
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking the potential of organopalladium complexes for high-grade serous ovarian cancer therapy†
Thomas
Scattolin
 *a,
Enrico
Cavarzerani
b,
Dario
Alessi
a,
Matteo
Mauceri
a,
Eleonora
Botter
b,
Giovanni
Tonon
b,
Isabella
Caligiuri
c,
Ombretta
Repetto
d,
Urska
Kamensek
e,
Simona Kranjc
Brezar
e,
Maria
Dalla Pozza
f,
Stefano
Palazzolo
c,
Maja
Cemazar
e,
Vincenzo
Canzonieri
cg,
Nicola
Demitri
h,
Steven P.
Nolan
i,
Gilles
Gasser
f,
Fabiano
Visentin
*b and
Flavio
Rizzolio
*bc
*a,
Enrico
Cavarzerani
b,
Dario
Alessi
a,
Matteo
Mauceri
a,
Eleonora
Botter
b,
Giovanni
Tonon
b,
Isabella
Caligiuri
c,
Ombretta
Repetto
d,
Urska
Kamensek
e,
Simona Kranjc
Brezar
e,
Maria
Dalla Pozza
f,
Stefano
Palazzolo
c,
Maja
Cemazar
e,
Vincenzo
Canzonieri
cg,
Nicola
Demitri
h,
Steven P.
Nolan
i,
Gilles
Gasser
f,
Fabiano
Visentin
*b and
Flavio
Rizzolio
*bc
aDipartimento di Scienze Chimiche, Università degli Studi di Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: thomas.scattolin@unipd.it
bDipartimento di Scienze Molecolari e Nanosistemi, Università Ca’ Foscari, Campus Scientifico Via Torino 155, 30174 Venezia-Mestre, Italy. E-mail: fvise@unive.it; flavio.rizzolio@unive.it
cPathology Unit, Centro di Riferimento Oncologico di Aviano (C.R.O.) IRCCS, via Franco Gallini 2, 33081, Aviano, Italy
dImmunopathology and Cancer Biomarkers, Centro di Riferimento Oncologico di Aviano (CRO), IRCCS, via Franco Gallini 2, 33081, Aviano, Italy
eDepartment of Experimental Oncology, Institute of Oncology Ljubljana, Ljubljana, 1000, Slovenia
fChimieParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, Laboratory for Inorganic Chemical Biology, 75005, Paris, France
gDepartment of Medical, Surgical and Health Sciences, Università degli Studi di Trieste, Strada di Fiume 447, Trieste, Italy
hArea Science Park, Elettra-Sincrotrone Trieste, S.S. 14 Km 163.5, Basovizza, 34149, Trieste, Italy
iDepartment of Chemistry and Centre for Sustainable Chemistry, Ghent University Krijgslaan 281, S-3, 9000 Ghent, Belgium
First published on 14th February 2025
Abstract
High-Grade Serous Ovarian Cancer (HGSOC) is the most common and lethal subtype of ovarian cancer, known for its high aggressiveness and extensive genomic alterations. Typically diagnosed at an advanced stage, HGSOC presents formidable challenges in drug therapy. The limited efficacy of standard treatments, development of chemoresistance, scarcity of targeted therapies, and significant tumor heterogeneity render this disease incurable with current treatment options, highlighting the urgent need for novel therapeutic approaches to improve patient outcomes. In this study we report a straightforward and stereoselective synthetic route to novel Pd(II)-vinyl and -butadienyl complexes bearing a wide range of monodentate and bidentate ligands. Most of the synthesized complexes exhibited good to excellent in vitro anticancer activity against ovarian cancer cells. Particularly promising is the water-soluble complex bearing two PTA (1,3,5-triaza-7-phosphaadamantane) ligands and the Pd(II)-butadienyl fragment. This compound combines excellent cytotoxicity towards cancer cells with substantial inactivity towards non-cancerous ones. This derivative was selected for further studies on ex vivo tumor organoids and in vivo mouse models, which demonstrate its remarkable efficacy with surprisingly low collateral toxicity even at high dosages. Moreover, this class of compounds appears to operate through a ferroptotic mechanism, thus representing the first such example for an organopalladium compound.
Introduction
Ovarian cancer (OC), especially its most common and aggressive histotype, high-grade serous ovarian cancer (HGSOC), exhibits molecular diversity and complex clinical profiles.1 Genomic features include prevalent TP53 mutations, homologous recombination repair defects, and widespread copy-number variations. About 25% of HGSOC cases have alterations in breast cancer (BRCA) genes and disruptions in tumor suppressor and cell-cycle-regulating genes.2This heterogeneity contributes to the non-curative nature of the disease and the challenges in formulating an effective therapeutic plan. Significant advances in therapy have been made in the past years, particularly with molecular targeted drugs such as the anti-VEGF antibody bevacizumab (VEGF = Vascular Endothelial Growth Factor) and PARP inhibitors (PARP = Poly ADP Ribose Polymerase) like olaparib, rucaparib, and niraparib.3 However, given the rarity of mismatch repair-deficient Epithelial Ovarian Cancer (EOC) and the limited efficacy demonstrated by immunotherapies, with a low response rate in OC, the development of new targeted drugs and biomarkers remains critical.4 In fact, the primary treatment for HGSOC patients is still surgery and platinum-based chemotherapy, but there is a high relapse rate of 70–80% and growing treatment resistance.4,5 Given HGSOC's genetic variability, a multitarget approach could be more effective. The therapeutic landscape for OC is evolving with the emergence of antibody–drug conjugates (ADCs) such as raludotatug deruxtecan, an ADC consisting of a humanized IgG1 antibody against cadherin 6 (CDH6), is currently in a Phase 1 trial for OC therapy6 and mirvetuximab soravtansine, which targets FRα (FRα = Folate Receptor α) showing efficacy and safety in the MIRASOL trial.7 Trastuzumab deruxtecan has also demonstrated significant results in HER2-positive solid tumors (HER2 = human epidermal growth factor receptor 2).8 These ADCs deliver potent cytotoxic agents directly to cancer cells, optimizing efficacy and minimizing side effects,9 suggesting that potent multitarget agents could be an effective strategy in OC therapy. The efficacy of treatments is often hindered by mutations in cancer cells that activate compensatory pathways, contributing to the high mortality rate from drug resistance, which is implicated in up to 90% of cancer-related deaths.10,11 Chemo-resistant cancer cells often result from enhanced DNA repair mechanisms or dysfunctional apoptosis pathways.12 Therefore, strategies that bypass the resistance to cell death, like ferroptosis, can be highly effective in treating HGSOC, which lacks specific targets and shows significant genetic variability and high recurrence rates.
The recognition of regulated cell death (RCD) in the 1960s opened the door to the concept of modulating cell death processes.13 For many years, the induction of caspase-dependent apoptosis represented the cornerstone of anti-cancer therapies.14 As research has advanced, it is now well-established that cancer cells may become resistant to apoptosis, prompting a shift in research to other types of RCD mechanisms.15 Current strategies in cancer treatments are evolving to address these resistances by harnessing non-apoptotic pathways to eliminate cancer cells more effectively.
Among the few non-apoptotic mechanisms, particularly interesting is ferroptosis. Cells undergoing this process display unique features such as increased mitochondrial membrane density and reduced mitochondrial volume.16,17 Importantly, some drug candidates operating via this mechanism have proven effective against several apoptosis-resistant cancer cells.18 Hence, the advent of ferroptosis paves the way for a novel research direction in the pursuit of potent cancer therapies.
However, most potential anticancer agents that act according to this mechanism are purely organic molecules.16,17 Far fewer contributions are present in the literature regarding organometallic compounds inducing ferroptosis,16a with only one example (an iridium complex) concerning OC and limited to in vitro studies on A2780 cells (cisplatin-sensitive).19
It is well-established that organometallic anticancer drugs possess some distinct advantages compared to purely organic counterparts, primarily due to their unique chemical properties that enable precise targeting of cancer cells while minimizing damage to healthy tissues.20 Indeed, their metal-containing cores offer diverse coordination geometries, facilitating interactions with specific cellular targets and overcoming resistance mechanisms. Moreover, organometallic compounds often exhibit enhanced stability and tunable reactivity, allowing for tailored optimization of therapeutic efficacy and reduced side effects.20
Some recent classes of organometallic compounds that have provided encouraging in vitro results towards OC cells contain palladium as the metal center.21 It has to be noted that palladium complexes are considerably less explored as potential antitumor agents compared to platinum, ruthenium, and gold complexes, which have passed or are still involved in clinical trials for cancer therapy.22 The main reason for this lies in the high reactivity of palladium complexes in biological media, which significantly increases the speciation and toxicity of this metal.23 However, it is possible to substantially mitigate this issue by employing ligands strongly anchored to the metal center, which suppress the hydrolysis of the complex, allowing it to reach the biotarget.24 In this context, the best option is usually the utilization of at least one metal–carbon bond (organometallic fragment), which provides the complex with high stability in solution, even under physiological conditions.25,26
Since the pioneering studies conducted by Köpf and Köpf-Maier in the early 1980s on the promising antitumor activity of metallocene complexes,27 many research groups have focused on the synthesis and the study of therapeutic effects of compounds bearing a wide range of organometallic fragments. The nature of the organometallic portion highly influences the hydrophilicity/hydrophobicity of the complexes, with a profound impact on the main cellular target and cellular uptake.28 Furthermore, given that most metallodrugs are prodrugs, the organometallic fragment as well as the metal center are crucial for modulating the reactivity of the complex towards nucleophiles or reducing agents present in the biological environment.29 These processes may involve the other ligands present in the coordination sphere of the metal (e.g. ligand substitution) as well as the organometallic fragment (e.g. attack on coordinated ligand, insertion or ligand substitution).
