
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphine/sulfoxide-carbone, a ligand with a flexible bonding mode for early to late transition metals†
Sophie
Hameury
 ab,
Laura
Bousquet
a,
Nathalie
Saffon-Merceron
c,
Antoine
Baceiredo
a,
David
Madec
a and
Eddy
Maerten
*a
ab,
Laura
Bousquet
a,
Nathalie
Saffon-Merceron
c,
Antoine
Baceiredo
a,
David
Madec
a and
Eddy
Maerten
*a
aUniversité de Toulouse, UPS, and CNRS, LHFA UMR 5069, 118 route de Narbonne, 31062 Toulouse, France. E-mail: eddy.maerten@univ-tlse3.fr
bUniversité de Poitiers, IC2MP, UMR CNRS 7285, 1 Rue Marcel Doré, CEDEX 9, 86073 Poitiers, France
cUniversité de Toulouse, UPS, and CNRS, ICT UAR 2599, 118 route de Narbonne, 31062 Toulouse, France
First published on 31st January 2025
Abstract
In recent years, carbones have emerged as a new exciting class of carbon-based ligands. We report here a series of organometallic complexes demonstrating the versatility in coordination mode of phosphine/sulfoxide carbone 1. Indeed, 1 is able to chelate both early and late transition metals, in a mono- or bidentate fashion depending on the oxyphilic character of metal center thanks to the presence of the sulfoxide moiety. All complexes have been fully characterized by X-ray diffraction analysis and by NMR spectroscopy (except hafnium complex because of its extreme insolubility). It is noteworthy that silver(I) and zirconium(IV) complexes are efficient transmetalating reagents toward copper(I) complexes.
Introduction
The discovery of the first stable carbenes more than 30 years ago1 shed light on divalent carbon chemistry.2 Since then, carbon-based ligands have undergone considerable development to the point of becoming essential tools in homogeneous transition metal catalysis.3 Carbenes naturally occupy a prominent place, but bis-ylides, also named “carbones”, have recently been attracting a great attention of many research groups.4 Indeed, these species I initially described by Ramirez in 1961,5 present a peculiar electronic environment with a divalent carbon atom bearing two lone pairs (Fig. 1).6 The central carbon being extremely electron-rich, they can be used as NHCs alternative as ligand, their electron-donating ability was even shown to be superior.7 Thanks to the presence of the two lone pairs on the central carbon atom, they are excellent ligands for the synthesis of a wide range of transition metal complexes. It has been shown that carbones I–IV (Fig. 1) can act as either two- or four-electron-donors8,9 and give access to homo- and hetero-bimetallic complexes.10,11 Nevertheless, their use in catalysis remains scarce when compared to NHCs12 probably due to the different electronic situation of central carbon atom. Indeed, NHCs are known to bind tightly to metal centers thanks to a strong C–metal bond with some π-back donation from the metal to the vacant orbital of carbon center.13 In the case of carbones, without an empty orbital available at the carbon center, the stabilization effect of π-back donation is not possible. Thus, we have shown that a carbone ligand V,14 featuring aminophosphine and sulfide moieties, despite a strong nucleophilic character could not lead to stable organometallic complexes. In contrast, the introduction of a sulfoxide moiety instead of sulfide dramatically alters the coordination behavior and with carbone 1 several stable organometallic complexes have been prepared.15 Indeed, the presence of sulfoxide function allows a better stabilization of one of the two lone pairs at the carbon center as indicated by a shorten C–S bond length.15a Therefore, as a part of our studies on low valent species,16 we have decided to evaluate in depth the ligand efficiency of phosphine/sulfoxide carbone 1 towards various metallic centers. | ||
| Fig. 1 Selected examples of carbones. | ||
Results and discussion
We have initiated our study with group 11 metals taking into account preliminary results obtained with gold(I) precursors. Indeed, we have previously established than 1 could act as a four electrons donor ligand towards gold and gem-aurated dinuclear complex VI was selectively formed by reaction of 1 with 2 equivalents of [AuCl(SMe2)] (Scheme 1, left).15a The use of 0.5 equivalent of metal precursor with respect to the ligand allowed the selective formation of the cationic bis-ligated Au(I) complex 2, which was isolated as a white powder in 80% yield (Scheme 1, right).17 In the 31P NMR spectrum, 2 displays a singlet signal at δ = 42.3 ppm, while the central carbon atom appears as a doublet at δ = 40.7 ppm (1JCP = 80.3 Hz) in the 13C NMR spectrum. | ||
| Scheme 1 Reactivity of 1 with [AuCl(SMe2)]. | ||
