
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-directed assembly in MOFs with linear trinuclear cobalt(II) nodes: formation of inverted Figaro chains†
Oluseun
Akintola
 ,
Helmar
Görls
and
Winfried
Plass
*
,
Helmar
Görls
and
Winfried
Plass
*
Institut für Anorganische und Analytische Chemie, Friedrich-Schiller-Universität, Jena, Humboldtstrasse 8, 07743 Jena, Germany. E-mail: sekr.plass@uni-jena.de; Fax: +49 (0)3641948132; Tel: +49 (0)3641 948130
First published on 26th March 2025
Abstract
Four new cobalt(II) coordination networks based on the nitrilotribenzoic acid ligand (H3ntb) with the formula [Co3(ntb)2(solv)2]n (where solv = dmf, def, dma, and EtOH), denoted as JUMP-4(solv), are reported. The trinuclear cobalt(II) clusters that constitute the three-dimensional network are coordinated by the six carboxylate groups of two bridging, deprotonated ntb3− ligands. These clusters have two flexible coordination sites on the two terminal cobalt ions, which are partially saturated by the condensation of these clusters, leading to unprecedented inverted Figaro chains. Three of the frameworks were synthesized by combining cobalt(II) chloride and the ligand in a mixture of acetonitrile and the corresponding amide (dmf, def, and dma) in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, while the ethanol analogue could only be obtained by single-crystal-to-single-crystal transformation starting from the dmf and dma derivatives. The difference in the size of the solvents coordinated to the terminal cobalt(II) ions results in a pore-partitioning effect, as reflected in the argon sorption behavior of the networks. Magnetic measurements reveal antiferromagnetic interactions between the anisotropic cobalt(II) ions along the alternating inverted Figaro chains.
:1 ratio, while the ethanol analogue could only be obtained by single-crystal-to-single-crystal transformation starting from the dmf and dma derivatives. The difference in the size of the solvents coordinated to the terminal cobalt(II) ions results in a pore-partitioning effect, as reflected in the argon sorption behavior of the networks. Magnetic measurements reveal antiferromagnetic interactions between the anisotropic cobalt(II) ions along the alternating inverted Figaro chains.
Introduction
It has been over three decades since the study of metal–organic frameworks (MOFs) entered mainstream coordination chemistry, and even today they continue to fascinate chemists with the possibilities that can be achieved with these materials. Over time, this has led to the development of compounds for a wide variety of applications, ranging from the more common ones such as gas sorption, gas separation,1–3 and catalysis,4–6 to others such as magnetism7,8 and drug delivery,9,10 to less known and quite recent applications such as qubits11 and electrodes for batteries and supercapacitors.12–15The majority of the networks have been generated through the well-known solvothermal route with slight modifications such as usage of templates.16–18 Sometimes changes, such as using a different solvent, may have varying impacts on their structures, ranging from slight to severe.19–22 In turn, these alterations could further influence the properties of the resultant product.23,24 A coordinating solvent can also offer the advantage of being post-synthetically removable, exposing coordination sites on the metal ion that can then be exploited for catalysis25,26 or gas adsorption and separation.27–29 In addition, the removal or replacement of coordinated solvent molecules can influence the behavior of the coordinated metal ions, which can lead to a change in their magnetic properties.30–32
Cobalt(II) ions, known for their significant intrinsic magnetic anisotropy, are excellent candidates for magnetic applications.33,34 In coordination polymers with carboxylate linkers, cobalt(II) ions typically form oligonuclear clusters bridged by these linkers, facilitating magnetic exchange interactions within the clusters.35,36 The nature of these interactions is influenced not only by the bridging ligands but also by additional coordinating solvent molecules.31,37,38 It is interesting to note that, in cases where one-dimensional chains are formed instead of isolated clusters, an intriguing cooperative effect can be observed, leading to single-chain magnetic behavior.39–43
Herein, we report the design and synthesis of a series of new MOFs based on nitrilotribenzoic acid (H3ntb) and cobalt(II) ions. We previously reported the synthesis of two anionic frameworks using the same ligand with dimethylformamide (dmf) as the solvent.44 Replacement of the majority of dmf by acetonitrile (nine parts out of ten) results in a new neutral network (JUMP-4(dmf)), in which dmf is coordinated at the constituent trinuclear cobalt clusters. Its analogs with diethylformamide (def) and dimethylacetamide (dma) (JUMP-4(def) and JUMP-4(dma)) can likewise be obtained by simply changing the amide component in the solvent mixture at the point of synthesis from dimethylformamide (dmf) to diethylformamide (def) and dimethylacetamide (dma), while retaining the acetonitrile. However, the fourth network (JUMP-4(EtOH)) is only obtainable through a single-crystal-to-single-crystal (SCSC) transformation of JUMP-4(dmf) or JUMP-4(dma) by immersion in ethanol. The structures of all four compounds have been established using single-crystal X-ray diffraction, with bulk purity confirmed by powder XRD. Sorption measurements suggest that all four materials exhibit permanent porosity, with an apparent influence of the coordinating solvents. Magnetic susceptibility measurements also provide evidence of the influence of coordinating solvent molecules on the magnetic properties of the constituting trinuclear cobalt(II) clusters.
Results and discussion
Synthesis
The ligand was obtained through an adjusted combination of several reported procedures. The 4,4′-iminodibenzonitrile was synthesized from p-fluorobenzonitrile and p-aminobenzonitrile using potassium tert-butoxide, following a high-yield method previously reported by Gorvin.45 The resulting product was then purified and used to synthesize the 4,4′,4′′-ntirilotrisbenzonitrile by reacting it with additional p-fluorobenzonitrile, using cesium fluoride as base, in procedure combining two reported methods.46,47 The nitrile product was subsequently hydrolyzed with ethanol and potassium hydroxide under reflux, followed by acidification with concentrated hydrochloric acid, yielding the pure, light brown product in high yield.The synthesis of the JUMP-4 derivatives was achieved through a solvothermal reaction of the ligand H3ntb with cobalt(II) chloride hexahydrate in a solvent mixture of acetonitrile and either dmf, def, or dma, yielding JUMP-4(dmf), JUMP-4(def), and JUMP-4(dma), respectively. All reaction mixtures were heated to 140 °C for three days under solvothermal conditions and subsequent cooling (5 °C h−1) led to the formation of violet crystalline material in moderate yields. These conditions are within the temperature range used for previous syntheses of the related MOFs JUMP-1, JUMP-3, and MIL-144 (130–150 °C). The same stoichiometry and absolute amount of reactants were used for the synthesis of all three compounds.
By testing various solvent ratios, the optimal proportion was determined to be 9:1 of acetonitrile to the respective amide. Further increasing the acetonitrile content in the mixture resulted in the formation of the same material but with insufficient crystallinity. The analytical composition of the three synthesized JUMP-4 frameworks was determined through a combination of elemental and thermogravimetric analyses (see Fig. S1–S4†), yielding the formulae {[Co3(ntb)2(dmf)2]}n (JUMP-4(dmf)), {[Co3(ntb)2(def)2]}n (JUMP-4(def)), and {[Co3(ntb)2(dma)2]}n (JUMP-4(dma)). All three networks contained additional solvent molecules within their pores.
