
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA picolinamide iridium catalyst immobilized on an aluminum-hydride anchor for the selective dehydrogenation of neat formic acid†
Lujain
Alrais
a,
Indranil
Dutta
 a,
Amol
Hengne
b,
Priyanka
Chakraborty
a,
Edy
Abou-Hamad
c,
Shibo
Xi
b,
Mohammad Misbahur
Rahman
a,
Jia
Zhang
d,
Benjamin W. J.
Chen
*d,
Jean-Marie
Basset
*ae and
Kuo-Wei
Huang
*ab
a,
Amol
Hengne
b,
Priyanka
Chakraborty
a,
Edy
Abou-Hamad
c,
Shibo
Xi
b,
Mohammad Misbahur
Rahman
a,
Jia
Zhang
d,
Benjamin W. J.
Chen
*d,
Jean-Marie
Basset
*ae and
Kuo-Wei
Huang
*ab
aCenter for Renewable Energy and Storage Technologies (CREST) and Division of Physical Science and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia. E-mail: jeanmarie.basset@kaust.edu.sa; hkw@kaust.edu.sa
bInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), Singapore 138634, Singapore
cCore Labs, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
dInstitute of High-Performance Computing (IHPC), Agency for Science, Technology, and Research (A*STAR), Fusionopolis Way, #16–16 Connexis, Singapore 138632, Singapore. E-mail: benjamin_chen@ihpc.a-star.edu.sg
eEcole National Supérieure de Chimie de Paris (ENSCP), 75231 Cedex 05 Paris, France
First published on 14th February 2025
Abstract
The dehydrogenation of formic acid can provide an efficient pathway for hydrogen generation in the presence of a suitable catalyst. Homogeneous catalysts have been extensively studied and utilized for highly active and selective processes compared to conventional heterogeneous catalysis, which often shows lower reactivity and selectivity. However, the latter is preferred for practical applications, considering its easy separation and recyclability. By incorporating a homogeneous organometallic complex on an appropriate support, the unique features of both catalysts can be combined and utilized effectively. Herein, we investigate the immobilization of an iridium picolinamide complex (1) supported on 3D fibrous modified silica that demonstrates high accessibility. The support involves a tetracoordinate aluminum hydride site featuring a strong Lewis acidic nature. A study of the interaction and coordination sites around the surface fragment was conducted via various techniques, including elemental analysis, FT-IR, solid-state NMR, XAS, and first-principles calculations, which provided informative data. We explored the use of solid additives in a solvent-free reaction medium and avoided utilizing volatile bases to achieve process feasibility with a high TOF of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1.
000 h−1.
Introduction
Formic acid (FA) dehydrogenation presents a promising approach for on-demand hydrogen production,1–5 with low lifecycle fugitive hydrogen losses.6,7 The reaction involves the use of suitable catalysts to selectively decompose FA into carbon dioxide (CO2) and hydrogen (H2), of which the latter can be utilized for clean energy production.8–11 Organometallic complexes such as ruthenium12–17 and iridium18–24 based catalysts have been extensively studied as homogeneous catalysts for effective and efficient FA dehydrogenation.25 In contrast, heterogeneous catalysis has demonstrated limited activity and selectivity.4 However, most industrially applied processes operate using a system based on heterogeneous catalysis, which is well known to offer advantages such as ease of separation, potential recyclability, and stability. Research continues to improve existing techniques and develop new catalytic approaches for more efficient and cost-effective operation. The goal is to synthesize catalysts that exhibit high activity and demonstrate good stability, recyclability, and selectivity toward hydrogen production.26 Immobilization of organometallic complexes on a suitable support could provide a powerful strategy. Elements of organometallic chemistry and surface science are combined to rationally design catalysts.27,28 Immobilized catalysts have shown promising results in selective FA dehydrogenation.29–34 However, reported immobilized catalysts mostly include an inert support, commonly silica, which demonstrates insignificant electronic impacts. In immobilized catalysis, the surface is considered an ancillary ligand that affects the supported complex. This has driven interest in designing isolated, molecularly defined active sites in which the support has a positive electronic influence on the catalytic system.35Surface organometallic chemistry (SOMC) can provide a practical strategy for surface preparation and processing.36–39 SOMC is concerned with the investigation into the structure, reactivity, and properties of the surface fragments. This approach allows us to understand the reactions of organometallic complexes with the surface of a support to prepare single-site catalysts on well-defined surfaces with several advantages such as improved stability and resistance to aggregation.
In this regard, the utilization of tetracoordinate aluminum hydride [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O–Si)(Si–O–)2Al–H] [Al-H]40 as an anchor site for organometallic complexes has shown interesting results, and its Lewis acidic nature can alter complex performance.41,42
Si–O–Si)(Si–O–)2Al–H] [Al-H]40 as an anchor site for organometallic complexes has shown interesting results, and its Lewis acidic nature can alter complex performance.41,42
Nowadays, researchers are advancing toward greener and more cost-effective methods for FA dehydrogenation by focusing on additive-free and solvent-free procedures43,44 because using solvents often leads to waste generation and can pose environmental hazards. Conducting the reaction under neat conditions and eliminating the solvents from the reaction medium would lead to a straightforward process with fewer purification steps and further reduce the operational cost. However, ensuring effective catalyst-to-substrate interaction without solvents, especially when using heterogeneous catalysts, can be challenging.