Among the numerous scenarios involving organometallic compounds with potential biological activity, summarized in a recent review by Sadler and co-workers (Scheme 1),30 the metal-vinyl and metal-butadienyl complexes are almost unexplored.
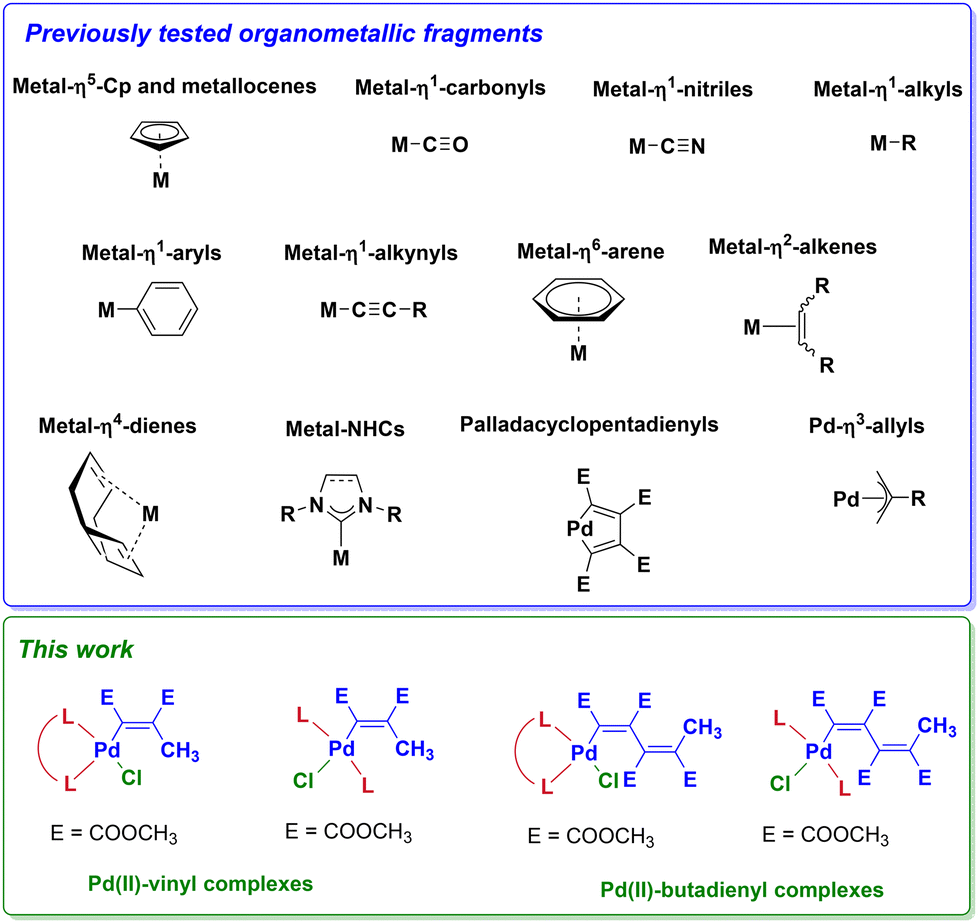 | ||
| Scheme 1 Overview of organometallic fragments with promising biological activity. | ||
In this context, we now report the synthesis of novel Pd(II)-vinyl and Pd(II)-butadienyl complexes and an in-depth study of their in vitro, ex vivo and in vivo antitumor activity on HGSOC models as well as some hypothesis about their mechanism of action, which seems to involve an unprecedent ferroptotic pathway, thus marking a significant step forward in targeted OC therapy. As mentioned above, the ferroptosis mechanism has been observed only in a very limited number of organometallic compounds with potential anticancer activity16a and, to the best of our knowledge, there are no examples of organopalladium compounds that act through this mode of action. These latter usually target DNA, interacting with it through either covalent or non-covalent bonds,31 or induce the death of tumor cells through an apoptotic process involving an early mitochondrial damage, often resulting from the inhibition of key proteins such as thioredoxin reductase (TrxR).32
Results and discussion
Synthesis of Pd(II)-vinyl and -butadienyl complexes bearing N–N, P–P and C–C bidentate ancillary ligands
Since Pd(II)-vinyl and Pd(II)-butadienyl complexes play a crucial role as intermediates in numerous cross-coupling, oligomerization, and polymerization processes of alkynes,33 some previous studies have highlighted that such organometallic species can be isolated through oxidative addition of Pd(0) complexes,34 addition of halogens/organohalides on palladacyclopentadienyl complexes,35 or by insertion of an alkyne into a Pd–C bond.36We believe that this latter synthetic strategy, which appears to involve the formation of a Pd(II)-η2-alkyne intermediate,37–39 is most suitable for the purposes of this work. This route involves a stereoselective insertion of an electron-deficient alkyne such as dimethyl-2-butynoate (DMAD) onto a Pd–methyl bond. Interestingly, in a previous work published by our group, we found that a particular N–S bidentate ligand, namely 2-methyl-6-(phenylthiomethyl)pyridine, allows to surprisingly control the number of alkyne molecules involved in the insertion (Scheme 2).36
 | ||
| Scheme 2 (A) General mechanism of formation of Pd(II)-vinyl complexes via insertion; (B) 2-methyl-6-(phenylthiomethyl)pyridine enable the controlled mono- and bis-insertion. | ||
Conversely, the use of P–P and N–N bidentate ligands leads to mixtures of mono- and poly-insertion products or hinders the reaction from proceeding beyond the mono-insertion product, respectively.40–44
The 2-methyl-6-(phenylthiomethyl)pyridine ligand, in addition to facilitating and making the alkyne insertion process controllable, has also another indisputable advantage. In fact, due to the distorting effect of the methyl substituent on the chelate ring, this pyridylthioether ligand can be effortlessly replaced by other ligands.45 With this approach, we have an easy access to numerous palladium vinyl and butadienyl complexes which cannot be obtained by direct insertion.
With this valuable information in hand, we have successfully reacted the Pd(II)-vinyl and -butadienyl precursors 1a–b with one equivalent of three different bidentate ligands (1,10-phenanthroline, neocuproine and dppp = 1,3-bis(diphenylphosphino)propane), and the corresponding complexes 2a–b, 3a–b and 4a–b were obtained in high yields and purity (Scheme 3). With a similar procedure, the dppe (dppe = 1,2-bis (diphenylphosphino)ethane) derivatives 5a–b were previously synthesized.36a Notably, complex 3a was previously synthesized with a different procedure.36b All reactions reported in Scheme 3 proceed at room temperature within 30 minutes, with the only exception of complex 3b which requires about 6 days to complete the replacement of the pyridylthioether ligand with neocuproine. This is most likely due to the simultaneous presence of the methyl groups in the ancillary ligand and to the higher steric demand of the butadienyl moiety compared to the vinyl one.
