
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal complexes with a twist: modified rhodamines as a promising theranostic approach for combating cancer
Fábio
Martins
 a,
Maria G. P. M. S.
Neves
b and
Ana M. G.
Silva
*c
a,
Maria G. P. M. S.
Neves
b and
Ana M. G.
Silva
*c
aLAQV-REQUIMTE, Department of Chemistry and Biochemistry (DQB), Faculty of Sciences, University of Porto (FCUP), 4169-007 Porto, Portugal
bLAQV-REQUIMTE, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
cLAQV-REQUIMTE, School of Medicine and Biomedical Sciences, University of Porto, 4050-313 Porto, Portugal. E-mail: ana.silva@icbas.up.pt
First published on 23rd April 2025
Abstract
Rhodamines have been recognized for their exceptional optical properties, making them suitable for detection, imaging, and disease diagnosis. However, their use as photosensitizers in Photodynamic Therapy (PDT) has been limited by their low singlet oxygen production and limited tissue penetration. The development of rhodamine-metal complexes has overcome these limitations, offering a promising new approach for cancer treatment. These complexes in combination with structural and optical tuning of rhodamines, have been engineered to enhance tumour cell selectivity, improve reactive oxygen species (ROS) generation, and mitochondrial-targeted delivery. Notably, a variety of metal ions, including iridium(III), ruthenium(II) and platinum(II/IV) can form complexes with bright rhodamines with excellent optical responses and remarkable ROS generation. These breakthroughs have the potential to improve cancer diagnosis and therapeutic applications. Photophysical properties, photostability, and targeting agents, particularly in the near-infrared (NIR) range, will be discussed, with a focus on their applications in cancer detection, localization, and cytotoxicity.
Introduction
Combating cancer has become a worldwide challenge. According to the World Health Organization (WHO)'s cancer agency, in 2022 approximately 20 million new cases of cancer were diagnosed and nearly 9.7 million lives were lost due to this disease.1 These numbers are extremely worrying and are projected to worsen considerably. The WHO predicts that by 2050, new cancer cases may increase to over 35 million, representing a 77% increase from the estimated 20 million cases in 2022.2,3 The increasing cancer burden is largely attributed to several factors, which include (i) population aging (longer lifespans increase cancer risk); (ii) population growth and (iii) higher exposure to risk factors such as air pollution, tobacco, alcohol, and obesity just to name a few.4Over the past few decades, the scientific community has made tremendous efforts in developing new and improved diagnostic and treatment solutions to combat cancer.5 This concerted effort has led to significant advances across multiple disciplines, with special emphasis on: (i) integration of AI in supporting cancer detection and diagnosis,6,7 (ii) development of new pharmaceuticals for targeted and effective cancer treatments,8–10 (iii) state-of-the-art instrumentation, including advanced imaging and diagnostic tools,11,12 and (iv) innovative therapeutic modalities that allow the expansion of available options of treatment.13
Among recent advances, photodynamic therapy (PDT) has gained attention as a powerful alternative to traditional cancer treatments. This approach uses light-sensitive compounds called photosensitizers (PS) that, when irradiated by light and in the presence of dioxygen (3O2), produce highly energetic species called reactive oxygen species (ROS). These species are capable of destroying cancer cells while minimizing damage to surrounding healthy tissue. Unlike chemotherapy and radiotherapy, which often have serious side effects, PDT offers a targeted method, reducing side effects.14–16
In the PDT process, after light absorption the PS is excited from the ground state (S0) to an electronically excited state (usually the first singlet excited state S1) (Fig. 1). At this stage, the PS can return to its ground state through fluorescence emission (S1 → S0) or can transition to a longer-lived triplet excited state (T1) through a nonradiative intersystem crossing (ISC). PS excited in the triplet state can then undergo two types of reaction pathways: photoinduced electron transfer to form radical intermediates (Type I) or energy transfer to dioxygen to form singlet oxygen (Type II).17–20 As a result, oxidative damage to the cell membrane and organelles can occur, potentially leading to cell death through necrosis, apoptosis, autophagy, or ferroptosis.21,22
 | ||
| Fig. 1 Simplified Jablonski diagram, depicting the electronic transition states and energy transfer between the photosensitizer and dioxygen in PDT, that leads to oxidative cell damage. | ||
It is generally considered that the effectiveness of a PS is determined by three main factors: (i) the PS should preferentially accumulate in the target tissue or cells, thereby minimizing damage to healthy surrounding areas (ii) ideally, the PS should absorb light in the “therapeutic window” (600–800 nm) for optimal tissue penetration and (iii) the PS should have high intersystem crossing (ISC) efficiency, i.e., should exhibit a good ability to transition from its first singlet excited state to its triplet state, a crucial step in the generation of reactive oxygen species, particularly singlet oxygen (1O2), which is the primary cytotoxic agent in PDT process.23
The field of PDT has long been dominated by porphyrin-based compound due to their unique therapeutic and diagnostic capabilities. These compounds demonstrate significant phototherapeutic effects, including photodynamic and photothermal therapies, along with impressive imaging functionalities such as near-infrared fluorescent (NIRF) imaging and magnetic resonance imaging (MRI), just to name a few.24 However, it is crucial to explore and develop alternative PSs to enhance the versatility and effectiveness of this therapy.25,26
Despite the vast number of fluorophores, there is one family of synthetic compounds that rises above all other. Xanthene dyes, among the oldest synthetic pigments ever created, are constituted by a series of substructures, all derived from the xanthene scaffold, which is a heterocyclic aromatic compound traditionally containing an oxygen bridge. This particular backbone can lead to a series of pigments with different chemical and optical properties that includes: fluoresceins, rhodamines and hybrid structures, such as rhodols.27,28
Recently, rhodamines have emerged as promising alternatives to the previously described PSs. In addition to their excellent, highly modular light-absorbing properties and photostability, their cationic nature provides better solubility in aqueous media, as well as faster clearance from the body, potentially reducing side effects and minimizing prolonged skin photosensitivity, which are common drawbacks of generic PSs.29 Furthermore, rhodamines have a unique ability to selectively target not only tumour cells but also subcellular localization in mitochondria,30 endoplasmic reticulum31 and lysosomes.32 This ability is crucial for enhancing the efficacy of PDT, because of the key role they play in cell function. It has been demonstrated that mitochondria-targeted PS, including those based on rhodamine, can induce higher levels of cellular stress, leading to more efficient cell death compared to non-organelle-targeting PSs.33,34 This specific accumulation in organelles maximizes the therapeutic effect while minimizing damage to surrounding tissues, making PDT more precise and effective in combating malignant cells.