More importantly, reports on the neat FA dehydrogenation by immobilized catalysis are scarce. Our previous investigation has provided insight into the positive influence of the [Al-H] surface species, in which the immobilized iridium PN3P-pincer complex has shown superior activity and stability compared to its homogeneous analog.45 In the quest for an alternative catalytic system, we evaluated the amine linkage (NH) effect by employing a picolinamide iridium complex 1 supported on [Al-H]. We investigated the impact of changes at the coordination sites around the surface fragment and the effect on reactivity. The FT-IR, solid-state NMR, XAS, and first-principles density functional theory (DFT) calculations provide informative data to characterize the obtained immobilized complex 2. The immobilized catalyst has been tested under solvent-free/neat conditions, showing unprecedented reactivity in FA dehydrogenation with a high TOF of 40000 h−1.
Results and discussion
Complex 1 was characterized by 1H and 13C NMR analysis (Fig. S1 and S2†), single crystal characterization (Fig. 1), and IR analysis (Fig. S3†), confirming the structure depicted in Scheme S1.† The 1H NMR spectra collected in CDCl3 at room temperature showed the characteristic peaks of methyl protons at δ 1.61 ppm, and aromatic protons were observed in the region of δ 6.78–7.06 ppm. The Ir complex crystallizes in the monoclinic P21/n space group. It was found during refinement that there are two different crystallographic units per unit cell in the structure. They are assigned as Uni-I and Uni-II. Only Uni-II has been shown in the manuscript (Fig. 1); however, both units are shown in ESI Fig. S4 and S5†. In both units, it was found that the Ir center is coordinated to a Cp* ligand and one pyridine N atom (N1 or N5) from the substituted pyridine ring. The other two coordination sites come from one imine N atom (N3 or N7) and chloride atoms. The presence of the anion outside confirms the +3 oxidation state of the Ir center. The FT-IR spectrum of complex 1, shown in Fig. S3,† displayed a set of peaks at 3150–2830 cm−1 and 1350–1235 cm−1, which belonged to the stretching and bending vibrations of C–H, respectively. The broadband at 3530 cm−1 was attributed to the bending vibration of N–H. These observations constitute the framework structure of complex 1. | ||
| Fig. 1 Crystal structure of unit-II of Ir complex 1′. Selected bond lengths (Å) and angles (°): C8–O1 1.272(12); C8–N3 1.340(12); C9–N3 1.446(11); C9–C10 1.518(13); C10–N4 1.492(13); Cl1–Ir2 2.428(2); Ir2–N1 2.051(8); Ir2–N3 2.084(8); N1–Ir2–Cl1 85.7(3); N1–Ir2–N3 76.5(3); N3–Ir2–Cl1 84.6(2). Several hydrogen atoms and solvent molecule(s) have been omitted for clarity. | ||
Immobilized catalyst 2 was synthesized (details in the ESI†) and the obtained pale-yellow material was stored in a glovebox for further characterization (Scheme 1). Throughout the gas analysis, it was found that H2 was released during the immobilization reaction (Fig. S7†). Elemental analysis of 2 suggested an iridium loading of 4.1 wt% corresponding to 0.2 mmol g−1. The carbon and nitrogen values of C/Ir and N/Ir were found to be 22.4 and 3.9, respectively, in close agreement with the theoretical values of 20 and 4 (Table 1).
| Element | wt% | mmol g−1 | M/Ir |
|---|---|---|---|
| a Carbon value after subtracting the residual support carbon. | |||
| Ir | 4.1 | 0.2 | |
| Al | 3.7 | 1.3 | 6.5 |
| C | 5.3 | 4.5 | 22.5 |
| N | 1.1 | 0.78 | 3.9 |
 | ||
| Scheme 1 Synthesis of complex 2. | ||
The FT-IR spectra of the immobilized catalyst 2 and the support [Al-H] were recorded (Fig. 2). The FT-IR spectrum of the support (a) showed a small peak at 3743 cm−1, which was attributed to the remaining silanol groups Si–OH. Signals of Si–H and Si–H2 were observed at 2252–2170 cm−1, which remained intact after the immobilization. The Al-hydride signal was detected at 1943 cm−1, confirming the successful preparation of the support. The spectra of complex 2 (b) indicated a reduction in the Al-H signal, and the emergence of all characteristic peaks corresponded to the successful immobilization of the organometallic complex on the support. Furthermore, the set of bands in the range of 3060–2800 cm−1 and 1490–1360 cm−1 were ascribed to the stretching and bending vibrations of the C–H bond of the alkyl and aryl groups. The broad peak at 3430 cm−1 was attributed to the vibration of the N–H fragment.
 | ||
| Fig. 2 The FT-IR spectra of (a) the [Al-H] support. (b) The immobilized catalyst 2. (c) Subtraction of b − a. | ||
The peak at 1690 cm−1 was assigned to the vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, whereas the signals between 1600 and 1520 cm−1 resulted from the vibration of the CC and CN bonds within the homogeneous complex.
O bond, whereas the signals between 1600 and 1520 cm−1 resulted from the vibration of the CC and CN bonds within the homogeneous complex.