Solvent directing effects
In the absence of acetonitrile, the resulting product corresponds to the previously reported MIL-144.44,48 This observation suggests that acetonitrile plays a directing role in the formation of the new compounds JUMP-4(dmf), JUMP-4(def), and JUMP-4(dma), consistent with previous findings on the significant role of solvents in regulating structure in MOF synthesis.49,50 Nevertheless, it should be noted that while the additional solvent influences the network structure, no structural differences are observed when dmf is replaced with either def or dma. Interestingly, the presence of an additional modulator in dmf solution leads to another previously reported MOF, JUMP-3 (see Scheme 1).51 Conversely, adding terephthalic acid as a linear dicarboxylate linker to the reaction mixture yields the pillared-layer MOF JUMP-1,52 where the coordination sites at the peripheral cobalt(II) centers of the trinuclear cluster are saturated by the carboxylate groups of the additional linker ligand. | ||
| Scheme 1 MOF synthesis showing the influence of the solvent used. | ||
All three MOFs, namely MIL-144, JUMP-1, and JUMP-3, share the characteristic that the coordination sites at the peripheral cobalt(II) centers of the trinuclear cluster are fully saturated by donor ligands present in the reaction mixture. In contrast, when the availability of donor ligands in the reaction medium is reduced by using acetonitrile as the primary solvent with only a minor amide-based component, such as dmf, def, or dma in the synthesis of the JUMP-4 derivatives, the coordination environment of the peripheral cobalt centers is only partially saturated. This leads to a Co2O2 bridging arrangement, where one of the coordination sites of the peripheral cobalt centers is coordinated by a carboxylate donor from a neighboring trinuclear cluster.
Single-crystal-to-single-crystal transformation
Attempts to synthesize an ethanol-coordinated derivative of JUMP-4 by directly replacing the amide component in the solvent mixture with ethanol were unsuccessful. To explore an alternative approach, the conversion of the formamide-based networks JUMP-4(dmf), JUMP-4(def), and JUMP-4(dma) into their ethanol-coordinated analogue (JUMP-4(EtOH)) via single-crystal-to-single-crystal (SCSC) transformation was investigated. For this purpose, the compounds were immersed in ethanol for seven days, with the solvent refreshed daily.Immersion of JUMP-4(dmf) and JUMP-4(dma) in ethanol resulted in an immediate and noticeable color change from deep violet to dark pink (see Fig. 1). This transformation was attributed to the replacement of the coordinated dmf or dma molecules with ethanol, leading to the formation of JUMP-4(EtOH). IR spectroscopy provided supporting evidence for this process, as the characteristic amide I band of dmf at 1660 cm−1 gradually disappeared during the transformation. However, due to overlap in the spectral region of the corresponding amide band of dma, confirmation of the transformation in JUMP-4(dma) via IR spectroscopy was not possible (see Fig. S5†).
 | ||
| Fig. 1 Images for JUMP-4(dmf) (top row) and JUMP-4(def) (bottom row) under different conditions: as-synthesized material (left), ethanol-immersed (middle), and dichloromethane-soaked (right). | ||
In contrast, the immersion of JUMP-4(def) in ethanol under identical conditions did not produce any color change. IR spectra of JUMP-4(def) revealed the retention of the amide I band at 1650 cm−1, corresponding to def, indicating that the coordinated def in the framework was not replaced. This suggests that, unlike the readily substituted dmf and dma, def exhibits higher stability within the framework and resists replacement by ethanol. Interestingly, the substitution of ethanol in JUMP-4(EtOH) was found to be reversible. When JUMP-4(EtOH) was re-immersed in either dmf or dma, the ethanol molecules were replaced, as evidenced by the reappearance of the corresponding amide I band in the IR spectra. This reversal was accompanied by a reappearance of the original color, further confirming the reformation of the dmf- or dma-coordinated materials.
To further evaluate the stability of the coordinated amides, the samples were immersed in dichloromethane, a solvent unlikely to coordinatively replace the amide ligands, for seven days. IR spectra of the dichloromethane-treated materials showed the persistence of the amide I band at 1650–1660 cm−1, albeit with reduced intensity compared to the original samples (see Fig. S5†). This finding supports the retention of the coordinated def molecules in both the dichloromethane- and the ethanol-treated material of JUMP-4(def). Additionally, no color change was observed in any sample after dichloromethane immersion, corroborating the lack of ligand substitution (see Fig. 1).
In summary, the formation of JUMP-4(EtOH) via SCSC transformation was successful for JUMP-4(dmf) and JUMP-4(dma), facilitated by the ease of replacement of the coordinated dmf and dma molecules with ethanol. In contrast, the conversion of JUMP-4(def) into JUMP-4(EtOH) was hindered by the stability of the coordinated def, which resisted substitution under identical conditions. These observations highlight the crucial role of the identity of the amide ligand in determining the feasibility of the SCSC transformation.
Crystal structures
Single-crystal X-ray diffraction measurements were performed on the crystalline products of all four frameworks. The data revealed that the frameworks are isostructural and crystallize in the monoclinic space group C2/c. Crystallographic data, along with details of the structure solution and refinement, are summarized in Table S1.†The asymmetric unit in all four structures consists of two independent cobalt(II) ions—one (Co2) occupying a special position (Wyckoff: 4a)—along with one ntb3− ligand and one coordinated dmf, def, dma, or EtOH as a co-ligand in JUMP-4(dmf), JUMP-4(def), JUMP-4(dma), and JUMP-4(EtOH), respectively (Fig. S6–S9†). This arrangement results in neutral three-dimensional networks. In the structure of JUMP-4(dmf), partial replacement of the coordinated dmf by a water molecule is observed, with fractional occupancies of 0.6 and 0.4, respectively. Moreover, additional solvent molecules are present in the structures of JUMP-4(dmf) (dmf and water), JUMP-4(def) (acetonitrile), JUMP-4(dma) (acetonitrile), and JUMP-4(EtOH) (EtOH and water). Bond lengths and angles for all four networks are summarized in Tables S2–S9.†
The three-dimensional network in all four compounds is constructed from trinuclear clusters serving as secondary building units (SBUs). These clusters possess an inversion center, with the central Co2 ion occupying the associated special position and two symmetry related peripheral Co1 ions (see Fig. 2). The central Co2 ion exhibits an almost ideal octahedral coordination geometry, with all six oxygen donor atoms being provided by distinct ntb3− ligands. In contrast, the peripheral Co1 ions are coordinated by six oxygen donor atoms from two distinct groups: (i) five from ntb3− ligands and (ii) one from either a coordinated amide or ethanol molecule, resulting in a distorted octahedral environment. Such trinuclear arrangements are commonly observed in cobalt(II) coordination networks, particularly those where all three cobalt(II) ions adopt octahedral geometries.51,53,54
 | ||
| Fig. 2 View of the trinuclear cobalt cluster SBU, illustrating the coordination environments around the cobalt(II) ions in JUMP-4(dmf), representative for all four networks. The connection to the neighboring trinuclear SBUs is indicated by broken lines to the corresponding symmetry related Co1 ion shown as transparent ball. For clarity, only carboxylate and dmf groups are shown; hydrogen atoms are omitted. Symmetry codes: A = 1 − x, 1 − y, 1 − z; B = x, 1 − y, −1/2 + z; C = −1/2 + x, 3/2 − y, −1/2 + z; D = 1 − x, y, 3/2 − z; E = 3/2 − x, −1/2 + y, 3/2 − z; F = 3/2 − x, 1/2 + y, 3/2 − z; G = −1/2 + x, 1/2 − y, −1/2 + z; H = 1 − x, 2 − y, 1 − z; I = x, −1 + y, z. | ||
Within these clusters, two carboxylate bridging modes can be identified (see Fig. 2): (i) a distorted bidentate syn–syn-μ1,3 mode (O1/O2 and O5/O6) and (ii) a monodentate μ1,1 mode (O3). The Co1⋯Co2 separations in all four compounds fall within a narrow range of 342–346 pm, while the Co1–O–Co2 bridging angles at the monodentate μ1,1-carboxylate bridge range from 107–109° (see Table S10†).
