
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium(II) and copper(II) polyamine complexes as promising antitumor agents: synthesis, characterization, and biological evaluation†
Yoel
Garrosa-Miró
abc,
Laura
Muñoz-Moreno
d,
Gerardino
D'Errico
 e,
Matilde
Tancredi
e,
M. Jose
Carmena
d,
M. Francesca
Ottaviani
f,
Paula
Ortega
*abc and
Javier
de la Mata
*abc
e,
Matilde
Tancredi
e,
M. Jose
Carmena
d,
M. Francesca
Ottaviani
f,
Paula
Ortega
*abc and
Javier
de la Mata
*abc
aUniversidad de Alcalá, Department of Organic and Inorganic Chemistry, and Research Institute in Chemistry “Andrés M. del Río” (IQAR), Madrid, Spain. E-mail: paula.ortega@uah.es
bNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Spain
cInstitute “Ramón y Cajal” for Health Research (IRYCIS), Spain
dUniversidad de Alcalá, Department of Biology of Systems, Biochemistry and Molecular Biology Unit, Madrid, Spain
eDepartment of Chemical Sciences, University of Naples Federico II, Via Cintia 4, Complesso Universitario di Monte Sant'Angelo, I-80126 Naples, Italy
fDepartment of Pure and Applied Sciences, University of Urbino “Carlo Bo”, Via Saffi 2, I-61029 Urbino, Italy
First published on 15th April 2025
Abstract
Ruthenium or copper complexes have emerged as some of the most promising alternatives for the treatment of many types of cancer. They have enhanced activity, greater selectivity and reduced side effects compared to their predecessors, cisplatin and its analogues. On the other hand, polyamine metabolism is often deregulated in cancer, leading to increased intracellular concentrations of polyamines that promote cell proliferation, differentiation, and tumorigenesis. In the present work, we report the synthesis and characterization of a family of mono- and binuclear Ru(II) and Cu(II) complexes functionalized with polyamine ligands derived from norspermine. The computer-aided analysis of the electron paramagnetic resonance (EPR) spectra provided magnetic and dynamic parameters, which helped to identify prevalent Cu–N2 coordination in a partially distorted square planar geometry of the Cu(II) complexes and the flexibility of the complexes in solution, slowed down by both the complex size and the hydrophobic interactions between chains. In vitro studies focused on advanced prostate cancer have demonstrated that these new metal complexes present a high level of cytotoxicity against PC3 cells. Furthermore, these metallic compounds exhibit the ability to inhibit cell adhesion and migration while reducing intracellular reactive oxygen species levels, which are key factors of metastasis.
1. Introduction
Prostate cancer is one of the most frequent malignant tumors in men, together with colorectal and lung cancer.1,2 Among the different prostate cancers, non-metastatic castration-resistant prostate cancer (nmCRPC) has no curative therapies, making the search for new drug treatments a vital necessity.3,4The use of metal complexes in cancer treatment started after the discovery of the antitumor activity of cisplatin.4,5 The advantage of these compounds over conventional, mainly organic pharmaceuticals is the ability of the metal centers to exhibit different oxidation states, number of charges, coordination index and redox properties, giving them unique structural, physical and chemical properties.6,7 However, there are challenges associated with the use of cisplatin such as high cytotoxicity, due to its lack of selectivity, and the development of tumor resistance. These drawbacks drive the search for more sophisticated organometallic systems capable of addressing these issues. Alternatives to cisplatin are Ru(II) and Cu(II) complexes.8,9 Although the mechanism of action of ruthenium(II) complexes is unclear, their anticancer properties might be linked to their ability to induce apoptosis via mitochondrial, autophagy or ROS species-dependent apoptosis, as well as their capacity to interact with DNA and different proteins.10,11 Regarding Cu(II) complexes, their antitumor activity is due to their ability to form non-covalent adducts with DNA and inhibit the proteosomal function, as well as the activity of topoisomerase I and II tumor cells.12,13 The nature of the ligand(s) present on the coordination sphere of the metal center determines its potential as an anticancer drug, playing an important role in its solubility and ability to cross the plasma membrane (lipophilic/hydrophilic balance).14,15 As an example, the presence of arene-type ligands in the Ru(II) coordination sphere imparts amphipathic properties to the complexes, where the hydrophobic aromatic ring shields the hydrophilicity of the metal center, facilitating the internalization of the complex in the cells.16
Biogenic amines and polyamines are essential growth factors in eukaryotic organisms, bacteria, and viruses.17 The coordination of polyamine ligands, in particular spermine,18 to metal centers such as Pd and Pt has led to the development of new organometallic compounds with anticancer activity as an alternative to cisplatin. This type of ligand provides flexible linkers and increases the hydrophobic character of the molecule, which is important for drug uptake and interaction with DNA.19,20 Some studies have shown that the coordinating capacity of these alkylamines can be utilized to coordinate metal ions such as Pt(II) and Pd(II), and their anticancer activities have been tested.21–23 In these complexes, polyamine ligands are thought to promote the internalization of the metal complexes into tumor cells since the cell recognises them as natural polyamines. Therefore, they are readily taken up and accumulated at high levels.
Based on previous studies of our research group, which demonstrated that carbosilane dendritic backbones favour the cell internalization of metallodrugs due to their hydrophobic nature,24,25 the present study focuses on the design, characterization, and biological evaluation of novel Ru(II) and Cu(II) complexes with norspermine-derived ligands with the aim to evaluate their biological properties, particularly against the prostate cancer cell line PC3. The inclusion of a carbosilane moiety in polyamine ligands will also help us in the future to use these compounds as references for the preparation of carbosilane metallodendrimers functionalized with these types of ligands.
Since the coordination structure and geometry of the complexes, and their interacting ability are key factors for their use in cancer treatment, electron paramagnetic resonance (EPR) studies were performed on the Cu(II) complex solutions. EPR has already been shown in previous studies from this group to provide structural and dynamical information of Cu(II)-carbosilane dendrimer complexes.26
2. Experimental part
2.1. Syntheses
All reactions were carried out under an inert atmosphere and solvents were obtained from MBraun's MB-SPS purification system. All the reagents for the different synthetic routes described were used as received from commercial companies without prior purification (see the ESI† for experimental details and methodologies of biological assays).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–); 8.63 (m, 2H, CHAr). 13C {1H}-NMR (CDCl3): δ 3.4 (ppm) ((H3CCH2)3Si–); 7.6 ((H3CCH2)3Si–); 11.5 (–SiCH2CH2CH2CH2N–); 22.0 (–SiCH2CH2CH2CH2N–); 28.3 (–N(CH2CH2CH2NCPh(o-N))2); 31.4 (–SiCH2CH2CH2CH2N–); 51.9 (–N(CH2CH2CH2NCPh(o-N))2); 53.9 (–SiCH2CH2CH2CH2N–); 59.7 (–N(CH2CH2CH2NCPh(o-N))2); 121.3, 124.7, 136.6 and 149.5 (CHAr); 154.7 (Cipso); 162.0 (–NCH–). Elemental analysis (%). Calc.: C, 70.10; H, 9.45; N, 14.60. Exp.: C, 70.75; H: 10.25; N:15.25. MS (ESI-Q): m/z [M + H+] = 480.35 uma.
N): 1638.2 cm−1.
N): 1645.6 cm−1.
