
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new class of ligand derived from the reactions of bis(dialkylphosphino)amines and 9-diazofluorene: preparation, structure and reactivity†
Matthew J. J.
Laprade
 ab,
Katherine N.
Robertson
a,
Jason A. C.
Clyburne
*a and
Christopher M.
Kozak
*b
ab,
Katherine N.
Robertson
a,
Jason A. C.
Clyburne
*a and
Christopher M.
Kozak
*b
aDepartment of Chemistry, Saint Mary's University Halifax, Nova Scotia, B3H 3C3, Canada. E-mail: jason.clyburne@smu.ca
bDepartment of Chemistry, Memorial University of Newfoundland, St John's, Newfoundland A1C 5S7, Canada. E-mail: ckozak@mun.ca
First published on 8th July 2025
Abstract
The reaction of tetra(isopropyl)diphosphazane (iPr-PNP) or tetraphenyldiphosphazane (Ph-PNP) with 9-diazofluorene (9-DAF) affords new chelating phosphazene ligands [F(NN)P(R)2NP(R)2(NHN)F] where F = fluorenylidene and R = iPr (H[1a]) or Ph (H[1b]). H[1b] has been shown to react with ZnEt2 and AlMe3 giving mononuclear organometallic complexes ZnEt[1b] (4) and AlMe2[1b] (5), respectively, while the reaction with NaH generates the chelated sodium complex Na(THF)2[1b] (6). Complexes 4, 5, and 6 have been fully characterized, and the solid-state structures of all complexes in this study have been authenticated using single-crystal X-ray diffraction. Complexes 4 and 5 catalyze the ring-opening copolymerization (ROCOP) of cyclohexene oxide (CHO) and phthalic anhydride (PA) giving polyesters in the presence of bis(triphenylphosphine)iminium chloride (PPNCl) as a nucleophilic co-catalyst, yielding moderate molar masses with narrow dispersities.
Introduction
Organometallic chemistry is dominated by the strategic use of robust ligands, whether neutral, anionic, or cationic. Examples of “traditional” neutral ligands include amines, phosphines, and N-heterocyclic carbenes (NHCs).1,2 Cationic ligands, such as the bis(arylamino)phosphenium cation, are rarer but recent work shows they can be paired with metals typically used in catalysis.3,4 The toolbox of anionic ligands is vast, ranging from those generally considered to be spectator ligands to those with well-established redox activity or so-called non-innocent behaviour.5,6 Two of the most notable families of N-donor, bidentate chelating anionic ligands are the 1,3-diketiminates (NacNac) and the bis(oxazolines) (BOX)7 shown in Fig. 1. Some of the present co-authors and others have worked on various related systems, most notably with the electron rich β-diketiminate (NacNac) ligands (Me2N)2NacNac.8–10 | ||
| Fig. 1 Commonly used N-based chelating ligands. | ||
Another extensive class of potentially monoanionic ligands are the electron-rich phosphorus ligands with P–N–P or N–P–N backbones, known as phosphazenes, phosphazenates, or iminophosphonamides11–20 and the related P–C–P backbone ligands, bis(phosphinimino)methanides.21 Phosphazenes generally have either cyclic or linear structures where the backbone is composed of alternating covalently bonded nitrogen and phosphorus atoms. These ligands, which include the ambidentate bis(diorganophosphino)amines iPr-PNP and Ph-PNP (Fig. 1), have gained attention recently due to their diverse coordination modes (monodentate, bridging, chelating, etc.).
Bis(diphenylphosphino)amine (or tetraphenyldiphosphazane) Ph-PNP is one of the most common short-bite angle R-PNP ligands,11–18,22–28 and its isopropyl analogue, bis(diisopropylphosphino)amine (or tetraisopropyldiphosphazane) iPr-PNP is also extensively studied.11–15,17,18,22–25,27,28Ph-PNP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 and iPr-PNP29 can be synthesized in similar manners through reaction of the substituted chlorophosphine with bis(trimethylsilyl)amine. The steric effects of the substituents are different for iPr-PNP and Ph-PNP and influence the structural properties of their derivatives, as will be discussed below.
26 and iPr-PNP29 can be synthesized in similar manners through reaction of the substituted chlorophosphine with bis(trimethylsilyl)amine. The steric effects of the substituents are different for iPr-PNP and Ph-PNP and influence the structural properties of their derivatives, as will be discussed below.
The R-PNP protons can be easily removed with base, creating a delocalized anion across the PNP bonds leading to monoanionic bidentate or tridentate coordination modes when bonded to metals.12–16,18 For example, deprotonation of iPr-PNP or Ph-PNP and reaction with BiCl3 yields catenated bismuth complexes (Fig. 2, i).27 The neutral R-PNP can also serve as a narrow bite-angle chelating bis(phosphine) ligand (Fig. 2, ii).16,30Ph-PNP has been shown to react with organic azides (RN3), undergoing loss of N2 to yield amino-bis(phosphazene)s (Fig. 2, iii), which have been used as ligands in metal complex formation.12–14,16,18,31–36 Though there are numerous examples of R-PNP reactivity with RN3, azo, azole, and imine reagents,11–16,18,31–36 to our knowledge there are no reported reactions of R-PNP with diazo (RN2) compounds.
 | ||
| Fig. 2 Metal complexes of bis(dialkylphosphino)amines, adapted from their respective works.27,30,34,42,43,47 | ||
Over the last two decades, complexes utilizing R-PNP type ligands have displayed extensive catalytic properties.16,37–41R-PNP-containing complexes, however, have primarily been utilized for the synthesis of dienes and polyolefins (Fig. 2, ii, iv and v).16,30,34,37–45 There are few examples of R-PNP compounds being used as catalysts for ring-opening polymerization (ROP), and most of those use cyclic phosphazene structures as nucleophiles for the ROP of cyclic esters.13,16,46 Homoleptic lanthanide bis(phosphanyl)amide complexes (Fig. 2, vi) produce poly(ε-caprolactone) of Mn up to 26.2 kg mol−1.16,47 Additionally, metal-free cyclic phosphazenes have been shown to perform ring-opening copolymerization (ROCOP) of epoxides and cyclic anhydrides.13,48,49
Due to the activity demonstrated by PNP-containing complexes for catalyzing polymerization, we investigated the use of R-PNP complexes for the ROCOP of epoxides and cyclic anhydrides. ROP and ROCOP are promising methods for the synthesis of polyesters and polycarbonates from a broad range of monomers to yield polymers with specifically tuned properties.50–59 Various metal complexes, including Zn and Al compounds, have been used as catalysts for the preparation of polyesters via ROCOP of epoxides and cyclic anhydrides.52,56–83 Although PNP-derived complexes of Zn and Al have also been reported,11–16,18,23,25,31,32,84 they have not been used as catalysts for ROCOP reactions. Here we describe the reactions of iPr-PNP and Ph-PNP with 9-diazofluorene (9-DAF) resulting in the isolation of two new “NNPNPNN” derivatives containing only group 15 elements in the backbone. These new phosphazenes were used as ligands for Al, Zn and Na, and the ROCOP activity of the organoaluminium and organozinc products was investigated.
Results and discussion
Preparation of compounds H[1a] and H[1b]
iPr-PNP or Ph-PNP was mixed with two equivalents of 9-DAF in dichloromethane (Scheme 1). This resulted in a decolourization of the dark red 9-DAF solutions to clear, yellow solutions. There was no evidence of gas evolution, as would be anticipated if N2 was being lost from 9-DAF. A yellow solid was isolated upon workup, and yellow crystals were obtained after recrystallization from CH2Cl2/hexane solutions. The solid-state structures of H[1a] and H[1b] belong to a new class of phosphazene compounds. These phosphazenes are easily prepared and are air stable, portending their utility in coordination chemistry and catalysis. H[1a] and H[1b] exhibit diagnostic high-resolution mass spectra (HRMS) with [M + H]+ peaks at m/z 634.3225 and 770.2581, respectively, consistent with their molecular formulae. Absorptions characteristic of a secondary amine, ν(N–H) at ∼3300 cm−1, were not observed in the infrared spectra, but broad peaks are observed in the 1H NMR spectra of H[1a] and H[1b], which could be ascribed to N–H moieties. The appearance of these peaks is strongly concentration and solvent dependant. In the solid state, as shown by single crystal X-ray diffraction, both H[1a] and H[1b] exhibit intramolecular hydrogen bonding (see below). These echo observations reported for a related ArNPNPN(H)Ar compound (iii in Fig. 2).31 The synthesis of H[1a] and H[1b] could be monitored by infrared spectroscopy by following the disappearance of the very strong ν(NN) peak at ∼2050 cm−1 as 9-DAF was consumed (Fig. S58 and S63 in ESI†). | ||
| Scheme 1 Reaction of iPr-PNP or Ph-PNP with 9-DAF to give H[1a] and H[1b], respectively. | ||
Our previous work with Ph-PNP showed the presence of the phenyl substituents on phosphorus promotes the formation of crystalline products.28 We also note that the starting material Ph-PNP is more easily prepared than iPr-PNP because the halophosphine (diphenylchlorophosphine) is more easily handled and is more affordable than the isopropyl derivative (diisopropylchlorophosphine). Additionally, while the formation of side products during the preparation of H[1b] was never observed, the preparation of H[1a] sometimes affords the hydrochloride salt H[1a]·HCl, 2a, as a minor product (Fig. 3 and Fig. S14†), as determined crystallographically. For these reasons, metalation reactions were carried out using H[1b] as the substrate.
 | ||
| Fig. 3 Phosphazenes and their metal complexes described in this work. | ||
Synthesis of compounds 2b and 3
Compounds 2b and 3 (Fig. 3) were isolated as side products during the exploration of the reactivity of H[1b]. The unsuccessful metalation reaction of H[1b] and NiCl2·6H2O in acetonitrile under atmospheric conditions produced a small amount of crystalline 2b. Later, 2b was also obtained from the reaction of 6 with NiCl2(DME) in incompletely dried THF. The formation of 2b is the result of H[1b] reacting with HCl, which was formed in situ under these conditions. Following these observations, the direct synthesis of the chloride salt 2b from H[1b] was attempted (Scheme 2). The solvent from the resulting solution was removed without additional workup, and pale-yellow crystals of 2b were obtained after recrystallization from THF/hexane solution. These crystals are spectroscopically and crystallographically identical to those of 2b obtained in the presence of metal halide salts. | ||
| Scheme 2 Direct synthesis of complex 2b. | ||
An oxidation product of H[1b] was also sometimes observed when metalation reactions were performed under benchtop conditions. This was attributed to adventitious oxygen that, in the presence of metals, led to decomposition of H[1b] producing the phosphine oxide 3, which was characterized spectroscopically and by single crystal X-ray diffraction (Fig. S30 in ESI†). Compound 3 was not generated from the direct addition of water to a room temperature solution of H[1b].