Single crystals of 2 were grown from a saturated dichloromethane/diethyl ether solution at 4 °C and the structure has been established by X-ray diffraction analysis (80% yield, Fig. 2). Complex 2 adopts a nearly linear geometry with a C1–Au1–C28 angle of 177.4°. Both C1 and C28 are in a quasi-planar planar environment (∑° = 357.5° and 355.7° respectively), the P–C and S–C bond lengths are longer than those in 1 but shorter than those in VI, indicating that only one lone pair is involved in the Au–C bond whereas the second one is delocalized towards phosphorus and sulfur atoms (Table 1). The Au1–C1 and Au1–C28 bond lengths of 2.043(2) and 2.051(2) Å are in the range of the previously observed values for carbone–Au complexes (2.01–2.08 Å)7d,10a,b,12a and slightly longer than those observed in the case of NHC ligated cationic gold(I) complexes (1.95–2.02 Å).18
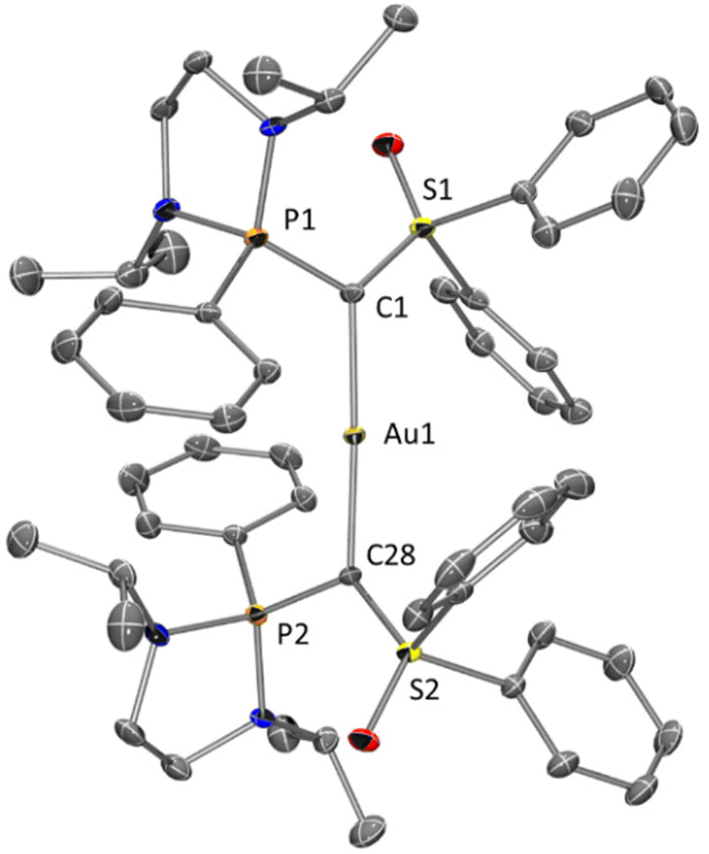 | ||
| Fig. 2 Molecular structure of 2. Thermal ellipsoids represent 30% probability. H, disordered atoms, solvent molecule (dichloromethane) and counter ion (Cl−) were omitted for clarity. Selected bond lengths [Å] and angles [°]: Au1–C1 2.043(2), Au1–C28 2.051(2), P1–C1 1.706(3), P2–C28 1.698(3), S1–C1 1.649(3), S2–C28 1.644(2), C1–Au1–C28 177.41(11), S1–C1–P1 116.26(15), S2–C28–P2 117.44(15), P1–C1–Au1 120.40(14), P2–C28–Au1 115.87(13), S1–C1–Au1 120.86(14), S2–C28–Au1 122.37(14). | ||
| 1 | 2 | VI | |
|---|---|---|---|
| P–C | 1.656(2) | 1.706(3) | 1.781(4) |
| 1.698(3) | |||
| S–C | 1.593(1) | 1.649(3) | 1.737(4) |
| 1.644(2) | |||
| P–C–S | 120.7(1) | 116.3(1) | 110.2(2) |
| 117.4(2) |
Using the same methodology 1 reacts with 0.5 equivalent of [AgOTf] leading to the cationic silver(I) complex 3 in 90% yield (Scheme 2).
 | ||
| Scheme 2 Formation of cationic silver complex 3 and transmetalation reaction with CuCl affording copper complex 4. | ||
In the 31P NMR spectrum, complex 3 shows a characteristic doublet signal at δ = 41.4 ppm (2JAgP = 5.9 Hz), which confirms unambiguously the coordination of the ligand to the silver atom. Moreover, in the 13C NMR spectrum, the central carbon atom appears as two sets of doublets of doublets at δ = 30.9 ppm and δ = 30.7 ppm. Indeed, in addition to the classical 1JCP coupling, 1JCAg coupling with both 107/109Ag isotopes are observed (1JCAg = 137.8 Hz, 1JCAg = 140.7 Hz, 1JCP = 60.0 Hz).19 The identification of the respective coupling constants was ensured by a 13C{1H;31P} spectrum (see ESI†).
Similarly to NHC–Ag(I) complexes,20 silver complex 3 is an excellent transmetalation agent toward copper (Scheme 2). The addition of copper chloride to 3 in CD2Cl2, monitored by 31P NMR spectroscopy, lead to a complete conversion in 16 h, as indicated by a new singlet signal at δ = 44.0 ppm corresponding to the new cationic complex 4. In the 13C NMR spectrum, the central carbon is slightly downfield shifted at δ = 31.2 ppm and the silver–carbon coupling constants have disappeared. Noteworthy, complex 4 can also be directly obtained from the direct reaction between carbone 1 and 0.5 equivalent of [CuCl] (77% yield).
The molecular structures of 3 and 4 were confirmed by X-ray diffraction analysis (Fig. 3). As complex 2, both cationic complexes 3 and 4 display an almost perfect linear geometry with a C1–Ag1–C2 angle of 179.3° for 3 and a C1–Cu1–C2 angle of 178.3° for 4 respectively. Again, for both complexes, the P1–C1, P2–C2, S1–C1 and S2–C2 distances are longer than in 1 indicating that each ligand acts as a two-electron donor. Those values together with C1–Ag1 and C2–Ag1 bond lengths (2.110(3) and 2.114(3) respectively) in 3 are in good agreement with those reported by Fujii for analogous silver complexes coordinated by iminosulfide/phosphine- and iminosulfide/sulfide-carbones.8f,10d–f
 | ||
| Fig. 3 Molecular structures of 3 and 4. Thermal ellipsoids represent 30% probability. H atoms, solvent (THF) and a triflate molecule (as counter ion) were omitted for clarity in 3. H atoms and a triflate molecule (as counter ion) were omitted for clarity in 4. Selected bond lengths [Å] and angles [°] for 3: Ag1–C2 2.110(3), Ag1–C1 2.114(3), P1–C1 1.695(3), P2–C2 1.694(3), S1–C1 1.636(3), S2–C2 1.634(3), C1–Ag1–C2 179.31(11), S1–C1–P1 115.58(16), S2–C2–P2 116.42(18), S1–C1–Ag1 118.59(15), S2–C2–Ag1 119.01(16), P1–C1–Ag1 120.38(16), P2–C2–Ag1 122.65(15). | ||
Selected bond lengths [Å] and angles [°] for 4: Cu1–C2 1.919(2), Cu1–C1 1.918(2), P1–C1 1.702(2), P2–C2 1.706(2), S1–C1 1.638(2), S2–C2 1.641(2), C1–Cu1–C2 178.34(9), S1–C1–P1 114.70(11), S2–C2–P2 114.49(11), S1–C1–Cu1 121.66(11), S2–C2–Cu1 121.44(11), P1–C1–Cu1 119.31(10), P2–C2–Cu1 118.16(11).