Further characterization was conducted by implementing multinuclear solid state (SS) NMR. The 1H-MAS SS-NMR spectrum of 2 exhibited significant broad signals at δ 1.0 and δ 1.7 ppm arising from the protons of the alkyl fragments δH(CH3) and δH(CH2) within the structure of the complex. The peak at 4.4 ppm was assigned to δH(N–H, Al-H, and Si–Hx). The aromatic protons’ δH(HCC) signal appeared at δ 7.82 ppm (Fig. 3a). The 13C CP/MAS solid-state NMR spectra (Fig. 3b) showed the characteristic peaks of the immobilized catalyst at δ 7.0 and δ 22.8 ppm, which correspond to various alkyl groups (C–H3 and C–H2) bonded to the ligand. These peaks correlate with the proton signal at δ 1.7 ppm observed in the 1H–13C HETCOR solid-state NMR spectra (Fig. S8†). The signal at δ 38.2 ppm was assigned to the methyl groups linked to the amine ((CH3)2–N) fragment. The peaks at δ 85.5 and δ 111.0 ppm were attributed to the aromatic carbons (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C) and (N–CC) within the pyridine and methylcyclopentadienyl groups. Signals overlapping at δ 146.5 ppm were assigned to (CN) and (C–N) and the peak at δ 154.4 ppm was attributed to the (N–CO) moiety. These results were consistent with the homogeneous complex 1, confirming the successful immobilization of 1 on the [Al-H] support.
C) and (N–CC) within the pyridine and methylcyclopentadienyl groups. Signals overlapping at δ 146.5 ppm were assigned to (CN) and (C–N) and the peak at δ 154.4 ppm was attributed to the (N–CO) moiety. These results were consistent with the homogeneous complex 1, confirming the successful immobilization of 1 on the [Al-H] support.
 | ||
| Fig. 3 (a) 1H MAS solid-state NMR spectra of 2. (b) 13C CP/MAS solid-state NMR spectra of 2. | ||
Additionally, the 1H–1H DQ solid-state NMR spectra (Fig. S9†) showed an autocorrelation on the diagonal of proton resonances from the different alkyl groups and aromatic protons. In other words, at least two protons of these fragments are in close proximity to each other. Outside the diagonal, a strong correlation at δ 5.4 ppm was attributed to the protons near the CH2–N–H group, [5.4 ppm in F1: δH(NH) + δH(CH) = 4.4 + 1.0]. The 1H–H1 TQ solid-state NMR spectra of 2 (Fig. S9†) were also recorded; an autocorrelation at 5 ppm in F1 corresponded to the methyl groups. These observations offer valuable insights, and to the best of our understanding, the proposed structure of 2 in Scheme 1 is the most suitable option based on available data.
To further investigate the coordination environment between complex 1 and the support, 27Al solid-state NMR was conducted to study the coordination state of the Al site on the surface. Fig. 4 displays a broad signal at δ 40.16 ppm, corresponding to a distorted tetra-coordination arrangement. A small shoulder at δ 6.87 ppm attributed to the penta-coordinated Al site was also observed, suggesting an acid–base interaction between the surface species and complex 1.
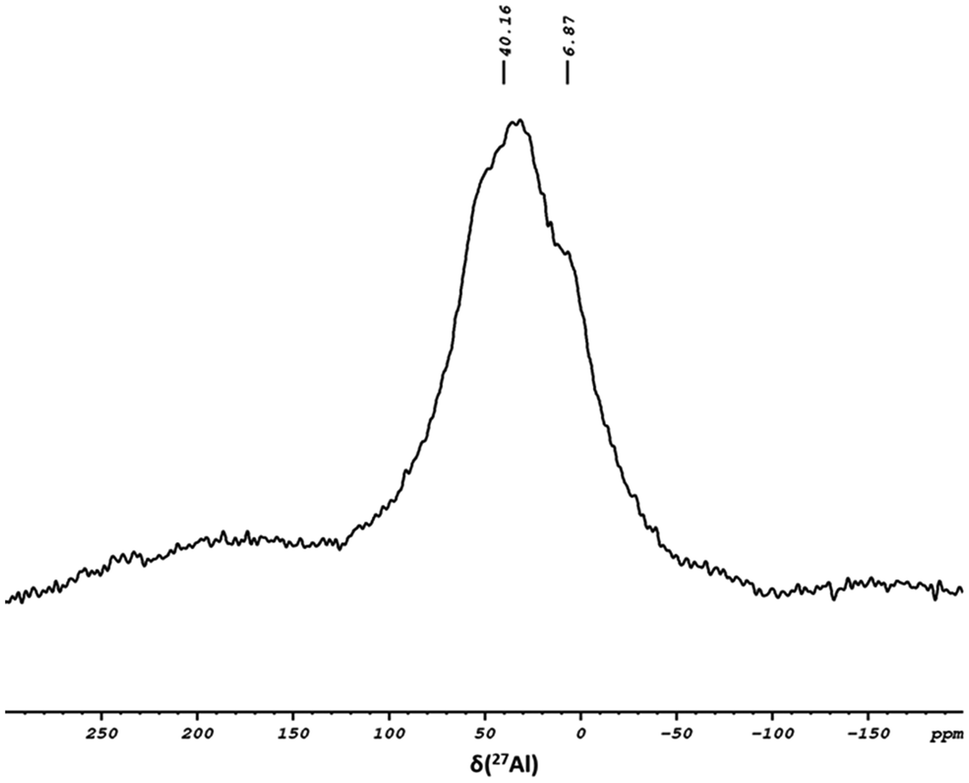 | ||
| Fig. 4 The 27Al solid-state NMR spectrum of 2. | ||
A notable broadband in the region of δ 150–200 ppm was associated with Al-Alkyl, remaining on the support surface, arising from the incomplete transformation of Al-isobutyl to Al-hydride during the support preparation procedure.40,46,47 The high-angle annular dark field imaging (HAADF) TEM analysis (Fig. 5) confirmed the preservation of the fibrous 3D-mesophase of the support and demonstrated a well and uniform distribution of the elements on the support surface, as evidenced by EDS elemental mapping. Additionally, the analysis revealed the presence of iridium at 3.27 wt%, which closely matches the results obtained through elemental analysis (Fig. S10†). To elucidate the electronic structure and coordination environment at the atomic level, X-ray absorption spectroscopy measurement was conducted. The X-ray absorption near-edge structure (XANES) spectra of 2 and the reference compound at the Ir L-edge were recorded (Fig. S11†). IrO2 was used as a reference for the +4 oxidation state, while IrCl3 served as a reference for the +3 oxidation state. The EXANS energies for 2 and the reference IrCl3 were found to be identical and equal to 11218.17 eV, suggesting an average oxidation state of +3 for the supported system. Subsequently, analysis of the extended X-ray fine structure (EXAFS) Fourier transforms at the iridium L-edge for 2 (Fig. S12†) showed spectral features corresponding to those of the homogeneous Ir complex, thus suggesting the successful formation of the desired Ir complex on the support surface.