CH–); 9.36 (m, 2H, CHAr). 13C {1H}-NMR (CD3OD): δ (ppm) 4.3 ((H3CCH2)3Si–); 7.8 ((H3CCH2)3Si–); 11.7 (–SiCH2CH2CH2CH2N–); 18.8 (–CCH3cim); 20.3 (–SiCH2CH2CH2CH2N–); 21.5 (–CH(CH3)2cim); 24.7 (–N(CH2CH2CH2NCPh(o-N))2); 28.0 (–SiCH2CH2CH2CH2N–); 32.1 (–CH(CH3)2cim); 50.8 (–N(CH2CH2CH2NCPh(o-N))2); 53.4 (–SiCH2CH2CH2CH2N–); 65.2 (–N(CH2CH2CH2NCPh(o-N))2); 86.2–88.6 (CHcim); 129.8, 130.2, 141.1 and 157.0 (CHAr); 169.4 (–NCH–). Elemental analysis (%): calc.: C, 52.79; H, 6.74; N, 6.41. Exp.: C, 52.69; H: 6.47; N: 6.47. ICP-OES (%): calc.: Ru, 18.5; exp.: 18.7. MS (QTOF): m/z ([M + H+] – 2 Cl) = 1021.3121 uma.
CH–); 8.63 (m, 2H, CHAr). 13C {1H}-NMR (CDCl3): δ 3.4 (ppm) ((H3CCH2)3Si–); 7.6 ((H3CCH2)3Si–); 11.5 (–SiCH2CH2CH2CH2N–); 22.0 (–SiCH2CH2CH2CH2N–); 28.3 (–N(CH2CH2CH2NCPh(o-N))2); 31.4 (–SiCH2CH2CH2CH2N–); 51.9 (–N(CH2CH2CH2NCPh(o-N))2); 53.9 (–SiCH2CH2CH2CH2N–); 59.7 (–N(CH2CH2CH2NCPh(o-N))2); 121.3, 124.7, 136.6 and 149.5 (CHAr); 154.7 (Cipso); 162.0 (–NCH–). Elemental analysis (%). Calc.: C, 70.10; H, 9.45; N, 14.60. Exp.: C, 70.75; H: 10.25; N:15.25. MS (ESI-Q): m/z [M + H+] = 480.35 uma.
N): 1638.2 cm−1.
N): 1645.6 cm−1.
CH–); 9.36 (m, 2H, CHAr). 13C {1H}-NMR (CD3OD): δ (ppm) 4.3 ((H3CCH2)3Si–); 7.8 ((H3CCH2)3Si–); 11.7 (–SiCH2CH2CH2CH2N–); 18.8 (–CCH3cim); 20.3 (–SiCH2CH2CH2CH2N–); 21.5 (–CH(CH3)2cim); 24.7 (–N(CH2CH2CH2NCPh(o-N))2); 28.0 (–SiCH2CH2CH2CH2N–); 32.1 (–CH(CH3)2cim); 50.8 (–N(CH2CH2CH2NCPh(o-N))2); 53.4 (–SiCH2CH2CH2CH2N–); 65.2 (–N(CH2CH2CH2NCPh(o-N))2); 86.2–88.6 (CHcim); 129.8, 130.2, 141.1 and 157.0 (CHAr); 169.4 (–NCH–). Elemental analysis (%): calc.: C, 52.79; H, 6.74; N, 6.41. Exp.: C, 52.69; H: 6.47; N: 6.47. ICP-OES (%): calc.: Ru, 18.5; exp.: 18.7. MS (QTOF): m/z ([M + H+] – 2 Cl) = 1021.3121 uma.
3. Results and discussion
3.1. Synthesis and characterisation of organic ligands
Norspermidine (NSPD), a polyamine ligand and chemical homologue of spermidine (SPD), was selected for the synthesis of new Ru(II) and Cu(II) derivatives due to its anti-tumor activity.27To favor the cellular uptake of organometallic compounds, the NSPD was modified with a carbosilane moiety, Br(CH2)4Si(CH2CH3)3, by N-alkylation of the secondary amines present in the NSPD ligand with the primary amino groups protected with phthalimide28 to obtain Et3Si(CH2)4N[(CH2)3NPht]2 (1). The alkylation was confirmed through 1H and 13C-NMR where the resonances of the newly formed CH2–N group were observed at 2.33 ppm along with the signals corresponding to the aromatic ring of the phthalimide group at 7.64 and 7.74 ppm. The next step was the deprotection of the amino groups in compound 1 with an excess of hydrazine, heated under reflux with MeOH as the solvent, resulting in the formation of compound 2, Et3Si(CH2)4N[(CH2)3NH2]2. The displacement of the signal from 3.66 to 2.92 ppm in CDCl3 for the methylene –CH2NPht group and the disappearance of the aromatic signals in the 1H-NMR spectrum confirmed the deprotection reaction. Furthermore, the presence of free amines was corroborated by the Kaiser test, and by infrared spectroscopy (FTIR), where the bands at 2927 and 2890 cm−1 corresponding to the νNH tension were observed.
The presence of free amine groups in compound 2 allowed us to carry out a condensation reaction to obtain Schiff bases, which have demonstrated excellent coordination capabilities for metal complexes. Therefore, the reaction of compound 2 with 2-pyridincarbaldehyde under an inert atmosphere gave rise to the formation of N,N-chelating ligand Et3Si(CH2)4N[(CH2)3NCPh(p-N)]2 (3). The reaction was carried out in the presence of MgSO4 to remove the water generated during the reaction.
Compounds 1–3 were characterized by mono- and bi-dimensional multinuclear NMR spectroscopy (1H, 13C{1H}), mass spectrometry and elemental analysis. The analytical and spectroscopic data reported in the Experimental section and the ESI† are consistent with the proposed structures (Scheme 1).
 | ||
| Scheme 1 Synthesis of organic ligands. | ||
3.2. Synthesis and characterisation of metallic complexes
[RuCl2(p-cymene)]2 as well as CuCl2 and Cu(NO3)2, were used as precursors to obtain the ruthenium and copper derivatives, respectively.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in methanol as the solvent to obtain Et3Si(CH2)4N[(CH2)3NH2]2CuCl2 (4) and Et3Si(CH2)4N[(CH2)3NH2]2Cu(NO3)2 (5). The presence of Schiff bases in the structure of compound 3 opened up the possibility of coordinating more than one metal center. Therefore, when the reaction of the copper salts with ligand 3 was carried out at a stoichiometric 2:1 ratio (metal/ligand), this led to the formation of bimetallic derivatives Et3Si(CH2)4N[(CH2)3NCPh(o-N)][CuCl2·H2O]2 (6) and Et3Si(CH2)4N[(CH2)3NCPh(o-N)][Cu(NO3)2·H2O]2 (7). In all cases, the incorporation of the metal complex into the structure was confirmed by FTIR spectroscopy and UV-vis spectrophotometry. For compounds 6 and 7, FTIR spectroscopy showed the shift of the νCN strain band from 1600 cm−1 to 1650 cm−1. UV-vis spectra showed a band at around λ = 290 nm that can be ascribed to a transition band (MLCT or LMCT).29 The d–d transfer band could only be identified as a broad band of low intensity at around 650 nm (Fig. 1). In addition, the percentages of copper in the new complex were determined by ICP-OES, resulting in 14.53%, 14.62%, 16.52% and 14.25% for 4, 5, 6 and 7, respectively and confirming the presence of 1 or 2 copper atoms per molecule.