In addition to carbophilic metals from group 11, the coordination behavior of 1 toward oxophilic metals was also studied. In this context, group 4 metals were chosen to favor the coordination of the sulfoxide to the metal center. Our study was initiated with the reactivity of 1 toward titanium precursor [TiCl4(THF)2] with the aim of synthesizing complex 5 in which 1 could act as a (C,O)-chelating ligand (Scheme 3). Surprisingly, instead of complex 5, product 6 was formed, resulting from the chlorination of the central carbon of 1 and reduction of the sulfoxonium moiety into the corresponding sulfonium.21
 | ||
| Scheme 3 Reactivity of 1 with titanium(IV) precursor. | ||
The molecular structure of 6 was confirmed by X-ray diffraction analysis (Fig. 4). Monitoring the reaction by 31P NMR spectroscopy, chlorinated compound 6 was formed with 90% selectivity together with 10% hydrolysis product. Product 6 can also be obtained by reacting C2Cl6 with phosphine/sulfide-carbone 7 (Scheme 3).
 | ||
| Fig. 4 Molecular structure of 6. Thermal ellipsoids represent 30% probability. H atoms and a triflate molecule (as counter ion) were omitted for clarity. Selected bond lengths [Å] and angles [°]: P1–C1 1.710(3), S1–C1 1.684(3), Cl1–C1 1.748(3), S1–C1–P1 120.3(2), P1–C1–Cl1 118.0(2), S1–C1–Cl1 119.0(2). | ||
The formation of 6 was assigned to the high reactivity of [TiCl4] and its tendency to form titanium oxide, which formation was confirmed by XRD analysis. With less reactive titanium precursors, such as [Ti(Cp)2Cl2] or [Ti(Cp)2(Me)2], no reaction was observed. In contrast, 1 reacts with [ZrCl4] affording a new cationic complex 9 (90% selectivity) together with 10% of phosphonium/sulfoxonium salts 10 (Scheme 4). Colorless crystals of 9, suitable for X-ray diffraction analysis, were obtained from a saturated THF solution (Fig. 5).
 | ||
| Scheme 4 Reactivity toward zirconium(IV). | ||
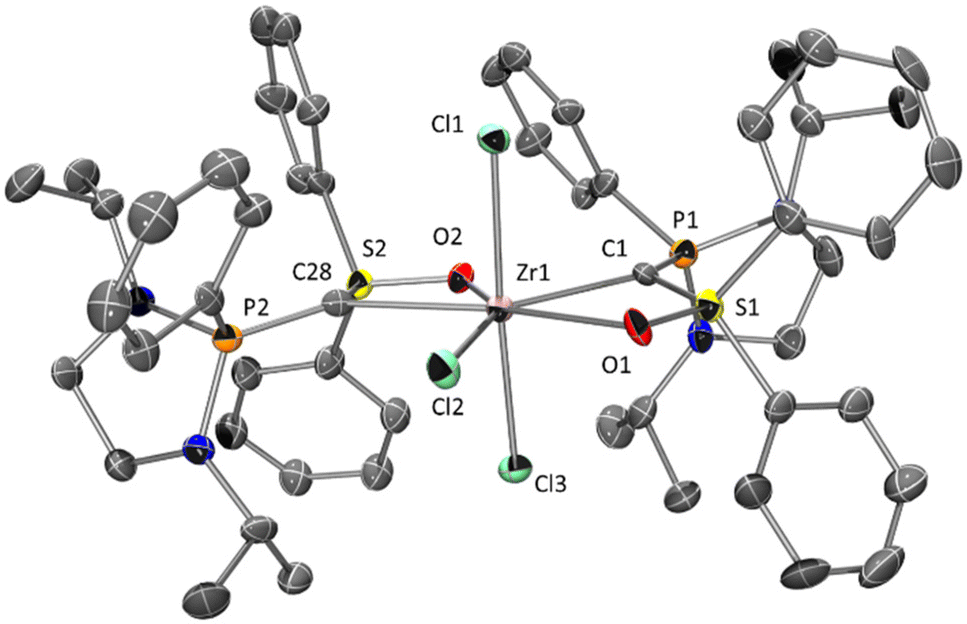 | ||
| Fig. 5 Molecular structure of 9. Thermal ellipsoids represent 30% probability. H atoms, disordered atoms, anion (ZrCl5−) and solvent molecules (THF) were omitted for clarity. Selected bond lengths [Å] and angles [°]: P1–C1 1.689(15), P2–C28 1.726(13), S1–C1 1.619(14), S2–C28 1.602(12), Zr1–C1 2.438(13), Zr1–O1 2.295(9), Zr1–C28 2.383(12), Zr1–O2 2.244(8), Zr1–Cl1 2.491(3), Zr1–Cl2 2.446(4), Zr1–Cl3 2.453(4), S1–C1–P1 126.4(8), S2–C28–P2 124.2(7) S1–C1–Zr1 91.6(6), S2–C28–Zr1 92.3(5), P1–C1–Zr1 140.2(7), P2–C28–Zr1 141.4(6). | ||
Instead of the expected carbone–ZrCl4 complex 8, a cationic complex 9 featuring two carbone ligands was observed (Scheme 4). Indeed, even though the overall stoichiometry of the reaction was respected, two carbone ligands act as (C,O)-bidentate hybrid ligand chelating a single [ZrCl3]+ moiety. The electroneutrality of the complex is ensured by [ZrCl5(THF)]− anion resulting from a halide abstraction thus allowing the formation of the cationic counterpart.