The colour of a DMSO solution of H[1b] changed from yellow to dark orange upon heating to 160 °C, suggesting thermal decomposition. After 4 h at this temperature, the solution turned bright red. IR spectroscopy does not show a band attributable to the diazo group of 9-DAF, which was expected as a strong signal at 2052 cm−1 (Fig. S64 in ESI†). The 1H NMR spectrum, however, indicates the presence of 9-fluorenone hydrazone in the sample, as a resonance is observed at 3.54 ppm, which is attributed to the R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-N
N-N![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2 group (Fig. S73†). The presence of new signals in the 1H, 13C{1H}, and 31P{1H} NMR spectra after heating the solution of H[1b] indicates partial thermal decomposition, as resonances of H[1b] are still observed as the major component of the mixture (Fig. S70–S75 in ESI†). The DMSO solution was cooled, diluted with CH2Cl2 and crystals of H[1b] were recovered in over 80% yield. No crystals of 9-DAF and 9-fluorenone hydrazone were obtained and there was no evidence for the formation of 3 as a product.
2 group (Fig. S73†). The presence of new signals in the 1H, 13C{1H}, and 31P{1H} NMR spectra after heating the solution of H[1b] indicates partial thermal decomposition, as resonances of H[1b] are still observed as the major component of the mixture (Fig. S70–S75 in ESI†). The DMSO solution was cooled, diluted with CH2Cl2 and crystals of H[1b] were recovered in over 80% yield. No crystals of 9-DAF and 9-fluorenone hydrazone were obtained and there was no evidence for the formation of 3 as a product.
Synthesis of compounds 4–6
Organometallic compounds 4 and 5 were prepared from the reaction of H[1b] with ZnEt2 or AlMe3, respectively, in anhydrous THF under an inert atmosphere (Scheme 3). 4 (ZnEt[1b]) and 5 (AlMe2[1b]) were isolated in excellent yield and fully characterized, including by single crystal X-ray diffraction. The most diagnostic spectroscopic feature of the isolated solids is the detection of the organometallic alkyl group in their respective NMR and IR spectra. In the IR spectrum of 4, bands attributed to the alkyl C–H groups appear as weak peaks at 2923, 2885, and 2850 cm−1. The IR spectrum of 5 shows weak peaks at 2926 and 2855 cm−1 that are attributed to the Me groups on Al. Additionally, peaks at 3306 and 1731 cm−1 are observed, which are possibly attributed to the solvating THF within the crystal of 5. When co-crystallized THF is observed in NMR spectra, the 1H integrations are always for fewer hydrogen atoms than anticipated based on the number of THF molecules observed in the single crystal X-ray data.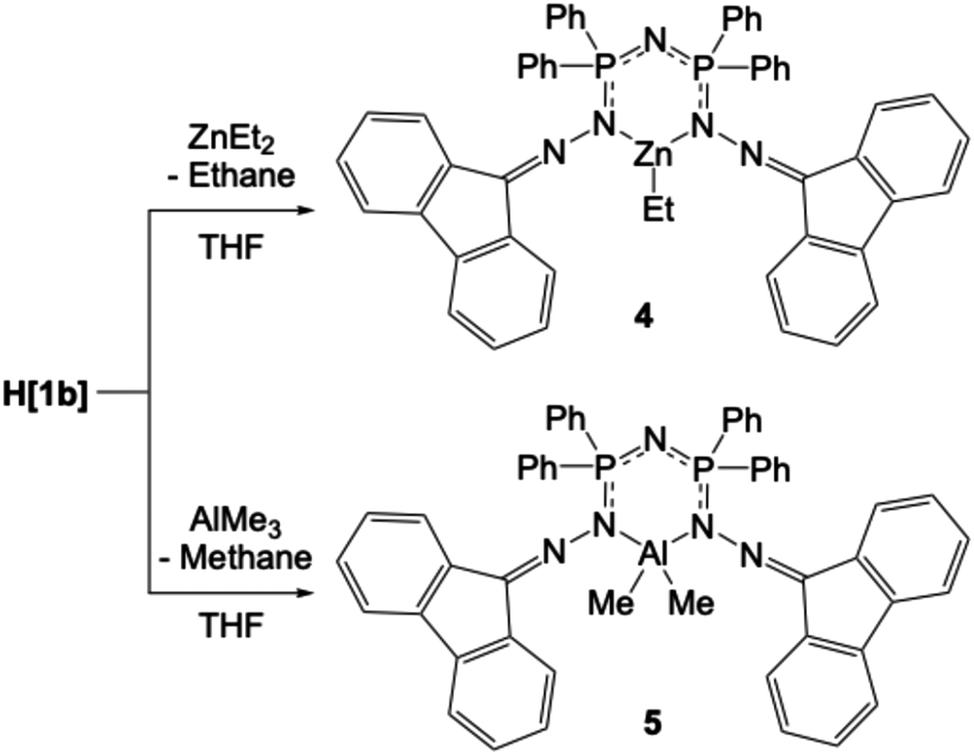 | ||
| Scheme 3 Synthesis of complexes 4 and 5. | ||
Compounds 4 and 5, like their ligand precursor H[1b], are stable when handled in air in the solid-state. However, our initial attempts to obtain NMR spectra in CD2Cl2 solutions show that both compounds decompose in this solvent within an hour. The complexes are more stable in C6D6, and the spectra show only minimal decomposition. In the 1H NMR spectrum of 4 in C6D6, the ethyl group gives rise to a triplet at 0.93 ppm and a quartet at 3.70 ppm (Fig. S90 and S91 in ESI†), while the 1H NMR spectrum of 5 shows a singlet at −0.53 ppm (Fig. S97 in ESI†). In both 1H NMR spectra, small peaks attributable to the free alkane are present; ethane at 0.80 ppm in the spectrum of 4, and methane at 0.16 ppm in the spectrum of 5. These indicate that slight decomposition still occurs in the C6D6 solutions.
The aluminum-bound methyl groups of 5 appear as a singlet at −5.75 ppm in the 13C{1H} NMR spectrum. The resonances for the Zn![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2H3 group in 4 appear at 2.42 and 11.76 ppm in the 13C{1H} NMR spectrum in C6D6. The 31P{1H} spectrum of 4 in C6D6 (Fig. S95 in ESI†) shows one resonance at 22.36 ppm, indicative of equivalent phosphorus environments. This is supported by the solid-state structure (Fig. S37 and S38 in ESI†). The 31P{1H} spectrum of 5 in C6D6 (Fig. S101 in ESI†) has a major resonance at 27.77 ppm attributed to the metalated ligand and a small peak at 22.39 ppm, consistent with some H[1b] being present from the decomposition.
H2H3 group in 4 appear at 2.42 and 11.76 ppm in the 13C{1H} NMR spectrum in C6D6. The 31P{1H} spectrum of 4 in C6D6 (Fig. S95 in ESI†) shows one resonance at 22.36 ppm, indicative of equivalent phosphorus environments. This is supported by the solid-state structure (Fig. S37 and S38 in ESI†). The 31P{1H} spectrum of 5 in C6D6 (Fig. S101 in ESI†) has a major resonance at 27.77 ppm attributed to the metalated ligand and a small peak at 22.39 ppm, consistent with some H[1b] being present from the decomposition.
The 27Al NMR spectrum of 5 (C6D6, Fig. S102 in ESI†) shows a single, broad resonance at ∼70 ppm, with a peak width at half-height, ω1/2 = 2573 Hz. Though such a broad peak would normally be atypical for 27Al NMR, values for complexes similar to 5 demonstrate peak widths at half-height of up to 13700 Hz in their 27Al NMR spectra.85,86 In fact, some complexes similar to 5 do not produce any signals in their 27Al NMR spectra due to low local symmetry around the aluminium I = 5/2 nucleus and quadrupolar broadening.85–89
Compound 6 (Na(THF)2[1b]) can be synthesized by two methods (Scheme 4). The direct reaction of H[1b] with NaH in THF led to the formation of 6 within minutes (Method A). The golden solution of H[1b] turned dark teal in colour and a torrent of small bubbles (hydrogen gas) was released during the reaction. For the indirect synthesis of 6, Ph-PNP was dissolved in a mixture of THF and toluene. Two equivalents of 9-DAF and excess NaH were added (Method B). This reaction proceeded identically to that of Method A, with the exception that evolution of small bubbles was only observed intermittently. A longer reaction time of 12 h was necessary for Method B, but formation of a dark teal solution was again ultimately observed. Yellow crystals of 6 grew from the dark teal solutions of both methods and were characterized by NMR spectroscopy. In the 1H NMR spectrum of 6 in CD2Cl2, two THF molecules are observed per Na (Fig. S104 in ESI†). The 31P{1H} NMR spectrum collected in THF shows one singlet at 17.6 ppm (with CD2Cl2 for lock) or 18.0 ppm (with C6D6 for lock) consistent with symmetric phosphorus environments (Fig. S108 in ESI†). In the IR spectrum of 6, weak alkyl bands at 2958, 2924, and 2864 cm−1 are attributed to the coordinating THF (Fig. S103 in ESI†).
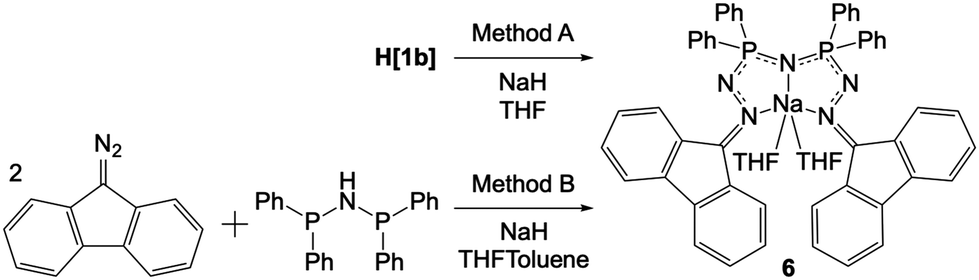 | ||
| Scheme 4 Two synthetic methods for preparation of 6. | ||
The strong absorbance by 6 in the visible region is consistent with observations made in other highly conjugated ligand systems such as those described by Barrett and co-workers,90 and Thompson and co-workers.91 A similar dark teal colour was observed when H[1b] was reacted with KH or nBuLi, however these products have not yet been isolated and thus are not discussed here.