:1 ratio in methanol as the solvent to obtain Et3Si(CH2)4N[(CH2)3NH2]2CuCl2 (4) and Et3Si(CH2)4N[(CH2)3NH2]2Cu(NO3)2 (5). The presence of Schiff bases in the structure of compound 3 opened up the possibility of coordinating more than one metal center. Therefore, when the reaction of the copper salts with ligand 3 was carried out at a stoichiometric 2:1 ratio (metal/ligand), this led to the formation of bimetallic derivatives Et3Si(CH2)4N[(CH2)3NCPh(o-N)][CuCl2·H2O]2 (6) and Et3Si(CH2)4N[(CH2)3NCPh(o-N)][Cu(NO3)2·H2O]2 (7). In all cases, the incorporation of the metal complex into the structure was confirmed by FTIR spectroscopy and UV-vis spectrophotometry. For compounds 6 and 7, FTIR spectroscopy showed the shift of the νCN strain band from 1600 cm−1 to 1650 cm−1. UV-vis spectra showed a band at around λ = 290 nm that can be ascribed to a transition band (MLCT or LMCT).29 The d–d transfer band could only be identified as a broad band of low intensity at around 650 nm (Fig. 1). In addition, the percentages of copper in the new complex were determined by ICP-OES, resulting in 14.53%, 14.62%, 16.52% and 14.25% for 4, 5, 6 and 7, respectively and confirming the presence of 1 or 2 copper atoms per molecule.
 | ||
| Fig. 1 UV-vis spectra for the compounds Et3Si(CH2)4N[(CH2)3NH2]2CuCl2 (4), Et3Si(CH2)4N[(CH2)3NH2]2Cu(NO3)2 (5), Et3Si(CH2)4N[(CH2)3NCPh(o-N)][CuCl2·H2O]2 (6) and Et3Si(CH2)4N[(CH2)3NCPh(o-N)][Cu(NO3)2·H2O]2 (7). | ||
Finally, to understand the Cu(II) coordination and geometry to polyamine ligands, EPR spectroscopy was employed. The Aii and gii values used to compute the spectra at low temperature were the same as those used to compute the spectra at room temperature, and this held for all samples as shown in Table S1 of the ESI.† The discrepancies between the experimental and the computed lines indicated the presence of further spectral components at low relative intensities, but the main component showed a good fit as shown in Fig. 2 and S13 (ESI†). For the monometallic complex 5 (Fig. 2), the parameters, compared with those in the literature, were indicative of a Cu–N2 coordination with two nitrogen sites (the amino groups) in a partially distorted square planar symmetry. Probably a third nitrogen site was also present during the complexation, but positioned at a greater distance from the copper atom. Some examples in the literature, which exhibit crystallographic data of similar complexes, show structures based on a tripodal coordination to the metal center, with the three nitrogen atoms of the norspermidine acting as donor atoms (see Scheme 2).30 However, EPR data show that in solution the coordination was different, differentiating the position of the amino groups with respect to the third nitrogen site.
 | ||
| Fig. 2 Experimental and computed spectra of the complex Et3Si(CH2)4N[(CH2)3NH2]2Cu(NO3)2 (5) in MeOH solution at 298 K (a) and at 120 K (b). The main parameters used for computations are shown in the figure legends. | ||
 | ||
| Scheme 2 Synthesis of mono- and bimetallic complexes of Ru(II) and Cu(II). The structures proposed are based on spectroscopic results obtained. | ||
Interestingly, at room temperature, a further parameter was needed for the computation, which renders the computation less accurate with respect to the magnetic and mobility parameters. This additional parameter was the spin exchange frequency (Wex), which was related to collisions between paramagnetic moieties occurring in a fluid medium. In this case, this was not due to condensation because the solution was clear, and the concentration was not high enough to lead to aggregation. Furthermore, the cupric ions cannot approach each other due to charge repulsion. We suspect that the presence of hydrophobic chains in the ligand, even if relatively short, favored chain–chain interactions, which led to Cu–Cu approaching in spite of the charge repulsion. The counterions played a significant role, as the presence of chloride ions (monometallic complex 4) instead of nitrate ions (monometallic complex 5) favored line broadening at low temperature in line with cupric ions sitting close to each other after charge neutralization, as expected for chloride ions, which much better interact with Cu(II) if compared to nitrate ions. Therefore, spectral computation was not possible for sample 4, but this was also informative of the properties of the complex.
For bimetallic complexes 6 and 7, the results obtained were similar to those of 4 and 5, but with a substantial difference: a significant fraction (about 50%) of Cu(II) was not complexed by ligand 3 and was interacting with the solvent. The Cu(II)–solvent complex was simply indicated as Cu–MeOH. The computed Cu–MeOH signals both at 298 K and 120 K are shown in Fig. S13 (ESI),† while the main parameters of computations are listed in Table S1.† These parameters are characteristic of a Cu–O4 coordination (square planar) unit of Cu(II) with 4 MeOH molecules, while two additional MeOH molecules are located at farther distances in the apical octahedral positions. We noted that the microviscosity of the Cu(II) environment, denoted by τ, was about half for the Cu–MeOH complex when compared to the Cu-ligand complexes, as expected for the steric bulk of the ligands. Also, we want to underline that we assumed a constant τ value (17 ns) for all spectral computations at 120 K, assuming almost rigid motion at that temperature. Computations of the spectra of 6 and 7 were performed after subtracting the Cu–MeOH signal. In this case too, the computations are shown in Fig. S1† and the main parameters of computation are listed in Table S1.† Similarly to the situation of monometallic 4 and 5, in this case too, the substitution of the nitrate counterion with the chloride one provoked line broadening, mainly at room temperature. The magnetic parameters of 6 and 7 indicated coordination similar to that of 5, but in a trigonally distorted structure. This means that polyamine ligand 3 generated a trigonally distorted square planar complex with Cu(II). The large fraction of Cu–MeOH complex suggested lower interacting ability of ligand 3 with respect to ligand 2.
In order to study in detail the coordination situations of Cu(II) in the new mono- and bimetallic derivatives, a computer-aided EPR analysis was carried out on the ligands 2 and 3 with increasing concentration of Cu(II). Fig. 3 shows the experimental spectra of the sample termed for simplicity 2-NO3 (2-NO3 indicates a sample containing the ligand 2 and the cupric nitrate salt in MeOH solution), for three different Cu(II) concentrations at 298 K (Fig. 3a) and at 120 K (Fig. 3b). Three different spectral components were identified in the spectra, whose minimum peaks are indicated with arrows in Fig. 3 and termed signal 1, signal 2, and Cu-MeOH. These components were extracted by subtracting the spectra from each other. This allowed us to evaluate the relative percentages of each component in the spectra. Then, the components were computed. We noted the presence of other minor spectral components, which, in some cases, were responsible for decreasing the quality of the fitting between experimental and computed signal 1 and signal 2. Fig. 4 shows experimental and computed signal 1 minima obtained for 3-NO3, selected as an example, at [Cu(II)] = 6.25 mM, that is, at almost the lowest copper concentration, at 298 K (a) and at 120 K (b).