ZrIV ions feature a coordination number of seven with two (C,O)-chelating carbones and three terminal chlorido ligands (pentagonal bipyramid), more classically observed with group 5 metals.22 The two carbones and a chlorido group constitute a quasi-planar pentagonal environment. The P–C and C–Zr bond lengths differ quite significantly for the two carbones (∼0.04 Å and ∼0.05 Å respectively, see Fig. 5). The apical positions are occupied by the last two chlorido groups, with typical Zr–Cl bonds lengths of 2.491(3) and 2.453(4) Å, slightly longer than the one in the pentagonal plan [2.442(4) Å]. If the P–C bond lengths in complex 9 are extremely similar to those described in the complexes 2, 3 or 4 (less than 1% variation), the S–C bond lengths are significantly shortened upon coordination of the sulfoxonium to the metal center (respectively 1.619 Å and 1.602 Å in 9 compared to 1.634–1.649 Å observed in 2, 3 or 4). The characterization by NMR spectroscopy in solution suffers from large signals that arise from fluxional behavior (see ESI†).23 Nevertheless, a large signal can be observed in the 31P NMR spectrum at δ = 42.7 ppm, in the same range of previous complexes.
Zirconium complexes being known to be efficient transmetalation agents,24 transfer of carbones from Zr to Cu was tested. Pleasingly, complex 9 smoothly reacts with [CuCl] at room temperature to afford quantitatively copper complex 4 (Scheme 5).
 | ||
| Scheme 5 Carbones transfer from Zr complex 9 to Cu complex 4. | ||
A heavier analogue of complex 9 was obtained by reacting 1 with [HfCl4] (Scheme 6). The formation of cationic complex 11 was evidenced by X-Ray diffraction analysis. The ligand coordination mode and complex geometry are strictly identical to complex 9 (see ESI† for X-Ray data). Unfortunately, no NMR characterization could be performed because of an extreme insolubility of 11 in all common organic solvent.
 | ||
| Scheme 6 Reactivity of 1 with hafnium(IV) chloride. | ||
Conclusions
A series of group 4 and 11 complexes coordinated by phosphine/sulfoxide-carbone 1 were successfully synthesized in good yields under mild conditions. The X-ray diffraction analyses of all the synthesized complexes have contributed to the elucidation of the solid-state coordination modes. For group 11, new cationic complexes were obtained when reacted with half an equivalent of the metal. A bimetallic complex could be obtained in the case of gold, when 1 was reacted with 2 equivalents of metal. For group 4 complexes, carbone 1 acts as a bidentate (C,O)-chelating ligand which are the first examples of such coordination mode. Silver and zirconium complexes 3 and 9 are excellent transmetallation agents toward copper(I) complexes. Taking in account the easy access of carbone 1, the mild conditions used to obtain the described complexes and their relative stability, their use in catalytic transformations should extend the scope of carbones as ligand.Experimental
General procedure
All manipulations were performed under an inert atmosphere of argon by using standard Schlenk techniques. Dry and oxygen-free solvents were used. 1H, 13C and 31P NMR spectra were recorded on Brucker Avance 500 or Avance 300 spectrometers. 1H NMR and 13C NMR chemical shifts are reported in parts per million (ppm) relative to Me4Si as external standard. 31P NMR downfield chemical are expressed in ppm relative to 85% H3PO4. 19F-chemical shifts were reported in ppm relative to CFCl3 as an external standard. Mass spectra were recorded on Hewlett Packard 5989A spectrometer. Powder X-Ray diffraction data were recorded at room temperature on a Rigaku MiniFlex600 (θ–2θ) diffractometer with Cu Kα1,Kα2 radiation (λ = 1.54059, 1.54442 Å). Data collection was performed over the angular range 5° < 2θ < 90° with a step size of 0.02°. All commercially available reagents were used without further purification otherwise noted. Ylide 1-HOTf15a and 7-HOTf14a were prepared following previously reported procedures.Synthetic procedures
31P{1H} NMR (CD2Cl2, 298 K, 202 MHz) δ = 42.3 (s). 1H NMR (CD2Cl2, 298 K, 500 MHz) δ = 7.88–7.80 (m, 8H, CHPh), 7.72–7.64 (m, 8H, CHPh), 7.61–7.53 (m, 10H, CHPh), 7.41–7.36 (m, 4H, CHPh), 3.01–2.82 (m, 12H, CH2bridge & CHiPr), 0.37 (d, JHH = 6.6 Hz, 12H, CH3iPr), 0.34 (d, JHH = 6.6 Hz, 12H, CH3iPr). 13C NMR (CD2Cl2, 298 K, 126 MHz) δ = 147.6 (d, JCP = 6.3 Hz, SCipso), 133.9 (d, JCP = 130.0 Hz, PCipso), 132.9 (s, CHPh), 132.8 (d, JCP = 2.9 Hz, CHPh), 132.5 (d, JCP = 10.1 Hz, CHPh), 129.4 (s, CHPh), 128.9 (d, JCP = 13.2 Hz, CHPh), 127.6 (s, CHPh), 44.9 (d, JCP = 5.3 Hz, CHiPr), 40.7 (d, JCP = 80.3 Hz, PCS), 38.5 (d, JCP = 7.2 Hz, CH2bridge), 19.5 (2 overlapping broad doublets, CH3iPr). HRMS (ES+): m/z [M]+ calculated for C54H66O2N4P2S2Au = 1125.3762, found = 1125.3793.