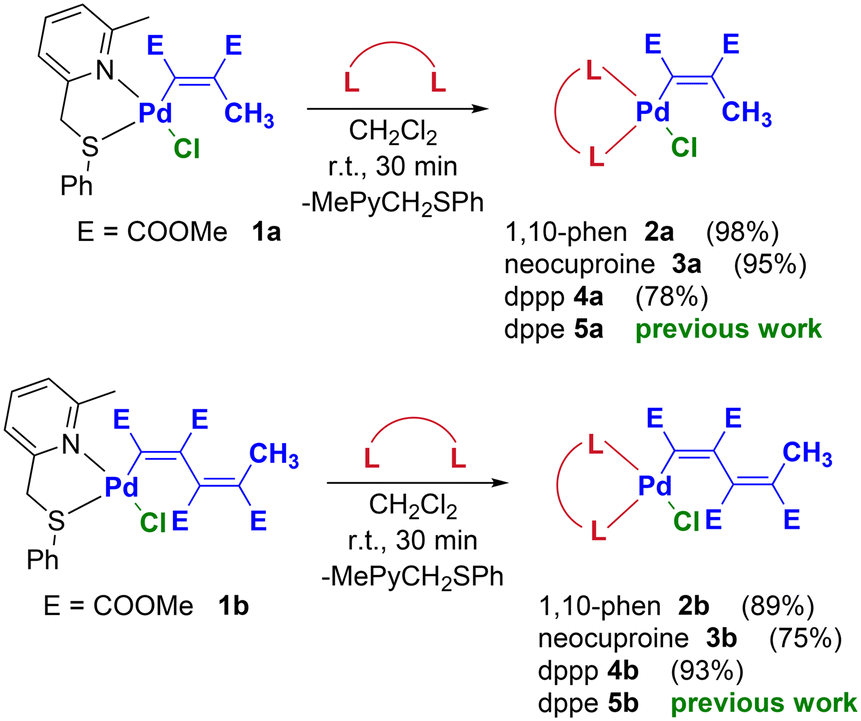 | ||
| Scheme 3 Synthetic procedure leading to Pd-vinyl and -butadienyl complexes bearing N–N (2a–b and 3a–b) and P–P bidentate ligands (4a–b and 5a–b). | ||
The products were exhaustively characterized by NMR, IR, elemental analysis and, in some cases, by single crystal X-ray diffraction (see Crystal structure determination section). In the 1H NMR and 13C NMR spectra of complexes 2a–b and 3a–b, the signals of coordinated 1,10-phenanthroline or neocuproine usually appear at higher chemical shifts with respect to those of the free ligand. On the other hand, the coordination of the vinyl or butadienyl moieties is evidenced by the presence in the 1H NMR spectra of the terminal methyl protons signal (2.1–2.6 ppm) and those ascribable to the ester protons OCH3 (3.2–3.9 ppm). Moreover, in the 13C NMR spectra the organopalladium fragment shows the following diagnostic signals: carbonyl carbons (161–174 ppm), vinyl carbons (127–168 ppm), ester carbons OCH3 (51–52 ppm) and the terminal methyl carbon (19–22 ppm).
In the case of the diphosphine complexes 4a–b and 5a–b, the 31P NMR spectra confirm the coordination of dppp and dppe. More in detail, it is possible to observe the presence of two doublets (−6 and 11–15 ppm with JP–P = 41–46 Hz for 4a–b; 53–56 and 40–45 ppm with JP–P = 19–24 Hz for 5a–b), significantly downshifted compared to uncoordinated dppp and dppe ligands. Similarly to the complexes bearing N–N bidentate ligands, all vinyl and butadienyl signals as well as the alkyl/aryl signals of the diphosphine ligand are present and assigned in the 1H and 13C NMR spectra. Particularly diagnostic is the signal ascribable to the vinyl carbon directly bound to palladium which resonates as a doublet at ca. 170 ppm (JC–P ≈ 120 Hz). Finally, in the IR spectra the stretching bands of the carbonyl groups at 1700 cm−1 are particularly worthy of mention.
The last class of bidentate ligands that we have considered in this work is that of diNHCs (NHCs = N-heterocyclic carbenes). Such ligands are strong σ-donors and generally efficiently stabilize late transition metal complexes.46 However, it must be remembered that, in some cases, their high trans-labilizing effect tends to labilize the fragment trans to the carbene carbon. It is therefore always necessary to modulate the steric and electronic characteristics of both the diNHC ligand and the other coordinated fragments to ensure the obtainment of the species of interest in a pure form and with sufficient stability. With these premises we carried out the reaction between the precursors 1a–b and the silver complex 6, which is equipped with a methylene bridge and benzyl wingtip substituents (Scheme 4).
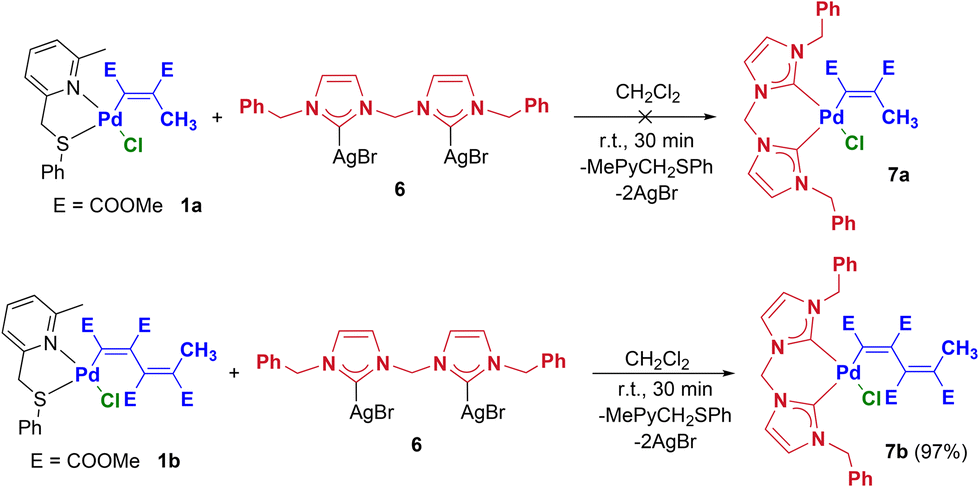 | ||
| Scheme 4 Synthetic procedure leading to Pd-vinyl and -butadienyl complexes bearing chelating diNHC ligands (7a–b and 3a–b). | ||
Although the transmetallation reaction took place in both cases, it was possible to isolate in a pure form and in good yield only the butadienyl derivative 7b. The reduced steric hindrance of the vinyl unit compared to the butadienyl one compromises the stability of 7a, generating a mixture of by-products that are difficult to identify and increase in percentage over time. The 1H NMR spectrum of 7b presents, in addition to the typical signals of the butadienyl fragment, all those attributable to the dicarbene ligand. In particular, the methylene protons NCH2Ph and NCH2N resonate as AB systems between 4.7 and 6.3 ppm, due to the blocked rotation about the NCH2–Ph bonds and the blocked conformational movement of the chelate ring, respectively. In the 13C NMR spectrum, the signals of the three methylene carbons (54–64 ppm) and those of the two carbene carbons (166 and 175 ppm) deserve to be highlighted.
Synthesis of Pd(II)-vinyl and -butadienyl complexes bearing monodentate ancillary ligands
The lability of 2-methyl-6-(phenylthiomethyl)pyridine also enables the introduction of two monodentate ligands into the coordination sphere of palladium. Therefore, by adding 2 equivalents of PPh3, PTA (PTA = 1,3,5-triaza-7-phosphaadamantane) or AsPh3 to the palladium precursors 1a–b it is possible to selectively obtain the complexes 8a–b, 9a–b and 10a–b in high yields (Scheme 5).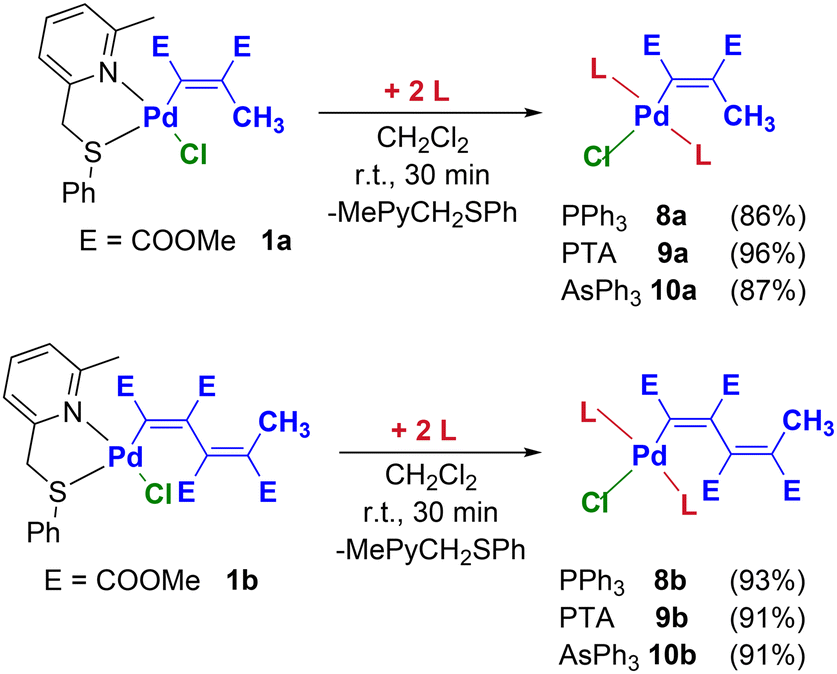 | ||
| Scheme 5 Synthetic procedure leading to Pd-vinyl and -butadienyl complexes bearing two monodentate ancillary ligands (8a–b, 9a–b and 10a–b). | ||
The formation of the trans isomers (thermodynamic products) is confirmed by the presence in the 31P NMR spectra of 8a–b and 9a–b of one singlet significantly downshifted with respect to the uncoordinated phosphine (Δδ ≈ 30–40 ppm). Consistently, in the 1H and 13C NMR spectra it is possible to identify a single set of aryl signals for complexes 8a–b/10a–b and, as regards the PTA derivatives 9a–b, a single signal attributable to the protons and methylene carbons (NCH2P and NCH2N). In particular, the NCH2P and NCH2N protons resonate as tight multiplets at 4.3 and 4.5 ppm, respectively, whereas the corresponding carbons are located at ca. 50 and 70 ppm. Finally, the vinyl carbon signal directly bound to palladium appears as a triplet at 157–170 ppm (JC–P ≈ 10 Hz), owing to the coupling with the two magnetically equivalent phosphorus nuclei in the complexes 9a–b.