 | ||
| Fig. 3 Experimental spectra of the 2-NO3 sample at 298 K (a) and at 120 K (b), and at three different Cu(II) concentrations; three different spectral components (signal 1, signal 2, and Cu-MeOH) are indicated with arrows in the figure. | ||
 | ||
| Fig. 4 Experimental and computed signal 1 obtained for the 3-NO3 sample at [Cu(II)] = 6.25 mM and at 298 K (a) and at 120 K (b). | ||
Other examples of computations of signal 1 are shown in the ESI (Fig. S13†). Signal 1 was almost the only signal that constituted the EPR spectra at the lowest Cu(II) concentrations. The magnetic parameters (summarized in Table 1) corresponded to a distorted square planar Cu–N4 coordination, implying that at least two ligands were involved in the complexation of each Cu(II) ion. An apparent contradiction occurred with respect to the variation of Azz, since we expected a higher Azz value (and lower gzz value) for ligand 2, bearing amino groups, with respect to 3, bearing pyridine groups, due to the higher electron-donor strength of the former ligand with respect to the latter one25,27Also, gxx values are lower for the Cu(II) complex formed with ligand 2 (Cu-2 complexes) than for the Cu(II) complex formed with ligand 3 (Cu-3), indicating a higher anisotropy for the Cu-2 ligand field. Furthermore, at 298 K the spectra showed a lower mobility (higher τ) for the Cu-3 complexes, in line with a higher steric hindrance. This indicates that, although the amino groups in ligand 2 exhibit higher affinity for Cu(II) compared to the nitrogen sites in ligand 3, the structure of ligand 2 leads to less stable Cu–N4 complexes than those formed with ligand 3. This is attributed to the bidentate nature of the iminopyridine ligand in compound 3, which forms stable chelate rings with the metal center, enhancing the stability of the Cu–N4 coordination mode. Finally, it is possible to analyze the influence of the counterions, NO3 or Cl, on the coordination of the metal center. The spectra show that in the case of ligand 3, the nature of the counterion does not affect the coordination mode, which in both NO3 and Cl cases is Cu–N4 coordination. Conversely, chloride counterions perturbed the Cu-2 binding more than nitrate counterions, due to the stronger Cu–Cl binding if compared to the Cu–NO3 one.
| Sample | g xx | g yy | g zz | A xx (G) | A yy (G) | A zz (G) | τ (ns) | W ex s−1 |
|---|---|---|---|---|---|---|---|---|
|
4 both 298 and 120 K (2-Cl 1:1) |
Broad and not resolved spectrum | |||||||
|
5 120 K (2-NO3 1:1) |
2.03 | 2.08 | 2.25 | 5 | 5 | 153 | 17 | |
| 5 298 K | 2.03 | 2.08 | 2.25 | 5 | 5 | 153 | 0.1 | 7 × 108 |
|
6 subtracted Cu-MeOH 120 K (3-Cl 1:2) |
2.038 | 2.11 | 2.29 | 25 | 35 | 149 | 17 | |
| 6 298 K | Intense Cu-MeOH signal, not subtractable, and broad signal | |||||||
|
7 subtracted Cu-MeOH 120 K (3-NO3 1:2) |
2.03 | 2.08 | 2.21 | 15 | 15 | 153 | 17 | |
| 7 subtracted Cu-MeOH 298 K | 2.03 | 2.08 | 2.21 | 15 | 15 | 153 | 0.1 | |
| Cu-MeOH 120 K | 2.0795 | 2.11 | 2.438 | 1 | 1 | 109 | 17 | |
| Cu-MeOH 298 K | 2.0795 | 2.11 | 2.438 | 1 | 1 | 109 | 0.05 | |
| 2-NO3 signal 1 120 K | 2.005 | 2.05 | 2.211 | 5 | 5 | 180 | 17 | |
| 2-NO3 signal 1 298 K | 2.005 | 2.05 | 2.211 | 5 | 5 | 180 | 0.09 | |
| 2-Cl signal 1 120 K | 2.005 | 2.053 | 2.21 | 10 | 10 | 178 | 17 | |
| 2-Cl signal 1 298 K | 2.005 | 2.053 | 2.21 | 10 | 10 | 178 | 0.1 | |
| 3-NO3 signal 1 120 K | 2.012 | 2.03 | 2.21 | 5 | 5 | 183 | 17 | |
| 3-NO3 signal 1 298 K | 2.012 | 2.03 | 2.21 | 5 | 5 | 183 | 0.25 | |
| 3-Cl signal 1 120 K | 2.012 | 2.03 | 2.21 | 5 | 5 | 183 | 17 | |
| 3-Cl signal 1 298 K | 2.012 | 2.03 | 2.21 | 5 | 5 | 183 | 0.25 | |
| 2-NO3 signal 2 120 K | 2.03 | 2.08 | 2.25 | 5 | 5 | 153 | 17 | |
| 2-NO3 signal 2 298 K | 2.03 | 2.08 | 2.25 | 5 | 5 | 153 | 0.17 | |
| 2-Cl signal 2 120 K | 2.038 | 2.13 | 2.265 | 5 | 5 | 150 | 17 | |
| 2-Cl signal 2 298 K | 2.038 | 2.13 | 2.265 | 5 | 5 | 150 | 0.1 | 8 × 108 |
| 3-NO3 signal 2 120 K | 2.038 | 2.05 | 2.26 | 5 | 5 | 170 | 17 | |
| 3-NO3 signal 2 298 K | 2.038 | 2.085 | 2.26 | 5 | 5 | 170 | 0.2 | |
| 3-Cl signal 2 120 K | 2.038 | 2.09 | 2.265 | 5 | 5 | 173 | 17 | |
| 3-Cl signal 2 298 K | 2.038 | 2.09 | 2.265 | 5 | 5 | 173 | 0.1 | 8 × 108 |
Signal 2 formed in an intermediate range of Cu(II) concentrations, and reached maximum relative percentage when the Cu(II)/ligand molar ratio was about 1:2 (for ligand 2) and 1:4 (for ligand 3). This is evident in Fig. S12 of the ESI,† which shows the variations of the relative percentages of signal 1, signal 2 and Cu-MeOH as a function of [Cu(II)] for the various samples. Fig. 5 shows experimental and computed signal 2 peaks obtained after the subtraction of signal 1 from the spectrum of 2-NO3 at [Cu(II)] = 0.05 M and 298 K (a) and at 120 K (b). The main parameters of computation are shown in the figure legends. Other examples of computations of signal 2 for the other samples are shown in Fig. S13 (ESI).† The main parameters of computations are reported both in the figure legends and in Table S1.† Interestingly, the magnetic parameters (gii and Aii) of signal 2 for 2-NO3, shown in Fig. 5, were the same as the magnetic parameters of the spectrum of the monometallic complex 5, shown in Fig. 2. This means that these two samples have the same coordination and geometry, being in both cases Cu(II) ions complexed by the 2 amino groups of the ligand 2-NO3 (coordination Cu–N2) in a distorted square planar (octahedral) coordination. The other ligands were solvent molecules, but a more distant nitrogen site and counterions are probably included in the Cu(II) coordination sphere.