31P{1H} NMR (CD2Cl2, 298 K, 121 MHz) δ = 41.4 (d, JAgP = 5.9 Hz). 1H NMR (CD2Cl2, 298 K, 300 MHz) δ = 7.89 (d, JHH = 7.3 Hz, 8H, CHPh), 7.74–7.54 (m, 18H, CHPh), 7.46–7.36 (m, 4H, CHPh), 3.06–2.70 (m, 12H, CH2bridge & CHiPr), 0.38 (d, JHH = 6.3 Hz, 24H, CH3iPr). 13C NMR (CD2Cl2, 298 K, 75 MHz) δ = 148.9 (d, JCP = 8.2 Hz, SCipso), 148.8 (d, JCP = 8.3 Hz, SCipso), 134.9 (d, JCP = 130.0 Hz, PCipso), 132.9 (d, JCP = 3.0 Hz, CHPh), 132.8 (s, CHPh), 132.3 (d, JCP = 9.7 Hz, CHPh), 129.7 (s, CHPh), 129.3 (d, JCP = 13.1 Hz, CHPh), 127.3 (s, CHPh), 121.7 (q, JCF = 321.6 Hz, CF3), 45.1 (d, JCP = 5.2 Hz, CHiPr), 38.6 (d, JCP = 6.9 Hz, CH2bridge), 30.8 (ddd, JCAg = 137.9 Hz, JCAg = 138.1 Hz, JCP = 60.0 Hz, PCS), 19.8 (d, JCP = 4.6 Hz, CH3iPr), 19.6 (d, JCP = 3.0 Hz, CH3iPr). 19F{1H} NMR (CD2Cl2, 298 K, 282 MHz): δ = −78.7 (s).
31P{1H} NMR (CD2Cl2, 298 K, 121 MHz) δ = 44.0 (s). 1H NMR (CD2Cl2, 298 K, 300 MHz) δ = 7.89 (d, JHH = 7.4 Hz, 8H, CHPh), 7.77–7.58 (m, 18H, CHPh), 7.50–7.40 (m, 4H, CHPh), 3.05–2.70 (m, 12H, CH2bridge & CHiPr), 0.37 (d, JHH = 6.5 Hz, 12H, CH3iPr) 0.33 (d, JHH = 6.5 Hz, 12H, CH3iPr). 13C NMR (CD2Cl2, 298 K, 75 MHz) δ = 147.7 (d, JCP = 7.7 Hz, SCipso), 134.8 (d, JCP = 127.5 Hz, PCipso), 133.0 (d, JCP = 3.1 Hz, CHPh), 132.9 (s, CHPh), 132.2 (d, JCP = 10.0 Hz, CHPh), 129.6 (s, CHPh), 129.3 (d, JCP = 13.0 Hz, CHPh), 127.4 (s, CHPh), 121.6 (q, JCF = 321.5 Hz, CF3), 44.9 (d, JCP = 5.6 Hz, CHiPr), 38.6 (d, JCP = 7.1 Hz, CH2bridge), 31.2 (d, JCP = 64.5 Hz, PCS), 19.8 (d, JCP = 4.8 Hz, CH3iPr), 19.6 (d, JCP = 3.0 Hz, CH3iPr). 19F{1H} NMR (CD2Cl2, 298 K, 282 MHz): δ = −78.7 (s). Elemental analysis for 4 calculated for C55H66CuF3N4O5P2S3: C: 57.86; H: 5.83; N: 4.91. Found: C: 57.12; H: 5.84; N: 4.82.
31P{1H} NMR (CDCl3, 298 K, 121 MHz) δ = 49.8 (s). 1H NMR (CDCl3, 298 K, 300 MHz) δ = 7.83–7.69 (m, 3H, CHPh), 7.68–7.52 (m, 12H, CHPh), 3.45–3.33 (dsept, JHH = 6.6 Hz, JPH = 9.0 Hz, 2H, CHiPr), 3.45–3.33 (m, CH2, 2H), 3.29–3.17 (m, CH2, 2H), 1.03 (d, JHH = 6.6 Hz, 6H, CH3iPr), 0.98 (d, JHH = 6.6 Hz, 6H, CH3iPr). 13C NMR (CDCl3, 298 K, 75 MHz) δ = 135.0 (d, JCP = 3.0 Hz, CHPh), 133.1 (s, CHPh), 132.5 (d, JCP = 10.4 Hz, CHPh), 130.8 (s, CHPh), 130.5 (d, JCP = 3.8 Hz, SCipso), 130.3 (d, JCP = 13.4 Hz, CHPh), 129.1 (s, CHPh), 123.7 (d, JCP = 128.3 Hz, PCipso), 121.1 (q, JCF = 321.1 Hz, CF3), 45.5 (d, JCP = 5.6 Hz, CHiPr), 38.9 (d, JCP = 9.8 Hz, CH2), 37.5 (d, JCP = 158.0 Hz, PCS), 20.6 (d, JCP = 4.8 Hz, CH3iPr), 20.2 (d, JCP = 3.1 Hz, CH3iPr).
31P{1H} NMR (CD2Cl2, 298 K, 121 MHz) δ = 42.7 (broad s). 1H NMR (CD2Cl2, 298 K, 300 MHz) δ = 8.40–7.90 (m, 5H, CHPh), 7.86–7.51 (m, 7H, CHPh), 7.49–7.05 (m, 2H, CHPh), 4.60–4.38 (coord THF), 3.95–3.41 (m, 4H, CHiPr + THF), 3.24–2.73 (m, CH2, 4H), 2.11–1.63 (m, coord. THF + THF, 4H), 0.73–0.28 (m, 6H, CH3iPr), 0.20 (d, JHH = 6.2 Hz, 6H, CH3iPr). 13C NMR (CD2Cl2, 298 K, 75 MHz) δ = 140.9 (broad s, CHPh), 135.3 (s, CHPh), 133.1 (d, JCP = 3.0 Hz, CHPh), 132.0 (d, JCP = 128.4 Hz PCipso), 130.9 (s, CHPh), 130.0 (s, CHPh), 128.6 (s, CHPh), 128.5 (overlapping d, JCP could not be calculated, CHPh),76.7 (s, CH2-coord THF), 68.5 (s, CH2-THF), 45.2 (broad s, CHiPr), 38.9 (d, JCP = 7.9 Hz, CH2bridge), 26.1 (s, CH2-THF), 20.5 (d, JCP = 5.6 Hz, CH3iPr), 18.9 (broad s, CH3iPr). The signal for the central carbon was not observed in this analysis but was detected in another sample [δ = 33.1 (d, JCP = 135.1 Hz, PCS)].