Solid-state structures
The structures of eight compounds were obtained by single crystal X-ray diffraction. Crystallographic information is given in Table S1 in the ESI.† The following discussion focuses on the neutral proligands H[1a] and H[1b], their corresponding organometallic complexes 4 and 5, and the sodium derivative 6. Discussions of the crystal structures for the two HCl adducts, 2a and 2b, and for the oxidized product, 3, can be found in the ESI.†Tables 1 and 2 contain some of the relevant bond lengths and angles for the new products and for their previously reported starting materials, which are provided for comparison.24,26| Bond | iPr-PNP24 | Ph-PNP26 | H[1a] | H[1b] | 4 |
5a |
6b |
|---|---|---|---|---|---|---|---|
| a Values of both M–Ci are included. Values of 5 are taken from only one molecule in the asymmetric unit. b Values for M–N2 and M–N4, are for M–N3 and M–N5, respectively. All values are taken from the major component. | |||||||
| P1–N1 | 1.706(4) | 1.692(2) | 1.551(4) | 1.5646(15) | 1.582(3) | 1.5980(14) | 1.5848(13) |
| P2–N1 | 1.705(4) | 1.692(2) | 1.606(4) | 1.6054(15) | 1.585(3) | 1.5931(14) | 1.5821(13) |
| P1–N2 | 1.682(4) | 1.67228(16) | 1.648(3) | 1.6519(14) | 1.6332(14) | ||
| P2–N4 | 1.649(4) | 1.6267(15) | 1.649(3) | 1.6660(14) | 1.6327(13) | ||
| N2–N3 | 1.370(5) | 1.395(2) | 1.406(4) | 1.4344(19) | 1.3627(19) | ||
| N4–N5 | 1.368(5) | 1.372(2) | 1.414(4) | 1.3944(19) | 1.3611(18) | ||
| M–Ci | 1.974(4) | 1.9680(19) 1.9670(17) | NaN1 = 2.3824(14) | ||||
| M–N2 | 1.995(3) | 1.9663(15) | 2.7770(15) | ||||
| M–N4 | 1.994(3) | 1.9511(15) | 2.5872(15) | ||||
| Angle | iPr-PNP24 | Ph-PNP26 | H[1a] | H[1b] | 4 |
5a |
6b |
|---|---|---|---|---|---|---|---|
| a Values of both M–Ci are included. Values for 5 are taken from only one molecule in the asymmetric unit. b Value of N2–M–N4 is instead for N3–M–N5. All values are taken from the major component. | |||||||
| P1–N1–P2 | 121.2(2) | 118.9(2) | 134.7(2) | 133.38(10) | 132.5(2) | 123.51(9) | 147.06(9) |
| N1–P1–N2 | 109.9(2) | 111.00(8) | 109.58(15) | 109.90(7) | 116.35(7) | ||
| N1–P2–N4 | 109.4(2) | 122.14(8) | 110.52(15) | 108.96(7) | 114.30(7) | ||
| P1–N2–N3 | 117.0(3) | 113.86(12) | 116.1(2) | 110.57(10) | 113.21(11) | ||
| P2–N4–N5 | 109.6(3) | 112.51(11) | 112.5(2) | 106.17(10) | 114.45(10) | ||
| N2–N3–Ci | 118.3(4) | 117.8(14) | 113.1(3) | 116.82(14) | 114.68(13) | ||
| N4–N5–Cii | 115.5(4) | 114.27(15) | 115.0(3) | 123.11(14) | 116.29(13) | ||
| N2–M–N4 | 99.35(11) | 101.74(6) | 140.77(5) | ||||
| N1–M–N3 | 69.87(5) | ||||||
| N1–M–N5 | 70.97(5) | ||||||
Compounds H[1a] and H[1b]
The single-crystal molecular structures of H[1a] and H[1b] are shown in Fig. 4. The bond lengths and angles along the backbones of these molecules are different from those in their respective PNP starting materials. In both structures, all the angles at P and N widen (relative to the PNP starting compounds) as is expected from the presence of increased steric crowding and tetrahedral rather than pyramidal phosphorus centres. From the experimental parameters, particularly the bond lengths shown in Table 1, the Lewis structure shown in Scheme 1 is appropriate, with shorter lengths corresponding to localized double bonds and longer lengths corresponding to single bonds.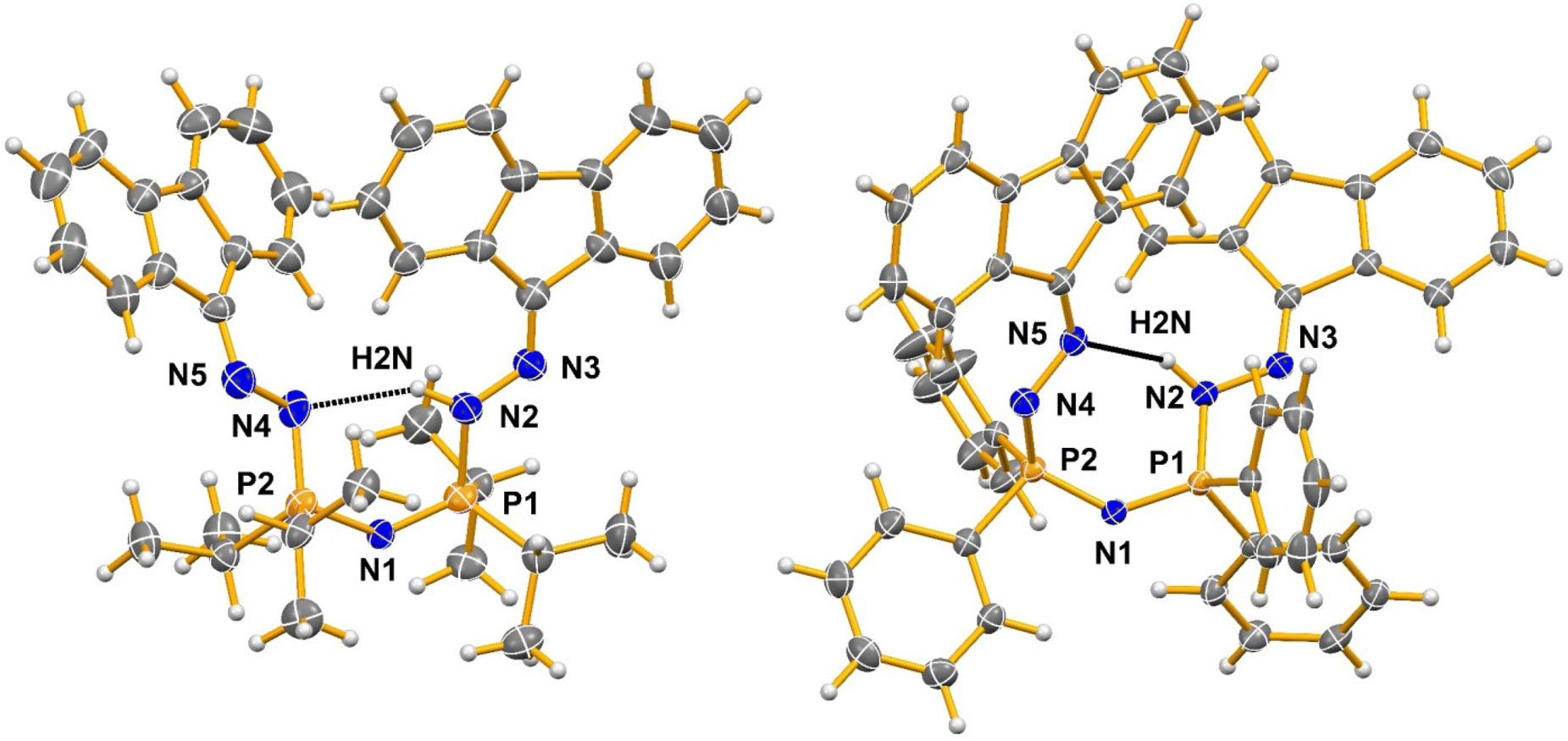 | ||
| Fig. 4 Comparison of the N–H⋯N hydrogen bonding in compounds H[1a] (left) and H[1b] (right) (see Table S2 in ESI† for values). The co-crystallized molecule of hexane in the structure of H[1a] and the disordered molecule of CH2Cl2 present in the structure of H[1b] have been removed for clarity. Only the H(N)-atoms and the heteroatoms in the backbones have been labelled. Thermal ellipsoids are drawn at the 50% probability. | ||
Molecules H[1a] and H[1b] are no longer symmetrical when compared to the PNP starting materials, wherein the delocalized PN bond lengths are equivalent, and angles are similar within the molecule even if they are not required to be so by symmetry. In H[1a] and H[1b], there is a distinct alternation of single and double bonds along the chain, with the H(N) atom preventing resonance delocalization. The hydrogen atom on N2 is uniquely located in each structure (they are visible in the Fourier maps and can be refined isotropically); they are fully occupied and always on a single atom (designated N2) in the crystal structures.
A variety of intermolecular interactions are present in these structures. In H[1b], the CH2Cl2 participates in the network of interactions formed. Intramolecular N–H⋯N hydrogen bonding is observed in both H[1a] and H[1b], but the atoms participating in these bonds differ between the two molecules. In H[1a] the hydrogen bond donor N2 is the N–H group bonded to one of the P sites and the hydrogen bond acceptor is the corresponding N bonded to the other phosphorus atom, N4, leading to a 6-membered ring system. The bonding is characterized by the parameters N2–H2N⋯N4, with d[N2–H2N] = 0.89(5) Å, d[H2N⋯N4] = 2.16(5) Å, d[N2⋯N4] = 2.954(6) Å and the angle N2–H2N⋯N4 = 148(4)°. In H[1b] the hydrogen bond donor is the NH group bonded to one of the phosphorus sites, N2, as in H[1a], but the hydrogen bond acceptor is the N atom bonded to the fluorene group, N5, leading to the formation of a 7-membered ring. This bonding has the corresponding distances of d[N2–H2N] = 0.91(2), d[H2N⋯N5] = 2.39(2) Å, d[N2⋯N5] = 3.154(2) Å and the angle N2–H2N⋯N5 = 142.3(18)°. The hydrogen bonding in the protonated hydrogen chloride salts (2a and 2b; Fig. 5, see the ESI† for details) differs from that in H[1a] and H[1b], respectively, in that the hydrogen bonds in 2a and 2b involve two H(N) groups per cation and the chloride anion is the acceptor for both.
 | ||
| Fig. 5 Comparison of the N–H⋯Cl hydrogen bonding in compounds 2a (left) and 2b (right). (see Table S2 in ESI† for values). Only the atoms involved in the hydrogen bonding and the heteroatoms in the backbone have been labelled. Thermal ellipsoids are drawn at 50% probability. | ||
The difference in the hydrogen bonding between H[1a] and H[1b] possibly arises due to differences in the steric encumbrance of the two phenyl groups compared to the two isopropyl groups on the phosphorus atoms. The conformational changes required to bring the DAF groups on neighboring molecules in the packing of H[1b] into close enough proximity to form short stacking contacts likely gives rise to the different hydrogen bonding arrangements in the two structures. In H[1b] the distance between the centroids of the two closest central rings of fluorene (C13, C14, C19, C20 and C25) is 3.6330(12) Å. This occurs between DAF groups on two different molecules (the symmetry of the second molecule is 1 − x, 1 − y, −z). On both molecules, the same ring is involved, namely the ring on the side of the molecule where the NH group is also present. Stacking must be important as it is maintained in the other phenyl group-containing compounds reported here (2b and 3). In H[1a] there are no such close stacking contacts observed between rings, likely due to the disposition of the isopropyl groups. Instead, the stacking is replaced by the formation of additional C–H⋯ring intermolecular contacts (with C–H perpendicular to the plane of the interacting ring).
