMed.
- (a) A. M. Khenkin, G. Leitus and R. Neumann, J. Am. Chem. Soc., 2010, 132, 11446 CrossRef CAS PubMed; (b) B. Nohra, H. E. Moll, L. M. R. Albelo, P. Mialane, J. Marrot, C. Mellot-Draznieks, M. O'Keeffe, R. N. Biboum, J. Lemaire, B. Keita, L. Nadjo and A. Dolbecq, J. Am. Chem. Soc., 2011, 133, 13363 CrossRef CAS PubMed; (c) J.-S. Qin, D.-Y. Du, W. Guan, X.-J. Bo, Y.-F. Li, L.-P. Guo, Z.-M. Su, Y.-Y. Wang, Y.-Q. Lan and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 7169 CrossRef CAS PubMed; (d) J. Lehmann, A. Gaita-Arino, E. Coronado and D. Loss, Nat. Nanotechnol., 2007, 2, 312 CrossRef CAS PubMed; (e) V. Prabhakaran, B. L. Mehdi, J. J. Ditto, M. H. Engelhard, B. Wang, K. D. D. Gunaratne, D. C. Johnson, N. D. Browning, G. E. Johnson and J. Laskin, Nat. Commun., 2016, 7, 11399 CrossRef CAS PubMed; (f) N. Kawasaki, H. Wang, R. Nakanishi, S. Hamanaka, R. Kitaura, H. Shinohara, T. Yokoyama, H. Yoshikawa and K. Awaga, Angew. Chem., Int. Ed., 2011, 50, 3471 CrossRef CAS PubMed; (g) H. Tanaka, M. Akai-Kasaya, A. TermehYousefi, L. Hong, L. Fu, H. Tamukoh, D. Tanaka, T. Asai and T. Ogawa, Nat. Commun., 2018, 9, 2693 Search PubMed; (h) Y.-R. Wang, Q. Huang, C.-T. He, Y. Chen, J. Liu, F.-C. Shen and Y.-Q. Lan, Nat. Commun., 2018, 9, 4466 CrossRef PubMed.
- (a) L. Pettersson, I. Andersson and L.-O. Öhman, Acta Chem. Scand., Ser. A, 1985, 39, 53 CrossRef; (b) L. Pettersson, I. Andersson and L.-O. Öhman, Inorg. Chem., 1986, 25, 4726 CrossRef CAS; (c) L. Pettersson, Acta Chem. Scand., Ser. A, 1975, 29, 677 Search PubMed; (d) G. Johansson, L. Pettersson and N. Ingri, Acta Chem. Scand., Ser. A, 1978, 32, 681 CrossRef; (e) O. W. Howarth, L. Petterson and I. Andersson, J. Chem. Soc., Dalton Trans., 1991, 1799 RSC.
- (a) J. Bulis, D. Garay-Ruiz, M. Segado-Centallas, E. Petrus and C. Bo, Chem. Sci., 2024, 15, 14218 RSC; (b) J. Build, D. Garay-Ruiz, E. Petrus, M. Segado-Centallas and C. Bo, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-r2lsq.
- S. Himeno, M. Hashimoto and T. Ueda, Inorg. Chim. Acta, 1999, 284, 237 CrossRef CAS.
- (a) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed; (b) J. A. R. van Veen, O. Sudmeijer, C. A. Emeis and H. de Wit, J. Chem. Soc., Dalton Trans., 1986, 1825 RSC; (c) L. Pettersson, I. Andersson and L.-O. Öhman, Inorg. Chem., 1986, 25, 4726 CrossRef CAS; (d) C. Marchal-Roch, E. Ayrault, L. Lisnard, J. Marrot, F.-X. Liu and F. Sécheresse, J. Cluster Sci., 2006, 17, 283 Search PubMed; (e) F. Li and L. Xu, Dalton Trans., 2011, 40, 4024 Search PubMed.
- C. Li, N. Mizuno, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2019, 141, 7687 CrossRef CAS PubMed.
- (a) C. Li, A. Jimbo, K. Yamaguchi and K. Suzuki, Chem. Sci., 2021, 12, 1240 RSC; (b) C. Li, K. Yamaguchi and K. Suzuki, Chem. Commun., 2021, 57, 7882 Search PubMed.
- (a) C. Li, K. Yamaguchi and K. Suzuki, Angew. Chem., Int. Ed., 2021, 60, 6960 Search PubMed; (b) A. Jimbo, C. Li, K. Yonesato, T. Ushiyama, K. Yamaguchi and K. Suzuki, Chem. Sci., 2023, 14, 10280 Search PubMed; (c) H. Sun, A. Jimbo, C. Li, K. Yonesato, K. Yamaguchi and K. Suzuki, Chem. Sci., 2024, 15, 9281 Search PubMed.
- L. A. Combs-Walker and C. L. Hill, Inorg. Chem., 1991, 30, 4016 CrossRef CAS.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019 CrossRef CAS PubMed.
- D. Augustin-Nowacka and L. Chmurzyñski, Anal. Chim. Acta, 1999, 381, 215 CrossRef CAS.
- D. Qu, X. Liu, F. Duan, R. Xue, B. Li and L. Wu, Dalton Trans., 2020, 49, 12950 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Tables S1–S5 and Fig. S1–S14. CCDC 2408026–2408031. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00252d |
| This journal is © The Royal Society of Chemistry 2025 |