Crystallography
The data of the structures for 2, 3, 4, 6, 9 and 11 were collected at 193 K on a Bruker-AXS APEX II CCD Quazar diffractometer equipped with a 30 W air-cooled microfocus source (3, 6, 9 and 11), or on a Brucker-AXS D8-Venture diffractometer equipped with a CMOS Area detector (2 and 4) with MoKα radiation (wavelength = 0.71073 Å) by using phi- and omega-scans. The data were integrated with SAINT, and an empirical absorption correction with SADABS was applied.25 The structures were solved using an intrinsic phasing method (ShelXT)26 and refined using the least-squares method on F2 (ShelXL-2014).27 All non-H atoms were treated anisotropically. All H atoms attached to C atoms were fixed geometrically and treated as riding on their parent atoms with C–H = 0.95 Å (aromatic), 0.98 Å (CH3), 0.99 Å (CH2) or 1.0 Å (CH) with Uiso(H) = 1.2Ueq(CH, CH2) or Uiso(H) = 1.5Ueq(CH3).Most of the structures were found to be strongly disordered, especially the solvent molecules and the counter anions. Several restraints (SAME, SADI, SIMU, DELU, RIGU, ISOR) and equal xyz and Uij constraints EXYZ and EADP (for 11) were applied to refine some moieties of the molecules and to avoid the collapse of the structures during the least-squares refinement by the large anisotropic displacement parameters. Some bond lengths were restrained with DFIX and DANG to suitable target values (9 and 11).
Author contributions
Conceptualization: E.M., S.H.; investigation: S.H. except the synthesis of 2 which was performed by L.B.; validation: E.M.; data curation: E.M.; X-ray structural studies: N. S. M.; supervision: E.M., D.M.; writing—original draft preparation: S.H., E.M.; writing—review and editing: all authors.; All authors have read and agreed to the published version of the manuscript.Data availability
Additional data supporting this article have been included as part of the ESI† (additional experimental information, NMR spectra and crystallographic data).CCDC 2366566–2366571 (2, 3, 4, 6, 9, 11) contain the supplementary crystallographic data for this paper.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thanks the Ministère de l'Enseignement Supérieur et de la Recherche, the CNRS and the Université de Toulouse (UPS) for financial support. Dr Marie Fustier-Boutignon is greatly acknowledged for fruitful discussions. The NMR, the mass spectroscopy and S. Mallet-Ladeira from X-ray services from the ICT are also acknowledged.References
- (a) A. Igau, H. Grützmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463–6466 CrossRef CAS; (b) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- (a) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed; (b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (c) N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed; (d) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed; (e) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314–325 CrossRef CAS PubMed; (f) P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2014, 53, 1485–1487 CrossRef CAS PubMed.
- (a) F. Glorius, in N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, Berlin, Heidelberg, 2006, vol. 21, p. 1 Search PubMed; (b) T. Kato, E. Maerten and A. Baceiredo, in Transition Metal Complexes of Neutral η1-Carbon Ligands, ed. R. Chauvin and Y. Canac, Springer, Berlin, 11th edn, 2010, vol. 30, p. 131 Search PubMed; (c) E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed.
- (a) M. Fustier-Boutignon, N. Nebra and N. Mézailles, Chem. Rev., 2019, 119, 8555–8700 CrossRef CAS PubMed; (b) F. Krischer and V. H. Gessner, JACS Au, 2024, 1709–1722 CAS.
- F. Ramirez, N. B. Desai, B. Hansen and N. McKelvie, J. Am. Chem. Soc., 1961, 83, 3539–3540 CrossRef CAS.
- (a) R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2006, 45, 8038–8042 CrossRef CAS PubMed; (b) R. Tonner and G. Frenking, Angew. Chem., Int. Ed., 2007, 46, 8695–8698 CrossRef CAS PubMed; (c) R. Tonner and G. Frenking, Chem. – Eur. J., 2008, 14, 3260–3272 CrossRef CAS PubMed; (d) R. Tonner and G. Frenking, Chem. – Eur. J., 2008, 14, 3273–3289 CrossRef CAS PubMed; (e) D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2013, 53, 370–374 CrossRef PubMed; (f) G. Frenking, Angew. Chem., Int. Ed., 2014, 53, 6040–6046 CrossRef CAS PubMed; (g) D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 6047–6048 CrossRef CAS PubMed.
- (a) V. Lavallo, Y. Canac, A. DeHope, B. Donnadieu and B. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 7236–7239 CrossRef CAS PubMed; (b) S. Marrot, T. Kato, H. Gornitzka and A. Baceiredo, Angew. Chem., Int. Ed., 2006, 45, 2598–2601 CrossRef CAS PubMed; (c) C. A. Dyker, V. Lavallo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206–3209 CrossRef CAS PubMed; (d) A. Fürstner, M. Alcarazo, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 47, 3210–3214 CrossRef PubMed; (e) V. Lavallo, C. A. Dyker, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 5411–5414 CrossRef CAS PubMed; (f) F. Lavigne, A. El Kazzi, Y. Escudié, E. Maerten, T. Kato, N. Saffon-Merceron, V. Branchadell, F. P. Cossio and A. Baceiredo, Chem. – Eur. J., 2014, 20, 12528–12536 CrossRef CAS PubMed.