 | ||
| Fig. 5 Experimental and computed Signal 2 obtained after subtraction of Signal 1 from the spectrum of 2-NO3 at [Cu(II)] = 0.05 M and at 298 K (a) and at 120 K (b). The main parameters of computation are listed in the figure legend. | ||
The inclusion of the counterions (nitrate or chloride ions) in the coordination sphere was proved by the difference between the magnetic parameters of signal 2 for 2-NO3 and 2-Cl: as already underlined, Cl perturbs the ligand field strength of Cu(II) with the nitrogen ligands leading to a decrease of Azz (from 153 G for 2-NO3 to 150 G for 2-Cl) and an increase of gzz (from 2.25 for 2-NO3 to 2.265 for 2-Cl). However, a significant difference was evident by comparing Fig. 2 and 5, mainly for the spectra at 298 K. The spectrum of monometallic complex 5 at 298 K (Fig. 2(b)) needed a high Wex value in the computation, while signal 2 at 298 K was computed without including the Wex parameter. This last event probably happened because 40% of Cu(II) ions were engaged in forming the Cu–N4 coordination with 2-NO3 (signal 1), as shown in Fig. S12 (ESI†). This impeded complexes like 5 from approaching each other, chain-to-chain. Similarly, the spectra of complex 6 could not be computed because they were completely unresolved, while signal 2 of 2-Cl, obtained after subtraction of signal 1, was broad at 120 K, and partially resolved (Fig. S13, ESI†) allowing us to compute it and obtain reliable main parameters of computation. Interestingly, Signal 2 of 2-Cl needed a high Wex in the computation (Table S1†), again related to the electrostatic binding of Cl to the Cu complex which favors chain-to-chain interactions in solution. The parameters of signal 2 for ligand 3 were almost those of ligand 2, with the exception of Azz, which is higher than expected for the Cu–N2 coordination described above. Again the bidentate nature of the iminopyridine moiety in ligand 3 played a role in increasing the electron spin density on the Cu(II) nucleus in the perpendicular direction.
:1 ratio (metal/ligand). Metal center coordination was corroborated by 1H-NMR, where the signal attributed to the methylene group attached to the nitrogen of the secondary amine was shifted to 3.65 ppm, while for the free ligand it was located at 2.92 ppm (Fig. 6). However, the signal corresponding to methylene groups attached to the central tertiary amine was unaffected by the coordination of the metal center. Based on these spectroscopic data, a novel coordination mode was proposed where the primary amine groups would chelate the Ru(II) complex stabilizing the metal center without involving the central ligand nitrogen (see Scheme 2). This proposed monometallic structure was in agreement with the ICP-OES and elemental analysis data, which corroborated a metal/ligand ratio of 1:1 as mentioned above.
 | ||
| Fig. 6 1H and 13C{1H} NMR spectra of Et3Si(CH2)4N[(CH2)3NH2]2Ru(η6-p-cymene)Cl2 (8). | ||
A Ru(II) bimetallic complex, Et3Si(CH2)3[N[[(CH2)3NCPh(o-N)]Ru(η6-p-cymene)Cl2]2] (9), was also synthesized using a 1:1 ratio (metal/ligand 3). Again, 1H-NMR allowed for the determination of the mode of coordination of the metal center by the displacement of the signal corresponding to the iminic hydrogen, which appeared at 8.30 ppm, while in the free ligand it was observed at 9.39 ppm. Additionally, the splitting of the signal corresponding to the methylene group attached to the nitrogen atom of the imine group, CH2NCPh, indicated the formation of the metallic complex (Fig. 7) Moreover, the amount of Ru determined by ICP-OES in the compound (18.70%) was in agreement with the expected results for bimetallic complexes. Moreover, compound 9 was further characterized by Q-Toff mass spectrometry, which showed the peak corresponding to the di-cationic compound after the loss of the two chloride ions present in the coordination sphere (theoretical mass = 1021.27 uma).
 | ||
| Fig. 7 1H and 13C{1H} NMR spectra of Et3Si(CH2)3[N[[(CH2)3NCPh(o-N)]Ru(η6-p-cymene)Cl2]2] (9). | ||
3.3. Evaluation of biological properties as antitumoral agents
 | ||
| Fig. 8 Cyclic voltammograms of compounds 4, 5, 6 and 7 (a). CV experiments were recorded in 0.1 M PBS, using 0.1 V s−1 as the scan rate. The black dotted line corresponds to the background scan. (b) Expansion of the area where the anodic peak (Ea) is located. | ||
In the case of Ru(II) derivatives, the literature has reported the stability of similar triethylsilane iminopyridine ligands acting as chelates, where only one chlorine atom remains coordinated to the metal center. These complexes, exhibiting the same coordination mode as compound 9, demonstrated high stability in D2O, PBS-D2O, or neat DMSO-d6 after 72 hours in solution.33 In the case of compound 8, we have proposed, based on spectroscopic data, a novel coordination mode where the primary amine groups will chelate with the Ru(II) complex, stabilizing the metal center without involving the central ligand nitrogen. The stability of compound 8 was evaluated by 1H-NMR spectroscopy in D2O and PBS-D2O. In this case, the appearance over time (0–24 h) of new signals at 2.40 ppm was observed, probably attributed to the methylene group directly bonded to the amino group (–CH2NH2), suggesting a possible de-coordination, at least of one of the amino groups of the polyaminated ligand from the metal center (see ESI Fig. S18†). This behavior aligns with previously studied ruthenium compounds bearing nitrogen ligands.33
Firstly, the IC50 of the new metallic compounds was determined by MMT assays; the obtained values are presented in Table 3. All the tested compounds induced a significant decrease in cell viability, even at low concentrations. The presence of two metal centers in the molecules generally implied a reduction of the IC50 values decreasing from 16.8 to 13.6 μM for the ruthenium complexes (8 and 9). For the iminopyridine Ru(II) complex, despite bearing half of the metal centers, it is important to note that the cytotoxicity observed (IC50 = 13.6 ± 1.5) is quite similar to first-generation carbosilane metallodendrimers based on arene ruthenium(II) complexes that contain four metallic centers in the prostatic cell line (IC50 = 7.8 ± 1.4).34 In the case of ruthenium derivatives, the low IC50 observed for the monometallic derivative suggests that the therapeutic activity could be due not only to the presence of a single metal atom but probably also the presence of free amino groups due to the instability of the complex in solution, as evidenced by 1H-NMR spectroscopy.
The cytotoxicity of copper complexes was consistent with previously reported organometallic copper(II) chelate complexes containing N,N-ligands, which inhibit the growth of PC3 cells.26 No clear trends were observed regarding the different ligands—chloride or nitrate—in the coordination sphere or the nature of the ligand, 2 (polyamino) and 3 (iminopyridine), in copper complexes. In the monometallic copper complexes, the difference in activity observed where the most active complex was complex 5 (10.6 ± 0.5 μM) containing a nitrate ligand, could be attributed to its greater solubility in aqueous medium compared to the chloride derivative, which creates a more hydrophobic environment and exhibits lower activity (16.0 ± 0.9 μM).24 However, in the case of bimetallic complexes 6 and 7 the tendency was unclear indicating that the nature of the counterion is crucial in the biological outcome of these derivatives, in agreement with the EPR results. This difference in activity among the copper complexes bearing different ligands—chloride or nitrate—in the coordination sphere, has been previously reported in the literature for dendritic carbosilane systems decorated with the same iminopyridine complexes. These studies revealed that replacing the nitrate ion with chloride increased both the quantity and strength of dendrimer interactions with membrane models, with the chloride-containing dendrimer exhibiting the strongest interactions. However, excessive stabilization within the cell membrane could sometimes lead to a reduced cytotoxic effect or even affect the internalization mechanism of the dendritic species within the cell.25
Next, we focused on two processes, cell migration and adhesion, intimately related to tumor invasion and metastasis processes.35,36
Cell migration occurs throughout the carcinogenesis cascade and is particularly important during invasion, which is the first step towards metastasis. After chemotactic migration, a cancer cell infiltrates the adjacent tissue and local vasculature. The ability of the compounds to inhibit cell migration was evaluated using a wound closure assay. Fig. 9A and B shows that all the compounds caused a significant decrease in cell migration. The percentage (%±ESM) of wound closure after 24 h of treatment with respect to the untreated control (100%) was as follows: (4), 39 ± 8; (5), 35 ± 7; (8), 35 ± 9; (6), 38 ± 10; (7), 46 ± 10; (9), 54 ± 10. Most of the compounds showed great capacity to prevent closure, with no significant differences between them, with only the bimetallic Ru(II) 9 complex showing capacity of wound closure lower than 50% with respect to the control at 24 hours.