The characterization of compounds H[1a] and H[1b] confirm the preparation of a new class of ligands having chains of seven group 15 atoms running through the molecules. Reaction of two DAF molecules with one R-PNP molecule (R = iPr or Ph) gives chains of the form F(NN)P(R)2NP(R)2(NHN)F, where F represents the fluorenylidene group. There are few DAF-containing molecules with similar structures described in the literature and the longest chains found involve only 3 atoms. In 1992, Woolstencroft and co-workers reported the structure of the condensation product of triphenylphosphine and 9-DAF, a molecule containing a P(NN)F 3-atom chain.92 The geometry of this fragment is similar to that portion of the non-hydrogen containing chain in H[1b]. More recently, Stephan and co-workers reported the phosphinoboration of diazomethanes, which included the structure resulting from the condensation of 9-DAF with Ph2PB(Mes)2.93 In this molecule, the geometry of the 3-atom chain P(NN)F differs substantially from H[1b], primarily because in the former the phosphorus centre is 3-coordinate.
Compounds 4 and 5
Crystal structure determinations were carried out on the zinc and aluminum compounds obtained after the reaction of ZnEt2 and AlMe3 with H[1b] (Fig. 6). In 4, the zinc centre is 3-coordinate (two bonds to nitrogen and one to the carbon atom of the ethyl group) in a distorted trigonal planar geometry. The Et group is coplanar with Zn and most of the core ring, as seen from the N2–Zn1–C51–C52 and N4–Zn1–C51–C52 torsion angles of −2.43° and 177.51° respectively. The bond angles at Zn show a distortion from an ideal trigonal planar geometry with a N–Zn–N angle of 99.35(11)°. In contrast, compound 5 contains a 4-coordinate distorted tetrahedral Al centre with two Al–N and two Al–C bonds. The Me groups lie above and below the central ring plane. There are two crystallographically distinct molecules in the unit cell and the N–Al–N angles are 101.74(6)° and 99.88(6)° in molecules 1 and 2, respectively, like that in the Zn structure. | ||
| Fig. 6 Structures of compounds 4 (left) and 5 (right), with selected atoms labelled. The disordered THF solvent present in the structure of 5 has been removed for clarity. Thermal ellipsoids have been drawn at 50% probability. | ||
In compound 4, the 6-membered ZnNPNPN core is puckered with the mean deviation from the plane being 0.210 Å. However, the Zn atom lies only 0.024 Å out of this plane. The entire central core being relatively planar should facilitate delocalization of the electron density around the ring. In contrast, the central 6-membered rings in the two independent molecules within the Al structure have boat conformations, with Al and the PNP nitrogen atom lying out of the ring plane in the same relative direction. In the structure of 5, molecule 1 has a mean deviation from the 6-atom ring plane of 0.270 Å and Al1 lies 0.361 Å out of this plane. For molecule 2, the same calculations give 0.296 Å for the mean deviation and Al2 is 0.413 Å out of the plane. The Al atoms lie much further out of their ring planes than does the Zn atom, which could reduce electron delocalization around the Al metallocycle ring systems.
The two Zn–N bonds in 4 are of equal length, 1.994(3) and 1.995(3) Å, but are not required to be so by symmetry. The Zn–C bond is slightly shorter at 1.974(4) Å. In 5, the Al–N and Al–C bonds are of similar lengths with the Al–N bonds being slightly shorter than the Al–C bonds. The bonds of the same type lying on opposite sides of the central ring are now similar in length in both the Zn and Al structures, unlike what was observed in the starting compound H[1b]. Loss of the HN hydrogen atom allows electron delocalization throughout the ring with an average bond order of 1.5 as shown in the Lewis structures for 4 and 5 in Scheme 3.
The central cores of compounds 4 and 5 have similar bond lengths and angles, however, the disposition of the side groups in the Al and Zn structures are different. The two metal–N bonds in the two Al molecules are less symmetrical than those in the Zn molecule, where they are essentially identical. For the rest of the core ring, the bond lengths and angles are more similar in the two structures. For the central core of the molecules, the interior angles at N2 and N4 are close to 120°, while the interior ring angles at P1 and P2 are generally 110°, all as expected. The P1–N1–P2 angle of the Zn compound, 132.5(2)°, is the only one not as would be predicted based on hybridization of the central atom. It is not very different from the angle in H[1b], where it is 133.38(10)°. In the Al structure, the corresponding angles are somewhat smaller, P2–N1–P1 = 123.51(9)° and P3–N6–P4 = 124.19(9)°.
The intermolecular interactions of the metal compounds differ significantly from those observed in the parent compound H[1b]. There are no longer any close stacking contacts between the fluorene rings in either structure. There is one new intramolecular stacking contact involving two of the phenyl rings in the Al compound; one such contact occurs in each independent molecule (Fig. S47 in ESI†).
There are few other reports of comparable structures to 4 and 5. Stalke and co-workers prepared and structurally characterized [N{P(NMe2)2NSiMe3}2ZnN(SiMe3)2] (vii in Fig. 7).18,94 The zinc centre of vii is also 3-coordinate forming a similar 6-membered ring as 4 in the central core of the molecule. The N–Zn bonds within the ring of vii are equivalent by symmetry and similar in length to those in 4. All the bonds of the NPNPN backbone are also of similar lengths. The angle at Zn of vii is smaller in 4, possibly because the ethyl group is less bulky than the N(SiMe3)2 group. The central ring of vii was described as slightly twisted, just as observed in 4. Overall, the geometry and conformation of the central ring is similar for vii and 4.
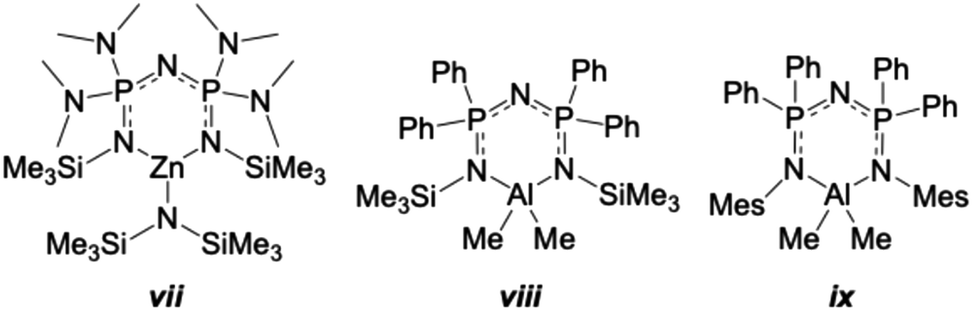 | ||
| Fig. 7 Structurally related Zn and Al complexes to 4 and 5.31,84,94 | ||
Two structures related to compound 5 have been reported, N(PPh2NSiMe3)2Al(Me)218,84 and [{N(Ph2PN(2,4,6-Me3C6H2))2}AlMe2]18,31 (viii and ix, respectively, in Fig. 7). Both viii and ix have the same core 6-membered ring as found in 5 and the geometries and conformations of the rings are broadly similar in all three structures. The bond lengths in the P–N–Al–N–P portion of the ring are slightly shorter in 5 compared to the previously reported structures viii and ix. The slight asymmetry of the Al–N bond lengths observed in 5 is also evident in ix, which has Al1–N1 = 1.936(2) Å and Al1–N3 = 1.923(2) Å. The N–Al–N angles are the smallest in 5, which likely occurs because the planar fluorene groups are less sterically demanding than either the N-SiMe3 groups of viii, or the N-(2,4,6-trimethylphenyl) groups of ix.
Compound 6
The crystal structure of 6 contains one molecule of disordered THF in the asymmetric unit. In addition to the free THF molecule there are two THF molecules, one of which is disordered, coordinated to the sodium atom. One of the DAF groups in the main molecule is also disordered, with either face of the fluorene oriented “up” in the two-part model employed. The main component refines to an occupancy of 95% with the minor component contributing only 5%. A result of this disorder is that the sodium atom bonds to N3A in the main component but to N2B in the minor one because the N2 groups of the ligand cross in the disordered arrangement (Fig. 8). | ||
| Fig. 8 The two components of the disordered model used to refine 6, with selected atoms labelled. The uncoordinated THF molecule has been removed for clarity. The major contribution (part A) is shown on the left and the minor component (part B) is shown on the right. Thermal ellipsoids have been drawn at 50% probability. | ||
In total, the sodium atom of 6 bonds to two oxygen atoms of THF groups, and to three nitrogen atoms from a deprotonated H[1b] ligand. In addition to N2B/N3A and N5 of the two DAF groups, it also bonds to N1 of the PNP portion of the molecule. The Na–O bond lengths are the shortest, but amongst the Na–N bond lengths the Na–N1 distance is significantly shorter than the others (over 0.2 Å). All the Na–N bonds are, however, longer than the Zn–N or Al–N bonds in the previous metal complexes reported.
Ignoring the minor component of the disorder, the formation of the novel Na–N1 bond results in the formation of two 5-membered rings: Na1–N3A–N2A–P1–N1 and Na1–N5–N4–P2–N1. The first of the 5-membered rings has a mean deviation from its plane of 0.325 Å (larger because of the disorder) while in the second ring it is 0.209 Å. The angle between these two planes is 31.7°. The sodium atoms do not lie in the ring planes, being 0.192 Å and 0.300 Å out of the planes 1 and 2, respectively. The Na atom can be described as having a very distorted trigonal bipyramidal geometry with the oxygen atoms of THF in the axial positions and N1, N3 and N5 in the equatorial plane.