Despite these developments, several challenges in rhodamine-based PSs needed to be addressed, such as the low singlet oxygen production, the predominance of less effective Type I photochemical reactions, and the limited tissue penetration due to light absorption in the visible range, which is typical of generic rhodamine derivatives.35 To overcome these drawbacks and thanks to the highly modular nature of rhodamines, there has been a growing interest in recent years in the development of new rhodamine modifications and conjugations.
In this Frontier article, we critically assess recent advancements in the development of rhodamine-metal complexes for cancer treatment, particularly in the context of PDT. The article is organized into different sections: firstly, we provide an overview of the rhodamine structural modifications that have significantly impacted their photophysical properties. This is followed by an analysis how the combination of rhodamines with various metal ions, such as iridium(III), ruthenium(II), and platinum(II/IV), has successfully addressed the recurring challenges faced by rhodamines. A final section is dedicated to a critical discussion of important aspects for PDT applications. This includes how the structural and optical tuning of complexes, affects the photophysical and photochemical properties namely ROS generation, tumour cell selectivity, cellular-targeted delivery among others. We conclude by exploring future perspectives expected for this research line.
Exploring rhodamine modifications
With more than a century of advances, extensive structural modifications to rhodamines have significantly impacted their photophysical and photochemical properties (Fig. 2).27 These modifications have fine-tuned their absorption and emission properties from the green region of the visible spectrum (515 nm) to the NIR region (750 nm), improving vital optical properties such as fluorescence quantum yield (ΦF) and molar absorptivity (ε). This shift to the NIR range of the electromagnetic spectrum, improves tissue penetration bringing rhodamines a step closer to PDT applications. | ||
| Fig. 2 Structure of rhodamine and main modification strategies aiming to improve the photophysical and photochemical properties. | ||
From the high number of modifications performed on the basic rhodamine skeleton, several structural trends have been identified that provide insight into the photophysical properties of rhodamines. These trends include: (a) the alkylation of the amine groups at the 3rd and 6th positions of the xanthene ring, increasing rigidity through the fusion of the amine and xanthene ring,23 and (b) the introduction of heavy atoms via bromination or iodination, or by replacing the oxygen atom in the rhodamine backbone with other heteroatoms (e.g. Si, B, C, S, Ge, Sn, N, P and Te), thereby leading to improvements in the triplet excited state population.36–38 All these modifications allow a significant shift in light absorption towards the NIR range of the spectrum, with the latter modifications yielding the best results without a significant loss of solubility, fluorescence intensity, or fluorescence quantum yield (ΦF). This shift enhances their suitability for bioimaging and PDT applications due to reduced light scattering, deeper tissue penetration, and lower autofluorescence in the NIR region.27,36
While the introduction of bromine and iodine substituents effectively enhances ISC efficiency and promotes triplet state formation,39,40 it may also affect the lipophilic/hydrophilic character of cationic rhodamines, conditioning their selective phototoxicity toward tumour cells.41
Looking for heavy atom-free triplet photosensitizers with strong absorption of visible light and efficient ISC, alternative strategies have been introduced. One of the most interesting examples involves acid-responsive triplet PSs using rhodamine-C60 dyads.42 These dyads take advantage of the spirocyclic balance of rhodamines (Scheme 1), which can be activated in the presence of an acid (open form of rhodamine) and deactivated in the presence of a base (closed form). Thus, the rhodamine moiety acts as an acid-activated visible light-harvesting antenna, while the C60 moiety is the singlet energy acceptor and the spin converter, allowing the production of the triplet state that is enhanced in the presence of acid.42 Other strategies involve: (a) combining rhodamine B or rosamine with porphyrins, and also with Zn(II) phthalocyanines, to create conjugates with effective singlet oxygen production;43–45 and (b) combine rhodamine with stable radicals, such as nitroxide radical TEMPO, to improve the ISC via radical enhancement.46
 | ||
| Scheme 1 Acid controllable spirocyclic balance of rhodamines. | ||
Nevertheless, a novel strategy is being introduced to enhance photodynamic activity, based on the combination of rhodamines with transition-metal complexes including iridium(III), ruthenium(II) and platinum(II/IV). So far, rhodamines have proven to be excellent chelates, forming stable complexes with various metal ions.47 This property facilitates their easy complexation with metals, resulting in versatile rhodamine-metal complexes with relevant optical properties for sensing various analytes. Some of these complexes exhibit high selectivity for cations, and other biologically important species. Key examples include (1) a rhodamine-appended Fe(III) catecholate complex serving as a dual-modal MRI and optical imaging agent for detecting nitric oxide and acidic pH,48 (2) a rhodamine-Tb(III) complex for selective detection of hypochlorous acid in lysosomes of live cells,49 (3) a ytterbium porphyrinate complex combined with a rhodamine B derivative for detection of Hg(II) with responsive emission in the visible and near-IR region,50 and (4) a rhodamine-BODIPY/dansyl conjugate for intracellular monitoring of Hg(II) and Au(III) via differential fluorescence responses.51,52
Rhodamine-Ir(III)-complexes
Metal-based compounds can offer a wide range of biological and chemical diversity that is distinct from metal-free organic molecules, making them highly attractive as bioactive compounds for various therapeutic applications. Due to their unique properties, Ir(III) complexes stand out as a promising generation of theranostic agents, providing new avenues in medical diagnostics, precision imaging with single-cell resolution, and targeted anticancer therapy.53Table 1 summarizes contributions from several groups on rhodamine-Ir(III) complexes, highlighting key photophysical and photochemical parameters namely absorption (λabs) and emission (λem) wavelengths, molar absorption coefficient (ε), fluorescence quantum yield (ΦF), lifetime (τ) and singlet oxygen quantum yield (ΦΔ).