The choice of PTA is mainly due to the high solubility in water of this particular phosphine and its transition metal complexes.47 This feature, together with the stability of PTA to oxidation, is the reason for its success in the development of new generations of metal-based anticancer agents.48 Furthermore, since PTA is a less encumbered phosphine than the classical triphenylphosphine (cone angles = 102° and 145°, respectively), its derivatives 9a–b might be significantly different from a steric point of view compared to their triphenylphosphine congeners 8a–b. On the contrary, the electronic characteristics of these two phosphines are known to be similar.
Interestingly, any attempt to introduce two monodentate NHC ligands resulted in mixtures of unidentified products and significant decomposition to metallic palladium, suggesting poor stability of the desired complexes. Two examples of such reactions are reported in Scheme 6.
 | ||
| Scheme 6 Reaction between 1a–b and 2 equiv. of silver–NHC complex 11. | ||
For completeness, we conducted a classical study of organometallic reactivity involving Pd(II)-vinyl complexes and [(PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)Sn(n-Bu)3]. This study dealing with the vinyl fragment extrusion, which is reported in detail in the ESI,† allowed the isolation of an unprecedented palladium complex containing both Pd(II)-vinyl and Pd(II)-phenylacetylide fragments. Complex 13, owing to its unique structure and high stability in solution, was subsequently considered for biological tests (Scheme 7).
C)Sn(n-Bu)3]. This study dealing with the vinyl fragment extrusion, which is reported in detail in the ESI,† allowed the isolation of an unprecedented palladium complex containing both Pd(II)-vinyl and Pd(II)-phenylacetylide fragments. Complex 13, owing to its unique structure and high stability in solution, was subsequently considered for biological tests (Scheme 7).
 | ||
| Scheme 7 Synthetic procedure leading to complex 13. | ||
Stability of Pd(II)-vinyl and -butadienyl complexes
Before carrying out the biological tests, we have monitored the stability of the synthesized complexes in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 D2O/DMSO-d6 solution by NMR spectroscopy. After 48 hours no significant changes of the spectra are detectable, indicating that the complexes retain their structural integrity. Taking advantage of the solubility in water of PTA derivatives, we have additionally monitored the stability of complex 9b in a 1:1 culture medium/D2O solution by 31P NMR (Fig. S1 in ESI†). The complex was stable even under these conditions, without releasing the chloride ligand and/or the other fragments bound to palladium.
:1 D2O/DMSO-d6 solution by NMR spectroscopy. After 48 hours no significant changes of the spectra are detectable, indicating that the complexes retain their structural integrity. Taking advantage of the solubility in water of PTA derivatives, we have additionally monitored the stability of complex 9b in a 1:1 culture medium/D2O solution by 31P NMR (Fig. S1 in ESI†). The complex was stable even under these conditions, without releasing the chloride ligand and/or the other fragments bound to palladium.
Crystal structure determination
2a, 2b, 3b, 5a and 13 crystalline forms bear one crystallographically independent palladium complex each (Fig. 1). All the molecules are neutral and the corresponding palladium centers show square planar coordination spheres, with geometries in agreement with previously published data (Tables S2–7†). Significant longer bond lengths can be clearly highlighted replacing the phenanthroline based ligands with a bidentate phosphine. Monodentate PTA moieties in 13 allow P ligands to adopt a trans configuration and relaxed Pd angles with neighbour ligands (i.e. closer to “ideal value” of 90°). 1,10-Phenantroline based complexes (2a, 2b, 3b) show perfectly superimposable Pd coordination spheres in 2a and 2b and a notable ligand planarity distortion in 3b, due to steric hindrance introduced by methyl groups. This effect allows also a deep conformational rearrangement of butadiene ligand which can form an intramolecular stacking interaction with the dimethyl-phenanthroline (the average angle between the phenanthroline and the peripheric olefine mean planes is ∼22° in 3b, while it's ∼72° in 2b). Equivalent methyl maleate dimethyl ester conformation is present in complexes 2a, 13 and 5a (R.M.S.D. ∼1.1 Å, mostly due to –COOCH3 rotations) with an average angle between the phenanthroline and the olefin mean planes of ∼85°. | ||
| Fig. 1 X-ray molecular structures of complexes 2a, 2b, 3b, 5a and 13 are presented, showing thermal displacement ellipsoids at the 50% probability level with hydrogen atoms and solvent molecules omitted for clarity. | ||
Crystal packing show hydrophobic contacts among neighbour molecules, involving intermolecular π⋯π stacking among phenanthroline moieties and CH⋯π interactions, involving peripheral methoxy groups. Solvent molecules (chloroform) have been found in the crystal packing of 2a, 13 and 5a and they are bound to ligand heteroatoms through polar contacts (with shortest dCH⋯O = 3.204(2) Å in 2a, dCH⋯N = 3.189(3) Å in 13, dCH⋯O = 3.180(3) Å in 5a).
Anticancer activity on human cancer and non-cancerous cell lines and patient derived tumor organoids grown from patient tissues or ascites fluids
With the aim of investigating the potential anticancer activity of the Pd(II)-vinyl and -butadienyl complexes, a panel of four different human tumor cell lines (OC A2780, with its cisplatin resistant clone A2780cis, triple-negative breast cancer MDA-MB231 and colon cancer DLD1) and MRC-5 non-cancerous cells (human lung fibroblasts) were treated for 96 hours with our compounds and cisplatin (positive control). In Fig. 2A are reported the resulting half inhibitory concentrations (IC50) values. | ||
| Fig. 2 (A) IC50 values after 96 h of incubation. Stock solutions in DMSO for all complexes; stock solutions in H2O for cisplatin. A2780 and A2780cis (cisplatin-sensitive and cisplatin-resistant OC cell lines), MDA-MB231 (triple-negative breast cancer cell line), DLD1 (colon cancer cell line) and MRC-5 (normal lung fibroblasts); (B) dose–response curves and (C) IC50 values of patient-derived and mouse-derived organoids after 96 h of incubation. | ||
Based on the IC50 values obtained, it is possible to make some important considerations. As far as concerned the two OC cell lines (A2780 and A2780cis), all the compounds examined, except for those containing triphenylarsine (10a and 10b), have comparable or lower IC50 values than cisplatin, especially in the cisplatin-resistant cell line (A2780cis). Most of the compounds exhibit comparable activity between these two OC lines, suggesting a different mechanism of action from that of cisplatin. For the latter, there are in fact almost two orders of magnitude between the IC50 values obtained in the two cell lines (0.4 μM vs. 27 μM).
As for the triple-negative breast cancer cells MDA-MB231, all compounds, except those containing triphenylarsine (10a and 10b), show a marked anticancer activity compared to cisplatin. In the colon cancer line (DLD-1), only compounds 3a–b, 8a and 9a–b are more active than cisplatin. In the same line, compounds 2a–b, 4a–b, 5a–b and 8b show good cytotoxicity, while complexes 7b, 10a–b and 13 are practically inactive.
Comparing the cytotoxicity of compounds containing the same ancillary ligands (monodentate or bidentate), there is not a clear difference in activity between the vinyl derivatives and their butadienyl congeners.
Interestingly, the analyses on MRC-5 non-cancerous cells show that most compounds that exhibited antitumor activity, except for those bearing N–N bidentate ligands (2a–b and 3a–b), are at the same time poorly cytotoxic towards non-cancerous cells. In this context, compounds 8a, 9a and 9b are particularly promising, since they show excellent antitumor activity on all cancer cell lines examined and reduced cytotoxicity towards human lung fibroblasts. It should be noted that the selectivity of the compounds containing the PTA ligand has already been observed in the past with Pd(II)-allyl,32a,48d palladacyclopentadienyl21b and Pd(0)-olefin45e complexes.