- (a) W. C. Kaska, D. K. Mitchell and R. F. Reichelderfer, J. Organomet. Chem., 1973, 47, 391–402 CrossRef CAS; (b) H. Schmidbaur, C. E. Zybill, G. Müller and C. Krüger, Angew. Chem., Int. Ed. Engl., 1983, 22, 729–730 CrossRef; (c) W. Petz, F. Weller, J. Uddin and G. Frenking, Organometallics, 1999, 18, 619–626 CrossRef CAS; (d) W.-C. Chen, Y.-C. Hsu, C.-Y. Lee, G. P. A. Yap and T.-G. Ong, Organometallics, 2013, 32, 2435–2442 CrossRef CAS; (e) W.-C. Chen, J.-S. Shen, T. Jurca, C.-J. Peng, Y.-H. Lin, Y.-P. Wang, W.-C. Shih, G. P. A. Yap and T.-G. Ong, Angew. Chem., Int. Ed., 2015, 54, 15207–15212 CrossRef CAS PubMed; (f) T. Morosaki, R. Iijima, T. Suzuki, W.-W. Wang, S. Nagase and T. Fujii, Chem. – Eur. J., 2017, 23, 8694–8702 CrossRef CAS PubMed; (g) T. Troadec, T. Wasano, R. Lenk, A. Baceiredo, N. Saffon-Merceron, D. Hashizume, Y. Saiton, N. Nakata, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 15207–15212 CrossRef PubMed; (h) A. Kroll, H. Steinert, L. T. Scharf, T. Scherpf, B. Mallick and V. H. Gessner, Chem. Commun., 2020, 56, 8051–8054 RSC.
- J. Sundermeyer, K. Weber, K. Peters and H. G. von Schnering, Organometallics, 1994, 13, 2560–2562 CrossRef CAS.
- (a) H. Schmidbaur and O. Gasser, Angew. Chem., Int. Ed. Engl., 1976, 15, 542–543 CrossRef; (b) J. Vicente and A. R. Singhal, Organometallics, 2002, 21, 5887–5900 CrossRef CAS; (c) M. Alcarazo, C. W. Lehmann, A. Anoop, W. Thiel and A. Fürstner, Nat. Chem., 2009, 1, 295–301 CrossRef CAS PubMed; (d) T. Morosaki, W.-W. Wang, S. Nagase and T. Fujii, Chem. – Eur. J., 2015, 21, 15405–15411 CrossRef CAS PubMed; (e) T. Morosaki, T. Suzuki and T. Fujii, Organometallics, 2016, 35, 2715–2721 CrossRef CAS; (f) B. S. Aweke, C.-H. Yu, J.-S. Shen, S. Wang, G. P. A. Yap, W.-C. Chen and T.-G. Ong, Inorg. Chem., 2023, 62, 12664–12673 CrossRef CAS PubMed.
- (a) C. Reitsamer, W. Schuh, H. Kopacka, K. Wurst and P. Peringer, Organometallics, 2009, 28, 6617–6620 CrossRef CAS; (b) K. Kubo, H. Okitsu, H. Miwa, S. Kume, R. G. Cavell and T. Mizuta, Organometallics, 2017, 36, 266–274 CrossRef CAS.
- (a) R. Corberan, S. Marrot, N. Dellus, N. Merceron-Saffon, T. Kato, E. Peris and A. Baceiredo, Organometallics, 2009, 28, 326–330 CAS; (b) M. J. Goldfogel, C. C. Roberts and S. J. Meek, J. Am. Chem. Soc., 2014, 136, 6227–6230 CAS; (c) Y.-C. Hsu, J.-S. Shen, B.-C. Lin, W.-C. Chen, Y.-T. Chan, W.-M. Ching, G. P. A. Yap, C.-P. Hsu and T.-G. Ong, Angew. Chem., Int. Ed., 2015, 54, 2420–2424 CrossRef CAS PubMed; (d) C. C. Roberts, D. M. Matias, M. J. Goldfogel and S. J. Meek, J. Am. Chem. Soc., 2015, 137, 6488–6491 CrossRef CAS PubMed; (e) C. Pranckevicius, L. Fan and D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 5582–5589 CrossRef CAS PubMed; (f) J. S. Marcum, C. C. Roberts, R. S. Manan, T. N. Cervarich and S. J. Meek, J. Am. Chem. Soc., 2017, 139, 15580–15583 CrossRef CAS PubMed; (g) A. L. Liberman-Martin and R. H. Grubss, Organometallics, 2017, 36, 4091–4094 CrossRef CAS; (h) C. R. Aversa-Fleener, D. K. Chang and A. L. Liberman-Martin, Organometallics, 2021, 40, 4050–4054 CrossRef CAS.
- (a) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS; (b) H. Jacobsen, A. Correa, C. Costabile and L. Cavallo, J. Organomet. Chem., 2006, 691, 4350–4358 CrossRef CAS; (c) N. Ségaud, C. Johnson, A. Farre and M. Albrecht, Chem. Commun., 2021, 57, 10600–10603 RSC.
- (a) N. Dellus, T. Kato, X. Bagán, N. Saffon-Merceron, V. Branchadell and A. Baceiredo, Angew. Chem., Int. Ed., 2010, 49, 6798–6801 CrossRef CAS PubMed; (b) N. Dellus, T. Kato, N. Saffon-Merceron, V. Branchadell and A. Baceiredo, Inorg. Chem., 2011, 50, 7949–7951 CrossRef CAS PubMed.