Overall, the central Co2 ion in the JUMP-4 derivatives exhibits an almost ideal octahedral geometry, as reflected in its bond lengths and angles, whereas the Co1 ion exhibits a significant geometric distortion (see Tables S2–S9†). The distinct differences in geometric distortion between the two cobalt(II) centers were further quantified using continuous shape measures (CSM),55 yielding S(Oh) values of about 0.1 for the octahedral Co2 center and a range of 2.6 to 4.0 for the distorted Co1 center in the JUMP-4 derivatives (see Table S11 and Fig. S10†).
Along the crystallographic [010] direction, each trinuclear cluster is connected to another through an additional μ1,1-carboxylate bridge (O6 in Fig. 2) with an angle of about 97°. The distance between the peripheral Co1 centers linking adjacent clusters in the JUMP-4 derivatives ranges from 311 to 315 pm, forming infinite rod-like, one-dimensional inverted Figaro-type substructures running along the [010] direction (see Scheme 2). Similar arrangements have been reported in a few other MOFs, all characterized by broad one-dimensional open channels.56–58 This structural motif also results in two distinct polyhedral connection modes along the inverted Figaro chain: (i) vertex-sharing within the cluster and (ii) edge-sharing between adjacent clusters (see Fig. 3). Notably, the Co1⋯Co2⋯Co1 axis of the trinuclear clusters in all JUMP-4 derivatives deviates from the crystallographic [010] direction by about 18° (17.9° to 18.5°), resulting in a zigzag arrangement of the SBUs along this axis. A hinge angle of approximately 119° is observed between the trinuclear SBUs at the peripheral cobalt(II) ions along the inverted Figaro chain (see Fig. 3).
 | ||
| Scheme 2 Depiction of normal Figaro chains with clusters consisting of two short links and one longer link in between (above). Inverted Figaro chains in JUMP-4 networks, showing clusters with two longer links (Co1⋯Co2: 342–346 pm) and one shorter link (Co1⋯Co1: 311–315 pm) in between. | ||
 | ||
| Fig. 3 Representation of two linear trinuclear cobalt(II) SBUs, illustrating the combination of vertex- and edge-sharing octahedra that form the one-dimensional inverted Figaro in the networks of all four JUMP-4 derivatives. Plum and turquoise octahedra represent the alternating trinuclear clusters. The black line indicates the crystallographic [010] direction. Pertinent distances and angles: Co1⋯Co1, 311–315 pm; Co1⋯Co2, 342–346 pm; Co1⋯Co1⋯Co2, 117.8–119.7°. | ||
The inverted Figaro chains are exclusively connected via the triphenylamine ligands, which establish the three-dimensional framework (Fig. 4 and Fig. S11, S12†). This results in a two-dimensional void space in the crystallographic (110) plane within the framework of the JUMP-4 derivatives, as depicted in Fig. 5 (cf. Fig. S13–S15†). The network-accessible helium volumes, assuming full occupancy of the coordination sites at the Co1 ions with dmf, def, dma, and EtOH as co-ligands, are 1637, 1265, 1271, and 1737 Å3 (29.4, 23.0, 23.3, and 31.4%), respectively. In contrast, assuming full occupancy of these sites with water molecules, the network-accessible helium volumes increase to 2502, 2518, 2476, and 2576 Å3 (45.1, 45.9, 45.3, and 46.6%), respectively. These differences are attributed to steric effects introduced by the different coordinating co-ligands, which are expected to significantly influence the porous properties.
 | ||
| Fig. 4 Three-dimensional framework of JUMP-4(dmf) viewed along the crystallographic [010] direction. The linear trinuclear cobalt(II) SBUs are arranged into one-dimensional chains along the [010] direction and interconnected by ntb3− ligands. Plum colored polyhedra represent cobalt(II) centers, and the unit cell is outlined by black lines. | ||
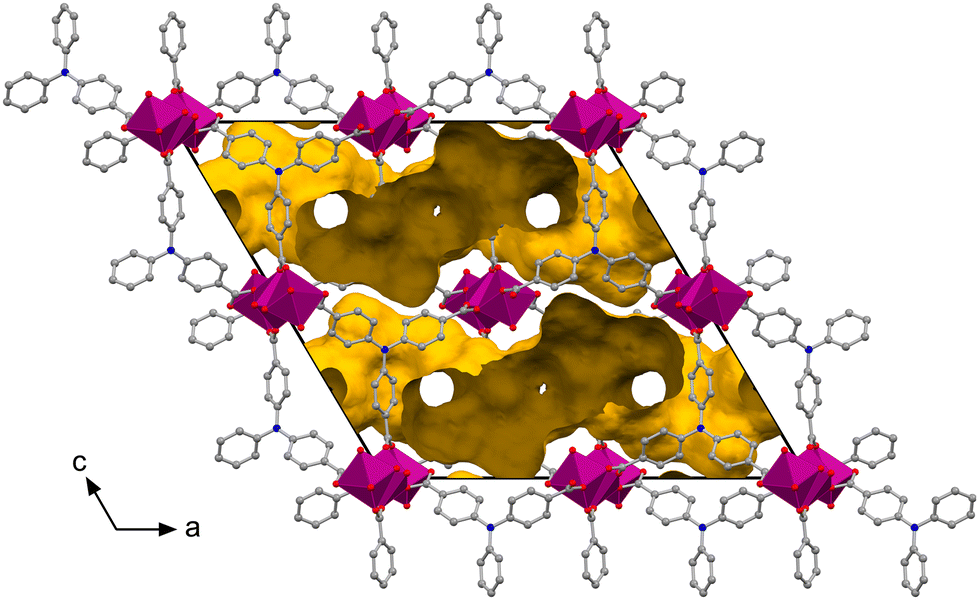 | ||
| Fig. 5 Representation of the two-dimensional void space in the crystal structure of JUMP-4(dmf) as yellow surface within the unit cell, viewed along the crystallographic direction [010]. The linear trinuclear cobalt(II) SBUs are arranged into one-dimensional chains (plum colored polyhedra) along the direction [010] and interconnected by ntb3− ligands. | ||
To further characterize the frameworks, topological analysis using TOPOS 5.5 was performed.59–61 Treating the trinuclear SBUs as nodes, each connects to six ligands and two neighboring nodes, resulting in an eight-connected node. The second node, represented by the central nitrogen atom of the ntb3− ligand, is three-connected. This analysis reveals that the frameworks adopt a three-dimensional, binodal, 3,8-connected net with a yet unknown topology and point symbol (42·5)2(44·56·610·75·8.92) (see Fig. 6, Fig. S16, S17, and Tables S12, S13†).