Based on these encouraging biological data, we selected compound 9b for further studies on more complex biological systems such as tumoroids extracted from OC patients (ex vivo tests) as well as on animal models (in vivo tests).
As shown in Fig. 2C, compound 9b was evaluated across five patient-derived tumor organoids (PDTOs) originating from both primary tumors and ascites fluid in patients diagnosed with HGSOC (PDTO-2, 3, 4, 5) and one low-grade serous OC (LGSOC) (PDTO-1), and one mouse-derived liver organoid (MDO-liver). Surprisingly, compound 9b proved to be effective in three out of the five PDTOs, including two that are resistant to carboplatin (PDTO-1 and PDTO-2). Notably, the sensitivity of the PDTOs to carboplatin (the reference compound for clinical standard therapy) is representative of the actual clinical response of the patient from whom these PDTOs were derived. This suggests that compound 9b might represent a novel therapeutic option for a subset of patients that show resistance to carboplatin, the standard first-line chemotherapy for ovarian cancer. Additionally, the toxicity profile of 9b was evaluated in MDO-liver, demonstrating no toxicity in comparison to carboplatin.
Mechanism of cell death
After observing promising cytotoxic effects in OC PDTOs, we decided to investigate how 9b acts in these cells by carrying out specific assays to unravel the mechanisms by which this compound might lead to cell death. Before proceeding with the elucidation of the mechanism of action, it is crucial to underline that cell death is not exclusively associated with malignancy; rather, it is a vital process regulated by complex molecular mechanisms that are essential for maintaining bodily homeostasis and facilitating normal development. This understanding sets the stage for exploring the specific pathways through which therapeutic agents may induce cell death in cancer cells, allowing us to distinguish between pathological and physiological cell death processes.49 It is important to note that a variety of cell death mechanisms such as apoptosis, necroptosis, pyroptosis, and autophagy have been identified and each of them have its unique mechanism.50 Therefore, experiments aimed at deciphering the cellular death mechanism were conducted. As demonstrated by subsequent treatments with compound 9b in A2780 and OVCAR5 (IC50 value is reported in Fig. S2A in ESI†) cells at different time points up to 96 hours, there was no observed increase in the expression of apoptosis-related proteins such as cytochrome C (Fig. S2B and E†) and caspases 3/7 (Fig. S2C and D in ESI†). Intriguingly, there was a statistically significant increase in reactive oxygen species (ROS) in both A2780 and OVCAR5 cells after treatment with compound 9b (Fig. 3A and B). Summarizing, as reported in Fig. 3C, the lack of caspase 3/7 and Cytochrome C activation excludes apoptotic cell death, while absence of nuclear atypia and cellular swelling/blebbing excludes necrosis cell death (Fig. S4 in ESI†), suggesting the potential involvement of ferroptosis.51 The potential implication of ferroptosis in these observations is underscored by its distinction in morphological and biochemical profiles from established programmed cell death pathways, such as apoptosis, necroptosis, pyroptosis, and autophagy.52 Characterized by a disruption in cellular redox homeostasis, ferroptosis manifests through elevated intracellular reactive oxygen species (ROS) and a reduction in NADPH levels, without the typical features of plasma membrane blebbing, DNA fragmentation, or Caspase-3 activation. Additionally, the lack of activation in the pro-apoptotic proteins Bax and Bak suggests that apoptosis inhibitors do not thwart this mode of cell death.53 | ||
| Fig. 3 ROS production in OVCAR5 (A) and A2780 (B) cells treated with compound 9b (1, 25, 50 μM) and Doxorubicin (1 μM as positive control) at 24, 48, and 96-hour intervals; (C) summary of the assays performed to identify the mechanism of 9b in A2780 and OVCAR-5: the lack of apoptotic mechanism due to non-activation of caspase 3/7 and Cytochrome-C, and the generation of ROS leads to the hypothesis of a ferroptosis mechanism; (D) proteomic profile: graphical representation of the abundance (Ab.) of proteins in 1 μM treated OVCAR5 versus NT groups (Padjusted < 0.05). | ||
To further explore the molecular mechanisms underlying the cell death, we proceeded to profile the proteome of OVCAR5 cells following treatment with compound 9b. From around 2000 proteins studied, label-free LC-MS/MS analysis identified a group of proteins significantly differing in abundance between 1 μM 9b-treated OVCAR5 versus NT cells (Padjusted < 0.05, Fig. S3 in ESI†). As shown in Fig. 3D this analysis highlighted a statistically significant reduction in the expression of Phospholipid hydroperoxide glutathione peroxidase (GPX4), Ferritin light chain (FTL) and Glutaredoxin-1 (GLRX). It is well-established that the inhibition of GPX4 constitutes a key step in the induction of ferroptosis.54 This understanding has led us to propose the hypothesis that the cytotoxic effects of compound 9b observed in OC cell systems could be ascribed to ferroptosis. Ferroptosis induction is closely linked to GPX4 inhibition, which mitigates lipid peroxidation by reducing peroxides using glutathione, thereby safeguarding cells from oxidative stress. Impaired GPX4 function results in ROS build-up and subsequent cell death (Fig. 4).55 Additionally, the ferroptotic mechanism can be facilitated by blocking the synthesis of glutathione (GSH), such as through the inhibition of GLRX, or by the increase in Fe3+, a substrate of the Fenton reaction, caused by an inhibition of FTL (FTL = Formate Tetrahydrofolate Ligase).56
 | ||
| Fig. 4 An illustration showing the role of GLRX, GPX4 and FTL suppression, operated by compound 9b, in predisposing therapy-resistant cancer cells to ferroptosis. The image was generated with DAVID software (https://david.ncifcrf.gov/tools.jsp) and subsequently customized. | ||
In vivo experiments on an ovarian cancer mouse model
Finally, to test the efficacy of compound 9b in an in vivo model, mice were treated intravenously up to 100 mg kg−1. Surprisingly, the body weight of mice does not change (Fig. 5A). Following, we compared the effect of compound 9b to cisplatin in a subcutaneous model of OC. Compound 9b significantly inhibits the growth of OVCAR5 tumor mass like cisplatin (Fig. 5B and C). Even the overall survival of the mice increases significantly compared to untreated mice (Fig. 5D). As confirmation, the analyses of body weight of mice do not reveal signs of toxicity (Fig. 5A). | ||
| Fig. 5 Therapeutic effectiveness (intravenously) in OVCAR5 tumors after treatment with cisplatin or complex 9b (n = 6); (A) animal body weight after treatment with cisplatin or complex 9b, data represent the mean and standard deviation; (B) tumor growth curves, data represent the mean and standard error of the mean. p-Values were calculated using two-way ANOVA * p ≤ 0.05; (C) tumor growth delay of tumors compared to untreated tumors (CTRL), data represent the mean and standard error; (D) Kaplan–Meier survival curve illustrating the percentage of survival over 35 days for a control group, a group treated with cisplatin (cis), and a group treated with compound 9b. Both treatment groups exhibit improved survival rates over the control, p-values calculated by log-rank test * p ≤ 0.05. | ||
Conclusions
We have described the synthesis of novel Pd(II)-vinyl and -butadienyl complexes using a simple and stereoselective synthetic route that involved the controlled mono- or bis-insertion of an electron-poor alkyne, followed by the exchange of the pyridyl-thioether ligand with a wide range of mono- and bidentate ligands. All compounds were characterized by NMR, IR, elemental analyses, and, in some cases, by X-ray diffraction.Most of the synthesized complexes exhibited good to excellent in vitro anticancer activity against ovarian, breast, and colon cancer cell lines. Specifically, the performance of the vinyl complexes was comparable to that of their butadienyl congeners, with IC50 values in the micro- and sub-micromolar range, often significantly lower than those of cisplatin. Particularly interesting are compounds 8a, 9a, and 9b, which showed excellent activity against all examined tumor lines and reduced cytotoxicity towards MRC-5 non-cancerous cells. Among these derivatives, compound 9b was selected for further studies on ex vivo and in vivo models.
Experiments on PDTO derived HGSOC revealed a generally higher cytotoxicity of 9b compared to carboplatin (standard therapy), even in these more complex biological models. Interestingly, the selectivity of 9b towards cancer tissues was also confirmed in ex vivo models, as it was found to be substantially inactive towards non-cancerous MDO.
This organopalladium compound has proven to be exceedingly effective not only in vitro and ex vivo (PDTOs) models but has also delivered excellent results in an animal model. It was found to be non-toxic at high dosages and effective in reducing tumor growth in an animal model of HGSOC.
Even more interestingly, a detailed analysis of the mechanism of action of compound 9b suggests that this organopalladium derivative operates through a ferroptotic mechanism. To the best of our knowledge, this represents the first case of an organopalladium compound inducing tumor cell death through such a biological pathway. In the few cases studied, organopalladium compounds typically operate through apoptotic pathways involving DNA or mitochondrial proteins as the main biotargets.