 | ||
| Fig. 9 (A) Wound closure assay. Images were obtained at 0 h and 24 h after the wounds. (B) Cell migration under treatment with compounds 4–9 after 24 h. (C) Cell adhesion under treatment with compounds 4–9. Values are mean ± SEM representative of three independently performed experiments are shown. ***p < 0.001 with respect to control values. | ||
Cell adhesion is a critical component in cancer progression and in the development of resistance in cancer treatment. Through this mechanism, tumor cells are able to invade the tissues bordering the primary tumor and extravasate during metastasis. Regarding cell adhesion assays, the capacity of PC3 cells to adhere to a type I collagen layer was analyzed at two different times, 0 and 80 minutes after treatment of the cells with the synthesized compounds. The obtained results were compared with their respective untreated controls and are shown in Fig. 9C. All the complexes had the capacity to reduce cell adhesion, with the monometallic complexes 4, 5 and 8 with primary amine type ligands being more relevant, which achieved even significant reductions immediately after the addition of the respective compounds with no significant difference in the nature of the metal core. On the other hand, bimetallic complexes with iminopyridine-type ligands, 6, 7 and 9, were able to reduce adhesion at short times, while, at longer times, they showed a lower efficiency with respect to monometallic derivatives.
In any case, the fact that all complexes have the capacity to reduce cell adhesion and migration is an encouraging result, taking into account that advanced prostate cancer tends to metastasize more frequently to the bone, whose matrix is mainly composed of collagen.37 Therefore, at this point it could be affirmed with the adhesion and migration results that the complexes could have antimetastatic activity in vitro.
Anti-cancer therapies aim to eliminate cancer cells through various mechanisms of destruction, including metabolic death, apoptosis, and clonogenic death.38 Apoptosis is a form of programmed cell death that results in nuclear DNA fragmentation,39 while clonogenic death is a result of the loss of a cancer cell's ability to proliferate indefinitely.
The effect of metallic compounds on decreasing the number of tumor cell colonies formed from individualized cells was analyzed by a clonogenic assay that measures the capability of a single cell to form a colony. Fig. 10 shows the colonies of PC3 cells before (control) and after the treatments. In the case of the copper derivatives, no cell colonies were observed on the plate. This suggests that complexes 4–7 have the highest antiproliferative activity. In the case of the ruthenium derivatives 8 and 9, some colonies were observed, but their numbers were minimal with respect to the control (untreated) cells. Consequently, the newly generated complexes are able to prevent the uncontrolled growth of small colonies that would eventually constitute the tumor.40
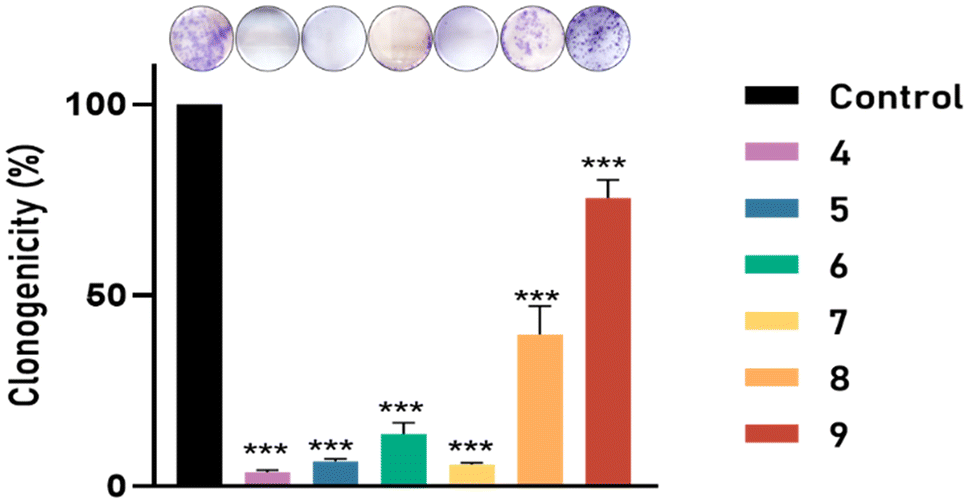 | ||
| Fig. 10 Effect of compounds 4–9 on the clonogenicity in PC3. The results are expressed as a percentage of the control value. Data are mean ± SEM of three independent experiments; ***p < 0.001, with respect to control values. | ||
Other important parameters in tumorigenesis is cell cycle regulation, and therapeutic agents designed to regulate the cell cycle have beneficial effects on tumorigenesis.41,42 The arrest of the cell cycle allows damaged cells to be repaired, and prevents the progression of DNA alterations. If this repair is unsuccessful, it may lead to the apoptosis of damaged cells.
Analysis of the cell cycle was carried out to discover any change in the PC3 cell cycle that may be related to the reduction of cell proliferation (Fig. 11). Analysis of the cell cycle allowed us to classify cells in different phases: G1 (cell growth), S (DNA duplication), G2/M (cell growth and mitosis), and SubG0 (resting phase, that could lead to apoptosis). Table 2 shows that, after treatment of PC3 cells with all compounds the number of cells in the G2/M phase decreased to 13.05 ± 0.9%, whereas cells in the S-phase increased up to 15.49 ± 1.76% after treatment with compounds 4, 5, 6 and 7, indicating S-phase arrest. Moreover, the treatment with compounds 4 and 5 led to apoptosis, leading to 9.60 ± 1.13% cells in this phase. These results indicate that our complexes could produce cyclic arrest, and apoptosis would reduce the progression of cancer cells. These results are consistent with the literature for different types of Ru(II) and Cu(II) metal complexes, where it is postulated that death by apoptosis may occur by different pathways mediated by death receptors, mitochondria and/or endoplasmic reticulum stress.43–45
 | ||
| Fig. 11 Analysis of the cell cycle in PC3 cells after treatment with polyamine ligand complexed by ruthenium(II) or copper(II). The table shows the results as a percentage of cells in each phase of the cycle compared to untreated control cells. Data in the table are the mean ± SEM of four independent experiments; *p < 0.05; **p < 0.01; ***p < 0.001 vs. control. | ||
| Compound | E a (V)a | i a (μA)b | E c (V)c | i c (μA)d | ΔE° (V)e |
|---|---|---|---|---|---|
| a Anodic peak potential. b Anodic intensity. c Cathodic peak potential. d Cathodic intensity. e Peak-to-peak separation. | |||||
| (4) | −0.225 | −42.0 | 0.132 | 9.8 | 0.357 |
| (5) | −0.203 | −44.4 | 0.142 | 13.8 | 0.345 |
| (6) | −0.150 | −62.1 | −0.102 | 23.1 | 0.048 |
| (7) | −0.160 | −56.6 | −0.098 | 24.1 | 0.062 |
| Monometallic complex | IC50 (μM) ± SEM | Bimetallic complex | IC50 (μM) ± SEM | |
|---|---|---|---|---|
| Cu(II) | 4 | 16.0 ± 0.9 | 6 | 9.6 ± 0.5 |
| 5 | 10.6 ± 0.5 | 7 | 15.1 ± 0.8 | |
| Ru(II) | 8 | 16.8 ± 1.1 | 9 | 13.6 ± 1.5 |
Furthermore, a large number of studies have shown that reactive oxygen species (ROS) play an important role in almost all types of cancer. Many aspects of tumor development and progression are favored by elevated levels of ROS in these cancers. Nevertheless, the presence of antioxidant proteins in the tumor microenvironment has also been described, suggesting that a delicate balance of intracellular ROS levels is required to allow cancer cells to have a viable life. Fine-tuning intracellular ROS signaling to block tumor-promoting effects and enhance ROS-driven apoptosis is a key challenge for new therapies.46
We assessed the effect of new metallic complexes on the levels of ROS generated after treatment with bis(2-ethylhexyl)-3,4,5,6-tetrabromophthalate (TBPH). The cells were incubated with complexes for 24 h and then treated for 30 min with the probe CMH2DCFDA. The effect of TBPH on cell levels of endogenous ROS was studied with a pulse. As shown in Fig. 12, a significant increase in intracellular ROS production was achieved in cells treated with TBPH. Pre-treatment with all compounds significantly decreased the endogenous levels of ROS induced by this pulse of TBPH indicating a potential antioxidant effect of all complexes in PC3 cells. Therefore, our complexes could limit ROS levels, inhibiting protumoral and prometastatic signaling pathways.