| Photosensitizer | λ abs (nm) | λ em (nm) | ε (M−1 cm−1) | Φ F (a.u) | τ (μs) | Φ Δ (%) | Targeting (Ref.) |
|---|---|---|---|---|---|---|---|

|
564 | 598 | 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
2.9 × 10−1 | n.d. | 0.3 | Mitochondria (54) |

|
575 | 629 | 87500 |
1.4 × 10−2 | 0.82 | 43.0 | Mitochondria (54) |

|
578 | 635 | 68100 |
7.0 × 10−4 | 9.73 | 72.6 | Endoplasmic reticulum (55) |

|
580 | 635 | 67500 |
7.0 × 10−4 | — | 70.0 | — (56) |
|
576 | 635 | 91050 |
3.0 × 10−4 | 36.3 | 71.8 | — (57) |
|
700 | 728 | 90000 |
1.3 × 10−1 | — | — | Mitochondria (58) |

|
713 | 744 | 40000 |
2.0 × 10−2 | — | 69.0 | Mitochondria (58) |
To combine the advantages of rhodamines with those of luminescent transition-metal complexes, Liu et al. developed a series of transition metal complexes using Re(I), Ir(III), Rh(III), and Pt(II), appended to bipyridine substituted rosamine bpyRos (Table 1).54 The results were particularly noteworthy for the IrRos-1 complex. Although a reduction of the emission intensities was observed, which is dependent on the metal coordinated and related with the IL-excited state of rhodamine, the IrRos-1 complex demonstrated high efficiency in producing 1O2 (evaluated through 1O2 emission at about 1270 nm upon excitation at 532 nm), indicating that IrRos-1 was the most effective producer of this cytotoxic species. The in vitro and in vivo biological evaluations in the human breast carcinoma cells (MCF-7) confirmed that all rhodamine metal complexes retained the mitochondria-targeting ability of rhodamine, with IrRos-1 standing out as the most photocytotoxicity derivative. These findings highlight that the balance between the positive charge and lipophilicity of these derivatives is likely responsible for their improved ability to target mitochondria, thereby confirming their potential for PDT, particularly for IrRos-1.
To further enhance performance in PDT, Zhou et al. introduced a strategic modification by utilizing an extended π-conjugated 2,3-diphenylquinoxaline cyclometalation ligand, which features a lower intraligand (IL) state, leading to the development of the IrRos-2 derivative (Table 1).55IrRos-2 exhibits an outstanding ability to generate 1O2 in both solution and intracellular media. This superior performance is attributed to the efficient sensitization of the triplet state in the rhodamine component, achieved either through triplet–triplet energy transfer (TTET) from the triplet state of the Ir(III) component or via ISC from the rhodamine singlet state (S1).
This process is energetically favourable due to the lower energy of the rhodamine triplet excited state (T1) compared to the iridium-based T1′ state (1.70 eV versus 1.89 eV). The T1 state lifetime in IrRos-2 is 10 times longer than that of IrRos-1 (9.73 μs versus 0.82 μs), which is aligned with its superior performance in generating 1O2. Additionally, IrRos-2 preferentially localizes in the endoplasmic reticulum (ER), a key organelle in eukaryotic cells involved in biosynthesis, sensing, and signalling. Studies confirmed that IrRos-2 accumulates at the tumour site and significantly reduces tumour growth.
Considering that cyclooxygenase-2 (COX-2) is typically expressed at low levels in normal cells but is significantly upregulated in several types of tumours, such as stomach, colorectal, and pancreatic cancers, a promising synthetic strategy was proposed to target this enzyme. This strategy involves incorporating an indomethacin moiety, known for its inhibitory activity towards COX-2.56 Following this idea, Liu et al. prepared IrRos-3 by conjugating IrRos-2 with indomethacin (Table 1).56 However, the studies revealed that the excitation wavelength of IrRos-3 is in the visible region, leading to poor tissue penetration. This limitation significantly hinders the potential of IrRos-3 for treating deep-seated tumours, thus restricting its therapeutic applications.
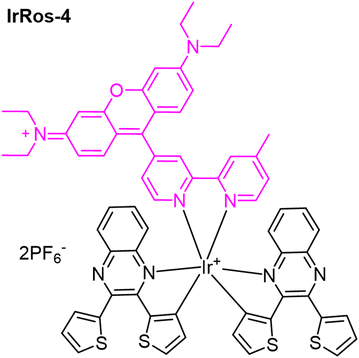
The possibility of modulating the type of ROS produced by hybrid systems based on Ir(III) complexes, which include bipyridine-substituted rhodamines bpyRos, was achieved by varying the type of ligand.57 In this study, ten ligands analogous to 2,3-diphenylquinoxaline were selected, and the results from the corresponding Ir(III) complexes showed that the efficiency and type of ROS produced are strongly dependent on the energy gap between the Ir(III)-based triplet metal-to-ligand charge transfer (3MLCT) state and the rhodamine singlet state (S1). A narrower energy gap between the 3MLCT and S1 states results in higher 1O2 generation efficiency. Additionally, the introduction of electron-donating moieties into the cyclometalating ligands can increase the generation of toxic radicals. In particular, IrRos-4 (Table 1), which features thiophene units in its cyclometalating ligand, demonstrated, within the studied series, the highest ROS generation efficiency.57 It effectively produced toxic radicals through a Type I PDT mechanism. The biological results for IrRos-4 were highly promising. This complex exhibited excellent biocompatibility and safety in vivo. Besides directly destroying cancer cells, IrRos-4 also activated M1 macrophages, even in hypoxic conditions, and enhanced T-cell infiltration into tumours. This immune activation contributed to a potent antitumour effect.
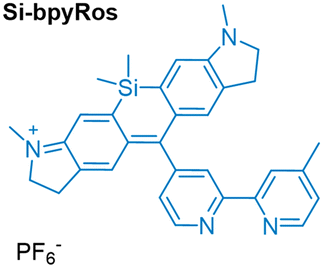
More recently, the complex IrRos-5, which feature iridium tethered to a silane-modified rhodamine (Table 1), was developed and found to be particularly responsive to laser stimulation at approximately 808 nm.58IrRos-5 demonstrates a high 1O2 quantum yield, with a value of 0.69. In vitro PDT results indicate that IrRos-5 exhibits low dark toxicity and excellent photocytotoxicity against 4T1 cells. Importantly, in vivo PDT studies for deep tumour therapy confirm the superiority of using a near-infrared (NIR) light source with IrRos-5 compared to IrRos-2. This approach shows great potential for developing PS capable of effectively utilizing NIR light for deep PDT applications.