The completely novel mechanism of action for this class of organopalladium derivatives suggests that it holds considerable promise for future studies in the field of HGSOC, offering a new frontier in the targeted treatment of this challenging cancer subtype.
Author contributions
Thomas Scattolin: conceptualization, investigation, methodology formal analysis and validation of analytical data, visualisation, writing – original draft; Enrico Cavarzerani: conceptualization, investigation, methodology formal analysis and validation of analytical data, visualisation, writing – original draft; Dario Alessi: investigation, methodology formal analysis and validation of analytical data; Matteo Mauceri: investigation, methodology formal analysis and validation of analytical data; Eleonora Botter: investigation, methodology formal analysis and validation of analytical data; Giovanni Tonon: investigation, methodology formal analysis and validation of analytical data; Isabella Caligiuri: investigation, methodology formal analysis and validation of analytical data; Ombretta Repetto: investigation, methodology formal analysis and validation of analytical data; Urska Kamensek: investigation, methodology formal analysis and validation of analytical data; Simona Kranjc Brezar: investigation, methodology formal analysis and validation of analytical data; Maria Dalla Pozza: investigation; Stefano Palazzolo: investigation, methodology formal analysis and validation of analytical data; Maja Cemazar: investigation, methodology formal analysis and validation of analytical data; Vincenzo Canzonieri: project administration, resources; Nicola Demitri: data curation, formal analysis, validation and visualisation of X-ray single crystal data; Steven P. Nolan: project administration, writing – review and editing; Gilles Gasser: project administration, writing – review and editing; Fabiano Visentin: conceptualization, investigation, methodology formal analysis and validation of analytical data, supervision, project administration, writing – original draft; Flavio Rizzolio: conceptualization, investigation, methodology formal analysis and validation of analytical data, supervision, project administration, writing – original draft, resources.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the BOF (starting and senior grants) and the FWO to SPN for financial support. FR was financially supported by Fondazione AIRC per la Ricerca sul Cancro (Grant AIRC IG23566) and VC from Ministero della Salute – Ricerca Corrente.References
- G. C. Jayson, E. C. Kohn, H. C. Kitchener and J. A. Ledermann, Lancet, 2014, 384, 1376–1388 CrossRef PubMed.
- D. T. Le, J. N. Durham, K. N. Smith, H. Wang, B. R. Bartlett, L. K. Aulakh, S. Lu, H. Kemberling, C. Wilt, B. Luber, F. Wong, N. S. Azad, A. A. Rucki, D. Laheru, R. C. Donehower, A. Zaheer, G. A. Fisher, T. S. Crocenzi, J. J. Lee, T. F. Greten, A. G. Duffy, K. K. Ciombor, A. Eyring, B. Lam, A. K. Joe, S. P. Kang, M. Holdhoff, L. Danilova, L. Cope, C. F. Meyer, S. Zhou, R. M. Goldberg, D. K. Armstrong, K. M. Bever, A. N. Fader, J. M. Taube, F. Housseau, D. Spetzler, N. Xiao, D. M. Pardoll, N. Papadopoulos, K. W. Kinzler, J. R. Eshleman, B. Vogelstein, R. A. Anders and L. A. Díaz, Science, 2017, 357, 409–413 CrossRef CAS PubMed.
- U. A. Matulonis, R. Shapira-Frommer, A. D. Santin, A. Lisyanskaya, S. Pignata, I. Vergote, F. Raspagliesi, G. S. Sonke, M. J. Birrer, D. Provencher, J. Sehouli, N. Colombo, A. González-Martín, A. Oaknin, P. B. Ottevanger, V. Rudaitis, K. Katchar, H. Wu, S. M. Keefe, J. Ruman and J. A. Ledermann, Ann. Oncol., 2019, 30, 1080–1087 CrossRef CAS PubMed.
- S. Pignata, S. C. Cecere, A. Du Bois, P. Harter and F. Heitz, Ann. Oncol., 2017, 28, 51–56 CrossRef PubMed.
- L. A. Torre, B. Trabert, C. DeSantis, K. D. Miller, G. Samimi, C. D. Runowicz, M. M. Gaudet, A. Jemal and R. L. Siegel, Ca-Cancer J. Clin., 2018, 68, 284–296 CrossRef PubMed.
- D. Mauricio, S. Bellone, L. Mutlu, B. McNamara, D. Manavella, C. Demirkiran, M. S. Verzosa, N. Buza, P. Hui, T. M. P. Hartwich, J. Harold, Y. Yang-Hartwich, M. Zipponi, G. Altwerger, E. Ratner, G. S. Huang, M. Clark, V. Andikyan, M. Azodi, P. E. Schwartz and A. D. Santin, Gynecol. Oncol., 2023, 170, 38–45 CrossRef CAS PubMed.
- H. Suzuki, S. Nagase, C. Saito, A. Takatsuka, M. Nagata, K. Honda, Y. Kaneda, Y. Nishiya, T. Honda, T. Ishizaka, K. Nakamura, T. Nakada, Y. Abe and T. Agatsuma, Mol. Cancer Ther., 2024, 23, 257–271 CrossRef CAS PubMed.
- Y.-A. Heo, Drugs, 2023, 83, 265–273 CrossRef CAS PubMed.
- B. Feroz, C. Marth and A. G. Zeimet, Mag. Eur. Med. Oncol., 2024, 17, 130–134 Search PubMed.
- (a) Z. Liu, Y. Zhang, W. Lu, Y. Han, J. Yang, W. Jiang, X. You, Y. Luo, S. Wen, Y. Hu and P. Huang, Redox Biol., 2020, 36, 101652 CrossRef PubMed; (b) X. Song, X. Wang, Z. Liu and Z. Yu, Front. Oncol., 2020, 10, 597434 CrossRef PubMed; (c) N. Vasan, J. Baselga and D. M. Hyman, Nature, 2019, 575, 299–309 CrossRef CAS PubMed; (d) X. Wang, H. Zhang and X. Chen, Cancer Drug Resist., 2019, 2, 141–160 Search PubMed.
- (a) W. Dong, M. A. Keibler and G. Stephanopoulos, Metab. Eng., 2017, 43, 113–124 CrossRef CAS PubMed; (b) J. V. Voorde, T. Ackermann, N. Pfetzer, D. Sumpton, G. Mackay, G. Kalna, C. Nixon, K. Blyth, E. Gottlieb and S. Tardito, Sci. Adv., 2019, 5, eaau7314 CrossRef PubMed; (c) J. D. Weyandt, C. B. Thompson, A. J. Giaccia and W. K. Rathmell, Am. Soc. Clin. Oncol. Educ. Book, 2017, 825–832 Search PubMed.
- (a) J. Roh, E. H. Kim, H. J. Jang, J. Y. Park and D. Shin, Cancer Lett., 2016, 381, 96–103 CrossRef CAS; (b) M. J. Hangauer, V. S. Viswanathan, M. Ryan, D. Bole, J. K. Eaton, A. Matov, J. Galeas, H. D. Dhruv, M. E. Berens, S. L. Schreiber, F. McCormick and M. T. McManus, Nature, 2017, 551, 247–250 CrossRef CAS; (c) L. F. Ye, K. Chaudhary, F. Zandkarimi, A. Harken, C. J. Kinslow, P. S. Upadhyayula, A. Dovas, D. Higgins, H. Tan, Y. Zhang, M. Buonanno, T. J. C. Wang, T. K. Hei, J. N. Bruce, P. Canoll, S. K. Cheng and B. R. Stockwell, ACS Chem. Biol., 2020, 15, 469–484 CrossRef CAS.
- T. Hirschhorn and B. R. Stockwell, Free Radicals Biol. Med., 2019, 133, 130–143 CrossRef CAS PubMed.
- K. Schulze-Osthoff, D. Ferrari, M. Łos, S. Wesselborg and M. E. Peter, Eur. J. Biochem., 1998, 254, 439–459 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- (a) S. Li, H. Yuan, Y. Chen and Z. Guo, Fundam. Res., 2023, 3, 525–528 CrossRef CAS; (b) K. Peng, Y. Zheng, W. Xia and Z. Mao, Chem. Soc. Rev., 2023, 52, 2790–2832 RSC.
- H. Zhao, Y. Xu and H. Shang, Int. J. Med. Sci., 2022, 19, 1847–1855 CrossRef CAS PubMed.
- G. Lei, Z. Li and B. Gan, Nat. Rev. Cancer, 2022, 22, 381–396 CrossRef CAS PubMed.
- X. Wang, F. Chen, J. Zhang, J. Sun, X. Zhao, Y. Zhu, W. Wei, J. Zhao and Z. Guo, Sci. China: Chem., 2019, 63, 65–72 CrossRef.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 RSC.