 | ||
| Fig. 5 HAADF-STEM image of 2: (a) images and elemental mapping of aluminum (b), iridium (c), nitrogen (d), silicon (e), and superimposed elements (f). | ||
Catalytic test: selective FA dehydrogenation
The catalytic performance of catalysts 2 and 1 was examined for the dehydrogenation of FA under neat conditions. In this study, we highlighted the use of solid bases as additives and avoided using volatile bases such as Et3N to enhance process feasibility. Initially, we evaluated the activity of catalyst 2 in the presence of various solid bases, including NaOH, KOH, and KO2CH. The best conversion was recorded when NaO2CH (SF) was employed in the reaction medium (Table S3†). When the [Al-H] support was examined as a catalyst in the reaction, no gas was detected (Table 2, entry 1). No reaction was observed in the absence of an additive as well (Table 2, entry 2). Thus, the reaction was conducted with varying amounts of SF. The results indicated that a remarkable TOF of 37000 h−1 could be achieved at an FA/SF ratio of 2:1, with a 90% yield (Table 2, entry 3). When the FA/SF ratio was reduced to 1:1, a higher TOF of 40000 h−1 was observed; however, the yield decreased to 88% (Table 2, entry 4). Changing the ratio of FA/SF to 1:2 resulted in a reduction in the TOF of 17000 h−1, and an 81% yield was recorded (Table 2, entry 5). Under the same optimized reaction conditions, catalyst 1 achieved a TOF of up to 45000 h−1 and a yield of 93% (Table 2, entry 6). To obtain more information about the effect of FA concentration, the reaction was conducted using 91% and 95% FA under the same optimized reaction conditions (Table S4†). The results indicated that the reaction was faster in the case of 91% FA, and a yield of 98% was achieved. To gain insight into the reaction mechanism, a control experiment was conducted in which 2 reacted with an excess amount of FA for 1 h; the remaining FA was washed with dry pentane and the obtained material was dried overnight and subjected to solid-state NMR analysis. The 1H MAS solid-state NMR spectra (Fig. 6) exhibit similar peaks to those observed in the spectra of 2, with an additional peak at δ 8.6 ppm indicating the presence of formate species. 13C MAS solid-state NMR spectra revealed a peak at δ 173.8 ppm further supporting the presence of the formate fragment (Fig. S14†). Based on the information we have, a mechanistic pathway was proposed. The first step involves coordinating the formate moiety under the reaction conditions. This is followed by the release of CO2 gas and the generation of the hydride intermediate. Subsequently, H2 gas evolution would lead to the regeneration of the active site.
 | ||
| Fig. 6 1H-MAS solid-state NMR spectrum of the intermediate. | ||
a
| Entry | Catalyst | FA:SF |
TOF | Yield |
|---|---|---|---|---|
| a General conditions: FA (1.2 ml, 30 mmol), catalyst 2 (10 mg, 2 μmol), temperature (90 °C). b FA (1.2 ml, 30 mmol), [Al-H] (20 mg). c FA (0.6 ml, 15 mmol), catalyst 2 (10 mg, 2 μmol), temperature (90 °C). d Catalyst 1 (1.2 mg, 2 μmol), FA (1.2 ml, 30 mmol), temperature (90 °C). The maximum TOF was estimated based on the H2 production in the first 10 min. | ||||
| 1b | [Al-H] | 1:1 |
No reaction | 2% |
| 2 | 2 | 1:0 |
No reaction | 7% |
| 3 | 2 | 2:1 |
37000 h−1 |
90% |
| 4 | 2 | 1:1 |
40000 h−1 |
88% |
| 5c | 2 | 1:2 |
17000 h−1 |
81% |
| 6d | 1 | 1:1 |
45000 h−1 |
93% |
The optimized catalytic performance test of catalyst 2 was conducted under solvent-free/neat conditions, and a TON of 25000 was recorded. Despite our several attempts to optimize different parameters to increase the TON, it did not further improve and indicated limited stability of the immobilized catalyst under these harsh conditions. We examined the batch-wise process; however, catalyst 2 decomposes after only a few cycles of dehydrogenation upon accumulation of FA and an increase in the acidity of the medium. Likewise, an optimized catalytic performance test was conducted using the homogeneous complex. Catalyst 1 demonstrated limited stability and a TON of 16000 was achieved.