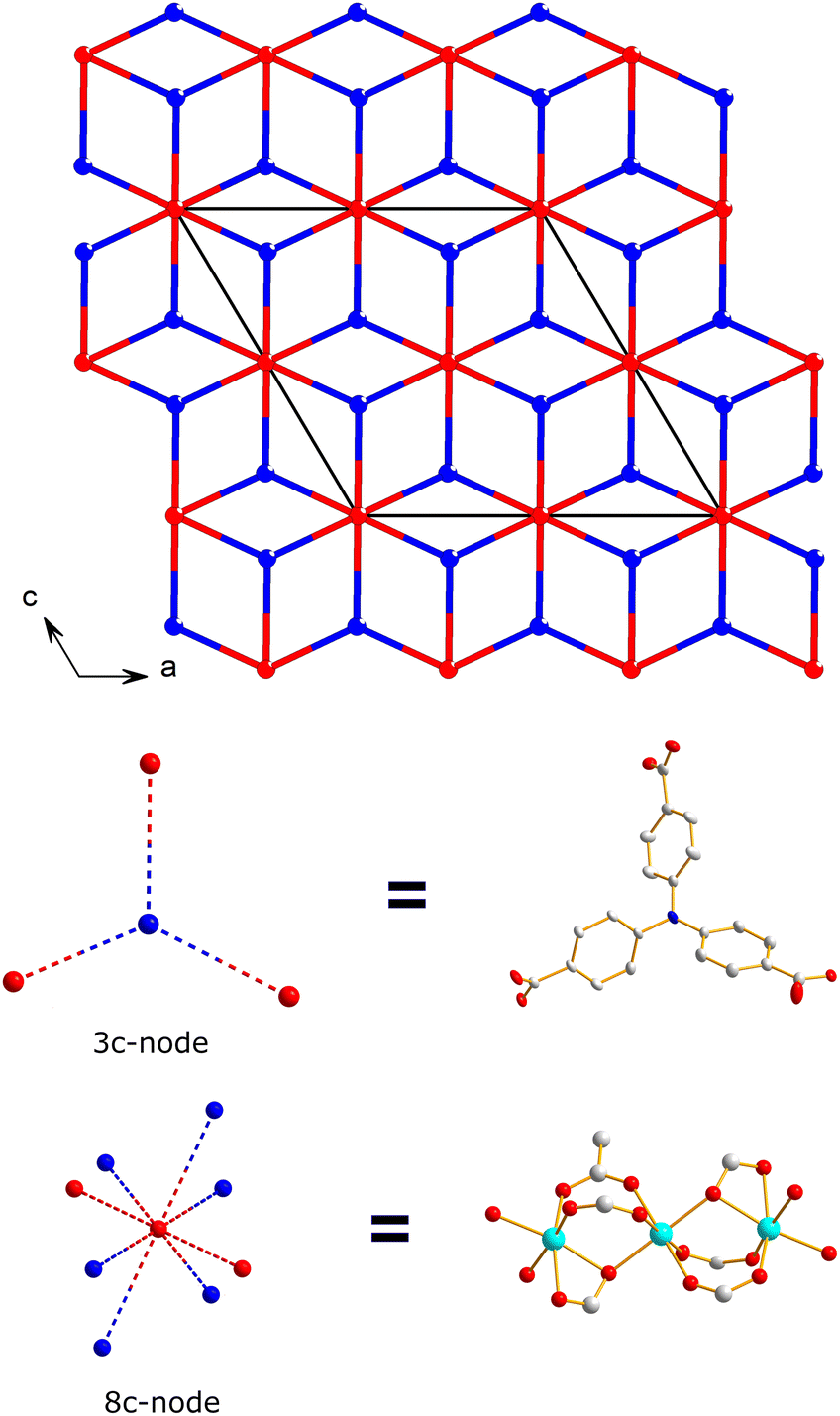 | ||
| Fig. 6 Representation of the 3,8-connected net of the JUMP-4 derivatives viewed along the crystallographic [010] direction. The red and blue spheres represent the Co2 ion of the trinuclear cobalt clusters (8-connected nodes) and the nitrogen atom of the ntb3− ligands (3-connected nodes), respectively. | ||
X-ray powder diffraction (XRPD) patterns were measured for all four networks to confirm the phase purity of the bulk materials. The experimental powder patterns closely match the simulated patterns derived from the respective single-crystal structure data, demonstrating the consistency between the single-crystal data and the bulk material (see Fig. S18–S22†).
Gas-sorption properties
The porosity of the activated samples of the JUMP-4 derivatives was investigated by measuring Argon isotherms, depicted in Fig. 7 (for a semi-logarithmic representation see Fig. S23†). A summary of the corresponding data is presented in Table 1, while consistency criteria for BET surface area calculations are detailed in Table S14.† | ||
| Fig. 7 Argon adsorption isotherms measured at 87 K for the JUMP-4 derivatives. Open circles denote the desorption process. | ||
| Sample | a BET/m2 g−1 | V pore/cm3 g−1 | V X-raypore (solv)/cm3 g−1 | V X-raypore (H2O)/cm3 g−1 | Pore width | |
|---|---|---|---|---|---|---|
| Modal/nm | Mean/nm | |||||
| JUMP-4(dmf) | 830 | 0.31 | 0.22 | 0.39 | 1.00 | 1.91 |
| JUMP-4(def) | 640 | 0.28 | 0.16 | 0.38 | 0.90 | 1.76 |
| JUMP-4(dma) | 840 | 0.33 | 0.17 | 0.38 | 0.96 | 1.51 |
| JUMP-4(EtOH) | 350 | 0.19 | 0.24 | 0.40 | 0.90 | 2.58 |
All four frameworks exhibit moderate to high BET surface areas. The highest values were observed for the dmf- and dma-based derivative with 830 and 840 m2 g−1, respectively, followed by the def-based framework (640 m2 g−1). The ethanol-based analogue shows a significantly lower value (350 m2 g−1). This trend is also reflected in the pore volumes of the materials (Table 1). This is consistent with the experimental pore volumes of the amide-based derivatives JUMP-4(dmf), JUMP-4(def), and JUMP-4(dma), which fall within the range expected for the MOF framework, assuming variations at the solvent-accessible coordination site of the Co1 ion in the trinuclear cobalt clusters—either occupied by the relevant amide or a water molecule as a ligand. In contrast, for the ethanol-exchanged derivative JUMP-4(EtOH), the experimental pore volume is found to be below this range, which is in accordance with its significantly lower BET surface area.