The most visible difference between the Na product and the previous Zn and Al compounds is the bonding mode of the metal. In 4 and 5 the ligand is bidentate, but in 6 it adopts a tridentate bonding mode, which agrees with previously reported observations by Sheldrick18,84 and Stalke.18,94 The bond lengths in 6 are similar to those in 4 and 5, suggesting similar hybridization of the atoms in the core of the molecule (Table 1). The NN bonds of the DAF residue are slightly shorter in the sodium compound relative to those in the Zn and Al compounds, and shorter even than those in H[1b]. The N–CF bond lengths are longer than in any of the other compounds reported here. There is no evidence for short π–π stacking or other intermolecular interactions in 6.
Two related 3-coordinate lithium derivatives have been reported, though the most comparable to 6 is [{N(Ph2PN(2,4,6-Me3C6H2))2}Li·OEt2] (x in Fig. 9),18,31 which contains a Li(OEt2) unit bonded to the multidentate ligand through the nitrogen atoms of the two NMes groups. In the structure of x, a puckered 6-membered N3P2Li ring is formed, with no bond observed between Li and the central nitrogen atom of the PNP group. The core ring adopts a slight boat conformation, with P1 and N3 rising above the near plane of the other atoms in this six-membered chelate.
 | ||
| Fig. 9 S-block metal complexes of NPNPN ligands that are structurally similar to 6.31,95,96 | ||
To our knowledge, there are no reported structures for tridentate, W-shaped NPNPN complexes with phenyl groups bonded to phosphorus. However, such complexes, including with alkali and alkaline earth metals, have been reported with other groups bonded to P. Such structures were first discussed as a distinct group in 1994 in a review by Witt and Roesky.18 A sodium compound, {NaN[P(NMe2)2NSiMe3]2}2 (xi in Fig. 9), exhibits a similar bonding mode as found in 6.95 The structure of xi contains two 4-coordinate Na atoms. One Na coordinates to the three core nitrogen atoms of an NPNPN ligand, and the fourth bond is to a terminal nitrogen atom of the second NPNPN ligand. This results in a W-shape that is more distorted than what is observed in 6. Structures that have core geometries more like 6 include M{N[P(NMe2)2NSiMe3]2}2, for M = Ca,95 and Ba96 (xii in Fig. 9). Both the Ca and Ba complexes of xii contain 6-coordinate metal centres, making three bonds to each of two symmetrically disposed and mutually perpendicular ligands. The bond to the central nitrogen of the core NPNPN group in xii is the shortest to both ligands resulting in a W-shape resembling that found in 6. Complexes of NPNPN ligands, therefore, show a wide variety of coordination geometries and bonding modes with alkali/alkaline earth and group 13 metals.
Electrostatic potential calculations
Of key interest was the electron distribution within the new ligand systems after they had been incorporated into the organometallic complexes. Localized bonding models based on structural data provided a starting point but the true nature of the charge distribution within the CNNPNPNNC backbone was unclear. Interpretation of the resonance structures was made more complicated because of the puckering observed within the large rings; this promotes electron localization rather than delocalization. Electrostatic surface potential (ESP) calculations were carried out to provide a visual display of the relative electron distributions in the complexes 4 and 5. Knowledge of the charge distributions can be used to understand how the molecules behave, especially with respect to their intermolecular interactions in the solid state. The surface maps give an indication of polarity and show nucleophilic/electrophilic regions, all important contributors to catalytic activity. The molecular computations were begun from the constrained heavy atom geometries determined in the X-ray analyses. The structures were optimized (using Spartan'20 software)97 at the DFT/6-31G* level of theory, and the optimized atomic coordinates can be found in the ESI.† Electrostatic potential values were then mapped onto a 0.002 au isosurface and plotted (Fig. 10). A legend is provided between the two maps which are presented on a common scale. | ||
| Fig. 10 (Top) Electrostatic potential plots of compounds 5 and 6. Electrostatic potential values are mapped onto a 0.002 au isosurface. A common scale is shown between the two maps. (Bottom) The molecular structures of the same two compounds drawn in the same orientation as shown in the electrostatic potential maps. | ||
The electrostatic potential plots clearly show that most of the relative positive regions are distributed onto the hydrogen atoms of the phenyl rings but to a lesser extent onto the hydrogen atoms of the fluorenyl rings. The nitrogen atoms within the rings all exhibit relative negative ESPs, as do the exocyclic nitrogen atoms of the DAF groups in some cases. There are also regions of negative electrostatic potential around the organometallic fragments, particularly on the alkyl groups. It is clear from the results of this study that simple resonance structures are not sufficient to predict the nuanced electronic distribution in these molecules.
Polymerization experiments
The catalysis of the ROCOP reactions of cyclohexene oxide (CHO) and phthalic anhydride (PA) using H[1b], 4, and 5 (Table 3), with and without the use of bis(triphenylphosphine)iminium chloride (PPNCl) as a co-catalyst (Scheme 5), were studied. Optimum conversions of PA were obtained when a co-catalyst was used at 80 °C giving effectively complete conversion to perfectly alternating copolymer within 4 h in neat CHO. Samples were immediately characterized by 1H NMR spectroscopy to determine the conversion to polyether or polyester, for the ROP of epoxide or ROCOP, respectively.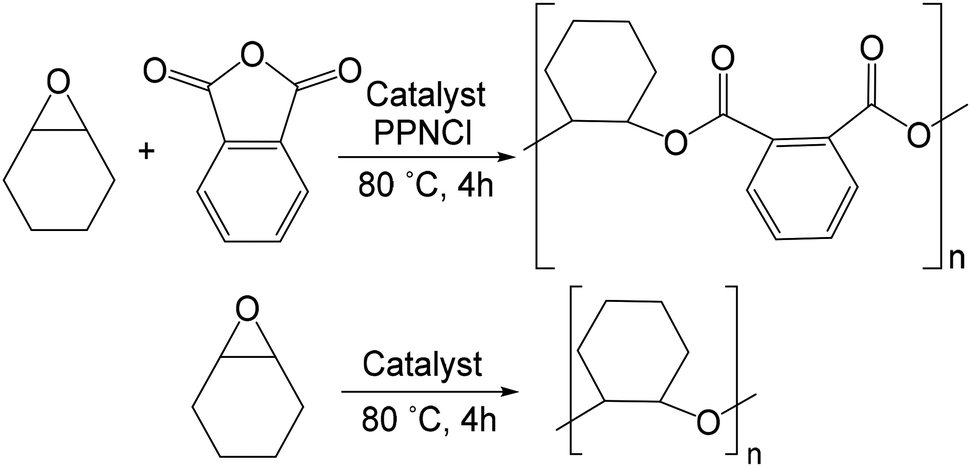 | ||
| Scheme 5 ROCOP of CHO and PA to form polyester (top) and ROP of CHO to form polyether (bottom). | ||
a
| Entry | Catalyst | Conversion% of PAb | Ester:ether linkagesb |
M
n Calc (kg mol−1) |
M
nc (kg mol−1) |
Đ (Mw/Mn)c | TONd |
|---|---|---|---|---|---|---|---|
|
a Reaction conditions unless otherwise stated: 1:1:100:500 catalyst:PPNCl:PA:CHO. 80 °C, 4 h. All reactions were carried out in neat epoxide. Crystalline form of catalyst used for each reaction.
b Determined by 1H NMR spectroscopy.
c
M
n Cal = ({M of PA + CHO} × [PA]/[cat] × conversion (%)). Mn and Đ (Mw/Mn) determined by gel permeation chromatography (GPC) in THF with universal calibration.
d TON, turnover number of PA to ester linkages = [(mol PA/mol catalyst) × conversion (%)].
e Reaction was performed without the use of a co-catalyst.
f ND = not determined.
g TON, turnover number of CHO to ether linkages = [(mol PA converted/mol catalyst) × (% ether linkage/% ester linkage)].
h Reaction was performed at 20 °C, over 3 h.
i Reaction was performed with only PPNCl co-catalyst.
|
|||||||
| 1 | H[1b] | >99 | 99:1 |
24.6 | 7.7 | 1.05 | 100 |
| 2 | H[1b]e | NDf | ND | ND | ND | ND | ND |
| 3 | 4 | >99 | 99:1 |
24.6 | 10.1 | 1.25 | 100 |
| 4 |
4e |
22 | 7:93 |
32.7 | 21.5 | 2.09 | 22 (292g) |
| 5 | 5 | >99 | 99:1 |
24.6 | 7.1 | 1.04 | 100 |
| 6 |
5e |
33 | 7:93 |
46.1 | 8.8 | 3.29 | 33 (438g) |
| 7 |
4h |
10 | 99:1 |
ND | ND | ND | ND |
| 8 |
4e,h |
5.6 | 24:76 |
1.8 | 52.5 | 1.74 | 5 (18g) |
| 9 |
5h |
12 | 99:1 |
ND | ND | ND | ND |
| 10 |
5e,h |
5 | 4:96 |
13.5 | 71.3 | 1.83 | 5 (120g) |
| 11i | N.A. | >99 | 87:13 |
24.6 | 12.2 | 1.34 | 100 |
Interestingly, all compounds H[1b], 4, and 5 displayed excellent catalytic utility for the polymerization of epoxide/cyclic anhydride to form copolymer. With PPNCl as a co-catalyst, H[1b], 4, and 5 showed complete conversion of PA with high selectivity for ester linkages under the conditions in Table 3, entries 1, 3, and 5. The observed polymer molecular masses were lower than the calculated masses, but the polymer mass dispersities were narrow. In the absence of the co-catalyst, no conversion of either PA or CHO was observed with H[1b] (Table 3, entry 2), while 4 and 5 displayed higher selectivity for the ROP of CHO (Table 3, entries 4 and 6) leading to polymers with a high ratio of ether-to-ester linkages. The poly(ether-co-ester)s obtained were more disperse than the completely alternating polyesters obtained in the presence of the co-catalyst.
The MALDI-TOF mass spectra of the polymers obtained for entries 3 and 4 in Table 3 are shown in the ESI.† The polyester produced in the presence of 4 (Table 3, entry 3) shows groups of three different repeating mass distributions that are separated by mass differences of 246 Da, which corresponds to a repeating unit of PA/CHO. Of the three repeating mass groups, the most intense isotopic pattern can be modeled as the expected polymer ion consisting of polyester with chloride and hydroxyl end groups charge balanced with a Na+ ion. The least intense isotopic pattern can be modeled as polyester possessing two hydroxyl end groups and a Na+ ion. The third isotopic pattern is from a polymer having chloride and hydroxyl end groups and a Na+ cation, but with the presence of both ester and ether linkages (Fig. S113 in ESI†). The polyester produced by 5 (Table 3, entry 5) exhibits a similar MALDI-TOF mass spectrum but with only two repeating sets of isotopic patterns. Here the polyester diol appears to be absent. Only the polyester with chloride and hydroxyl end groups cationized by a Na+ ion and the ether linkage containing polymer are present (Fig. S120 in ESI†).