To gain a deeper understanding, a control experiment was carried out at the end of the stability test. The reaction mixture was filtered, and the used solid catalyst was subjected to elemental analysis which revealed a significant decrease in iridium loading from 0.2 to 0.005 mmol g−1. On the other hand, the aluminum loading was found to be 1.2 mmol g−1, a negligible decrease compared to the fresh catalyst. The liquid phase, which contained the leached catalyst, was dried first to remove the residual FA and water. Afterward, it was dissolved in d-chloroform and analyzed using liquid-state NMR analysis. The spectrum of the leached catalyst displays a significant change compared to the homogeneous analog 1, and the ligand appears rather fragmented. As far as we can tell, this is potentially caused by the hydrolysis of the ligand backbone, leading to the rupture of the catalyst core (Fig. S17 and S18†).
First-principles modelling of FA decomposition
We performed first-principles periodic density functional theory (DFT) calculations to understand the reactivity of catalysts 1 and 2 for FA decomposition. Similar to our previous works,41,45 we modeled the [Al-H] support with an amorphous silica surface with a silanol group (Si–OH) density of 1.69 nm−2. To obtain realistic, ground state structures of the catalysts, which is especially challenging for the grafted catalyst 2 due to the low symmetry of the support, we performed machine learning (ML) accelerated simulated annealing molecular dynamics (SA-MD) simulations,48,49 followed by geometry optimization of the final structures of the trajectories (see the Methods section in the ESI†). The first-principles optimized catalyst structures are shown in Fig. 7a, where we can see good agreement of the structure of catalyst 1 with the experimental crystal structure in Fig. 1.
 | ||
| Fig. 7 First-principles modeling of FA decomposition over catalysts 1 and 2. (a) Atomistic models of the activated catalysts. The H* state is illustrated for simplicity. Ligands of the catalysts are depicted as sticks and O and H atoms of the adsorbates and spectator H2O molecules are shown in green and light blue, respectively, for clarity. (b) Potential energy diagrams for FA decomposition. Energies correspond to Gibbs free energies relative to the initial states. Insets show the illustrations of catalytic intermediates, with the adsorbed species highlighted in blue. Atomistic models of the intermediates are provided in Fig. S20 and S21† for catalysts 1 and 2, respectively. | ||
We model the FA dehydrogenation reactivity of catalysts 1 and 2 in their activated state, where Cl− is exchanged with HCOO−. This is consistent with earlier studies showing that similar catalysts undergo an activation process involving this ligand exchange.50,51 Once activated, the catalysts can undergo multiple turnovers with FA in the catalytic cycle (Fig. S19†). As water is always present in commercially available neat FA, we additionally considered 2 explicit spectator H2O molecules in the system. Our previous works showed that water can play key roles by significantly lowering the barriers of several elementary steps in the reaction via hydrogen bonding and proton shuffling.41,45 It should be noted that as CO is not produced in the reaction, we have only considered the formate (HCOO*) pathway and not the carboxyl (COOH*) pathway, as the decomposition of COOH* is known to easily generate CO*.52
From our calculated potential energy diagrams (PEDs), the reaction mechanism for FA dehydrogenation has been found to be similar for both catalysts (Fig. 7b). After activation, HCOO* is adsorbed on the catalysts. From this initial state, HCOO* then flips to form HCOOm*, where the C–H bond points downwards facing the Ir center. Deprotonation of HCOOm* releases CO2 and leaves behind a hydridic H* on the Ir center. Next, an HCOOH molecule binds to the catalyst via hydrogen bonding of its CO group with the –NH2+ group on the catalyst ligand. The acidic –OH group of HCOOH then reacts with the hydridic H* on Ir via proton shuffling mediated by a water molecule. This acid–base reaction releases H2 in a spontaneous process for 2 and via an intermediate 2H* state for 1. Finally, the initial state is regenerated as the HCOO− formed binds to the Ir vacancy created by the loss of H2.
The energetic spans of the computed reaction mechanisms over catalysts 1 and 2 are 0.90 and 0.94 eV, respectively (Fig. 7b). This is consistent with the slightly lower TOF over the immobilized catalyst 2. The rate-determining intermediate (RDI) is the H* species, which is likely bound with an HCOO− counterion. This agrees with the NMR analysis of dried catalyst 2 after the reaction with FA. The higher energetic span for 2 is attributed to the slightly higher barrier for H2 production from the H* species. However, the PEDs are in general similar, which is consistent with our finding that the calculated Bader charges of Ir on 1 and 2 are the same (+0.69 and +0.67|e−|, respectively). Overall, our simulations reveal that the reactivity of catalyst 1 is not significantly affected by grafting to the Al center as the atoms attached to Al are relatively far away from the active Ir center, allowing 2 to maintain a high TOF similar to 1.