The argon isotherms of all JUMP-4 derivatives display type Ia behavior (see Fig. 7), according to the IUPAC classification,62,63 which is characteristic of materials containing exclusively micropores with rigid pore structures.64,65 Pore-size distributions, estimated using the QSDFT approach implemented in Quantachrome Instrument software,66 are shown in Fig. 8. All materials exhibit pore sizes below 2 nm, consistent with the observed type Ia isotherms, with similar distributions across all derivatives, reflecting their structural similarities. Interestingly, the pore diameter range determined from the crystal structure data (0.3–0.6 nm) is notably smaller than the values derived from the QSDFT analysis, suggesting potential limitations in the underlying model assumptions.
 | ||
| Fig. 8 Pore size distribution data for activated samples of the JUMP-4 derivatives, fitted using argon at 87 K on carbon (cylindrical pores, QSDFT adsorption branch model). | ||
Notably, the ethanol-exchanged derivative JUMP-4(EtOH) shows significantly lower BET surface area and pore volume compared to the amide-based derivatives. This is attributed to framework destabilization, likely caused by breaking hydrogen-bonding interactions between ethanol guest molecules and the host during activation, which leads to partial pore collapse.
In contrast, the amide-based frameworks demonstrate remarkable stability under high-temperature activation, attributed to the rod-like one-dimensional chains running through the structure. These chains impose rigidity and minimize deformation during guest removal, thereby preserving the pore framework. XRPD measurements conducted after activation confirm retention of long-range order in these materials (see Fig. S24†).
Overall, the sorption behavior suggests that the size of the coordinated solvent molecules influences accessible porosity. Smaller ligands, such as dmf and dma, enable better pore accessibility compared to the larger def ligand. The ethanol-based framework, however, exhibits reduced porosity due to framework destabilization. The stability and preserved porosity of the amide-based networks after activation are attributed to the stabilizing role of the rod-shaped structural building units.57
Magnetic properties
The magnetic susceptibility data for all JUMP-4 derivatives were measured in the temperature range of 2–300 K with an applied field of 2 kOe. As an example, the data for JUMP-4(dmf) is shown in Fig. 9 as a temperature-dependent plot of χMT and χM−1. Data for the remaining derivatives are provided in Fig. S25–S27.† | ||
| Fig. 9 Temperature dependence of χMT (open circles) and χM−1 (filled circles) for JUMP-4(dmf) with solid red and blue lines, respectively, representing the best fits (see text for parameters). | ||
The room temperature χMT value for JUMP-4(dmf) of approximately 8.5 cm3 K mol−1 is significantly higher than the spin-only value for three magnetically isolated cobalt(II) ions (5.63 cm3 K mol−1S = 3/2, g = 2), indicating strong spin–orbit coupling.67 Upon cooling, the χMT value decreases gradually down to 20 K, with a slightly steeper slope below 100 K, followed by a pronounced drop to 4.3 cm3 K mol−1 at 2 K. This behavior can be attributed to spin–orbit coupling as well as antiferromagnetic exchange interactions between the cobalt(II) ions.68–70 The χMT vs. T data for all four JUMP-4 derivatives exhibit similar behavior. Interestingly, this behavior also mirrors that observed for the MOFs JUMP-152 and JUMP-3,51 even though the latter two contain isolated trinuclear cobalt(II) clusters, in contrast to the one-dimensional chains present in all JUMP-4 derivatives. Unfortunately, the magnetic data of the JUMP-4 derivatives cannot be simulated based on an appropriate spin Hamiltonian due to the 1D chain arrangement of the trinuclear clusters.
Nevertheless, the χM−1vs. T data of JUMP-4(dmf) confirm paramagnetic behavior above 50 K, with a Curie constant of 8.76 cm3 K mol−1 and a Weiss constant of −8.0 K (see Table 2). The negative Weiss constant is consistent with antiferromagnetic coupling. This again is consistent with earlier observations for the JUMP-152 and JUMP-351 MOF structures containing isolated trinuclear cobalt(II) clusters, indicating that magnetic exchange within the trinuclear clusters and along the 1D chain seem not to dominate the magnetic behavior.
| Sample | C (cm3 K mol−1) | θ (K) |
|---|---|---|
| JUMP-4(dmf) | 8.76 | −8.0 |
| JUMP-4(def) | 8.74 | −7.7 |
| JUMP-4(dma) | 8.74 | −8.6 |
| JUMP-4(EtOH) | 8.66 | −4.6 |
Also the field-dependent magnetization for all JUMP-4 derivatives was measured in a temperature range from 2 to 5 K and up to fields of 5 T (see Fig. S28–S31†). The data indicates that saturation has not been reached under the given conditions for all derivatives. The overall the data is consistent with strong magnetic anisotropy being present. In particular, the observed values are consistent with the presence with the presence of three octahedral cobalt(II) ions,71 as found in the crystal structures of the JUMP-4 derivatives. However, due to the 1D chain arrangement of the trinuclear clusters an exact treatment of the magnetization data is not feasible. To further address the magnetic properties of JUMP-4 derivatives alternating-current susceptibility measurements as well as zero-field cooled and field cooled magnetization measurements have been performed. However, neither a signature for slow magnetization dynamics nor any kind of ordering could be detected.
Conclusion
We have successfully synthesized derivatives of the new JUMP-4 framework using a solvent-directing effect. The solvothermal route, employing a mixture of an amide-based solvent (solv: dmf, def, or dma) and acetonitrile, directly yields the corresponding MOF systems JUMP-4(solv). An additional compound, JUMP-4(EtOH), is obtained via single-crystal-to-single-crystal transformation in the solid state. All four MOFs are three-dimensional networks containing one-dimensional chains of cobalt(II) ions arranged in an inverted Figaro-type configuration.These frameworks exhibit considerable thermal stability, with JUMP-4(dmf) being the most stable and its ethanol-converted analogue, JUMP-4(EtOH), the least stable. Notably, only JUMP-4(dmf) and JUMP-4(dma) (but not JUMP-4(def)) underwent single-crystal-to-single-crystal transformation to form the ethanol derivative JUMP-4(EtOH). This suggests a solvent-based discrimination mechanism, likely due to steric effects that prevent ligand exchange.