A different approach was envisioned by Ma et al. to develop a series of metal complexes using rhodamine B hydrazide or rhodamine 6G hydrazide as the starting scaffolds.59 This approach resulted in the synthesis of a significant number of rhodamine-modified fluorescent half-sandwich iridium or ruthenium complexes.32,59,60 All the bichromophoric cyclometalated Ir(III) complexes demonstrated strong photostability and lysosome-targeting ability. Some of these complexes exhibited anticancer activity, outperforming cisplatin. These complexes have promising applications in bioimaging and as anticancer agents, expanding their potential beyond their use as PSs in PDT.
Additionally, half-sandwich Ir(III) and Ru(II) complexes have been widely explored for their dual functionality in imaging and therapy. Ir(III) complexes, particularly those featuring Cp* or biphenyl ligands, have demonstrated selective uptake in cancer cells, leading to enhanced lysosomal targeting and ROS generation, which contributes to apoptosis.32 Similarly, Ru(II) complexes, owing to their photophysical properties and redox activity, have shown great potential in PDT and bioimaging. Ru(II) complexes demonstrated a slightly greater capacity to induce apoptosis in A549 cells, aligning with their superior anticancer activity.61
Rhodamine-Ru(II) complexes
Ru(II) polypyridyl complexes dominate transition metal research due to their superior photophysical properties, namely extended excited state lifetime, 1O2 generation, visible light absorption, cellular uptake, and two-photon excitation capability.61,62 However, since the MLCT band of most polypyridyl ruthenium complexes is located in the blue region, their phototherapeutic applications are limited due to the low tissue penetration of blue light. To address this limitation, several research groups have explored conjugates of polypyridyl ruthenium complexes with rhodamine, aiming to enable photoactivation with longer-wavelength photons while maintaining the complexes’ stability in the dark.In this context, Bahreman et al. reported the synthesis of the complex [RuRho-1]3+ (Fig. 3), where a rhodamine B unit is covalently linked to the 4′ position of the [Ru(terpy)(bpy)(Hmte)]2+ (where terpy is 2,2′;6′,2′′-terpyridine, bpy is 2,2′-bipyridine, and Hmte is 2-(methylthio)ethanol).63 This derivative exhibits a slower exchange of the Hmte ligand with water when activated by yellow light compared to blue light. The formation of aqua photoproducts is particularly appealing for phototherapeutic applications, as these species may interact with biomolecules and exhibit higher cytotoxicity than the parent complex. The data indicate that while yellow photons lack sufficient energy to populate the 1MLCT state of [RuRho-1]3+, their absorption still induces Hmte photosubstitution with nearly the same quantum efficiency as blue photons. This photosubstitution occurs even faster under yellow light irradiation than under blue light for the parent complex [Ru(terpy)(bpy)(Hmte)]2+. The fluorescence quenching of the rhodamine B moiety in [RuRho-1]3+ and the high efficiency of Hmte photosubstitution suggest that energy transfer from the rhodamine B moiety to the ruthenium centre is highly efficient. The subsequent nonradiative decay likely occurs primarily from the 3MLCT state of the ruthenium moiety rather than from the S1 excited state of the rhodamine B moiety.
 | ||
| Fig. 3 Rhodamine Ru(II) complexes for cancer therapy, with schematic representation of the acid-controlled switching of the triplet state of [RuRho-2]2+. | ||
As research progresses, the development of new and improved triplet state PSs and their capability to generate ROS through triplet state photochemical reactions have become the main focus of this research area. Within this promising research subject, the creation of a new generation of PSs with controllable switching (ON–OFF) of the triplet state and thereby their photochemical reactions have taken a front seat on PDT research. This can be achieved by controlling the population of the triplet state.
Several strategies have been proven successful, including changes in PS microenvironment, like pH, as demonstrated by McDonnell et al.64 The authors demonstrated that the 1O2 photosensitizing capability of bromo-azaBODIPY can be switched ON or OFF. This approach has been applied to develop targeted PDT reagents that can be selectively activated by the acidic microenvironment of tumour tissue. Despite this, many other photo-switching mechanisms are still left to explore.
As previously mentioned, rhodamine is well-known for its ability to reversibly switch between two tautomeric forms: the spirolactam structure and the open amide structure (Scheme 1). These tautomers display significantly different photophysical properties allowing for switchable optical properties.65
Recognizing the importance of controlling the triplet excited state in the development of new functional molecules—particularly for PDT, where the activation of the PS can be mediated by external stimuli—prompted Cui et al. to develop the Ru(II) tris(bipyridine)–rhodamine triad [RuRho-2]2+ (Fig. 3).66 The rationale behind this design was the possibility of controlling PET by ring-opening the rhodamine spirolactam structure under acidic conditions. In the absence of acid, a photoinduced electron transfer (PET) process (with the rhodamine moiety acting as the electron donor) was responsible for quenching the triplet metal-to-ligand charge-transfer (3MLCT) excited state (much shorter lifetime than of reference Ru complex, which is devoid of the rhodamine moiety: τT = 103.6 ns vs. τT = 1.58 μs). In the presence of acid, PET is inhibited by the ring-opening of the rhodamine spirolactam structure, leading to an increase in the triplet-state lifetime (5.70 μs) and the relocalization of the T1 state in the rhodamine unit. The authors highlighted that this was the first time the triplet localization and lifetime of an excited Ru(II) complex have been controlled simultaneously.66 The results confirmed the occurrence of intramolecular singlet–triplet energy transfer (STEnT), with the rhodamine unit acting as the singlet energy donor and the Ru 3MLCT state as the energy acceptor. Additionally, the results confirmed an exceptionally low triplet–triplet energy transfer (TTET) from the Ru(II) coordination centre (3MLCT) to the rhodamine unit (3IL), with the rhodamine unit acting as the energy acceptor. In the presence of adequate triplet energy acceptors like 9,10-diphenylanthracene and perylene, the conjugate was successfully used for acid-controllable triplet–triplet annihilation (TTA) upconversion processes.