Conclusion
In summary, this work demonstrated the immobilization of an iridium picolinamide complex supported on 3D fibrous modified silica. The support contains a tetracoordinate aluminum hydride as an anchor site, which features high Lewis acidity. We explored the use of solid additives in a solvent-free reaction medium and avoided using volatile bases to assess the feasibility of the process. Catalyst 2 achieved a high TOF of 40000 h−1, comparable to its homogeneous analog 1. Catalyst 2 demonstrated a higher TON compared to its counterpart, highlighting the positive role of the Al-H support.
DFT calculations revealed similar mechanisms of FA decomposition over catalysts 1 and 2. The results showed that the high TOF of 2 can likely be attributed to the grafting site being located away from the active Ir center, which prevents a decrease in the reactivity of 1. However, catalyst 2 demonstrated limited stability. As outlined in the proposed mechanism, which is supported by DFT calculations, the reaction pathway involved the protonation of the N–H side arm, consequently weakening the bond between complex 1 and the support and facilitating leaching from the surface.
Moreover, analysis of the deactivated catalyst after the optimized catalytic performance test indicated a significant reduction in the iridium loading. We hypothesized that this is potentially caused by the hydrolysis of the ligand backbone, leading to the rupture of the catalyst core. As the grafting anchoring point is positioned away from the Ir active site, it results in minimal changes to the coordination sites. The data presented here exemplify the importance of understanding the structure–reactivity relationship. Consequently, significant advancement can be made in the design of catalysts to tailor catalysts for specific applications.
Data availability
The data supporting this article have been included in the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the King Abdullah University of Science and Technology (KAUST) for financial support. B. W. J. C. is grateful for the support from the A*STAR SERC Central Research Fund award. B. W. J. C. and Z. J. are grateful for the high-performance computational facilities provided by the National Supercomputing Centre (NSCC) Singapore and the A*STAR Computational Resource Centre (A*CRC).References
- J. Eppinger and K.-W. Huang, Formic Acid as a Hydrogen Energy Carrier, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS
.
- S. Sharma and S. K. Ghoshal, Hydrogen the future transportation fuel: From production to applications, Renewable Sustainable Energy Rev., 2015, 43, 1151–1158 CrossRef CAS
- J. O. M. Bockris, The hydrogen economy: Its history, Int. J. Hydrogen Energy, 2013, 38, 2579–2588 CrossRef CAS
- I. Dutta, S. Chatterjee, H. Cheng, R. K. Parsapur, Z. Liu, Z. Li, E. Ye, H. Kawanami, J. S. C. Low, Z. Lai, X. J. Loh and K.-W. Huang, Formic Acid to Power towards Low-Carbon Economy, Adv. Energy Mater., 2022, 12, 2103799 CrossRef CAS
-
S. Chatterjee, I. Dutta and K.-W. Huang, in Power to Fuel, ed. G. Spazzafumo, Academic Press, 2021, pp. 169–210, DOI:10.1016/B978-0-12-822813-5.00006-0
- I. Dutta, R. K. Parsapur, S. Chatterjee, A. M. Hengne, D. Tan, K. Peramaiah, T. I. Solling, O. J. Nielsen and K.-W. Huang, The Role of Fugitive Hydrogen Emissions in Selecting Hydrogen Carriers, ACS Energy Lett., 2023, 8, 3251–3257 CrossRef CAS
- Y. Cai, S. Chatterjee, K. N. Salama, L.-J. Li and K.-W. Huang, Sensing fugitive hydrogen emissions, Nat. Rev. Electr. Eng., 2024, 1, 210–211 CrossRef
- G. M. Whitesides and G. W. Crabtree, Don’t Forget Long-Term Fundamental Research in Energy, Science, 2007, 315, 796–798 CrossRef CAS PubMed
- N. Armaroli and V. Balzani, The Hydrogen Issue, ChemSusChem, 2011, 4, 21–36 CrossRef CAS PubMed
- R. van Putten, T. Wissink, T. Swinkels and E. A. Pidko, Fuelling the hydrogen economy: Scale-up of an integrated formic acid-to-power system, Int. J. Hydrogen Energy, 2019, 44, 28533–28541 CrossRef CAS
- S. Chatterjee, I. Dutta, Y. Lum, Z. Lai and K.-W. Huang, Enabling storage and utilization of low-carbon electricity: power to formic acid, Energy Environ. Sci., 2021, 14, 1194–1246 RSC
- T. C. Johnson, D. J. Morris and M. Wills, Hydrogen generation from formic acid and alcohols using homogeneous catalysts, Chem. Soc. Rev., 2010, 39, 81–88 RSC
- Y. Himeda, S. Miyazawa and T. Hirose, Interconversion between Formic Acid and H2/CO2 using Rhodium and Ruthenium Catalysts for CO2 Fixation and H2 Storage, ChemSusChem, 2011, 4, 487–493 CrossRef CAS PubMed