- (a) T. Scattolin, E. Bortolamiol, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio, L. Cavallo, B. Dereli, M. V. Mane, S. P. Nolan and F. Visentin, Chem. Commun., 2020, 56, 12238–12241 RSC; (b) T. Scattolin, S. Giust, P. Bergamini, I. Caligiuri, L. Canovese, N. Demitri, R. Gambari, I. Lampronti, F. Rizzolio and F. Visentin, Appl. Organomet. Chem., 2019, 33, e4902 CrossRef; (c) T. Scattolin, I. Caligiuri, L. Canovese, N. Demitri, R. Gambari, I. Lampronti, F. Rizzolio, C. Santo and F. Visentin, Dalton Trans., 2018, 47, 13616–13630 RSC.
- (a) S. Y. Lee, C. Y. Kim and T. Nam, Drug Des., Dev. Ther., 2020, 14, 5375–5392 CrossRef CAS; (b) G. Moreno-Alcántar, P. Picchetti and A. Casini, Angew. Chem., Int. Ed., 2023, 62, e202218000 CrossRef PubMed; (c) M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev., 2019, 48, 447–462 RSC; (d) T. A. C. A. Bayrakdar, T. Scattolin, X. Ma and S. P. Nolan, Chem. Soc. Rev., 2020, 49, 7044–7100 RSC; (e) B. Bertrand and A. Casini, Dalton Trans., 2014, 3, 4209–4219 RSC; (f) S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Med. Res. Rev., 2010, 30, 550–580 CrossRef CAS PubMed; (g) S. Y. Lee, C. Y. Kim and T.-G. Nam, Drug Des., Dev. Ther., 2020, 14, 5375–5392 CrossRef CAS PubMed; (h) S. Q. Zhang, L.-H. Gao, H. Zhao and K.-Z. Wang, Curr. Med. Chem., 2020, 27, 3735–3752 CrossRef CAS PubMed.
- A. S. Abu-Surrah and M. Kettunen, Curr. Med. Chem., 2006, 13, 1337–1357 CrossRef CAS.
- J. Albert, J. Granell, J. A. Durán, A. Lozano, A. Luque, A. Mate, J. Quirante, M. K. Khosa, C. Calvis, R. Messeguer, L. Baldomà and J. Badía, J. Organomet. Chem., 2017, 839, 116–125 CrossRef CAS.
- T. Scattolin, V. A. Voloshkin, F. Visentin and S. P. Nolan, Cell Rep. Phys. Sci., 2021, 2, 100446 CrossRef CAS.
- R. Paprocka, M. Wiese-Szadkowska, S. Janciauskiene, T. Kosmalski, M. Kulik and A. Helmin-Basa, Coord. Chem. Rev., 2022, 452, 214307 CrossRef CAS.
- P. Köpf-Maier, W. Wagner and H. Köpf, Cancer Chemother. Pharmacol., 1981, 5, 237–241 CrossRef.
- (a) I. Romero-Canelón and P. J. Sadler, Inorg. Chem., 2013, 52, 12276–12291 CrossRef PubMed; (b) Z. Liu and P. J. Sadler, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS PubMed.
- E. Boros, P. J. Dyson and G. Gasser, Chem, 2020, 6, 41–60 CAS.
- P. Zhang and P. J. Sadler, J. Organomet. Chem., 2017, 839, 5–14 CrossRef CAS.
- (a) T. Scattolin, I. Caligiuri, N. Mouawad, M. El Boustani, N. Demitri, F. Rizzolio and F. Visentin, Eur. J. Med. Chem., 2019, 179, 325–334 CrossRef CAS PubMed; (b) A. R. Kapdi and I. J. Fairlamb, Chem. Soc. Rev., 2014, 43, 4751 RSC; (c) G. Kalaiarasi, S. Dharani, V. M. Lynch and R. Prabhakaran, Dalton Trans., 2019, 48, 12496–12511 RSC.
- (a) T. Scattolin, E. Bortolamiol, F. Visentin, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio and A. Togni, Chem. – Eur. J., 2020, 26, 11868–11876 CrossRef CAS PubMed; (b) T. Scattolin, I. Pessotto, E. Cavarzerani, V. Canzonieri, L. Orian, N. Demitri, C. Schmidt, A. Casini, E. Bortolamiol, F. Visentin, F. Rizzolio and S. P. Nolan, Eur. J. Inorg. Chem., 2022, e202200103 CrossRef CAS; (c) A. Madabeni, T. Scattolin, E. Bortolamiol, F. Visentin and L. Orian, Organometallics, 2024, 43, 954–962 CrossRef CAS; (d) T.-H. Fong, C.-N. Lok, C. Y.-S. Chung, Y.-M. E. Fong, P.-K. Chow, P.-K. Wan and C.-M. Che, Angew. Chem., Int. Ed., 2016, 55, 11935 CrossRef CAS PubMed.
- (a) J. H. Groen, C. J. Elsevier, K. Vrieze, W. J. J. Smeets and A. L. Spek, Organometallics, 1996, 15, 3445–3455 CrossRef CAS; (b) B. Cazes, Pure Appl. Chem., 1990, 62, 1867–1878 CrossRef CAS; (c) M. Ahmar, B. Cazes and J. Gore, Tetrahedron Lett., 1984, 25, 4505–4508 CrossRef CAS; (d) M. Ahmar, B. Cazes and J. J. Barieux, J. Gore Tetrahedron, 1987, 43, 513–526 CrossRef CAS.
- A. Jutand and S. Négri, Organometallics, 2003, 22, 4229–4237 CrossRef CAS.
- (a) R. Van Belzen, C. J. Elsevier, A. Didieu, N. Veldman and A. L. Speck, Organometallics, 2003, 22, 722–736 CrossRef CAS; (b) L. Canovese, F. Visentin, T. Scattolin, C. Santo and V. Bertolasi, J. Organomet. Chem., 2016, 808, 48–56 CrossRef CAS; (c) L. Canovese, F. Visentin, T. Scattolin, C. Santo and V. Bertolasi, Polyhedron, 2016, 113, 25–34 CrossRef CAS; (d) T. Scattolin, F. Visentin, C. Santo, V. Bertolasi and L. Canovese, Dalton Trans., 2016, 45, 11560–11567 RSC; (e) L. Canovese, F. Visentin, T. Scattolin, C. Santo and V. Bertolasi, Dalton Trans., 2015, 44, 15049–15058 RSC; (f) L. Canovese, C. Santo, T. Scattolin, F. Visentin and V. Bertolasi, J. Organomet. Chem., 2015, 794, 288–300 CrossRef CAS; (g) F. Visentin, C. Santo, T. Scattolin, N. Demitri and L. Canovese, Dalton Trans., 2017, 46, 10399–10407 RSC.
- (a) L. Canovese, F. Visentin, G. Chessa, P. Uguagliati, C. Santo and A. Dolmella, Organometallics, 2005, 24, 3297–3308 CrossRef CAS; (b) V. De Felice, M. E. Cucciolito, A. De Renzi, F. Ruffo and D. Tesauro, J. Organomet. Chem., 1995, 493, 1–11 CrossRef CAS.
- K. Moseley and P. M. Maitlis, J. Chem. Soc., Dalton Trans., 1974, 169–175 RSC.
- (a) J. Vicente, J. A. Abad, E. Martinez-Viviente, M. C. Ramirez de Arellano and P. G. Jones, Organometallics, 2000, 19, 5597–5607 CrossRef CAS; (b) A. M. La Pointe and M. Brookhart, Organometallics, 1998, 17, 1530–1537 CrossRef CAS; (c) J. Vicente, J. A. Abad, K. F. Shaw, J. Gil-Rubio, M. C. Ramirez de Arellano and P. G. Jones, Organometallics, 1997, 16, 4557–4566 CrossRef CAS.
- P. M. Maitlis, J. Organomet. Chem., 1980, 200, 161–176 CrossRef CAS.
- J. R. Clark and S. T. Diver, Org. Lett., 2011, 13, 2896–2899 CrossRef CAS PubMed.
- H. C. Clark, C. R. C. Milne and C. S. Wong, J. Organomet. Chem., 1977, 136, 265–279 CrossRef CAS.
- A. Yamamoto, in Organotransition Metal Chemistry, Wiley, New York, 1986 Search PubMed.
- C. Carfagna, G. Gatti, L. Mosca, P. Paoli and A. Guerri, Organometallics, 2003, 22, 3967–3970 CrossRef CAS.
- E. G. Samsel and J. R. Norton, J. Am. Chem. Soc., 1984, 106, 5505–5512 CrossRef CAS.