Compounds 4 and 5 also display catalytic activity for the polymerization of epoxide/cyclic anhydride at a reduced temperature of 20 °C, though the conversion is poor with respect to those done at 80 °C. The 1H NMR conversion suggests that these polymers (Table 3, entries 8 and 10) are most likely CHO polyethers. From the GPC analysis, the Mn is significantly higher than the respective trials performed at 80 °C, while the dispersities are lower: 52.5 kg mol−1, Đ = 1.74 vs. 21.5 kg mol−1, Đ = 2.09 (Table 3, entries 8 and 4, respectively) and 71.3 kg mol−1, Đ = 1.83 vs. 8.8 kg mol−1, Đ = 3.29 (Table 3, entries 10 and 6, respectively). PPNCl alone without added metal complex does exhibit some activity toward polymerization (entry 11), but the polymer obtained has poor selectivity for ester linkages and gives broader polymer mass dispersity than when used alongside the metal complexes (entries 3 and 5) or indeed the proligand and PPNCl (entry 1).
Conclusions
Compounds H[1a] and H[1b] and their derived complexes can be easily synthesized with high yields and are stable under benchtop conditions. Compound 2b (the hydrochloride salt of H[1b]) and 6 could be useful starting materials for future synthesis of metal complexes by protonolysis or salt metathesis routes, respectively. Initial studies of complexes 4 and 5 have shown they display good activity as catalysts for the ROCOP of CHO and PA to form polyester and for the ROP of CHO to form polyether. 4 and 5 show co-catalyst-dependant selectivity for polyester or polyether formation.Experimental
General experimental
All preparations and manipulations were carried out in a nitrogen-filled MBraun glove box or under Schlenk conditions. Trimethyl aluminum (1 M in heptane) was purchased from Sigma Aldrich and diethyl zinc (10 wt% in hexane) was purchased from Strem. The PNP precursors iPr-PNP, and Ph-PNP were prepared and purified according to literature methods.24,26 9-Diazofluorene was synthesized using literature methods and purified by crystallization.98–100 Dry THF was obtained by distillation from sodium/benzophenone ketyl under nitrogen. Other solvents were purified over activated alumina under dry nitrogen using an MBraun solvent purification system.High resolution mass spectra (HRMS) of H[1a], H[1b], 2a and 3 were recorded on a Bruker Daltonics microTOF instrument, and of 4, 5, and 6 on a Sciex 7600 ZenoTOF instrument. Positive and negative ionizations via electrospray ionization (ESI) were used. Melting points were determined using an Electrothermal IA9000 Series melting point apparatus and are uncorrected. 1H, 13C{1H}, and 31P{1H} NMR spectra were recorded on a Bruker 300 MHz spectrometer at 300.26 MHz, 75.50 MHz, and 121.54 MHz, respectively. Anhydrous deuterated solvents for NMR spectroscopy were purchased from Sigma Aldrich and used as supplied. 1H and 13C{1H} NMR spectral shifts are reported in relation to either known residual solvent peaks or TMS, when present. 31P{1H} spectral shifts are externally referenced to 85% H3PO4 in D2O. Data were processed using Bruker Topspin software. Values are reported as follows: chemical shift (δ, ppm), integration, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, dd = doublet of doublets, m = multiplet), coupling constant (Hz), and assignment. The infrared spectra of all solids were collected as neat samples using a Bruker Vertex 70 infrared spectrometer at room temperature. Details of the X-ray crystallography are provided in the ESI.†
Polymer molar masses and dispersities were determined by gel permeation chromatography (GPC) using an Agilent 1260 Infinity high-performance liquid chromatograph coupled to a Wyatt Technologies triple detector system (light scattering, viscometer, and refractive index) and equipped with two phenogel columns (103 Å and 104 Å, 300 mm × 4.60 mm covering mass ranges of 1–75 and 5–500 kDa, respectively). HPLC grade THF was used as the eluent at a flow rate of 0.3 mL min−1 at 25 °C. The GPC data were processed using the Astra 6 software package.
MALDI-TOF mass spectrometry on the polymers was performed using a Bruker ultrafleXtreme MALDI TOF/TOF analyzer with a Bruker smartbeam-II laser (up to 2 kHz, operating at 355 nm) for linear and reflectron mode. Mass spectra of 1000 shots were accumulated. A cationizing agent salt solution (sodium trifluoroacetate, NaTFA) in THF was used at a concentration of 1 M. Polymer was dissolved in THF to a concentration of 10 mg mL−1, then combined with the dihydroxybenzoic acid (DHBA) matrix and NaTFA in a ratio of 3:20:1 μL, respectively. Aliquots of 0.5 μL of these solutions were spotted and the solvent was allowed to evaporate. MALDI-TOF MS data were processed, and images prepared using MestReNova software with the mass analysis plug-in.
H3 of iPr), 27.32 (dd, JCP = 87.02, 4.04, H of iPr), 119.60 (d, JCP = 5.25 Hz), 120.20, 126.39, 127.19, 127.59, 128.08, 130.53, 138.28, 139.07, 139.60. 31P{1H} NMR (121 Hz, CDCl3, ppm): δ 51.01 (s, N![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) N). IR (ATR, cm−1): 3383 (w), 3309 (w), 3195 (w), 3051 (w), 2928 (w), 2052 (w), 1607 (w), 1574 (m), 1445 (m), 1185, (m), 777 (m), 726 (vs). HRMS calcd for [C38H46N5P2]+, [M + H]+: 634.3228; found: 634.3225.
N), 20.09 (d, O). IR (ATR, cm−1): 3056 (w), 2922 (w), 2851 (w), 1673 (w), 1618 (w), 1590 (w), 1436 (m), 1370 (s), 1234 (vs), 1171 (s), 1118 (s), 724 (vs), 690 (vs), 538 (s), 504 (s). HRMS calcd for [C37H30N3P2O]+, [M + H]+: 594.1864; found: 594.1872.
N, from H[1b] contaminant), 27.77 (s, NN). 27Al NMR (78 Hz, C6D6, ppm): δ 69.6 (s br, ω1/2 = 2573.0 Hz). IR (ATR, cm−1): 3306 (w), 3050 (w), 2927 (w), 2888 (w), 2857 (w), 1731 (w), 1645 (m), 1587 (m), 1532 (m), 1468 (m), 1448 (m), 1435 (m), 1374 (m), 1311 (w), 1227 (m), 1171 (m), 1111 (m), 1093 (m), 1060 (m), 1025 (s), 982 (s), 968 (s), 883 (s), 849 (m), 775 (s), 725 (vs), 688 (vs), 6354 (s), 596 (vs), 528 (vs), 514 (vs), 463 (vs), 416 (s). HRMS calcd for [C52H43N5P2Al]+, [M + H]+: 826.2809; found: 826.3108. Anal. calcd (found) for C52H42N5P2Al: 75.63 (74.13); H, 5.13 (5.30); N, 8.48 (8.12). Anal. calcd for C52H42N5P2Al(C4H8O)0.35(CH2Cl2)0.2: 74.16; H, 5.25; N, 8.07.
N). IR (ATR, cm−1): 3383 (w), 3309 (w), 3195 (w), 3051 (w), 2928 (w), 2052 (w), 1607 (w), 1574 (m), 1445 (m), 1185, (m), 777 (m), 726 (vs). HRMS calcd for [C38H46N5P2]+, [M + H]+: 634.3228; found: 634.3225.
N), 20.09 (d, O). IR (ATR, cm−1): 3056 (w), 2922 (w), 2851 (w), 1673 (w), 1618 (w), 1590 (w), 1436 (m), 1370 (s), 1234 (vs), 1171 (s), 1118 (s), 724 (vs), 690 (vs), 538 (s), 504 (s). HRMS calcd for [C37H30N3P2O]+, [M + H]+: 594.1864; found: 594.1872.
N, from H[1b] contaminant), 27.77 (s, NN). 27Al NMR (78 Hz, C6D6, ppm): δ 69.6 (s br, ω1/2 = 2573.0 Hz). IR (ATR, cm−1): 3306 (w), 3050 (w), 2927 (w), 2888 (w), 2857 (w), 1731 (w), 1645 (m), 1587 (m), 1532 (m), 1468 (m), 1448 (m), 1435 (m), 1374 (m), 1311 (w), 1227 (m), 1171 (m), 1111 (m), 1093 (m), 1060 (m), 1025 (s), 982 (s), 968 (s), 883 (s), 849 (m), 775 (s), 725 (vs), 688 (vs), 6354 (s), 596 (vs), 528 (vs), 514 (vs), 463 (vs), 416 (s). HRMS calcd for [C52H43N5P2Al]+, [M + H]+: 826.2809; found: 826.3108. Anal. calcd (found) for C52H42N5P2Al: 75.63 (74.13); H, 5.13 (5.30); N, 8.48 (8.12). Anal. calcd for C52H42N5P2Al(C4H8O)0.35(CH2Cl2)0.2: 74.16; H, 5.25; N, 8.07.
Method A. 1b (0.31 g, 0.40 mmol) was dissolved in anhydrous THF. To this deep yellow solution, an excess of sodium hydride (41.6 mg, 1.73 mmol) was added. Within one minute of stirring, a rapid evolution of gas bubbles was observed. The solution rapidly became dark teal in colour. After 5 min, the evolution of gas ceased. The solution was decanted to remove the excess unreacted sodium hydride. The decanted solution was layered with heptane and placed at −30 °C overnight. Upon warming the mother liquor to room temperature, yellow prismatic crystals began to form from the dark teal solution. Yield = 0.30 g (0.30 mmol, 73%).