- W. Gan, D. J. M. Snelders, P. J. Dyson and G. Laurenczy, Ruthenium(II)-Catalyzed Hydrogen Generation from Formic Acid using Cationic, Ammoniomethyl-Substituted Triarylphosphine Ligands, ChemCatChem, 2013, 5, 1126–1132 CrossRef CAS
- M. Czaun, A. Goeppert, J. Kothandaraman, R. B. May, R. Haiges, G. K. S. Prakash and G. A. Olah, Formic Acid As a Hydrogen Storage Medium: Ruthenium-Catalyzed Generation of Hydrogen from Formic Acid in Emulsions, ACS Catal., 2014, 4, 311–320 CrossRef CAS
- C. Guan, D.-D. Zhang, Y. Pan, M. Iguchi, M. J. Ajitha, J. Hu, H. Li, C. Yao, M.-H. Huang, S. Min, J. Zheng, Y. Himeda, H. Kawanami and K.-W. Huang, Dehydrogenation of Formic Acid Catalyzed by a Ruthenium Complex with an N,N′-Diimine Ligand, Inorg. Chem., 2017, 56, 438–445 CrossRef CAS PubMed
- S. Kushwaha, J. Parthiban and S. K. Singh, Ruthenium-Catalyzed Formic Acid/Formate Dehydrogenation and Carbon Dioxide/(bi)carbonate Hydrogenation in Water, Organometallics, 2023, 42, 3066–3076 CrossRef CAS
- Y. Himeda, Highly efficient hydrogen evolution by decomposition of formic acid using an iridium catalyst with 4,4′-dihydroxy-2,2′-bipyridine, Green Chem., 2009, 11, 2018–2022 RSC
- E. Fujita, J. T. Muckerman and Y. Himeda, Interconversion of CO2 and formic acid by bio-inspired Ir complexes with pendent bases, Biochim. Biophys. Acta, 2013, 1827, 1031–1038 CrossRef CAS PubMed
- A. Matsunami, Y. Kayaki and T. Ikariya, Enhanced Hydrogen Generation from Formic Acid by Half-Sandwich Iridium(III) Complexes with Metal/NH Bifunctionality: A Pronounced Switch from Transfer Hydrogenation, Eur. J. Chem., 2015, 21, 13513–13517 CrossRef CAS PubMed
- Z. Wang, S.-M. Lu, J. Li, J. Wang and C. Li, Unprecedentedly High Formic Acid Dehydrogenation Activity on an Iridium Complex with an N,N′-Diimine Ligand in Water, Eur. J. Chem., 2015, 21, 12592–12595 CrossRef CAS PubMed
- S. Cohen, V. Borin, I. Schapiro, S. Musa, S. De-Botton, N. V. Belkova and D. Gelman, Ir(III)-PC(sp3)P Bifunctional Catalysts for Production of H2 by Dehydrogenation of Formic Acid: Experimental and Theoretical Study, ACS Catal., 2017, 7, 8139–8146 CrossRef CAS
- M. Iguchi, H. Zhong, Y. Himeda and H. Kawanami, Kinetic Studies on Formic Acid Dehydrogenation Catalyzed by an Iridium Complex towards Insights into the Catalytic Mechanism of High-Pressure Hydrogen Gas Production, Eur. J. Chem., 2017, 23, 17017–17021 CrossRef CAS PubMed
- N. Lentz and M. Albrecht, A Low-Coordinate Iridium Complex with a Donor-Flexible O,N-Ligand for Highly Efficient Formic Acid Dehydrogenation, ACS Catal., 2022, 12, 12627–12631 CrossRef CAS
- C. Guan, Y. Pan, T. Zhang, M. J. Ajitha and K. W. Huang, An update on formic acid dehydrogenation by homogeneous catalysis, Chem. – Asian J., 2020, 15, 937–946 CrossRef CAS PubMed
- S. Hübner, J. G. de Vries and V. Farina, Why Does Industry Not Use Immobilized Transition Metal Complexes as Catalysts?, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef
- J. D. A. Pelletier and J.-M. Basset, Catalysis by Design: Well-Defined Single-Site Heterogeneous Catalysts, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS PubMed
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Homogeneous and Heterogeneous Catalysis: Bridging the Gap through Surface Organometallic Chemistry, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed
- X. Wang, E. A. P. Ling, C. Guan, Q. Zhang, W. Wu, P. Liu, N. Zheng, D. Zhang, S. Lopatin, Z. Lai and K.-W. Huang, Single-Site Ruthenium Pincer Complex Knitted into Porous Organic Polymers for Dehydrogenation of Formic Acid, ChemSusChem, 2018, 11, 3591–3598 CrossRef CAS PubMed
- G. H. Gunasekar, H. Kim and S. Yoon, Dehydrogenation of formic acid using molecular Rh and Ir catalysts immobilized on bipyridine-based covalent triazine frameworks, Sustainable Energy Fuels, 2019, 3, 1042–1047 RSC
- Y. Shen, Y. Zhan, C. Bai, F. Ning, H. Wang, J. Wei, G. Lv and X. Zhou, Immobilized iridium complexes for hydrogen evolution from formic acid dehydrogenation, Sustainable Energy Fuels, 2020, 4, 2519–2526 RSC
- D. A. Bulushev, Progress in Catalytic Hydrogen Production from Formic Acid over Supported Metal Complexes, Energies, 2021, 14, 1334 CrossRef CAS
- L. Tensi, A. V. Yakimov, C. Trotta, C. Domestici, J. De Jesus Silva, S. R. Docherty, C. Zuccaccia, C. Copéret and A. Macchioni, Single-Site Iridium Picolinamide Catalyst Immobilized onto Silica for the Hydrogenation of CO2 and the Dehydrogenation of Formic Acid, Inorg. Chem., 2022, 61, 10575–10586 CrossRef CAS PubMed