 | ||
| Fig. 12 Effect of compounds 4–9 on oxidative stress induced by TBPH in PC-3 cells. The results are expressed as mean fluorescence per cell. Data are mean ± SEM of three independent experiments; ***p < 0.001, with respect to control values. | ||
4. Conclusions
In summary, organometallic complexes of Ru(II) and Cu(II) were successfully synthesized and characterized, containing a norspermine ligand modified with a carbosilane moiety in the central amino group of their structure. We found that the chelating effect of the iminopyridine ligand stabilizes the metallodrug and promotes the formation of a bimetallic complex, in contrast to polyamino ligand 2, which tends to incorporate a single metal center. EPR studies of the copper complex provided valuable information about the behavior of the metallodrugs in solution and revealed a partially distorted square planar Cu–N2 coordination for the complexes with molar ratios of ligand:Cu(II) ranging from 1:1 to 2:1. Their mobility was reduced to half if compared to the ions in the absence of the ligands. However, a parallel study with increasing Cu(II) concentrations shows the formation of a Cu–N4 coordination at the lowest Cu(II) concentrations, but partially surviving at the 1:1 molar ratio, while, starting from the same 1:1 molar ratio, an increasing fraction of Cu(II) ions were confined outside the complexes, only interacting with the solvent. The chloride counterions play a significant role in modifying the parameters with respect to the nitrate counterions, showing that they participate in the coordination sphere of the Cu(II) ions.
In the study of antitumoral activity, we observed how increased metallodrug lipophilicity and the nature of the inorganic ligand (chloride or nitrate in the case of copper metallodrugs) produces a different cytotoxic effect. However, while the type of metal ion does not seem to be critical for antitumoral activity—since all the complexes evaluated exhibited similar IC50 values, all compounds induced apoptosis and arrested the cell cycle, prevented cell migration and cell adhesion and decreased the endogenous levels of ROS induced by external stimuli—it plays a crucial role in antiproliferative activity, with the copper derivatives demonstrating a greater effect.
Overall, the promising results obtained with Ru(II) and Cu(II) complexes, as described in this work, provide a solid base for the design of multivalent platforms, such as dendrimers. Furthermore, this approach helps to understand the effects of incorporating multiple Ru(II) or Cu(II) complexes per dendritic branch into a single scaffold and allows the study of the impact on their antitumoral activity. In addition, the monometallic Cu(II) derivative (4) stands out with the best in vitro activity of all the complexes studied. These results open the door for future in vivo experiments confirming the potential anticancer and antimetastatic use of 4 in different prostate cancers.
Data availability
The data supporting this article (various structural tables, spectroscopic and EPR figures for complexes 1–9) have been included as part of the ESI.†Author contributions
Conceptualization, P. O.; formal analysis, Y. G. M., P. O., G. D'E, L. M. and M. T.; investigation, Y. G. M., M. T. and G. D'E.; resources, P. O., F. J. d. l. M. and G. D'E.; data curation, Y. G. M., P. O., G. D'E, and L. M.; writing – original draft, P. O.; writing – review and editing, Y. G. M., M. F. O., P. O, L. M. and F. J. d. l. M.; supervision P. O., F. J. d. l. M., M. J. C., and M. F. O.; funding acquisition, P. O. and F. J. d. l. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by the Ministry of Science and Innovation (ref. PID2020-114891RB-I00), University of Alcala PIUAH21/CC-041 and EPU-INV-UAH/2021/002. The Junta de Comunidades de Castilla la Mancha (SBPLY/23/180225/000109), the Instituto de Salud Carlos III through the project FORT23/00046, the Program FORTALECE of the Ministerio de Ciencia e Innovación y Universidades of Spain. CIBER-BBN is an initiative funded by the VI National R&D&i Plan 2008–2011, Iniciativa Ingenio 2010, Consolider Program, CIBER Actions, and financed by the Instituto de Salud Carlos III with assistance from the European Regional Development Fund. Y. G. M. thanks the University of Alcalá and Comunidad de Madrid for the research grants ref. EPU-INV2020/014 (UAH CAM).References
- G. Rebez, M. Barbiero, F. A. Simonato, F. Claps, S. Siracusano, R. Giaimo, G. Tulone, F. Vianello, A. Simonato and N. Pavan, Diagnostics, 2024, 14(17), 1864 CrossRef PubMed.
- R. Osiecki, M. Kozikowski, B. Sarecka-Hujar, M. Pyzlak and J. Dobruch, Cancers, 2023, 15(5), 1372 CrossRef PubMed.
- A. Tartarone, R. Lerose and M. Tartarone, Med. Oncol., 2022, 39(5), 56 CrossRef PubMed.
- T. S. Reddy, S. H. Priver, R. Ojha, N. Mirzadeh, G. R. Velma, R. Jakku, T. Hosseinnejad, R. Luwor, S. Ramakrishna, D. Wlodkowic, M. Plebanski and S. K. Bhargava, Eur. J. Med. Chem., 2025, 281(5), 117007 CrossRef CAS PubMed.
- A. M. P. Romani, Biochem. Pharmacol., 2022, 206, 115323 CrossRef CAS PubMed.
- A. K. Singh, A. Kumar, H. Singh, P. Sonawane, P. Pathak, M. Grishina, J. P. Yadav, A. Verma and P. Kumar, Chem. Biodiversity, 2023, 20, e202300061 CrossRef PubMed.
- P. K. Omer, N. M. Aziz and R. A. Omer, Rev. Inorg. Chem., 2024, 44, 699–710 CrossRef CAS.
- S. Swaminathan, J. Haribabu and R. Karvembu, ChemMedChem, 2024, 19, e202400435 CrossRef CAS PubMed.