Method B. Bis(diphenylphosphino)amine (0.58 g, 1.50 mmol) was dissolved in 10 mL of a 50
:50 toluene/THF mixture. To this was added 2 eq. 9-diazofluorene (0.60 g, 3.12 mmol) forming a red solution. To this solution was added an excess of sodium hydride causing a lightening of the red colour and a slow evolution of gas bubbles. The mixture was stirred overnight resulting in a dark teal solution. The solution was decanted to remove the excess sodium hydride, layered with heptane and placed at −30 °C overnight. Upon warming to room temperature, yellow prismatic crystals formed from the dark teal solution. Yield = 0.36 g (0.36 mmol, 23%). 1H NMR (300 MHz, CD2Cl2, ppm): δ 1.70 (m, 8H), 3.51 (m, 8H), 6.72 (t, 1H, JHH = 7.55 Hz), 6.96 (t, 1H, JHH = 7.55 Hz), 7.06 (t, 1H, JHH = 7.55 Hz), 7.12–7.22 (m, 5H), 7.27 (t, 1H, JHH = 7.55 Hz), 7.32–7.56 (m, 15H), 7.67 (d, 1H JHH = 7.55 Hz), 7.74 (d, 1H, JHH = 7.55 Hz), 7.96–8.09 (m, 8H), 8.19 (d, 1H, JHH = 7.55 Hz), 8.69 (d, 1H, JHH = 7.55 Hz). 13C{1H} NMR (75 MHz, CD2Cl2, ppm): δ 25.86, 68.10, 118.84, 119.66 (d, JCP = 11.55 Hz), 119.81, 119.85 (d, JCP = 11.55 Hz), 120.49, 125.58, 126.47 (d, JCP = 9.35 Hz), 127.21, 127.58, 127.89 (d, JCP = 12.38 Hz), 128.40, 128.48, 128.65, 128.83, 130.74, 131.02, 132.06, 132.73, 132.79, 134.09, 134.94, 136.52, 137.84, 138.44, 138.79, 139.19, 140.30. 31P{1H} NMR (121 Hz, THF + C6D6, ppm): δ 18.0 (s). 31P{1H} NMR (THF + CD2Cl2, ppm): δ 17.6 (s). IR (ATR, cm−1): 3050 (w), 2925 (w), 2868 (w), 1610 (w), 1516 (m), 1480 (w), 1432 (m), 1340 (w), 1326 (w), 1278 (w), 1246 (m), 1225 (m), 1170 (s), 1095 (vs), 1049 (vs), 1029 (s), 972 (s), 890 (m), 870 (m), 842 (s), 787 (m), 773 (vs), 743 (m), 727 (vs), 690 (vs), 644 (m), 623 (m), 599 (m), 538 (m), 516 (vs), 478 (vs), 448 (m), 429 (m). HRMS calcd for [C58H52N5P2O2Na (THF coordinated complex)]+, [M + H]+: 936.3416; found: 936.3064. HRMS calcd (no THF coordinated) for [C50H37N5P2Na]+, [M + H]+: 792.2422; found: 792.2496. Anal. calcd (found) for C58H52N5P2NaO2: C, 74.43 (71.94); H, 5.60 (5.11); N, 7.48 (7.64). Anal. calcd for C50H36N5P2Na(C4H8O)2(CH2Cl2)0.4: C, 72.34; H, 5.47; N, 7.25.
Representative ring-opening copolymerization of cyclohexene oxide with phthalic anhydride
A mixture of compound 4 (6.1 mg, 7.38 μmol), phthalic anhydride (118.3 mg, 0.79 mmol), and cyclohexene oxide (377.7 mL, 3.85 mmol) were combined in a thick-walled crimp-seal vial containing a small magnetic stir bar at 22 °C in a glovebox and sealed with a crimped septum cap under a N2 atmosphere. The resulting yellow solution was stirred magnetically at 80 °C for the desired reaction time or until stirring became hindered by increased sample viscosity. The resulting orange solution was exposed to air to quench the reaction, and an aliquot was taken immediately for 1H NMR determination of the conversion to products. The remaining solution was dissolved in dichloromethane (∼5 mL) and the polymer was precipitated by addition of 5% (v/v with 1 M HCl) acidified methanol. The resulting off-white polymer was dried in vacuo for 2 h.Author contributions
Conceptualization: Clyburne, Kozak. Supervision: Clyburne, Kozak, Robertson. Funding acquisition: Clyburne, Kozak. Investigation and methodology: Laprade. Formal analysis: Laprade. Writing – original draft: Laprade. Writing – review and editing: Clyburne, Kozak, Robertson.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for H[1a], H[1b], 2a, 2b, 3, 4, 5, and 6 have been deposited at the Cambridge Crystallographic Data Centre under 2352790–2352797† respectively.Acknowledgements
We gratefully acknowledge financial support from Saint Mary's University, Memorial University of Newfoundland, the Natural Sciences and Engineering Research Council of Canada (NSERC) for Discovery Grants to JACC and CMK, the Canada Foundation for Innovation (CFI), the Nova Scotia Research and Innovation Trust Fund, and the Newfoundland and Labrador Department of Industry, Energy and Technology. We also acknowledge Patricia Granados of Saint Mary's University (Halifax, NS, Canada) for EA and LRMS acquisition, Xiao Feng of Dalhousie University (Halifax, NS, Canada) and Stefana Egli of Memorial University of Newfoundland (St John's, NL, Canada) for HRMS acquisition, and Memorial University's CREAIT Network for their support with this work. MJJL thanks the Memorial University Department of Chemistry for a Dr Liqin Chen Graduate Excellence Award.References
- R. J. Lundgren and M. Stradiotto, Key Concepts in Ligand Design, Ligand Design in Metal Chemistry, John Wiley & Sons, Ltd, 2016, pp. 1–14 Search PubMed.
- D. J. Durand and N. Fey, Chem. Rev., 2019, 119, 6561–6594 CrossRef CAS PubMed.
- A. E. R. Watson, M. J. Grant, P. D. Boyle, P. J. Ragogna and J. B. Gilroy, Inorg. Chem., 2022, 61, 18719–18728 CrossRef CAS PubMed.
- N. J. Hardman, M. B. Abrams, M. A. Pribisko, T. M. Gilbert, R. L. Martin, G. J. Kubas and R. T. Baker, Angew. Chem., Int. Ed., 2004, 43, 1955–1958 CrossRef CAS PubMed.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS PubMed.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- D.-R. Dauer and D. Stalke, Dalton Trans., 2014, 43, 14432–14439 RSC.
- M. A. Land, B. Huo, K. N. Robertson, K. E. O. Ylijoki, P. T. K. Lee, J. Areephong, D. Vidović and J. A. C. Clyburne, Dalton Trans., 2018, 47, 10195–10205 RSC.
- B. Murugesapandian, R. Ganguly, P. T. K. Lee, M. Petković, J. A. C. Clyburne and D. Vidović, Aust. J. Chem., 2020, 73, 1219 CrossRef CAS.
- D. C. H. Do, A. Keyser, A. V. Protchenko, B. Maitland, I. Pernik, H. Niu, E. L. Kolychev, A. Rit, D. Vidovic, A. Stasch, C. Jones and S. Aldridge, Chem. – Eur. J., 2017, 23, 5830–5841 CrossRef CAS PubMed.
- M. S. Balakrishna, V. S. Reddy, S. S. Krishnamurthy, J. F. Nixon and J. C. T. R. Burckett St. Laurent, Coord. Chem. Rev., 1994, 129, 1–90 CrossRef CAS.
- P. Bhattacharyya and J. D. Woollins, Polyhedron, 1995, 14, 3367–3388 CrossRef CAS.
- A. Chakraborty, N. Ahmed and V. Chandrasekhar, in Phosphazenes, Organophosphorus Chemistry, ed. L. J. Higham, D. W. Allen and J. C. Tebby, 2022, vol. 51, pp. 355–397 Search PubMed.
- V. Chandrasekhar and R. S. Narayanan, in Phosphazenes, Organophosphorus Chemistry, ed. D. W. Allen, D. Loakes and J. C. Tebby, 2017, vol. 46, pp. 342–417 Search PubMed.
- V. Chandrasekhar, P. Thilagar and B. M. Pandian, Coord. Chem. Rev., 2007, 251, 1045–1074 CrossRef CAS.
- C. Fliedel, A. Ghisolfi and P. Braunstein, Chem. Rev., 2016, 116, 9237–9304 CrossRef CAS PubMed.
- M. D. Ryabochenko, K. V. Shumikhin, O. A. Sinegribova and A. M. Chekmarev, Theor. Found. Chem. Eng., 2020, 54, 786–791 CrossRef CAS.
- M. Witt and H. W. Roesky, Chem. Rev., 1994, 94, 1163–1181 CrossRef CAS.
- A. Steiner, S. Zacchini and P. I. Richards, Coord. Chem. Rev., 2002, 227, 193–216 CrossRef CAS.
- B. Goswami and P. W. Roesky, Inorg. Chem. Front., 2025, 12, 3555–3581 RSC.
- A. Jain, H. Karmakar, P. W. Roesky and T. K. Panda, Chem. Rev., 2023, 123, 13323–13373 CrossRef CAS PubMed.
- D. A. Dickie, M. T. Barker, M. A. Land, K. E. Hughes, J. A. C. Clyburne and R. A. Kemp, Inorg. Chem., 2015, 54, 11121–11126 CrossRef CAS PubMed.
- D. A. Dickie, U. Chadha and R. A. Kemp, Inorg. Chem., 2017, 56, 7292–7300 CrossRef CAS PubMed.
- D. A. Dickie, E. N. Coker and R. A. Kemp, Inorg. Chem., 2011, 50, 11288–11290 CrossRef CAS PubMed.
- T. Q. Ly and J. D. Woollins, Coord. Chem. Rev., 1998, 176, 451–481 CrossRef CAS.
- H. Nöth and E. Fluck, Z. Naturforsch., B: J. Chem. Sci., 1984, 39, 744–753 CrossRef.
- R. D. Riley, D. A. Dickie, M. A. Land, R. A. Kemp, C. L. B. Macdonald, U. Werner-Zwanziger, K. N. Robertson and J. A. C. Clyburne, Chem. – Eur. J., 2020, 26, 7711–7719 CrossRef CAS PubMed.
- R. D. Riley, K. N. Robertson and J. A. C. Clyburne, Can. J. Chem., 2022, 100, 737–750 CrossRef CAS.
- J. S. Ritch, T. Chivers, K. Ahmad, M. Afzaal and P. O'Brien, Inorg. Chem., 2010, 49, 1198–1205 CrossRef CAS PubMed.
- A. Bollmann, K. Blann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. S. McGuinness, D. H. Morgan, A. Neveling, S. Otto, M. Overett, A. M. Z. Slawin, P. Wasserscheid and S. Kuhlmann, J. Am. Chem. Soc., 2004, 126, 14712–14713 CrossRef CAS PubMed.
- K. Jaiswal, B. Prashanth, D. Bawari and S. Singh, Eur. J. Inorg. Chem., 2015, 2015, 2565–2573 CrossRef CAS.
- K. Jaiswal, B. Prashanth and S. Singh, Chem. – Eur. J., 2016, 22, 11035–11041 CrossRef CAS PubMed.