- K. Sawahara, S. Tanaka, T. Kodaira, R. Kanega and H. Kawanami, Iridium Catalyst Immobilized on Crosslinked Polyethyleneimine for Continuous Hydrogen Production Using Formic Acid, ChemSusChem, 2024, 17, e202301282 CrossRef CAS PubMed
- R. J. Witzke, A. Chapovetsky, M. P. Conley, D. M. Kaphan and M. Delferro, Nontraditional Catalyst Supports in Surface Organometallic Chemistry, ACS Catal., 2020, 10, 11822–11840 CrossRef CAS
- J.-M. Basset, F. Lefebvre and C. Santini, Surface organometallic chemistry: some fundamental features including the coordination effects of the support, Coord. Chem. Rev., 1998, 178–180, 1703–1723 CrossRef CAS
-
J.-M. Basset, R. Psaro, D. Roberto and R. Ugo, Modern surface organometallic chemistry, John Wiley & Sons, 2009 Search PubMed
- M. K. Samantaray, E. Pump, A. Bendjeriou-Sedjerari, V. D'Elia, J. D. A. Pelletier, M. Guidotti, R. Psaro and J.-M. Basset, Surface organometallic chemistry in heterogeneous catalysis, Chem. Soc. Rev., 2018, 47, 8403–8437 RSC
- M. K. Samantaray, V. D'Elia, E. Pump, L. Falivene, M. Harb, S. Ould Chikh, L. Cavallo and J.-M. Basset, The Comparison between Single Atom Catalysis and Surface Organometallic Catalysis, Chem. Rev., 2020, 120, 734–813 CrossRef CAS PubMed
- B. Werghi, A. Bendjeriou-Sedjerari, A. Jedidi, E. Abou-Hamad, L. Cavallo and J.-M. Basset, Single-Site Tetracoordinated Aluminum Hydride Supported on Mesoporous Silica. From Dream to Reality!, Organometallics, 2016, 35, 3288–3294 CrossRef CAS
- L. Yaacoub, I. Dutta, B. Werghi, B. W. J. Chen, J. Zhang, E. A. Hamad, E. P. L. Ang, E. Pump, A. B. Sedjerari, K.-W. Huang and J.-M. Basset, Formic Acid Dehydrogenation via an Active Ruthenium Pincer Catalyst Immobilized on Tetra-Coordinated Aluminum Hydride Species Supported on Fibrous Silica Nanospheres, ACS Catal., 2022, 12, 14408–14417 CrossRef CAS
- B. Werghi, E. Pump, M. Tretiakov, E. Abou-Hamad, A. Gurinov, P. Doggali, D. H. Anjum, L. Cavallo, A. Bendjeriou-Sedjerari and J.-M. Basset, Exploiting the interactions between the ruthenium Hoveyda–Grubbs catalyst and Al-modified mesoporous silica: the case of SBA15 vs. KCC-1, Chem. Sci., 2018, 9, 3531–3537 RSC
- S. Kar, M. Rauch, G. Leitus, Y. Ben-David and D. Milstein, Highly efficient additive-free dehydrogenation of neat formic acid, Nat. Catal., 2021, 4, 193–201 CrossRef CAS PubMed
- J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, A prolific catalyst for dehydrogenation of neat formic acid, Nat. Commun., 2016, 7, 11308 CrossRef CAS PubMed
- L. Alrais, S. S. Gholap, I. Dutta, E. Abou-Hamad, B. W. J. Chen, J. Zhang, M. N. Hedhili, J.-M. Basset and K.-W. Huang, Highly efficient immobilized PN3P-pincer iridium catalyst for dehydrogenation of neat formic acid, Appl. Catal., B, 2024, 342, 123439 CrossRef CAS
- J. W. Akitt, Multinuclear studies of aluminium compounds, Prog. Nucl. Magn. Reson. Spectrosc., 1989, 21, 1–149 CrossRef
- M. Haouas, F. Taulelle and C. Martineau, Recent advances in application of 27Al NMR spectroscopy to materials science, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 94–95, 11–36 CrossRef CAS PubMed
- B. W. J. Chen, B. Wang, M. B. Sullivan, A. Borgna and J. Zhang, Unraveling the Synergistic Effect of Re and Cs Promoters on Ethylene Epoxidation over Silver Catalysts with Machine Learning-Accelerated First-Principles Simulations, ACS Catal., 2022, 12, 2540–2551 CrossRef CAS
- B. W. J. Chen, X. Zhang and J. Zhang, Accelerating explicit solvent models of heterogeneous catalysts with machine learning interatomic potentials, Chem. Sci., 2023, 14, 8338–8354 RSC
- H. Wen, N. Luo, Q. Zhu and R. Luo, Amide Iridium Complexes As Catalysts for Transfer Hydrogenation Reduction of N-sulfonylimine, J. Org. Chem., 2021, 86, 3850–3859 CrossRef CAS PubMed
- Z. Yang, Z. Zhu, R. Luo, X. Qiu, J.-T. Liu, J.-K. Yang and W. Tang, Iridium-catalyzed highly efficient chemoselective reduction of aldehydes in water using formic acid as the hydrogen source, Green Chem., 2017, 19, 3296–3301 RSC
- S. Bhandari, S. Rangarajan, C. T. Maravelias, J. A. Dumesic and M. Mavrikakis, Reaction Mechanism of Vapor-Phase Formic Acid Decomposition over Platinum Catalysts: DFT, Reaction Kinetics Experiments, and Microkinetic Modeling, ACS Catal., 2020, 10, 4112–4126 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2412317. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03521f |
| This journal is © The Royal Society of Chemistry 2025 |