- D. P. Dorairaj, P. Kumar, H. Rajasekaran, N. Bhuvanesh, S. C. N. Hsu and R. Karvembu, J. Inorg. Biochem., 2025, 262, 112759 CrossRef CAS PubMed.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z. S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- S. Thota, D. A. Rodrigues, D. C. Crans and E. J. Barreiro, J. Med. Chem., 2018, 61, 5805–5821 CrossRef CAS PubMed.
- C. Molinaro, A. Martoriati, L. Pelinski and K. Cailliau, Cancers, 2020, 12, 2863 CrossRef CAS PubMed.
- P. Ji, P. Wang, H. Chen, Y. Xu, J. Ge, Z. Tian and Z. Yan, Pharmaceuticals, 2023, 16, 234 CrossRef CAS PubMed.
- M. J. Waring, Bioorg. Med. Chem. Lett., 2009, 19, 2844–2851 CrossRef CAS PubMed.
- R. K. Ameta, K. Soni and A. Bhattarai, Colloids Interfaces, 2023, 7, 16 CrossRef CAS.
- A. F. Peacock and P. J. Sadler, Chem. – Asian J., 2008, 3, 1890–1899 CrossRef CAS PubMed.
- R. A. Casero and L. J. Marton, Nat. Rev. Drug Discovery, 2007, 6, 373–390 CrossRef CAS PubMed.
- A. E. Pegg, IUBMB Life, 2014, 66, 8–18 CrossRef CAS PubMed.
- M. Vojtek, S. Gonçalves-Monteiro, P. Šeminská, K. Valová, L. Bellón, P. Dias-Pereira, F. Marques, M. P. M. Marques, A. L. M. Batista de Carvalho, H. Mota-Filipe, I. Ferreira and C. Diniz, Biomedicines, 2022, 10, 210 CrossRef CAS PubMed.
- T. J. Carneiro, M. Vojtek, S. Gonçalves-Monteiro, J. R. Neves, A. Carvalho, M. P. M. Marques, C. Diniz and A. M. Gil, Pharmaceutics, 2022, 14, 259 CrossRef CAS PubMed.
- I. Lamego, M. P. Marques, I. F. Duarte, A. S. Martins, H. Oliveira and A. M. Gil, J. Proteome Res., 2017, 16, 1773–1783 CrossRef CAS PubMed.
- L. J. Teixeira, M. Seabra, E. Reis, M. T. Girão da Cruz, M. Conceição Pedroso de Lima, E. Pereira, M. A. Miranda and M. P. M. Marques, J. Med. Chem., 2004, 47, 2917–2925 CrossRef CAS PubMed.
- T. M. Silva, S. Oredsson, L. Persson, P. Woster and M. P. M. Marques, J. Inorg. Biochem., 2012, 108, 1–7 CrossRef CAS PubMed.
- N. Sanz del Olmo, A. M. Bajo, M. Ionov, S. García-Gallego, M. Bryszewska, R. Gómez, P. Ortega and F. J. de la Mata, Eur. J. Med. Chem., 2020, 199, 112414 CrossRef CAS PubMed.
- B. Canonico, R. Carloni, N. Sanz Del Olmo, S. Papa, M. G. Nasoni, A. Fattori, M. Cangiotti, F. J. de la Mata, M. F. Ottaviani and S. García-Gallego, Mol. Pharm., 2020, 17, 2691–2702 CrossRef CAS PubMed.
- N. Sanz del Olmo, M. Maroto-Díaz, R. Gómez, P. Ortega, M. Cangiotti, M. F. Ottaviani and F. J. de la Mata, J. Inorg. Biochem., 2017, 177, 211–218 CrossRef CAS PubMed.
- T. Nishio, Y. Yoshikawa, C.-Y. Shew, N. Umezawa, T. Higuchi and K. Yoshikawa, Sci. Rep., 2019, 9, 14971 CrossRef PubMed.
- B. Brycki, I. Kowalczyk, J. Werner, T. Pospieszny and A. Kozirog, Int. J. Org. Chem., 2017, 7, 106–139 CrossRef CAS.
- S. García-Gallego, M. J. Serramía, E. Arnaiz, L. Díaz, M. A. Muñoz-Fernández, P. Gómez-Sal, M. F. Ottaviani, R. Gómez and F. J. de la Mata, Eur. J. Inorg. Chem., 2011, 2011, 1657–1665 CrossRef.
- N. J. Lundin, I. G. Hamilton and A. G. Blackman, Polyhedron, 2004, 23, 97–102 CrossRef CAS.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anticancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS PubMed.
- M. Maroto-Díaz, B. T. Elie, P. Gómez-Sal, J. Pérez-Serrano, R. Gómez, M. Contel and F. Javier de la Mata, Dalton Trans., 2016, 45, 7049–7066 RSC.
- M. Maroto-Diaz, N. S. del Olmo, L. Muñoz-Moreno, A. M. Bajo, M. J. Carmena, R. Gómez, S. García-Gallego and F. J. de la Mata, Eur. Polym. J., 2019, 113, 229–235 CrossRef CAS.
- M. Janiszewska, M. C. Primi and T. Izard, J. Biol. Chem., 2020, 295, 2495–2505 CrossRef CAS PubMed.
- S. E. Le Devedec, K. A. Yan, H. de Bont, V. Ghotra, H. Truong, E. H. Danen, F. Verbeek and B. van de Water, Cell. Mol. Life Sci., 2010, 67, 3219–3240 CrossRef CAS PubMed.
- N. W. Clarke, C. A. Hart and M. D. Brown, Asian J. Androl., 2009, 11, 57–67 CrossRef CAS PubMed.
- I. B. Roninson, E. V. Broude and B. D. Chang, Drug Resist. Updates, 2001, 4, 303–313 CrossRef CAS PubMed.
- P. Singh and B. Lim, Curr. Oncol. Rep., 2022, 24, 273–284 CrossRef CAS PubMed.
- N. A. P. Franken, H. M. Rodermond, J. Stap, J. Haveman and C. van Bree, Nat. Protoc., 2006, 1, 2315–2319 CrossRef CAS PubMed.
- J. Bai, Y. Li and G. Zhang, Cancer Biol. Med., 2017, 14, 348–362 CrossRef CAS PubMed.
- Y. Sun, Y. Liu, X. Ma and H. Hu, Int. J. Mol. Sci., 2021, 22(13), 6923 CrossRef CAS PubMed.
- C. O. D'Sousa Costa, J. H. Araujo Neto, I. R. S. Baliza, R. B. Dias, L. F. Valverde, M. T. A. Vidal, C. B. S. Sales, C. A. G. Rocha, D. R. M. Moreira, M. B. P. Soares, A. A. Batista and D. P. Bezerra, Oncotarget, 2017, 8, 104367–104392 CrossRef PubMed.
- I. H. de Sousa, V. N. S. Campos, A. A. M. Vale, V. L. Maciel-Silva, C. M. Leite, A. J. O. Lopes, P. S. Mourão, F. das Chagas Alves Lima, A. A. Batista, A. P. S. de Azevedo dos Santos, M. A. P. Almeida and S. R. F. Pereira, Toxicol. in Vitro, 2020, 62, 104679 CrossRef PubMed.
- S. Biswas, A. Wasai, M. Ghosh, C. Rizzoli, A. Roy, S. Saha and S. Mandal, J. Inorg. Biochem., 2023, 247, 112314 CrossRef CAS PubMed.
- G. Y. Liou and P. Storz, Free Radical Res., 2010, 44, 479–496 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03377a |
| This journal is © The Royal Society of Chemistry 2025 |