Rhodamine-Pt(II/IV) complexes
Combining rhodamines with Pt(II) and Pt(IV) scaffolds has become a hot topic in cancer research, ranging from its use as intracellular spatial and temporal therapeutic monitoring tools67,68 to the development of novel selective69 treatments based on new prodrugs and PDT photosensitisers. Even after 60 years since its discovery, cisplatin remains one of the most important chemotherapy drugs currently in clinical use, with many new Pt-based treatments still being inspired by it today. Since then, multiple generations of Pt(II)-based complexes have been discovered, many of which are FDA-approved. Despite being one of the most widely used chemotherapeutic agents, its usability is highly limited by its non-specific mode of action, high toxicity of Pt(II), severe side effects and frequent occurrence of drug resistance.67–69Since then, some strategies have been explored to mitigate its drawbacks, such as the development of stable Pt(IV) complexes, as anticancer prodrugs. These depend on intracellular activation through the loss of their axial ligands and consequent reduction of the kinetically inert Pt(IV) [due to their low-spin d6 electronic configuration and saturated coordination sphere] into its highly cytotoxic Pt(II) counterpart.67,68,70,71 Such activation can be obtained by a reduction process mediated by internal (ascorbate and glutathione)72–75 or external (riboflavin)71 reductants, with the latter being the more common approach. However, the use of an external catalyst has inherent limitations, as most photocatalysts do not always efficiently co-localize with Pt(IV) prodrugs in cancer cells, which restricts their photocatalytic efficiency. Additionally, the intracellular complexity poses a challenge to Pt(IV) prodrugs, as some may be undesirably activated by internal reductants on non-pathogenic sites.
To solve the aforementioned limitations, Deng et al. have proposed a novel class of photoactivatable Pt(IV) prodrugs (PtRho-1 and PtRho-2) called rhodaplatins (Fig. 4).71 These rely on the oxidation potential of photoexcited Rhodamine B (Rho B+/Rho B*: ∼1.3 V) as an adequate photocatalyst to reduce most conventional Pt(IV) prodrugs. By covalently incorporating an internal rhodamine-based photoswitch, the co-localisation and efficient photoactivation of the drug occur, ensuring a significant improvement on the performance against cancer cells. The authors found the rhodaplatins under study to exhibit up to 4.8 × 104-fold improvement on the conversion of Pt(IV) to Pt(II) under white light irradiation (400–760 nm, 4 mW cm−2) when compared to the use of non-covalently linked reducing agents, like free riboflavin and free Rho B.71
 | ||
| Fig. 4 Rhodamine-Pt(II/IV) complexes for cancer therapy. | ||
Apart from the improved photoconversion of rhodaplatins, the introduction of the covalently linked rhodamine moiety leads to further advantages such as mitochondria targeting, which is an unconventional target of Pt-based complexes thereby solving some of the more common resistance problems, namely nucleotide excision repair (NER) and histone protection, the two main factors responsible for the resistance of cancer cells towards Pt drugs.71
As before discussed, by relaying on the heavy-atom effect, or more broadly spin–orbit coupling, these complexes can populate the triplet excited state of a chromophore via the S0 → S1 → T1 process. Thanks to the lack of light-harvesting moieties, most common Pt(II) complexes lack PDT applications. However, the combination of a light-harvesting molecule, such as rhodamines, with long lived T1 excited states typical of Pt(II) complexes can drastically improve the generation of ROS.76
As shown by Huang et al.,76 the incorporation of a rosamine into a Pt(II) acetylide complex (Fig. 4, PtRos-3), resulted in intense UV/Vis absorption at 556 nm but also in an extensively prolonged triplet excited state lifetime (τT = 83.0 μs). This study presents, for the first time, the successfully employment of a Ros-Pt complex as a sensitizer for triplet–triplet annihilation (TTA) upconversion, with an upconversion quantum yield of 11.2%. This means that PtRos-3 can leverage TTA upconversion to efficiently convert low-energy photons into higher-energy ones, enabling activation with NIR-to-Visible light. This allows for effective light utilization even in deep tissues where lower-energy light penetrates with higher efficiency, thereby overcoming the limitation of traditional PDT, which is typically restricted to treating surface tumours or those located on organ linings.77,78 Additionally, its long-lived triplet excited state enhances energy transfer to molecular oxygen, generating 1O2, a key cytotoxic ROS responsible for inducing cell damage and apoptosis in PDT.
As stated before, the implementation of a switchable triplet excited state has become of major importance for this area of research, as a consequence, Majumdar et al. moved away from the rosamines that lack the ability to undergo spirocyclic equilibrium in favour of the rhodamine variant (Fig. 4).70 As a consequence, they prepared two new Pt(II) complexes containing both rhodamine and a BODIPY as ligands (PtRho-4 and PtRho-5) with different coordination profiles.
The main findings revealed that the two complexes exhibited long-lived triplet excited states (35 μs and 423 μs in dichloromethane). Upon the addition of trifluoroacetic acid (TFA), the triplet state lifetime of PtRho-4 increased significantly from 35 μs to 80 μs, with the triplet state localized on the BODIPY moiety. For PtRho-5, the addition of acid induced a switch in the triplet state from being confined to the BODIPY moiety to a delocalized triplet state spanning both the BODIPY and rhodamine moieties. This demonstrated that both the singlet and triplet excited states of these Pt(II) complexes could be modulated by an external stimulus.70
Their results highlight the potential of rhodamine-containing Pt(II) complexes for designing external stimuli-responsive transition metal complexes. The use of Pt(II) compounds is particularly significant in medicinal chemistry, as Pt(II)-based drugs, have proven critical in cancer therapy. These findings suggest that tailoring the excited state properties of Pt(II) complexes through external stimuli could enhance their functionality in biological applications, such as PDT or bioimaging.