- (a) T. Scattolin, C. Santo, N. Demitri, L. Canovese and F. Visentin, Dalton Trans., 2020, 173, 114144 Search PubMed; (b) T. Scattolin, E. Bortolamiol, I. Caligiuri, F. Rizzolio, N. Demitri and F. Visentin, Polyhedron, 2020, 186, 114607 CrossRef CAS; (c) L. Canovese, F. Visentin, T. Scattolin, C. Santo and V. Bertolasi, Polyhedron, 2018, 144, 131–143 CrossRef CAS; (d) T. Scattolin, L. Canovese, F. Visentin, C. Santo and N. Demitri, Polyhedron, 2018, 154, 382–389 CrossRef CAS; (e) T. Scattolin, N. Pangerc, I. Lampronti, C. Tupini, R. Gambari, L. Marvelli, F. Rizzolio, N. Demitri, L. Canovese and F. Visentin, J. Organomet. Chem., 2019, 899, 120857 CrossRef CAS; (f) T. Scattolin, L. Canovese, N. Demitri, C. Santo and F. Visentin, Polyhedron, 2019, 173, 114144 CrossRef CAS.
- (a) T. Scattolin, A. A. Logvinov, N. V. Tzouras, C. S. J. Cazin and S. P. Nolan, Organometallics, 2023, 42, 2692–2730 CrossRef CAS; (b) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- J. Bravo, S. Bolaño, L. Gonsalvi and M. Peruzzini, Coord. Chem. Rev., 2010, 254, 555–607 CrossRef CAS.
- (a) C. Sonkar, S. Sarkar and S. Mukhopadhyay, RSC Med. Chem., 2022, 13, 22–38 RSC; (b) J. Quero, F. Ruighi, J. Osada, M. C. Gimeno, E. Cerrada and M. J. Rodriguez-Yoldi, Biomedicine, 2021, 9, 1848 CAS; (c) A. K. Renfrew, J. Karges, R. Scopelliti, F. D. Bobbink, P. Nowak-Sliwinska, G. Gasser and P. J. Dyson, ChemBioChem, 2019, 20, 2876–2882 CrossRef CAS PubMed; (d) T. Scattolin, E. Bortolamiol, F. Rizzolio, N. Demitri and F. Visentin, Appl. Organomet. Chem., 2020, 34, e5876 CrossRef CAS.
- (a) Y. Wu, D. Wang, W. Fang, F. Xiong, S. Zhang, Z. Gong, L. Shi, X. Li, B. Xiang, J. Ma, H. Deng, Y. He, Q. Liao, W. Zhang, X. Li, Y. Li, C. Guo, Z. Zeng, G. Li and W. Xiong, FASEB J., 2020, 34, 16205–16223 CrossRef CAS PubMed; (b) D. Wang, Z. Zeng, S. Zhang, F. Xiong, B. He, Y. Wu, W. Liu, L. Tang, W. Fang, B. Xiang, Z. Li, Y. Zhou, M. Zhou, X. Li, Y. Li, G. Li, W. Xiong and C. Guo, FASEB J., 2020, 34, 8012–8027 CrossRef CAS PubMed.
- (a) Y. Wang, Y. Mo, P. Miao, S. Zhang, Z. Gong, Q. Yan, Y. Tang, Y. He, Q. Liao, X. Li, X. Wu, B. Xiang, M. Zhou, Y. Li, G. Li, X. Li, Z. Zeng and C. Guo, Autophagy, 2021, 18, 240–253 CrossRef PubMed; (b) S. Bedoui, M. J. Herold and A. Strasser, Nat. Rev. Mol. Cell Biol., 2020, 21, 678–695 CrossRef CAS PubMed.
- (a) B. A. Carneiro and W. S. El-Deiry, Nat. Rev. Clin. Oncol., 2020, 17, 395–417 CrossRef PubMed; (b) Y. K. Dhuriya and D. Sharma, J. Neuroinflammation, 2018, 15, 199 CrossRef PubMed; (c) Q. Nie, Y. Hu, X. Yu, X. Li and X. Fang, Cancer Cell Int., 2022, 22, 12 CrossRef CAS PubMed; (d) J. Hou, R. Zhao, W. Xia, C. W. Chang, Y. You, J. M. Hsu, L. Nie, Y. Chen, Y. C. Wang, C. Liu, W. Wang, Y. Wu, B. Ke, J. L. Hsu, K.-B. Huang, Z. Ye, Y. Yang, X. Xia, Y. Li, C. W. Li, B. Shao, J. A. Tainer and M. Hung, Nat. Cell Biol., 2020, 22, 1264–1275 CrossRef CAS PubMed; (e) J. Shi, Y. Zhao, K. Wang, X. Shi, Y. Wang, H. Huang, Y. Zhuang, T. Cai, F. Wang and F. Shao, Nature, 2015, 526, 660–665 CrossRef CAS PubMed; (f) J. M. M. Levy, C. G. Towers and A. Thorburn, Nat. Rev. Cancer, 2017, 17, 528–542 CrossRef CAS PubMed.
- X. Yu and Y. Long, Sci. Rep., 2016, 6, 30033 CrossRef CAS PubMed.
- (a) J. P. F. Angeli, D. V. Krysko and M. Conrad, Nat. Rev. Cancer, 2019, 19, 405–414 CrossRef PubMed; (b) N. Jyotsana, K. T. L. Ta and K. E. DelGiorno, Front. Oncol., 2022, 12, 858462 CrossRef CAS PubMed; (c) S. Doll, B. Proneth, Y. Y. Tyurina, E. Panzilius, S. Kobayashi, I. Ingold, M. Irmler, J. Beckers, M. Aichler, A. Walch, H. Prokisch, D. Trümbach, G. Mao, F. Qu, H. Bayır, J. Füllekrug, C. Scheel, W. Wurst, J. Schick, V. E. Kagan, J. P. F. Angeli and M. Conrad, Nat. Chem. Biol., 2016, 13, 91–98 CrossRef PubMed; (d) S. Neitemeier, A. Jelinek, V. Laino, L. Hoffmann, I. Eisenbach, R. Eying, G. K. Ganjam, A. M. Dolga, S. Oppermann and C. Culmsee, Redox Biol., 2017, 12, 558–570 CrossRef CAS PubMed; (e) M. Zille, S. S. Karuppagounder, Y. Chen, P. J. Gough, J. Bertin, J. N. Finger, T. A. Milner, E. A. Jonas and R. R. Ratan, Stroke, 2017, 48, 1033–1043 CrossRef PubMed.
- (a) D. Wang, L. Tang, Y. Zhang, G. Ge, X. Jiang, Y. Mo, P. Wu, X. Deng, L. Li, S. Zuo, Q. Yan, S. Zhang, F. Wang, L. Shi, X. Li, B. Xiang, M. Zhou, Q. Liao, C. Guo, Z. Zeng, W. Xiong and Z. Gong, Cell Death Dis., 2022, 13, 544 CrossRef CAS PubMed; (b) J. Lee, J. H. You, D.-H. Shin and J. Roh, Theranostics, 2020, 10, 7775–7786 CrossRef CAS PubMed; (c) K. Kalishwaralal, C. K. Keerthana, M. Mohan, J. Arivalagan, J. R. S. S. Christyraj, M. A. Firer, M. H. A. Choudry, R. J. Anto and Y. J. Lee, J. Cell. Biochem., 2021, 123, 532–542 Search PubMed; (d) Q. Nie, Y. Hu, X. Yu, X. Li and X. Fang, Cancer Cell Int., 2022, 22, 2 CrossRef PubMed; (e) E. C. Cheung and K. H. Vousden, Nat. Rev. Cancer, 2022, 22, 280–297 CrossRef CAS PubMed; (f) Y. Liu and W. Gu, Cell Death Differ., 2022, 29, 895–910 CrossRef CAS PubMed; (g) S. Dolma, S. L. Lessnick, W. C. Hahn and B. R. Stockwell, Cancer Cell, 2003, 3, 285–296 CrossRef CAS PubMed.
- J. Y. Cao and S. J. Dixon, Cell. Mol. Life Sci., 2016, 73, 2195–2209 CrossRef CAS PubMed.
- (a) X. Yang, Y. Ding, L. Sun, M. Shi, P. Zhang, Z. Huang, J. Wang, A. He, J. Wang, J. Wei, M. Liu, J. Liu, G. Wang, X. Yang and R. Li, Redox Biol., 2022, 58, 102555 CrossRef CAS PubMed; (b) Y. Wang, S. Qiu, H. Wang, J. Cui, X. Tian, Y. Miao, C. Zhang, L. Cao, L. Ma, X. Xu, Y. Qiao and X. Zhang, Front. Cell Dev. Biol., 2021, 9, 719187 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data and spectra. CCDC 2343666–2343670. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00194c |
| This journal is © The Royal Society of Chemistry 2025 |