Furthermore, the coordinating solvent significantly influences the sorption behavior of the networks, which can also be attributed to steric differences arising from the solvent molecules coordinated to the outer cobalt(II) ions. The magnetic properties reflect strong magnetic anisotropy of the cobalt(II) ions within the frameworks, and show only minor variations between the derivatives, likely due to structural and electronic differences induced by the coordinating solvent molecules.
Experimental section
Materials
The starting materials p-fluorobenzonitrile (Alfa Aesar), p-aminobenzonitrile (Alfar Aesar), and cobalt(II) chloride hexahydrate (Aldrich) were obtained from commercial sources and used without further purification. All other chemicals were of AR grade.Physical measurements
Simultaneous TG/DTA analyses were performed under static air atmosphere using a Netzsch STA Luxx PC analyzer up to 1000 °C. The FT-IR spectra were measured on a VERTEX 70-IR spectrometer from Bruker Optics using the Specac Diamond ATR optional accessory. Mass spectrometry was performed on a MAT SSQ710 mass spectrometer made by Bruker. NMR spectra were recorded with an Bruker AVANCE 400 spectrometer. The elemental analyses were done on a VARIO EL III analyser. The magnetic susceptibility of the bulk, vacuum dried (rotary vane pump, typically 0.1–1 mbar) materials was measured over the temperature range of 2–300 K using a Quantum Design MPMS-5 superconducting SQUID magnetometer, with the data corrected for diamagnetic contributions. Powder X-ray diffraction measurements were performed on a Stoe powder diffractometer equipped with a Mythen 1K detector at room temperature. The measurements were conducted using capillary tubes in Debye–Scherrer scan mode with a 2θ scan type. A Cu long-fine-focus X-ray tube was used as the radiation source. The powdered samples were suspended in DMF within 0.5 mm glass capillaries and subsequently measured. Data were collected over a 2θ range of 2–50°, with a step size of 2.1° per 20 seconds.Ligand synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 149.83 (C–N), 131.19 (C[Ar]), 125.91 (C[Ar]), 123.73 (C[Ar]); Selected IR data
O), 149.83 (C–N), 131.19 (C[Ar]), 125.91 (C[Ar]), 123.73 (C[Ar]); Selected IR data  (cm−1): 1672s, 1591s, 1509m, 1417s, 1314s, 1275vs, 1173s, 1129m, 932m. EI-MS: m/z (relative intensity): 377 (100%).
(cm−1): 1672s, 1591s, 1509m, 1417s, 1314s, 1275vs, 1173s, 1129m, 932m. EI-MS: m/z (relative intensity): 377 (100%).
MOF syntheses
:1 ratio) in a Parr acid digestion bomb (23 mL volume) and heated at 140 °C for 72 h under autogenous pressure. It was then allowed to cool at a rate of 5 °C h−1. Violet crystals could be seen in the room temperature sample, which were then washed repeatedly with DMF (5 × 5 mL) and subsequently dried in vacuo (rotary vane pump, typically 0.1–1 mbar) leading to the further denoted as-synthesized material of JUMP-4(dmf).
 (cm−1): 1660m, 1591vs, 1556s, 1504m, 1382vs, 1314s, 1272s, 1174m, 1089w, 844w, 780vs, 705w, 675w, 518m. TGA mass losses (see Fig. S1†): up to 300 °C attributed to guest H2O as well as both guest and bound dmf molecules (exp. 24.5%, calcd 25.1%); residual mass based on CoO: exp. 18.1%, calcd 18.1%.
(cm−1): 1660m, 1591vs, 1556s, 1504m, 1382vs, 1314s, 1272s, 1174m, 1089w, 844w, 780vs, 705w, 675w, 518m. TGA mass losses (see Fig. S1†): up to 300 °C attributed to guest H2O as well as both guest and bound dmf molecules (exp. 24.5%, calcd 25.1%); residual mass based on CoO: exp. 18.1%, calcd 18.1%.
 (cm−1): 1650m, 1590vs, 1553s, 1506m, 1382vs, 1314s, 1268s, 1172m, 1104w, 845w, 780vs, 706w, 674m, 513s. TGA mass losses (see Fig. S2†): up to 360 °C attributed to guest CH3CN and H2O molecules as well as bound def molecules (exp. 22.1%, calcd 25.7%); residual mass based on CoO: exp. 18.5%, calcd 18.0%.
(cm−1): 1650m, 1590vs, 1553s, 1506m, 1382vs, 1314s, 1268s, 1172m, 1104w, 845w, 780vs, 706w, 674m, 513s. TGA mass losses (see Fig. S2†): up to 360 °C attributed to guest CH3CN and H2O molecules as well as bound def molecules (exp. 22.1%, calcd 25.7%); residual mass based on CoO: exp. 18.5%, calcd 18.0%.
 (cm−1): 1591vs, 1555m, 1504m, 1388vs, 1313s, 1266s, 1174m, 1103w, 844w, 780vs, 706w, 673w, 516m. TGA mass losses (see Fig. S3†): up to 360 °C attributed to free guest solvent molecules as well as coordinated dma molecules (exp. 22.9%, Calc. 25.2%); residual mass based on CoO: exp. 18.2%, calcd 18.2%.
(cm−1): 1591vs, 1555m, 1504m, 1388vs, 1313s, 1266s, 1174m, 1103w, 844w, 780vs, 706w, 673w, 516m. TGA mass losses (see Fig. S3†): up to 360 °C attributed to free guest solvent molecules as well as coordinated dma molecules (exp. 22.9%, Calc. 25.2%); residual mass based on CoO: exp. 18.2%, calcd 18.2%.
Synthesis for the EtOH derivatives of JUMP-4
 (cm−1): 3353 br, 1591vs, 1556m, 1505w, 1387vs, 1315vs, 1272s, 1173m, 1042w, 844w, 780vs, 706w, 675m, 523m. TGA mass losses (see Fig. S4†): up to 150 °C attributed to guest H2O and bound EtOH molecules (exp. 10.1%, calcd 10.6%); residual mass based on CoO: exp. 21.7%, calcd 21.7%.
(cm−1): 3353 br, 1591vs, 1556m, 1505w, 1387vs, 1315vs, 1272s, 1173m, 1042w, 844w, 780vs, 706w, 675m, 523m. TGA mass losses (see Fig. S4†): up to 150 °C attributed to guest H2O and bound EtOH molecules (exp. 10.1%, calcd 10.6%); residual mass based on CoO: exp. 21.7%, calcd 21.7%.
X-ray structure determination
The single crystal X-ray data for all four JUMP-4 derivatives were collected on a Nonius KappaCCD diffractometer, using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at 133(2) K. Data have been corrected for Lorentz and polarization effects, and absorption has been accounted for on a semi-empirical basis using multiple-scans.72–74 The structures were solved by direct methods (SHELXS)75 and refined by full-matrix least squares techniques against Fo2 (SHELXL-2018).76 All hydrogen atoms were included at calculated positions with fixed thermal parameters. All non-disordered, non-hydrogen atoms were refined anisotropically. For disordered groups in all structures, restraints on the distances and anisotropic displacement parameters were used (DFIX, EADP, FLAT, RIGU, SADI, and SIMU). In addition, the disordered moieties of the ethanole molecules of JUMP-4(EtOH) were introduced and refined using the program disordered structure refinement (DSR).77 Crystallographic data as well as structure solution and refinement details are summarized in Table S1.† Diamond 5.1.078 and Mercury 2024.3.179 were used for structure analysis and representations.
Sample pretreatment and sorption measurements
Data availability
Data for this article containing TGA data, structural details and figures, topological data, XRPD patterns, sorption data, and magnetic details have been included as part of the ESI.† Crystallographic data for JUMP-4(dmf), JUMP-4(def), JUMP-4(dma), and JUMP-4(ETOH) have been deposited at the CCDC under 2057554, 2057555, 2057556, and 2057557,† respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Sebastian Seidenath and Katrin Schröder for their synthetic contributions during their lab internship. We are grateful to Dr Michael Böhme for valuable discussions. This project was supported by the Deutsche Forschungsgemeinschaft (PL 155/25-1).References
- D. K. J. A. Wanigarathna, J. Gao and B. Liu, Mater. Adv., 2020, 1, 310–320 Search PubMed.
- Q.-M. Sun, J.-H. He and J.-M. Lu, Energy Sci. Eng., 2022, 11, 952–973 Search PubMed.
- F. A. Sahayaraj, J. H. Prabu, J. Maniraj, M. Kannan, M. Bharathi, P. Diwahar and J. Salamon, J. Inorg. Organomet. Polym., 2023, 33, 1757–1781 Search PubMed.
- I. I. Alkhatib, C. Garlisi, M. Pagliaro, K. Al-Ali and G. Palmisano, Catal. Today, 2020, 340, 209–224 Search PubMed.
- Q. Yao, X. Zhang, Z.-H. Lu and Q. Xu, Coord. Chem. Rev., 2023, 493, 215302 Search PubMed.
- S. Ma, W. Han, W. Han, F. Dong and Z. Tang, J. Mater. Chem. A, 2023, 11, 3315–3363 RSC.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 Search PubMed.
- A. E. Thorarinsdottir and T. D. Harris, Chem. Rev., 2020, 120, 8716–8789 Search PubMed.
- M. Pourmadadi, Z. Omrani, Z. Forootan, M. S. Ebadi and F. Yazdian, J. Drug Delivery Sci. Technol., 2023, 86, 104690 Search PubMed.
- M. Moharramnejad, A. Ehsani, M. Shahi, S. Gharanli, H. Saremi, R. E. Malekshah, Z. S. Basmenj, S. Salmani and M. Mohammadi, J. Drug Delivery Sci. Technol., 2023, 81, 104285 CrossRef.
- T. Yamabayashi, M. Atzori, L. Tesi, G. Cosquer, F. Santanni, M.-E. Boulon, E. Morra, S. Benci, R. Torre, M. Chiesa, L. Sorace, R. Sessoli and M. Yamashita, J. Am. Chem. Soc., 2018, 140, 12090–12101 Search PubMed.
- S. Wu, J. Liu, H. Wang and H. Yan, Int. J. Energy Res., 2019, 43, 697–716 Search PubMed.
- O. Akintola, P. Gerlach, C. T. Plass, A. Balducci and W. Plass, Front. Chem., 2022, 10, 836325 Search PubMed.
- C. Aphirakaramwong, O. Akintola, C. T. Plass, M. Sawangphruk, W. Plass and A. Balducci, RSC Adv., 2023, 13, 12277–12284 Search PubMed.
- N. Koralkar, S. Mehta, A. Upadhyay, G. Patel and K. Deshmukh, J. Inorg. Organomet. Polym., 2024, 34, 903–929 Search PubMed.
- Y. Liu, H. Li, Y. Han, X. Lv, H. Hou and Y. Fan, Cryst. Growth Des., 2012, 12, 3505–3513 Search PubMed.
- Z. Zhang and M. J. Zaworotko, Chem. Soc. Rev., 2014, 43, 5444–5455 RSC.
- N. Zhao, K. Cai and H. He, Dalton Trans., 2020, 49, 11467–11479 RSC.
- P. Howlader and P. S. Mukherjee, Isr. J. Chem., 2018, 59, 292–298 CrossRef.
- X.-M. Jing, L.-W. Xiao, L. Wei, F.-C. Dai and L.-L. Ren, Inorg. Chem. Commun., 2016, 71, 78–81 CrossRef.
- M. Chen, M. Hu, H. Zhao, J.-Y. Tian and C.-S. Liu, Z. Anorg. Allg. Chem., 2016, 642, 778–784 Search PubMed.
- J. Hungerford and K. S. Walton, Inorg. Chem., 2019, 58, 7690–7697 Search PubMed.
- Y. Wu, G.-P. Yang, Y. Zhao, W.-P. Wu, B. Liu and Y.-Y. Wang, Dalton Trans., 2015, 44, 3271–3277 Search PubMed.
- J. Hwang, R. Yan, M. Oschatz and B. V. K. J. Schmidt, J. Mater. Chem. A, 2018, 6, 23521–23530 RSC.
- J.-C. Wang, F.-W. Ding, J.-P. Ma, Q.-K. Liu, J.-Y. Cheng and Y.-B. Dong, Inorg. Chem., 2015, 54, 10865–10872 CrossRef PubMed.
- J. N. Hall and P. Bollini, React. Chem. Eng., 2019, 4, 207–222 RSC.
- B. Chen, M. O'Keeffe, O. M. Yaghi, M. Eddaoudi, N. L. Rosi and J. Kim, J. Am. Chem. Soc., 2005, 127, 1504–1518 Search PubMed.
- Y.-S. Bae, O. K. Farha, A. M. Spokoyny, C. A. Mirkin, J. T. Hupp and R. Q. Snurr, Chem. Commun., 2008, 4135–4137 Search PubMed.
- O. K. Farha, A. M. Spokoyny, K. L. Mulfort, M. F. Hawthorne, C. A. Mirkin and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 12680–12681 CrossRef PubMed.
- A. Das, F. J. Klinke, S. Demeshko, S. Meyer, S. Dechert and F. Meyer, Inorg. Chem., 2012, 51, 8141–8149 CrossRef PubMed.
- F. J. Klinke, A. Das, S. Demeshko, S. Dechert and F. Meyer, Inorg. Chem., 2014, 53, 2976–2982 Search PubMed.
- J. Vallejo, E. Pardo, M. Viciano-Chumillas, I. Castro, P. Amorós, M. Déniz, C. Ruiz-Pérez, C. Yuste-Vivas, J. Krzystek, M. Julve, F. Lloret and J. Cano, Chem. Sci., 2017, 8, 3694–3702 CAS.