Conclusions and future prospects
Rhodamine-metal complexes represent a transformative step forward in theranostics and photodynamic therapy (PDT), offering significant improvements in diagnostic and therapeutic capabilities of cancer tissues. Through strategic modifications of the rhodamine scaffold, such as introducing heavy atoms into the rhodamine core [like bromine (Br) or iodine(I), or replacing the oxygen atom with sulfur (S), selenium (Se), or tellurium (Te)], and its conjugation with transition metals such as Ir(III), Ru(II), and Pt(II/IV), researchers have effectively addressed key challenges traditionally associated with PDT. These include enhancing 1O2 generation, achieving deeper tissue penetration through near-infrared (NIR) light absorption, higher hydrophilicity and improved tumour selectivity through organelle-targeted delivery mechanisms.Among the highlights of this review, rhodamine-Ir(III) complexes have demonstrated exceptional photophysical properties improving ROS generation and targeting capabilities. Rhodamine-Ru(II) complexes have introduced new possibilities for switchable triplet-state photosensitizers, leveraging reversible tautomeric transitions to enable external control over photophysical properties. In fact, although widely explored for the development of OFF–ON sensing platforms, the reversibly interchangeable tautomeric forms of rhodamine, remain largely underdeveloped particularly in terms of switching of the triplet excited state, highlighting the need for further research in this area. Rhodamine-Pt(II/IV) complexes have addressed limitations in chemotherapy, such as co-localisation and intracellular reduction of the kinetically inert Pt(IV) into the cytotoxic Pt(II). However, the use of platinum-rhodamine compounds has been predominantly tied to chemotherapy, with very few examples linked to, less intrusive, novel treatment strategies like PDT. The effective photoactivation of the prodrugs under physiological conditions, could be crucial for overcoming challenges related to drug resistance.
Despite these advancements, significant challenges persist, with the future of rhodamine-metal complexes in PDT being dependent on addressing several critical areas. To start, the synthesis of these PDT complexes often involves intricate, multi-step and labour-intensive processes with low yields. Furthermore, limited light penetration in deep tissues of most developed rhodamine-metal complexes, and fluorescence quenching due to metal incorporation, remain a concern in terms of imaging. This highlights the need to extend light absorption into the NIR region (Fig. 5) and to balance, the diagnostic and treatment aspect of these new theranostic complexes by modulating the triplet excited state. The development of stimuli-responsive systems that activate selectively in the tumour microenvironment offers immense potential. Recent work on switchable triplet-state photosensitizers highlights the potential of acid-activated or redox-responsive designs. Further exploration into external stimuli could lead to more precise activation mechanisms.
 | ||
| Fig. 5 Spectral coverage of the emission of some rhodamine derivates and related metal complexes [Ir(III), Ru(II) and Pt(II)]. | ||
Acknowledging the original study by Rivas et al.,79 two Gd(III) and Tb(III) complexes bearing DO3A-rhodamine ligands have been developed as dual-modal imaging agents, effectively combining MRI and optical imaging capabilities. The design of these ligands takes advantage of the pH-sensitive nature of the rhodamine fragment, which enhances fluorescence in the acidic environments typical of tumour microenvironments. Future directions for imaging agents may involve further exploration of similar rhodamine-Gd(III) complexes,80 and other lanthanide(III) complexes, that offer improved specificity and sensitivity for clinical applications.
While several complexes demonstrate promising in vitro and in vivo results, there still is a lack of comprehensive clinical data validating their safety, efficacy and thereby a thorough assessment is still needed. Additionally, challenges such as hypoxic tumour environments pose severe limitations in oxygen deficient sites. Future research should focus on designing oxygen-independent phototherapies or integrating oxygen-carrying nanocarriers to enhance the efficacy of PDT in hypoxic conditions.
In conclusion, while the field of therapeutic applications has and continues to make remarkable advancements, realizing the full potential of rhodamine-metal complexes requires overcoming substantial challenges. Collaborative efforts across disciplines, including chemistry, biology, and materials science, will be essential to drive innovation and ensure the successful transition of these promising molecules from the laboratory to clinical practice. However, we remain optimistic on the promising role that rhodamine complexes may play in the future in the development of new and improved theranostic approaches for combating cancer.
Author contributions
All authors contributed equally to the writing, reviewing, and editing of the manuscript and have approved its final version.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received financial support from FCT/MCTES (UIDP/50006/2020 https://doi.org/10.54499/UIDP/50006/2020) through national funds. This work received financial support from the PT national funds (FCT/MECI, Fundação para a Ciência e Tecnologia and Ministério da Educação, Ciência e Inovação) through the project UID/50006 -Laboratório Associado para a Química Verde - Tecnologias e Processos Limpos. Fábio Martins acknowledges FCT (and ESF) for PhD grant UI/BD/154485/2022. The authors also thank the University of Aveiro.References
- Global cancer burden growing, amidst mounting need for services, https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing–amidst-mounting-need-for-services.
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, CA Cancer J. Clin., 2024, 74, 229–263 CrossRef PubMed.
- American Cancer Society, Global Cancer Facts & Figures, 5th edn, 2024 Search PubMed.
- GBD 2021 Forecasting Collaborators, Lancet, 2024, 403, 2204–2256 CrossRef PubMed.
- D. Sun, X. Feng, X. Zhu, Y. Wang and J. Yang, Coord. Chem. Rev., 2024, 500, 215546 CrossRef CAS.
- M. S. Alshuhri, S. G. Al-Musawi, A. A. Al-Alwany, H. Uinarni, I. Rasulova, P. Rodrigues, A. T. Alkhafaji, A. M. Alshanberi, A. H. Alawadi and A. H. Abbas, Pathol., Res. Pract., 2024, 253, 154996 CrossRef PubMed.
- H. E. C. da Silva, G. N. M. Santos, A. F. Leite, C. R. M. Mesquita, P. T. de S. Figueiredo, C. M. Stefani and N. S. de Melo, PLoS One, 2023, 18, e0292063 CrossRef PubMed.
- L. Cuicui and J. Kang, Curr. Med. Chem., 2024, 31, 1839–1873 CrossRef PubMed.
- D. Karati, S. Meur, S. Mukherjee and S. Roy, Coord. Chem. Rev., 2024, 519, 216118 CrossRef CAS.
- Z. Ye, Y. Bao, Z. Chen, H. Ye, Z. Feng, Y. Li, Y. Zeng, Z. Pan, D. Ouyang, K. Zhang, X. Liu and Y. He, Coord. Chem. Rev., 2024, 504, 215654 CrossRef CAS.
- T. Pan and D. Luo, Phys. Imaging Radiat. Oncol., 2024, 31, 100601 CrossRef PubMed.
- S. Khan and S. Vasudevan, Rev. Sci. Instrum., 2023, 94, 91502 CrossRef CAS PubMed.
- E. Atlihan-Gundogdu, D. Ilem-Ozdemir, M. Ekinci, E. Ozgenc, E. S. Demir, B. Sánchez-Dengra and I. González-Alvárez, J. Pharm. Invest., 2020, 50, 349–361 CrossRef.