- W. Rong, J. Cheng, Z. Mou, H. Xie and D. Cui, Organometallics, 2013, 32, 5523–5529 CrossRef CAS.
- W. Rong, D. Liu, H. Zuo, Y. Pan, Z. Jian, S. Li and D. Cui, Organometallics, 2013, 32, 1166–1175 CrossRef CAS.
- W. Rong, M. Wang, S. Li, J. Cheng, D. Liu and D. Cui, Organometallics, 2018, 37, 971–978 CrossRef CAS.
- T. Tannoux and A. Auffrant, Coord. Chem. Rev., 2023, 474, 214845 CrossRef CAS.
- T. Agapie, Coord. Chem. Rev., 2011, 255, 861–880 CrossRef CAS.
- J. T. Dixon, M. J. Green, F. M. Hess and D. H. Morgan, J. Organomet. Chem., 2004, 689, 3641–3668 CrossRef CAS.
- D. S. McGuinness, Chem. Rev., 2011, 111, 2321–2341 CrossRef CAS PubMed.
- P. W. N. M. van Leeuwen, N. D. Clément and M. J. L. Tschan, Coord. Chem. Rev., 2011, 255, 1499–1517 CrossRef CAS.
- D. F. Wass, Dalton Trans., 2007, 816–819 RSC.
- M. Chen and C. Chen, Angew. Chem., Int. Ed., 2018, 57, 3094–3098 CrossRef CAS PubMed.
- N. A. Cooley, S. M. Green, D. F. Wass, K. Heslop, A. G. Orpen and P. G. Pringle, Organometallics, 2001, 20, 4769–4771 CrossRef CAS.
- S. Härzschel, F. E. Kühn, A. Wöhl, W. Müller, M. H. Al-Hazmi, A. M. Alqahtani, B. H. Müller, N. Peulecke and U. Rosenthal, Catal. Sci. Technol., 2015, 5, 1678–1682 RSC.
- Y. Zhou, H. Wu, S. Xu, X. Zhang, M. Shi and J. Zhang, Dalton Trans., 2015, 44, 9545–9550 RSC.
- F. Guo, Y. Yin, Z. Li, Y. Xu, S. Cao, Z. Liu, Y. Shi, C. Li and K. Guo, Eur. Polym. J., 2025, 229, 113848 CrossRef CAS.
- P. W. Roesky, M. T. Gamer, M. Puchner and A. Greiner, Chem. – Eur. J., 2002, 8, 5265–5271 CrossRef CAS PubMed.
- L. Wang, J. Zhang, N. Zhao, C. Ren, S. Liu and Z. Li, ACS Macro Lett., 2020, 9, 1398–1402 CrossRef CAS PubMed.
- J. Zhang, L. Wang, S. Liu and Z. Li, J. Polym. Sci., 2020, 58, 803–810 CrossRef CAS.
- K. Ambrose, J. N. Murphy and C. M. Kozak, Inorg. Chem., 2020, 59, 15375–15383 CrossRef CAS PubMed.
- K. Ambrose, K. N. Robertson and C. M. Kozak, Dalton Trans., 2019, 48, 6248–6260 RSC.
- T. S. Anderson and C. M. Kozak, Eur. Polym. J., 2019, 120, 109237 CrossRef CAS.
- K. A. Andrea and F. M. Kerton, Polym. J., 2021, 53, 29–46 CrossRef.
- R. K. Dean, A. M. Reckling, H. Chen, L. N. Dawe, C. M. Schneider and C. M. Kozak, Dalton Trans., 2013, 42, 3504–3520 RSC.
- A. M. DiCiccio, J. M. Longo, G. G. Rodríguez-Calero and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 7107–7113 CrossRef CAS PubMed.
- M.-L. Lin and C.-Y. Tsai, Inorg. Chem., 2023, 62, 12298–12307 CrossRef CAS PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- C. W. Vos, J. Beament and C. M. Kozak, Polym. Chem., 2023, 14, 5083–5093 RSC.
- B. A. Abel, C. A. L. Lidston and G. W. Coates, J. Am. Chem. Soc., 2019, 141, 12760–12769 CrossRef CAS PubMed.
- T. Aida and S. Inoue, Macromolecules, 1981, 14, 1166–1169 CrossRef CAS.
- T. Aida, K. Wada and S. Inoue, Macromolecules, 1987, 20, 237–241 CrossRef CAS.
- C. G. Rodriguez, R. C. Ferrier Jr., A. Helenic and N. A. Lynd, Macromolecules, 2017, 50, 3121–3130 CrossRef CAS.
- M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- E. D. Cross, G. K. Tennekone, A. Decken and M. P. Shaver, Green Mater., 2013, 1, 79–86 CrossRef.
- S. Dagorne, M. Bouyahyi, J. Vergnaud and J.-F. Carpentier, Organometallics, 2010, 29, 1865–1868 CrossRef CAS.
- K. Endo, Y. Liu, H. Ube, K. Nagata and M. Shionoya, Nat. Commun., 2020, 11, 6263 CrossRef CAS PubMed.
- N. Ikpo, J. C. Flogeras and F. M. Kerton, Dalton Trans., 2013, 42, 8998–9006 RSC.
- N. C. Johnstone, E. S. Aazam, P. B. Hitchcock and J. R. Fulton, J. Organomet. Chem., 2010, 695, 170–176 CrossRef CAS.
- W. Li, H. Ouyang, L. Chen, D. Yuan, Y. Zhang and Y. Yao, Inorg. Chem., 2016, 55, 6520–6524 CrossRef CAS PubMed.
- W. Li, W. Wu, Y. Wang, Y. Yao, Y. Zhang and Q. Shen, Dalton Trans., 2011, 40, 11378 RSC.
- W. Li, Y. Yao, Y. Zhang and Q. Shen, Chin. J. Chem., 2012, 30, 609–615 CrossRef CAS.
- Y. Liu, L. N. Dawe and C. M. Kozak, Dalton Trans., 2019, 48, 13699–13710 RSC.
- N. Merle, K. W. Törnroos, V. R. Jensen and E. Le Roux, J. Organomet. Chem., 2011, 696, 1691–1697 CrossRef CAS.
- N. Nakata, Y. Saito and A. Ishii, Organometallics, 2014, 33, 1840–1844 CrossRef CAS.
- H. Plommer, I. Reim and F. M. Kerton, Dalton Trans., 2015, 44, 12098–12102 RSC.
- H. Plommer, L. Stein, J. N. Murphy, N. Ikpo, N. Mora-Diez and F. M. Kerton, Dalton Trans., 2020, 49, 6884–6895 RSC.
- A. Thevenon, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2015, 54, 11906–11915 CrossRef CAS PubMed.
- N. J. Van Zee, M. J. Sanford and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 2755–2761 CrossRef CAS PubMed.
- A. Virachotikul, N. Laiwattanapaisarn, P. Wongmahasirikun, P. Piromjitpong, K. Chainok and K. Phomphrai, Inorg. Chem., 2020, 59, 8983–8994 CrossRef CAS PubMed.
- R. Xu, L. Hua, X. Li, Y. Yao, X. Leng and Y. Chen, Dalton Trans., 2019, 48, 10565–10573 RSC.
- K. Yin, L. Hua, L. Qu, Q. Yao, Y. Wang, D. Yuan, H. You and Y. Yao, Dalton Trans., 2021, 50, 1453–1464 RSC.
- C.-Y. Yu, H.-J. Chuang and B.-T. Ko, Catal. Sci. Technol., 2016, 6, 1779–1791 RSC.
- R. Hasselbring, H. W. Roesky, A. Heine, D. Stalke and G. M. Sheldrick, Z. Naturforsch., B, 1994, 49, 43–49 CrossRef CAS.
- J. W. Akitt, Aluminum, Gallium, Indium and Thallium, in Multinuclear NMR, ed. J. Mason, Plenum Press, New York, 1987, pp. 259–292 Search PubMed.
- I. Krossing, H. Nöth, C. Tacke, M. Schmidt and H. Schwenk, Chem. Ber., 1997, 130, 1047–1052 CrossRef CAS.
- I. Krossing, H. Nöth and H. Schwenk-Kircher, Eur. J. Inorg. Chem., 1998, 1998, 927–939 CrossRef.
- B. Prashanth, M. Bhandari, S. Ravi, K. R. Shamasundar and S. Singh, Chem. – Eur. J., 2018, 24, 4794–4799 CrossRef CAS PubMed.
- E. Rivard, P. J. Ragogna, A. R. McWilliams, A. J. Lough and I. Manners, Inorg. Chem., 2005, 44, 6789–6798 CrossRef CAS PubMed.
- R. Riedel, A. G. Seel, D. Malko, D. P. Miller, B. T. Sperling, H. Choi, T. F. Headen, E. Zurek, A. Porch, A. Kucernak, N. C. Pyper, P. P. Edwards and A. G. M. Barrett, J. Am. Chem. Soc., 2021, 143, 3934–3943 CrossRef CAS PubMed.
- R. M. Diaz-Rodriguez, K. N. Robertson and A. Thompson, Chem. Commun., 2018, 54, 13139–13142 RSC.
- D. Bethell, M. P. Brown, M. M. Harding, C. A. Herbert, M. M. Khodaei, M. I. Rios and K. Woolstencroft, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 683–687 CrossRef.
- A. Trofimova, J. H. W. LaFortune, Z.-W. Qu, S. A. Westcott and D. W. Stephan, Chem. Commun., 2019, 55, 12100–12103 RSC.
- S. K. Pandey, A. Steiner, H. W. Roesky and D. Stalke, Inorg. Chem., 1993, 32, 5444–5446 CrossRef CAS.
- R. Hasselbring, K. Pandey, H. W. Roesky, D. Stalke and A. Steiner, J. Chem. Soc., Dalton Trans., 1993, 3447–3451 RSC.
- S. K. Pandey, A. Steiner, H. W. Roesky and D. Stalke, Angew. Chem., Int. Ed. Engl., 1993, 32, 596–598 CrossRef.
- Wavefunction Spartan 20, Wavefunction Inc, Irvine, California, USA, 2019 Search PubMed.
- M. Z. Barakat, M. F. Abdel-Wahab and M. M. El-Sadr, J. Chem. Soc., 1956, 4685–4687 CAS.
- P. J. Davis, L. Harris, A. Karim, A. L. Thompson, M. Gilpin, M. G. Moloney, M. J. Pound and C. Thompson, Tetrahedron Lett., 2011, 52, 1553–1556 CrossRef CAS.
- K. H. Lee and K. Y. Ko, Bull. Korean Chem. Soc., 2006, 27, 185–186 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2352790–2352797. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03402c |
| This journal is © The Royal Society of Chemistry 2025 |