- S. Gómez-Coca, D. Aravena, R. Morales and E. Ruiz, Chem. Soc. Rev., 2015, 289, 379–392 Search PubMed.
- M. H. Pohle, T. Lohmiller, M. Böhme, M. Rams, S. Ziegenbalg, H. Görls, A. Schnegg and W. Plass, Chem. – Eur. J., 2024, 30, e202401545 Search PubMed.
- O. Fabelo, L. Canadillas-Delgado, J. Pasan, F. S. Delgado, F. Lloret, J. Cano, M. Julve and C. Ruiz-Perez, Inorg. Chem., 2009, 48, 11342–11351 CrossRef CAS.
- X.-H. Jing, X.-C. Yi, E.-Q. Gao and V. A. Blatov, Dalton Trans., 2012, 41, 14316–14328 RSC.
- Z. Su, M. Chen, T. a. Okamura, M.-S. Chen, S.-S. Chen and W.-Y. Sun, Inorg. Chem., 2011, 50, 985–991 CrossRef CAS PubMed.
- W.-X. Zhang, W. Xue and X.-M. Chen, Inorg. Chem., 2011, 50, 309–316 CrossRef PubMed.
- A. Jochim, M. Rams, M. Böhme, M. Ceglarska, W. Plass and C. Näther, Dalton Trans., 2020, 49, 15310–15322 Search PubMed.
- M. Rams, A. Jochim, M. Böhme, T. Lohmiller, M. Ceglarska, M. M. Rams, A. Schnegg, W. Plass and C. Näther, Chem. – Eur. J., 2020, 26, 2837–2851 CrossRef PubMed.
- A. Jochim, T. Lohmiller, M. Rams, M. Böhme, M. Ceglarska, A. Schnegg, W. Plass and C. Näther, Inorg. Chem., 2020, 59, 8971–8982 Search PubMed.
- M. Böhme, A. Jochim, M. Rams, T. Lohmiller, S. Suckert, A. Schnegg, W. Plass and C. Näther, Inorg. Chem., 2020, 59, 5325–5338 Search PubMed.
- M. Böhme and W. Plass, Chem. Sci., 2019, 10, 9189–9202 Search PubMed.
- O. Akintola, D. Hornig, A. Buchholz, H. Görls and W. Plass, Dalton Trans., 2017, 46, 8037–8050 Search PubMed.
- J. H. Gorvin, J. Chem. Soc., Perkin Trans. 1, 1988, 1331–1335 RSC.
- C. Hua, J.-Y. Ge, F. Tuna, D. Collison, J.-L. Zuo and D. M. D'Alessandro, Dalton Trans., 2017, 46, 2998–3007 Search PubMed.
- S. Nandi, D. Chakraborty and R. Vaidhyanathan, Chem. Commun., 2016, 52, 7249–7252 RSC.
- C. Livage, N. Guillou, A. Castiglione, J. Marrot, M. Frigoli and F. Millange, Microporous Mesoporous Mater., 2012, 157, 37–41 CrossRef.
- T. Wang, C. Zhang, Z. Ju and H. Zheng, Dalton Trans., 2015, 44, 6926–6935 RSC.
- Q. Yang, X. Chen, Z. Chen, Y. Hao, Y. Li, Q. Lu and H. Zheng, Chem. Commun., 2012, 48, 10016–10018 Search PubMed.
- O. Akintola, A. Buchholz, H. Gorls and W. Plass, Eur. J. Inorg. Chem., 2021, 2266–2273 CAS.
- O. Akintola, S. Ziegenbalg, A. Buchholz, H. Görls and W. Plass, CrystEngComm, 2017, 19, 2723–2732 CAS.
- M.-H. Zeng, Y.-L. Zhou, M.-C. Wu, H.-L. Sun and M. Du, Inorg. Chem., 2010, 49, 6436–6442 Search PubMed.
- B. Xu, B. Liu, H.-M. Hu, Y. Cheng, Z. Chang and G. Xue, Polyhedron, 2015, 96, 88–94 Search PubMed.
- M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS PubMed.
- F. Luo, Y.-X. Che and J.-M. Zheng, Inorg. Chem. Commun., 2008, 11, 358–362 CrossRef CAS.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
- T. Devic, C. Serre, N. Audebrand, J. Marrot and G. Férey, J. Am. Chem. Soc., 2005, 127, 12788–12789 Search PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- O. Delgado-Friedrichs, M. O'Keeffe and O. Yaghi, Phys. Chem. Chem. Phys., 2007, 9, 1035–1043 Search PubMed.
- M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 Search PubMed.
- X. Gao, T. Xu, Z. Jiang, H. Yu, Y. Wang and Y. He, Dalton Trans., 2019, 48, 16793–16799 RSC.
- H. Miura, V. Bon, I. Senkovska, S. Ehrling, S. Watanabe, M. Ohba and S. Kaskel, Dalton Trans., 2017, 46, 14002–14011 Search PubMed.
- QUANTACHROME, Advantages of QSDFT for Pore Size Analysis of Carbons.
- O. Kahn, Molecular Magnetism, Wiley-VCH Inc., Weinheim, 1993 Search PubMed.
- M.-X. Yao, M.-H. Zeng, H.-H. Zou, Y.-L. Zhou and H. Liang, Dalton Trans., 2008, 2428–2432 RSC.
- L.-F. Ma, Y.-Y. Wang, L.-Y. Wang, D.-H. Lu, S. R. Batten and J.-G. Wang, Cryst. Growth Des., 2009, 9, 2036–2038 CAS.
- P. Kapoor, A. P. S. Pannu, G. Hundal, R. Kapoor, M. Corbella, N. Aliaga-Alcalde and M. S. Hundal, Dalton Trans., 2010, 39, 7951–7959 CAS.
- F. Lloret, M. Julve, J. Cano, R. Ruiz-García and E. Pardo, Inorg. Chim. Acta, 2008, 361, 3432–3445 CrossRef CAS.
- R. W. W. Hooft, COLLECT. Data Collection Software, Nonius BV, Delft, The Netherlands, 1998 Search PubMed.
- Z. Otwinowski and W. Minor, Processing of X-ray Diffraction Data Collected in Oscillation Mode, in Methods in Enzymology, ed. C. W. Carter and R. M. Sweet, Academic Press, 1997, vol. 276, pp. 307–326 Search PubMed.
- SADABS 2.10, Bruker-AXS Inc., Madison, Wisconsin, USA, 2002 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- D. Kratzert and I. Krossing, J. Appl. Crystallogr., 2018, 51, 928–934 Search PubMed.
- H. Putz and K. Brandenburg, Diamond - Crystal and Molecular Structure Visualization, Bonn, Germany, https://www.crystalimpact.com/diamond Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 160, 49–56 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2057554–2057557. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00149h |
| This journal is © The Royal Society of Chemistry 2025 |