- A. Master, M. Livingston and A. Sen Gupta, J. Controlled Release, 2013, 168, 88–102 CrossRef CAS PubMed.
- D. Aebisher, J. Szpara and D. Bartusik-Aebisher, Int. J. Mol. Sci., 2024, 25, 8258 CrossRef CAS PubMed.
- S. Kwiatkowski, B. Knap, D. Przystupski, J. Saczko, E. Kędzierska, K. Knap-Czop, J. Kotlińska, O. Michel, K. Kotowski and J. Kulbacka, Biomed. Pharmacother., 2018, 106, 1098–1107 CrossRef PubMed.
- H. Ali and J. E. van Lier, Chem. Rev., 1999, 99, 2379–2450 CrossRef CAS PubMed.
- S. Kwiatkowski, B. Knap, D. Przystupski, J. Saczko, E. Kędzierska, K. Knap-Czop, J. Kotlińska, O. Michel, K. Kotowski and J. Kulbacka, Biomed. Pharmacother., 2018, 106, 1098–1107 CrossRef PubMed.
- R. Bonnett, in Metal Complexes for Photodynamic Therapy, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. B. T.-C. C. C. I. I. MeyerPergamon, Oxford, 2003, pp. 945–1003 Search PubMed.
- M. Przygoda, D. Bartusik-Aebisher, K. Dynarowicz, G. Cieślar, A. Kawczyk-Krupka and D. Aebisher, Int. J. Mol. Sci., 2023, 24, 16890 CrossRef CAS PubMed.
- P. S. Maharjan and H. K. Bhattarai, J. Oncol., 2022, 2022, 7211485 Search PubMed.
- C. Donohoe, M. O. Senge, L. G. Arnaut and L. C. Gomes-da-Silva, Biochim. Biophys. Acta, Rev. Cancer, 2019, 1872, 188308 CrossRef CAS PubMed.
- E. Pang, S. Zhao, B. Wang, G. Niu, X. Song and M. Lan, Coord. Chem. Rev., 2022, 472, 214780 CrossRef CAS.
- X. Xue, A. Lindstrom and Y. Li, Bioconjugate Chem., 2019, 30, 1585–1603 Search PubMed.
- A. Master, M. Livingston and A. Sen Gupta, J. Controlled Release, 2013, 168, 88–102 CrossRef CAS PubMed.
- N. Fakudze, H. Abrahamse and B. George, Lasers Med. Sci., 2025, 40, 78 CrossRef PubMed.
- L. D. Lavis, Annu. Rev. Biochem., 2017, 86, 825–843 Search PubMed.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed.
- K. S. Davies, M. K. Linder, M. W. Kryman and M. R. Detty, Bioorg. Med. Chem., 2016, 24, 3908–3917 CrossRef CAS PubMed.
- C. Liu, L. Zhou, F. Wei, L. Li, S. Zhao, P. Gong, L. Cai and K. M.-C. Wong, ACS Appl. Mater. Interfaces, 2019, 11, 8797–8806 CrossRef CAS PubMed.
- L. Zhou, F. Wei, J. Xiang, H. Li, C. Li, P. Zhang, C. Liu, P. Gong, L. Cai and K. M.-C. Wong, Chem. Sci., 2020, 11, 12212–12220 RSC.
- W. Ma, X. Ge, Z. Xu, S. Zhang, X. He, J. Li, X. Xia, X. Chen and Z. Liu, ACS Omega, 2019, 4, 15240–15248 CrossRef CAS PubMed.
- Z. Kejík, J. Hajduch, N. Abramenko, F. Vellieux, K. Veselá, J. L. Fialová, K. Petrláková, K. Kučnirová, R. Kaplánek, A. Tatar, M. Skaličková, M. Masařík, P. Babula, P. Dytrych, D. Hoskovec, P. Martásek and M. Jakubek, Commun. Chem., 2024, 7, 180 CrossRef PubMed.
- H. Zhang, G. Ren, W. Hou, L. Wang, Y. Sun and J. Liu, Spectrochim. Acta, Part A, 2024, 308, 123688 CrossRef CAS PubMed.
- O. Karaman, G. A. Alkan, C. Kizilenis, C. C. Akgul and G. Gunbas, Coord. Chem. Rev., 2023, 475, 214841 CrossRef CAS.
- F. Deng and Z. Xu, Chin. Chem. Lett., 2019, 30, 1667–1681 CrossRef CAS.
- M. Beija, C. A. M. M. Afonso and J. M. G. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- L. Wang, W. Du, Z. Hu, K. Uvdal, L. Li and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 14026–14043 CrossRef CAS PubMed.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC.
- M. Yoshinaga and W. R. Rocha, J. Phys. Chem. B, 2021, 125, 8932–8943 CrossRef CAS PubMed.
- S. H. D. Lacerda, B. Abraham, T. C. Stringfellow and G. L. Indig, Photochem. Photobiol., 2005, 81, 1430–1438 CrossRef CAS PubMed.
- F. Wang, X. Cui, Z. Lou, J. Zhao, M. Bao and X. Li, Chem. Commun., 2014, 50, 15627–15630 RSC.
- E. J. Ngen, L. Xiao, P. Rajaputra, X. Yan and Y. You, Photochem. Photobiol., 2013, 89, 841–848 CrossRef CAS PubMed.
- C. Queirós, A. Leite, N. M. M. Moura, A. F. R. Cerqueira, V. V. Serra, M. G. P. M. S. Neves, A. C. Tomé and A. M. G. Silva, Dyes Pigm., 2023, 217, 111431 CrossRef.
- D. K. Muli, P. Rajaputra, Y. You and D. V. McGrath, Bioorg. Med. Chem. Lett., 2014, 24, 4496–4500 CrossRef CAS PubMed.
- W. Zhu, Y. Wu, Y. Zhang, A. A. Sukhanov, Y. Chu, X. Zhang, J. Zhao and V. K. Voronkova, Int. J. Mol. Sci., 2023, 24, 11220 CrossRef CAS PubMed.
- Q. Zhang and K. M.-C. Wong, Coord. Chem. Rev., 2020, 416, 213336 CrossRef CAS.
- D. Maheshwaran, T. Nagendraraj, T. Sekar Balaji, G. Kumaresan, S. Senthil Kumaran and R. Mayilmurugan, Dalton Trans., 2020, 49, 14680–14689 RSC.