- (a) M. L. González, L. Bousquet, S. Hameury, C. Alvarez Toledano, N. Saffon-Merceron, V. Branchadell, E. Maerten and A. Baceiredo, Chem. – Eur. J., 2018, 24, 2570–2574 CrossRef PubMed; (b) U. Authesserre, S. Hameury, A. Dajnak, N. Saffon-Merceron, A. Baceiredo, D. Madec and E. Maerten, Molecules, 2021, 26, 2005 CrossRef CAS PubMed.
- (a) F. Lavigne, E. Maerten, G. Alcaraz, N. Saffon-Merceron and A. Baceiredo, Chem. – Eur. J., 2014, 20, 297–303 CrossRef CAS PubMed; (b) A. Garduno-Alva, R. Lenk, Y. Escudié, M. Lozano González, L. Bousquet, N. Saffon-Merceron, C. Alvarez Toledano, X. Bagan, V. Branchadell, E. Maerten and A. Baceiredo, Eur. J. Inorg. Chem., 2017, 29, 3494–3497 CrossRef; (c) A. Dajnak, E. Maerten, N. Saffon-Merceron, A. Baceiredo and T. Kato, Organometallics, 2020, 39, 3403–3412 CrossRef CAS; (d) A. Dajnak, G. Altınbaş Özpınar, R. Lenk, N. Saffon-Merceron, A. Baceiredo, T. Kato, T. Müller and E. Maerten, Dalton Trans., 2022, 51, 1407–1414 RSC; (e) N. Lentz, A. Sodreau, A. Acuña, S. Ladeira, E. Maerten, J. Sotiropoulos, R. S. Rojas and D. Madec, Dalton Trans., 2023, 52, 6841–6846 RSC; (f) U. Authesserre, V. S. V. S. N. Swamy, N. Saffon-Merceron, A. Baceiredo, T. Kato and E. Maerten, Molecules, 2023, 28, 3295 CrossRef CAS PubMed; (g) A. Dajnak, L. Shi, G. Altınbaş Özpınar, R. Lenk, N. Saffon-Merceron, A. Baceiredo, T. Kato, T. Müller and E. Maerten, Dalton Trans., 2023, 52, 3052–3058 RSC; (h) A. Acuña, S. Mallet-Ladeira, J.-M. Sotiropoulos, E. Maerten, A. R. Cabrera, A. Baceiredo, T. Kato, R. S. Rojas and D. Madec, Molecules, 2024, 29, 325 CrossRef PubMed.
- Attempts to isolate the mono-ligated neutral Au–Cl complex with from equimolar reaction of 1 with [AuCl(SMe)2] in various conditions only resulted in the formation of 2 and VI in a 40/60 ratio .
- (a) P. de Frémont, E. D. Stevens, M. R. Fructos, M. M. Diaz-Requejo, P. J. Pérez and S. P. Nolan, Chem. Commun., 2006, 2045–2047 RSC; (b) P. de Frémont, N. Marion and S. P. Nolan, J. Organomet. Chem., 2009, 694, 551–560 CrossRef; (c) M. Bouhrara, E. Jeanneau, L. Veyre, C. Copéret and C. Thieuleux, Dalton Trans., 2011, 40, 2995–2999 RSC; (d) C. Zhang, C. Hemmert, H. Gornitzka, O. Cuvillier, M. Zhang and R. W.-Y. Sun, ChemMedChem, 2018, 13, 1218–1229 CrossRef CAS PubMed; (e) A. Cervantes-Reyes, F. Rominger, M. Rudolph and A. S. K. Hashmi, Adv. Synth. Catal., 2020, 362, 2523–2533 CrossRef CAS.
- (a) R. Eujen, B. Hoge and D. Brauer, Inorg. Chem., 1997, 36, 1464–1475 CrossRef CAS PubMed; (b) R. Eujen, B. Hoge and D. J. Brauer, Inorg. Chem., 1997, 36, 3160–3166 CrossRef CAS PubMed; (c) C. Hansen, S. R. Docherty, W. Cao, A. V. Yakimov and C. Copéret, Chem. Sci., 2024, 15, 3028–3032 RSC.
- (a) H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS; (b) S. Hameury, P. de Frémont and P. Braunstein, Chem. Soc. Rev., 2017, 46, 632–733 RSC.
- (a) T. L. Ho and C.-M. Wong, Synth. Commun., 1973, 3, 37–38 CrossRef CAS; (b) S. Kano, Y. Tanaka, E. Sugino and S. Hibino, Synthesis, 1980, 695–697 CrossRef CAS; (c) M. Shimizu, K. Shibuya and R. Hayakawa, Synlett, 2000, 1437–1438 CAS; (d) H. Kominami, K. Nakanishi, S. Yamamoto, K. Imamura and K. Hashimoto, Catal. Commun., 2014, 54, 100–103 CrossRef CAS; (e) A. Luján-Montelongo, J.-B. Mateus-Ruiz and E. M. Valdez-García, Eur. J. Org. Chem., 2023, e202201156 CrossRef.
- (a) M. Polamo and M. Leskelä, J. Chem. Soc., Dalton Trans., 1996, 4345–4349 RSC; (b) D. Chakraborty, B. Rajashekhar, M. Mandal and V. Ramkumar, J. Organomet. Chem., 2018, 871, 111–121 CrossRef CAS; (c) R. Mundil, C. Bravo, N. Merle and P. Zinck, Chem. Rev., 2024, 124(1), 210–244 CrossRef CAS PubMed.
- J. Becker and V. H. Gessner, Organometallics, 2014, 33, 1310–1317 CrossRef CAS.
- (a) S. W. Reilly, C. E. Webster, T. K. Hollis and H. U. Vallea, Dalton Trans., 2016, 45, 2823–2828 RSC; (b) X. Yan and C. Xi, Coord. Chem. Rev., 2017, 350, 275–284 CrossRef CAS.
- SADABS, Program for data correction, Bruker AXS Search PubMed.
- G. M. Sheldrick, SHELXT—Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2366566–2366571. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03372h |
| This journal is © The Royal Society of Chemistry 2025 |