- L. Tian, H. Ma, B. Song, Z. Dai, X. Zheng, R. Zhang, K. Chen and J. Yuan, Talanta, 2020, 212, 120760 CrossRef CAS PubMed.
- T. Zhang, C. Chan, R. Lan, W. Wong and K. Wong, Chem. – Eur. J., 2014, 20, 970–973 CrossRef CAS PubMed.
- P. Majumdar, X. Cui, K. Xu and J. Zhao, Dalton Trans., 2015, 44, 4032–4045 RSC.
- F. Martins, A. Granja, S. Reis, P. Gameiro, G. Barone, M. G. P. M. S. Neves and A. M. G. Silva, Spectrochim. Acta, Part A, 2025, 329, 125534 CrossRef CAS PubMed.
- P. Szymaszek, M. Tyszka-Czochara and J. Ortyl, Eur. J. Med. Chem., 2024, 276, 116648 CrossRef CAS PubMed.
- C. Liu, L. Zhou, F. Wei, L. Li, S. Zhao, P. Gong, L. Cai and K. M.-C. Wong, ACS Appl. Mater. Interfaces, 2019, 11, 8797–8806 CrossRef CAS PubMed.
- L. Zhou, F. Wei, J. Xiang, H. Li, C. Li, P. Zhang, C. Liu, P. Gong, L. Cai and K. M.-C. Wong, Chem. Sci., 2020, 11, 12212–12220 RSC.
- C. Liu, J. Xiang, C. Xiang and H. Li, Bioorg. Chem., 2021, 114, 105142 CrossRef CAS PubMed.
- F. Wei, F. Chen, S. Wu, M. Zha, J. Liu, K.-L. Wong, K. Li and K. M.-C. Wong, Inorg. Chem., 2024, 63, 5872–5884 CrossRef CAS PubMed.
- J. Liu, X. Yang, S. Wu, P. Gong, F. Pan, P. Zhang, C.-S. Lee, C. Liu and K. M.-C. Wong, J. Mater. Chem. B, 2024, 12, 3710–3718 Search PubMed.
- W. Ma, X. Ge, L. Guo, S. Zhang, J. Li, X. He and Z. Liu, Dyes Pigm., 2019, 162, 385–393 CrossRef CAS.
- W. Ma, L. Guo, Z. Tian, S. Zhang, X. He, J. Li, Y. Yang and Z. Liu, Dalton Trans., 2019, 48, 4788–4793 RSC.
- X. Y. Ng, K. W. Fong, L. V. Kiew, P. Y. Chung, Y. K. Liew, N. Delsuc, M. Zulkefeli and M. L. Low, J. Inorg. Biochem., 2024, 250, 112425 CrossRef CAS PubMed.
- J. Mo, N. P. Mai Le and R. Priefer, Eur. J. Med. Chem., 2021, 225, 113770 CrossRef CAS PubMed.
- A. Bahreman, J.-A. Cuello-Garibo and S. Bonnet, Dalton Trans., 2014, 43, 4494–4505 RSC.
- S. O. McDonnell, M. J. Hall, L. T. Allen, A. Byrne, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2005, 127, 16360–16361 CrossRef CAS PubMed.
- N. Lardon, L. Wang, A. Tschanz, P. Hoess, M. Tran, E. D'Este, J. Ries and K. Johnsson, J. Am. Chem. Soc., 2021, 143, 14592–14600 CrossRef CAS PubMed.
- X. Cui, J. Zhao, A. Karatay, H. G. Yaglioglu, M. Hayvali and B. Küçüköz, Eur. J. Inorg. Chem., 2016, 2016, 5079–5088 CrossRef CAS.
- D. Montagner, S. Q. Yap and W. H. Ang, Angew. Chem., Int. Ed., 2013, 52, 11785–11789 CrossRef CAS PubMed.
- J. X. Ong, J. Y. Yap, S. Q. Yap and W. H. Ang, J. Inorg. Biochem., 2015, 153, 272–278 CrossRef CAS PubMed.
- R. Mehder, E. de la Torre-Rubio, I. de la Cueva-Alique, C. O'Malley, A. Pérez-Redondo, L. Gude, E. Royo and L. Ronconi, Inorganics, 2024, 12, 91 CrossRef CAS.
- P. Majumdar, X. Cui, K. Xu and J. Zhao, Dalton Trans., 2015, 44, 4032–4045 RSC.
- Z. Deng, C. Li, S. Chen, Q. Zhou, Z. Xu, Z. Wang, H. Yao, H. Hirao and G. Zhu, Chem. Sci., 2021, 12, 6536–6542 RSC.
- V. Pichler, S. Göschl, E. Schreiber-Brynzak, M. A. Jakupec, M. S. Galanski and B. K. Keppler, Metallomics, 2015, 7, 1078–1090 CrossRef CAS PubMed.
- W. Zhong, Q. Zhang, Y. Yan, S. Yue, B. Zhang and W. Tang, J. Inorg. Biochem., 1997, 66, 159–164 CrossRef CAS PubMed.
- J. Carr, M. Tingle and M. McKeage, Cancer Chemother. Pharmacol., 2002, 50, 9–15 CrossRef CAS PubMed.
- V. Reshetnikov, S. Daum and A. Mokhir, Chem. – Eur. J., 2017, 23, 5678–5681 CrossRef CAS PubMed.
- L. Huang, L. Zeng, H. Guo, W. Wu, W. Wu, S. Ji and J. Zhao, Eur. J. Inorg. Chem., 2011, 2011, 4527–4533 CrossRef CAS.
- Y. Li and G. Chen, Adv. NanoBiomed Res., 2022, 2, 2200092 CrossRef CAS.
- H. Zhou, X. Zeng, A. Li, W. Zhou, L. Tang, W. Hu, Q. Fan, X. Meng, H. Deng, L. Duan, Y. Li, Z. Deng, X. Hong and Y. Xiao, Nat. Commun., 2020, 11, 6183 CrossRef CAS PubMed.
- C. Rivas, G. J. Stasiuk, J. Gallo, F. Minuzzi, G. A. Rutter and N. J. Long, Inorg. Chem., 2013, 52, 14284–14293 CrossRef CAS PubMed.
- A. G. Robertson, A. J. Hall, A. Marfavi and L. M. Rendina, Chem. – Eur. J., 2024, 30, e202402244 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |