
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly phosphorescent N^C^N platinum(II)-peptide nucleic acid conjugates: synthesis, photophysical studies and hybridization behaviour†
Rosa Maria
Dell'Acqua
a,
Francesco
Fagnani
 a,
Monika
Wojciechowska
b,
Daniele
Marinotto
c,
Graziano
Colombo
d,
Isabella
Dalle-Donne
d,
Joanna
Trylska
b,
Silvia
Cauteruccio
*a and
Alessia
Colombo
*a
a,
Monika
Wojciechowska
b,
Daniele
Marinotto
c,
Graziano
Colombo
d,
Isabella
Dalle-Donne
d,
Joanna
Trylska
b,
Silvia
Cauteruccio
*a and
Alessia
Colombo
*a
aDepartment of Chemistry, Università degli Studi di Milano, via C. Golgi 19, I-20133 Milan, Italy. E-mail: alessia.colombo@unimi.it; silvia.cauteruccio@unimi.it
bCentre of New Technologies, University of Warsaw, S. Banacha 2c, 02-097 Warsaw, Poland
cIstituto di Scienze e Tecnologie Chimiche “Giulio Natta”, Consiglio Nazionale delle Ricerche (CNR-SCITEC), via C. Golgi 19, I-20133 Milan, Italy
dDepartment of Biosciences, Università degli Studi di Milano, via G. Celoria 26, I-20133 Milan, Italy
First published on 15th January 2025
Abstract
The synthesis of novel highly phosphorescent N^C^N tridentate platinum(II)-complex-peptide nucleic acid (PNA) bioconjugates was accomplished through the solid-phase approach. Melting temperature measurements and circular dichroism spectroscopy studies demonstrated that these conjugates maintain the PNA ability to recognize complementary ssDNA and ssRNA, though the length of the spacer between the metal center and the PNA sequence affects their hybridization properties. Noteworthy, the conjugation of PNA to this family of Pt(II) complexes significantly enhanced the luminescent features of the organometallic moiety, leading to increased quantum yields (82.8%, 10–5 M), even in the presence of oxygen (48.6%, 10–5 M). An in vitro cytotoxicity study of Pt(II)-PNA conjugates on HeLa cells showed no significative effect on cell growth in the dark (1 μM for 72 h).
Introduction
Nucleic acid analogues have been proposed to mimic the chemical features of natural DNA and RNA and to improve their biological properties, including increased enzymatic resistance and binding affinity towards complementary strands.1 All these properties make nucleic acid analogues useful agents in molecular biology, clinical diagnosis, and medical applications (e.g., gene therapy). Among them, peptide nucleic acids (PNAs), proposed for the first time by P. Nielsen in 1991,2 belong to one of the most promising classes of xeno-nucleic acids in which the negatively charged (deoxy)ribose phosphodiester skeleton of (DNA)RNA is replaced by the achiral and neutral pseudopeptide backbone, composed of N-(2-aminoethyl)glycine (aeg) units covalently linked to the nucleobases through an amide bond. Because of their neutral and unnatural backbone, PNAs show high specificity and stability in interactions with complementary DNA or RNA strands,3 as well as great enzymatic stability towards nucleases and proteases.4 Moreover, the high chemical stability of PNAs allows their easy manipulation and modification to improve and integrate their biological properties (i.e., cell penetration, tissue specificity and solubility in aqueous media).5,6 Thus, the PNA chemistry takes on an important role in the development of therapeutic agents, biosensors, and materials sciences.7 In particular, the conjugation of PNAs to luminescent coordination complexes represent an interesting approach to develop multifunctional bioconjugates potentially with great interest for biomedicine.8 Indeed, luminescent transition metal complexes display excellent optoelectronic properties. Their use as tracers for bioimaging in cells and tissues is of particular interest,9 since their high emission efficiency and long lifetimes allow for distinguishing their phosphorescence from the background autofluorescence of biological systems, which is an important advantage over common organic compounds. In the past, several studies have been reported on modified PNAs covalently linked to d6 transition-metal complexes, including complexes of Re(I)10 and Ru(II),11 where the organometallic moieties act as low-toxicity luminescent markers for PNA, as carriers for the internalization by living cells and as promoters of specific PNA recognition mechanisms, such as the double-duplex invasion. Conversely, very few examples of PNAs containing d8 platinum(II) complexes are present in the literature,12 and they are mainly focused on the use of cis-diamminedichloroplatinum(II) (cis-platin). This is quite surprising, taking into account that some platinum complexes, like neutral N^C^N tridentate Pt(II) derivatives, prove to be optimal candidates for biological applications, especially as promising long-lived bio-imaging probes13a–c or photosensitizers for photodynamic therapy (PDT).13d,e Moreover, they generally display very high quantum yield and good cell permeability. The unique photophysical features of these complexes, especially those bearing a cyclometallated 1,3-di(2-pyridyl)benzene structure (Fig. 1), contribute to their distinctive phosphorescence, making them useful for both in-depth bioimaging and PDT. When irradiated with light of the proper wavelength, these complexes can produce singlet oxygen in situ, and consequently damage or kill cells. The dual exploitation as both a dye for bioimaging and a PDT sensitizer could lead to the development of multifunctional compounds with great potential in biomedicine. These properties, combined with the low cytotoxicity and ability to penetrate cells localizing mainly in the nuclear region proved by some of them, make N^C^N tridentate Pt(II) complexes a transformative force in developing light-based cancer treatments and advanced imaging techniques. Furthermore, these cyclometallated Pt(II) complexes can be easily prepared using straightforward and well-defined procedures. The possibility to selectively functionalize the ligand skeleton with different substituents allows both the modulation of their photophysical properties and covalent conjugation to relevant biomolecules, including peptides suitable for targeted tumor therapy and theranostic applications.13d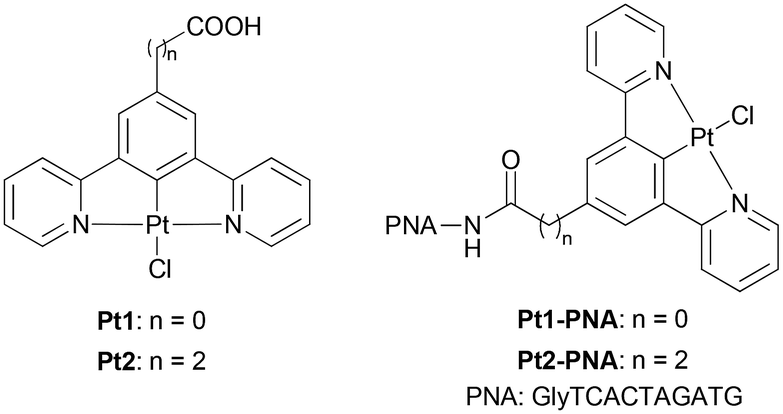 | ||
| Fig. 1 Structures of complexes Pt1 and Pt2 and of the corresponding Pt1-PNA and Pt2-PNA conjugates prepared in this study. | ||
In this context, the conjugation of N^C^N tridentate Pt(II) complexes to PNAs represents an unprecedented study toward an innovative bioorganometallic system. This system could be useful in diagnostics and gene therapy, thanks to the combination of the precise genetic targeting of PNAs with the aforementioned advantageous characteristics of platinum complexes.
Herein, we report the synthesis of two novel bioorganometallic dipyridyl benzene Pt(II)-PNA conjugates, Pt1-PNA and Pt2-PNA, formed by a model PNA decamer covalently linked to the corresponding Pt1 and Pt2 complexes (Fig. 1). An in-depth study of the hybridization of these conjugates with complementary DNA and RNA single strands has been performed using UV-monitored melting temperature measurements and circular dichroism spectroscopy. Limited data are present in the literature regarding the interaction of oligonucleotides with Pt(II) complexes featuring a cyclometallated tridentate ligand, since most studies have focused on platinum complexes similar to the cis-platin drug. Additionally, the photophysical properties of the Pt2-PNA conjugate have been investigated and compared with those of the Pt2 complex using absorption, excitation, and emission spectra. Finally, a preliminary in vitro evaluation of the Pt2-PNA toxicity in the dark has been performed using the MTT assay.
Results and discussion
Design and synthesis of the Pt(II)-PNA conjugates
Two Pt(II) complexes, Pt1 and Pt2, bearing benzoic and propanoic acid as anchoring groups, respectively, were selected for this study. These complexes differ in the length of the –(CH2)n– linker (n = 0 for Pt1 and n = 2 for Pt2) between the metal center and the PNA oligomer, allowing investigation of how the linker length affects the hybridization properties of the corresponding Pt-PNA conjugates. The carboxylic groups of Pt1 and Pt2 enabled covalent conjugation of the complexes to the PNA through the amide bond formation with the terminal amine group of the PNA sequence.Pt1 complex was prepared according to the literature,13d while Pt2 was synthesized in 72% yield by the complexation reaction between the tridentate ligand L (for the synthesis of L, see Scheme S1, ESI†) with K2PtCl4 in acetic acid at reflux (Scheme 1).
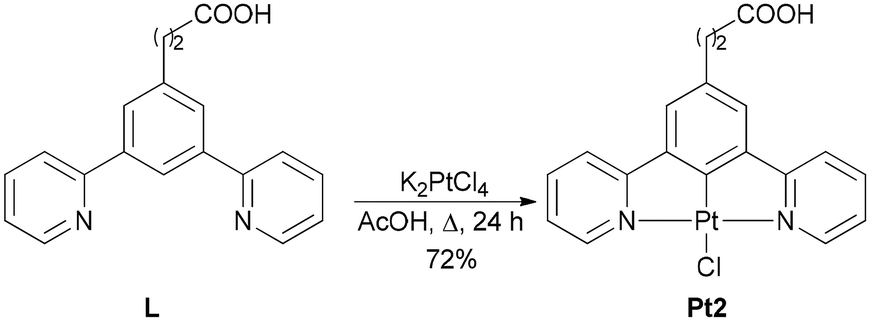 | ||
| Scheme 1 Synthesis of complex Pt2. | ||
Next, the model PNA decamer (NH2-GlyTCACTAGATG-H) was prepared through manual solid-phase synthesis using standard Fmoc/Bhoc (fluorenylmethyloxycarbonyl/benzhydryloxycarbonyl) chemistry.14 The Pt1-PNA and Pt2-PNA conjugates were synthesized by a three-step procedure starting from the resin-supported PNA decamer SP-PNA with complexes Pt1 and Pt2, respectively (Scheme 2).
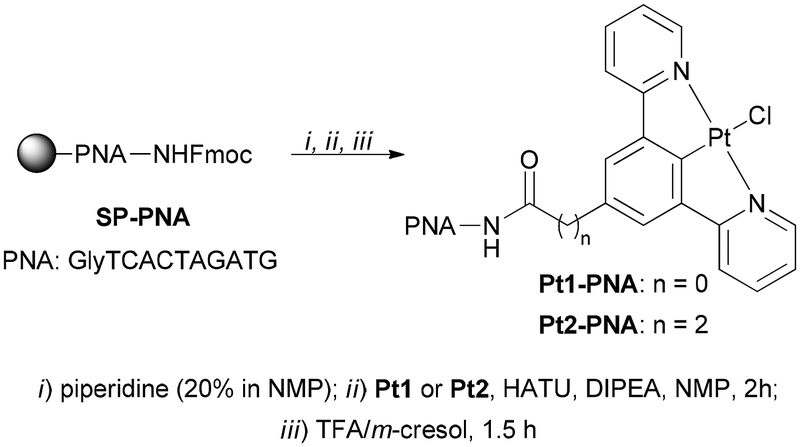 | ||
| Scheme 2 Synthesis of conjugates Pt1-PNA and Pt2-PNA. | ||
In particular, the N-terminal Fmoc protecting group was removed from SP-PNA with a solution of piperidine in 1-methyl-2-pyrrolidinone (NMP). The free amine group of SP-PNA was then reacted with the –COOH group of Pt1 or Pt2 to form the amide bond using O-(7-Aza-1H-benzotriazole-1-yl)-N,N,N,N-tetramethyluronium hexafluorophosphate (HATU) as condensing agent and N-ethyldiisopropylamine (DIPEA) as the base in NMP for 2 hours (Scheme 2). Finally, the cleavage of the conjugates from the resin under acidic conditions using trifluoroacetic acid (TFA) provided yellowish crude Pt1-PNA and Pt2-PNA conjugates, which were purified by reverse-phase high-performance liquid chromatography (RP-HPLC). The identity and purity of both conjugates were established by high-resolution electrospray ionization mass spectrometry (HRMS-ESI+) and RP-HPLC analyses.
Melting temperature and circular dichroism spectroscopy studies
We recorded melting profiles of platinum-PNA conjugates to examine how platinum(II) affects the thermal stability of duplexes formed between the PNA and the complementary DNA and RNA (Table 1 and Fig. S1†). The unconjugated PNA sequence, GlyTCACTAGATG, without the Pt(II) complex (unPNA), was included for comparison.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio). The Tm errors are SD from six measurements
:1 ratio). The Tm errors are SD from six measurements
| Oligonucleotides | T m/°C |
|---|---|
| unPNA | 41.5 ± 0.4 |
| unPNA/DNA | 51.8 ± 0.9 |
| unPNA/RNA | 54.8 ± 0.9 |
| Pt2-PNA | 70.2 ± 1.0 |
| Pt2-PNA/DNA | 60.8 ± 2.0 |
| Pt2-PNA/RNA | 49.1 ± 0.4 |
| Pt1-PNA | >80 |
| Pt1-PNA/DNA | Not determinable |
| Pt1-PNA/RNA | Not determinable |
| unPNA/Pt2 | 41.6 ± 1.2 |
| unPNA/DNA/Pt2 | 49.3 ± 2.3 |
| unPNA/RNA/Pt2 | 52.1 ± 2.3 |
The melting profiles of single-stranded DNA (ssDNA) and single-stranded RNA (ssRNA) confirmed the absence of self-duplexes (no Tm was observed). In contrast, unPNA exhibited a Tm of 41.5 ± 0.4 °C, which can be attributed to its partial self-complementarity and the known ability of PNA to form either anti-parallel or parallel duplexes with complementary sequences.15 The Tm values of unPNA with ssDNA and ssRNA aligned with the previously reported literature data (51.8 ± 0.9 °C and 54.8 ± 0.9 °C, respectively).16
An increase in Tm was observed for both Pt2-PNA and Pt1-PNA alone. Specifically, Pt2-PNA showed a Tm increase of 20.7 °C (Tm = 70.2 ± 1.0 °C), while Pt1-PNA exhibited an even greater increase of more than 30 °C (Tm > 80 °C). The conjugation of Pt2 to PNA led to higher Tm when Pt2-PNA formed a duplex with the complementary DNA strand (60.8 ± 2.0 °C) but slightly lower Tm with the RNA strand (49.1 ± 0.4 °C). For Pt1-PNA, determining the melting temperatures with DNA and RNA strands proved challenging, as the curves lacked an inflection point. This indicates that Pt1-PNA/DNA and Pt1-PNA/RNA duplexes do not adopt a dominant stable structure. To further understand the behavior of the platinum complexes in the presence of single-stranded PNA or PNA heteroduplexes with complementary oligonucleotides, melting temperatures were also measured with Pt2 alone. The results suggested that the platinum complex does not influence the thermal stability of unmodified PNA duplexes with DNA and RNA when it is not covalently connected to the PNA strand.
Overall, these results demonstrate that the platinum complexes covalently linked to the PNA strands affect the thermal stability of the duplexes formed with complementary oligonucleotides.
This effect likely stems from the planar geometry of the complexes, possibly enabling their intercalation between nucleobases and disrupting stacking. This notably impacts duplexes involving Pt1-PNA, likely because their direct connection to PNA forms a more rigid structure that restricts the conformational flexibility of Pt1. Conversely, in Pt2-PNA, the –(CH2)2– spacer makes the platinum complex more flexible and prone to intercalation. Intercalators are known to influence Tm by either increasing or decreasing thermal stability.17 Indeed, Pt2-PNA shows high duplex stabilization with DNA, though not with RNA. Furthermore, platinum complexes can cross-link DNA, coordinating with the N7 of purines replacing the labile Cl-atom, potentially affecting duplex stability through nucleobase bonding.18 A single example of a PNA conjugated to a cisplatin-based moiety has demonstrated that platinum-bearing PNA cross-linking with complementary DNA stabilizes the duplex.12a In the present study, given the variety of nucleobases available for platination, different products may be generated, each melting at distinct temperatures. This could explain why the Pt1-PNA duplexes lack an inflection point indicative of a single dominant complex between the Pt1-modified PNA and complementary oligonucleotide.
Interestingly, a subtle structural variation in the platinum-PNA conjugates, such as the addition of two methylene groups in the linker, led to markedly different thermal stabilities of PNA when paired with complementary DNA or RNA strands.
To delve deeper into the structural characteristics of the duplexes, circular dichroism (CD) spectra of the examined oligonucleotides were recorded (Fig. 2).
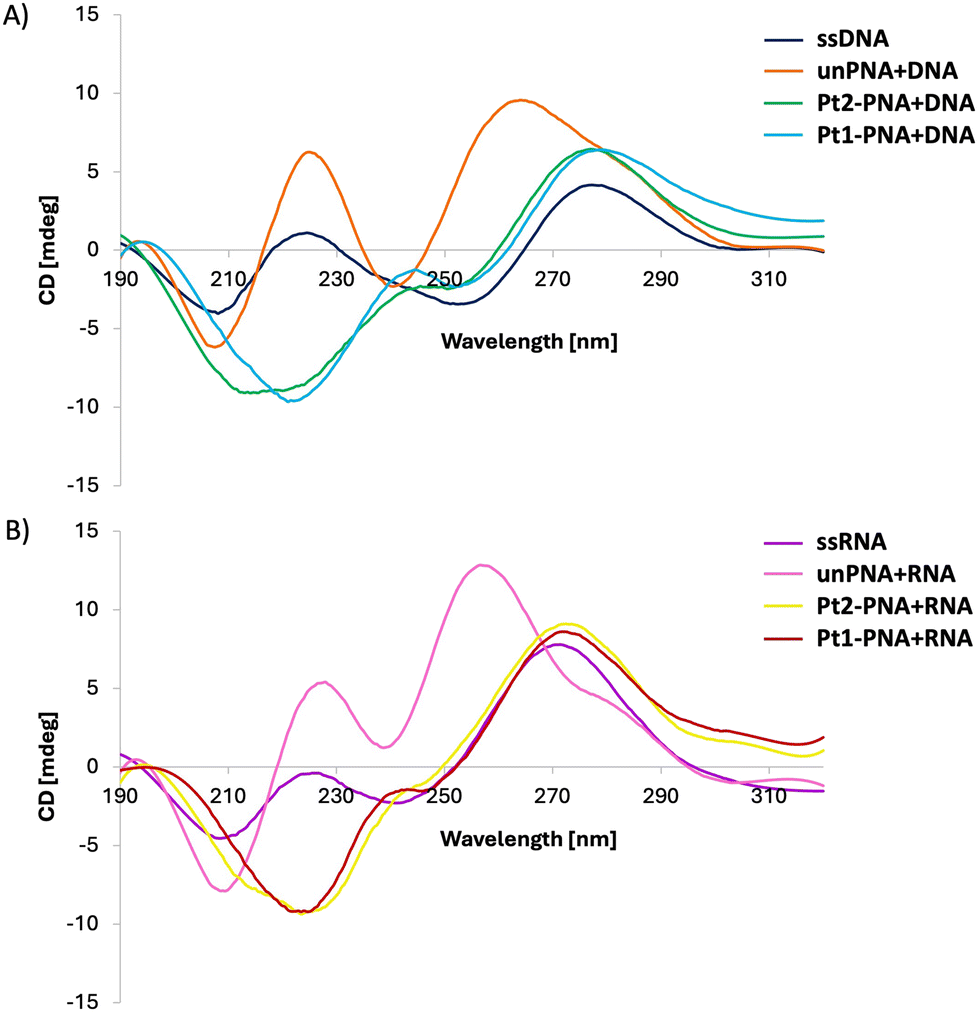 | ||
| Fig. 2 CD spectra of 4 μM solutions of either free DNA (A) and RNA (B) or with complementary PNAs and Pt-PNA conjugates in a 1:1 ratio. | ||
The CD spectra of ssDNA and ssRNA displayed characteristic bands for nucleic acids in helical forms, with positive bands appearing around 225 nm and 280 nm for DNA, and slightly shifted towards shorter wavelengths for ssRNA. The CD profiles of unPNA paired with complementary DNA and RNA aligned with those documented in the literature for the same sequences.19 The profile of the unPNA/DNA helix exhibited maxima at 225 nm, 265 nm, and 285 nm, and a minimum at 240 nm, while the unPNA/RNA duplex showed a shift of the band below 260 nm. The CD analysis of Pt2-PNA and Pt1-PNA with complementary DNA and RNA revealed a red-shift of the bands and a decrease in their amplitude, indicating a structural alteration of the helix in the presence of the platinum complexes. Specifically, cross-linking may perturb the overall geometry of the duplex and affect base stacking, leading to a reduced CD signal.20
The formation of cross-links may also perturb the helical structure, resulting in the red shift of the positive band at 265 nm.18c Thus, both Pt2-PNA and Pt1-PNA can form helical structures with their complementary nucleic acid strands, although in altered conformations compared to helices formed by unmodified PNA.
Photophysical characterization of Pt2 and Pt2-PNA conjugate
The photophysical properties of the promising Pt2-PNA conjugate have been deeply investigated by registering absorption, excitation, and emission spectra, and by measuring their absolute quantum yield (QY) and lifetimes in water solution, to compare them with those of Pt2 recorded in dichloromethane (CH2Cl2) solution. Although Pt2 is not soluble in water, it is important to underline that emission profile of N^C^N-Pt complexes recorded in CH2Cl2 and that obtained from DMSO/water solution shows negligible differences, thanks to a similar nature of the emitting excited state for the two compared conditions, as previously reported by some of us.9aThe absorption spectra of Pt2 and Pt2-PNA in CH2Cl2 and water solutions at different concentrations are shown in Fig. S2 and S10,† respectively, while Table 2 contains the salient photophysical data. Complex Pt2 shows an intense absorption band at 250–320 nm with a molar absorption coefficient (ε) between 17300 and 20700 M−1 cm−1, which can be attributed to intraligand 1π–π* transitions of the N^C^N ligand by analogy with related Pt(II) complexes.21 In addition, a less intense band (ε > 5700 M−1 cm−1) is observed at around 350–450 nm, which is associated with charge-transfer transitions concerning the cyclometallated ligand and the metal. Interestingly, as the concentration of the complex increases up to 1.3 × 10−4 M, we do not observe the formation of new bands due to aggregates of the complex, as also confirmed by the linearity in the absorbance vs. concentration graph, which follows the Lambert–Beer law, see Fig. S3.†
| Compound | λ abs nm−1 (ε/103 M−1 cm−1) | Concentration/M | λ max, em/nm | QYbefore FPT/% | QYafter FPT/% | τ av/μs |
|---|---|---|---|---|---|---|
| a λ ex = 374 nm → λem = 499 nm. b λ ex = 374 nm → λem = 492 nm. | ||||||
| Pt2 | 290 (17.3) | 1.0 × 10–6 | 499, 534 | 3.9 | 76.5 | 7.76 ± 0.03a |
| 380 (6.0) | 1.0 × 10–5 | 499, 534 | 3.0 | 55.0 | 6.42 ± 0.02a | |
| 410 (5.7) | 5.0 × 10–5 | 499, 534, 695 | 2.9 | 51.0 | 2.82 ± 0.01a | |
| Pt2-PNA | 260 (68.7) | 6.0 × 10–6 | 492, 523 | 54.6 | 87.1 | 16.9 ± 0.1b |
| 326 (7.7) | 1.0 × 10–5 | 492, 523, 640 | 48.6 | 82.8 | 16.8 ± 0.1b | |
| 380 (5.3) | 8.2 × 10–5 | 492, 526, 644 | 37.3 | 59.7 | 16.3 ± 0.2b | |
Complex Pt2-PNA displays an absorption spectrum characterized by a very intense absorption band at 260 nm (ε = 68700 M−1 cm−1), which can be mainly attributed to the absorption of the decamer moiety,22 and a weaker absorption band (ε = 5300 M−1 cm−1) at around 350–420 nm that is due to charge-transfer transitions of the cyclometallated ligand and the metal in analogy to the related Pt2 complex (Fig. 3).
 | ||
| Fig. 3 Normalized absorption (black) and emission (red, green, and blue) spectra in H2O for Pt2-PNA. Emission spectra with λex = 375 nm after the FPT cycles. | ||
An increase of the concentration of Pt2-PNA up to 1 × 10−5 M leads to the appearance of a new band at lower energy at 430–500 nm, ascribed to the formation of dimeric/aggregate species as confirmed by a deviation from the Lambert–Beer law (see Fig. S11†).
Emission spectra of compounds Pt2 and Pt2-PNA in deaerated CH2Cl2 and water solutions at different concentrations are shown in Fig. S4† and Fig. 3, respectively. At diluted concentrations, Pt2 shows an intense and highly structured band in the yellow-green region with emission maxima at 499 and 534 nm. Pt2-PNA displays similar highly structured bands, which are however slightly blue-shifted at 492 and 523 nm. These emission spectra originate from states of primarily ligand centered 3π–π* transitions of the monomeric species.
As the concentration of the solutions is increased, a new broad structureless emission band arises at lower energy with maxima at 695 and 644 nm for Pt2 and Pt2-PNA, respectively. The new bands at lower energy can be ascribed to the emission from bi-molecular emissive excited states (excimers and/or aggregates) of the N^C^N-Pt(II) moiety, as previously reported for related complexes.21b,23 In particular, the band at 695 nm of Pt2 originates from excimers emission, as evidenced by the recorded excitation spectra in which the profiles at 500 and 700 nm are perfectly superimposable (see Fig. S5†); since excimers do not have a ground state. Instead, the band at 644 nm of Pt2-PNA can be predominantly attributed to aggregate emission, although the presence of excimers cannot also be ruled out. The presence of aggregate has already been noted in the absorption spectra and is also confirmed by the different profiles in the excitation spectra at 492 and 640 nm.
The luminescence quantum yield (QY) of Pt2 and the conjugate Pt2-PNA in dilute deaerated solutions are respectively 76.5 and 87.1%. Even if the measurements were carried out in different solvents (CH2Cl2vs. H2O), it can be noticed that the QY is in any case higher for the Pt2-PNA than for the Pt2 alone (see Table 2). Interestingly, the QY value for Pt2-PNA surprisingly remains high even in the presence of O2 (54.6%), differently from Pt2 (3.9%) and previously reported complexes9a in which the QY in air-equilibrated solution was efficiently quenched. This difference is probably due to the steric hindrance provided by the PNA chain, which limits the interaction of the complex with oxygen. As the concentration of compounds Pt2 and Pt2-PNA is increased, the QY decreases. This behavior is related to the presence of excimers and/or aggregates characterized by less emissive excited states than the unimolecular ones.
Lifetime measurements of Pt2 and Pt2-PNA in deaerated CH2Cl2 and water solutions at different concentrations are shown in Fig. S13–S16 and S16–S20,† respectively.
For complex Pt2 in dilute solution, the excited state decay at the maximum emission wavelength (499 nm) follows a mono-exponential kinetic with τ = 7.76 ± 0.03 μs, which decreases at 2.82 ± 0.01 μs as the concentration is increased up to 5.0 × 10−5 M. At this concentration, the emission decay at 700 nm becomes bi-exponential due to the presence of excimers (see Fig. S9†), and following the article of J. B. Birks et al.,24 we have determined an excimer formation time of 1.79 ± 0.05 μs and an excimer decay time of 2.55 ± 0.06 μs.
The lifetime of Pt2-PNA at the maximum emission wavelength (492 nm) follows a mono-exponential kinetic only for the diluted solution with a τ = 16.9 ± 0.1 μs. By increasing the concentration, lifetimes become bi-exponential due to the presence of aggregates and excimers with a τav = 16.3 ± 0.2 μs for the most concentrated solution (8.2 × 10−5 M).
Preliminary in vitro cytotoxicity study of Pt2-PNA
The MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) is a widely used technique to assess cell viability and proliferation. This method is based on the reduction of yellow tetrazolium salt (MTT) to purple formazan by mitochondrial enzymes of metabolically active cells. The amount of formazan produced is proportional to the number of live cells. HeLa cells were seeded in a 24-well plate at the appropriate density (9.5 × 103 cells per well to ensure exponential growth for 4 days) and incubated for 24 hours at 37 °C with 5% CO2 to allow cell adhesion. After the initial incubation, cells were treated with Pt2-PNA (0–0.1 μM–1 μM) or relative vehicle (DMSO 0.05%–0.5%) for 24, 48 or 72 hours (Fig. 4). After incubation, 100 μl MTT solution (5 mg mL−1 in PBS) was added to each well, and cells were incubated for 3.5 hours at 37 °C with 5% CO2 to allow MTT reduction to formazan by live cells. After incubation, the supernatant was carefully removed and 500 μL of MTT solvent (4 mM HCl, 0.1% Nonidet P-40 in isopropanol) was added to each well and the plate was gently shaken for 15 minutes to ensure complete dissolution of the formazan. Absorbance at 590 nm was read using an EnSight Multimode Plate Reader (PerkinElmer). The cytotoxicity assay confirmed that the presence of the Pt-PNA conjugate does not affect the condition of eukaryotic cells. | ||
| Fig. 4 MTT assay on HeLa cells line after incubation with Pt2-PNA (0.1 μM–1 μM) for 24, 48 or 72 hours. | ||
Experimental section
Synthesis of complex Pt2
In a Schlenk tube, a solution of 3,5-dipyridin-2-ylphenylpropionic acid L (50 mg, 0.16 mmol) and K2PtCl4 (82.2 mg, 0.198 mmol, 1.2 eq.) in acetic acid (4 mL) was heated at reflux (120 °C, oil bath) for 48 hours under argon atmosphere. The reaction was then cooled to room temperature, and water was added to precipitate the complex. The solid was washed with methanol and subsequently with diethyl ether, affording Pt2 (62 mg, 72%) as a green powder.1H-NMR (400 MHz, DMSO-d6) δ: 12.11 (bs, 1H, –COOH), 9.11 (d, J = 5.8, J(195Pt) = 38 Hz, 2H, CH), 8.21 (ddd, J1 = 1.5 Hz, J2 = 7.7 Hz, J3 = 8.6 Hz, 2H, CH), 8.10 (d, J = 7.6 Hz, 2H, CH), 7.70 (s, 2H, CH), 7.55 (ddd, J1 = 1.4 Hz, J2 = 5.8 Hz, J3 = 7.7 Hz, 2H, CH), 2.87 (t, J = 7.4 Hz, 2H, CH2), 2.67 ppm (t, J = 7.4 Hz, 2H, CH2).
13C-NMR (100 MHz, DMSO-d6) δ:174.30 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 167.01 (Cq), 158.83 (Cq), 151.75 (CH), 140.84 (CH), 136.51 (Cq), 125.46 (CH), 124.50 (CH), 120.76 (CH), 118.57 (Cq), 35.75 (CH2), 31.49 ppm (CH2).
O), 167.01 (Cq), 158.83 (Cq), 151.75 (CH), 140.84 (CH), 136.51 (Cq), 125.46 (CH), 124.50 (CH), 120.76 (CH), 118.57 (Cq), 35.75 (CH2), 31.49 ppm (CH2).
HRMS (ESI−) m/z: [M − H]− calcd for C19H14N2O2Cl196Pt, 533.0393; found, 533.0399.
HRMS (ESI+) m/z: [M + Na]+ calcd for C19H15N2O2Cl196PtNa, 557.0364; found, 557.0378; [M − Cl − H + Na]+ calcd for C19H14N2O2195PtNa, 520.0601; found, 520.0614; [M − Cl]+ calcd for C19H15N2O2195Pt, 498.0781; found, 498.0796; [M − Cl − CH2COOH]+ calcd for C17H12N2195Pt, 439.0643; found, 439.0656.
General procedure for the synthesis of Pt1-PNA and Pt2-PNA conjugates
The resin-supported SP-PNA (40 mg, 0.2 mmol g−1, 8 μmol) was swollen in CH2Cl2 for 40 min, then the terminal N-Fmoc protecting group was removed by the treatment with a 20% v/v solution of piperidine in NMP (twice for 8 min). To a solution of the Pt1 or Pt2 (24 μmol, 3 eq.) and DIPEA (14 μL, 80 μmol, 10 eq.) in NMP a solution of HATU (9 mg, 24 μmol, 3 eq.) in NMP was added, and the resulting mixture was shaken for 2 min. The activated acid of Pt1 or Pt2 was then transferred to the resin and shaken for 2 h at room temperature. The resin was washed with NMP and CH2Cl2, and the Pt-PNA conjugates were cleaved from the resin treating with a mixture of TFA/m-cresol (9/1, v/v) for 1.5 h. The collected filtrate was concentrated before being precipitated with Et2O. The yellow solid was centrifugated, washed with Et2O, and dried under vacuum. The crude Pt-PNA conjugates were purified by RP-HPLC and characterized by HR-ESI+ mass spectrometry.Pt1-PNA conjugate: HRMS (ESI+) m/z: found (calculated) for C127H148N61O32Pt: 1618.5743 (1618.5828) [M − Cl + H]2+, 1630.0677 (1630.0712) [M − Cl + Na]2+, 1079.3855 (1079.3911) [M − Cl + 2H]3+, 809.5413 (809.5448) [M − Cl + 3H]4+. From the deconvolution of the signal: [M − Cl]+ calcd for C127H148N61O32195Pt, 3234.1477; found, 3234.1458. HR-ESI+ mass spectrum also displays signals related to multicharged dimers and trimers and their sodium adducts (2157.4307, 1623.5688, 1618.3242, 1387.6376, 1294.8596). RP-HPLC: tR = 18.3 min.
Pt2-PNA conjugate: HRMS (ESI+) m/z: found (calculated) for C129H152N61O32Pt: 1632.0919 (1632.0940) [M − Cl + H]2+, 1088.3956 (1088.3984) [M − Cl + 2H]3+, 1095.7225 (1095.7257) [M − Cl + H + Na]3+, 816.5493 (816.5506) [M − Cl + 3H]4+. From the deconvolution of the signal: [M + H]+ calcd for C129H152N61O32Pt, 3262.1790; found, 3262.1736. HR-ESI+ mass spectrum also displays signals related to multicharged dimers and trimers (2176.1160, 1958.9056, 1305.8730). RP-HPLC: tR = 17.1 min.
Conclusions
In conclusion, the conjugation of N^C^N tridentate Pt(II) complexes Pt1 and Pt2 to a model PNA decamer was accomplished through the amide bond formation on the solid phase. The corresponding Pt1-PNA and Pt2-PNA conjugates were isolated after the cleavage from the resin and the RP-HPLC purification, thanks to the chemical stability of these tridentate Pt(II) complexes under acidic cleavage conditions.Melting temperature studies of PNA and Pt(II)-PNA conjugates with complementary ssDNA and ssRNA confirmed duplex formation. However, the Pt(II) complexes influence PNA recognition to varying extents, depending on the linker length between Pt(II) and PNA. Circular dichroism spectra indicated that the Pt(II) complexes moderately affect the helical structure formed by PNA with DNA and RNA. Together, Tm and CD measurements provide insights into how platinum complexes bearing cyclometallated tridentate ligands interact with nucleic acids.
A detailed study of the photophysical properties of Pt2-PNA conjugate demonstrated that the intrinsic photophysical features of the platinum complex remained unaltered after the conjugation with PNA, thereby preserving its functional integrity. Notably, the quantum yield for Pt2-PNA was higher than that of Pt2 (QY equal to 83 vs. 55%, respectively), and remained high even in the presence of O2 probably due to the ability of PNA to protect the metal center by oxygen.
The in vitro preliminary MTT assay evaluating the cytotoxicity of Pt2-PNA showed no significative effect on cell growth after 3 days of incubation in the dark, both at 0.1 μM and 1 μM concentrations of the conjugate.
This work provides novel bioorganometallic PNA systems with diverse favorable features (e.g., high phosphorescence in solution, non-cytotoxicity) and opens many perspectives in various domains of PNA chemistry. Future work will focus on designing Pt(II)-PNA conjugates with dual-action capabilities for the synergistic treatment of cancer or bacterial infections, combining the precise genetic targeting of PNAs with the unique photophysical and biological features of N^C^N tridentate Pt(II) complexes. To achieve these goals, further investigation into cell uptake, internalization kinetics, and cellular distribution of this class of bioconjugates with specific biological targets will be needed to rationalize and design efficient therapeutic agents.
Author contributions
Rosa Maria Dell'Acqua: data curation, formal analysis, methodology, writing – review & editing. Francesco Fagnani: data curation, formal analysis, methodology, writing – review & editing. Monika Wojciechowska: data curation, formal analysis, methodology, writing – review & editing. Daniele Marinotto: data curation, formal analysis: equal; investigation, writing – review & editing. Graziano Colombo: data curation, formal analysis, methodology, writing – review & editing. Joanna Trylska: funding acquisition, supervision, writing – review & editing. Isabella Dalle-Donne: data curation, formal analysis, methodology, writing – review & editing. Silvia Cauteruccio: conceptualization, funding acquisition, supervision, writing – original draft. Alessia Colombo: conceptualization: equal; funding acquisition, supervision, writing – original draft.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. M. D. thanks Università degli Studi di Milano for the Ph.D. fellowship.This work was supported by Università degli Studi di Milano (PSR2022_DIP_005_PI_LCARL; PSR2022_DIP_005_PI_CGIAN).
Fondazione Cariplo and Regione Lombardia are acknowledged for the instrumentation bought during the SmartMatLab Centre project (2014).
Mass spectrometry analyses were performed at the Mass Spectrometry facility of the Unitech COSPECT at the University of Milan (Italy).
J. T. and M. W. thank National Science Centre Poland (UMO-2020/37/B/NZ1/02904) for funding.
References
- N. M. Bell and J. Micklefield, Chemical Modification of Oligonucleotides for Therapeutic, Bioanalytical and other Applications, ChemBioChem, 2009, 10, 2691–2703 CrossRef CAS.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide, Science, 1991, 254, 1497–1500 CrossRef CAS PubMed.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, PNA hybridizes to complementary oligonucleotides obeying the Watson–Crick hydrogen-bonding rules, Nature, 1993, 365, 566–568 CrossRef CAS PubMed.
- V. V. Demidov, V. N. Potaman, M. D. Frank-Kamenetskii, M. Egholm, O. Buchardt, S. H. Sönnichsen and P. E. Nielsen, Stability of peptide nucleic acids in human serum and cellular extracts, Biochem. Pharmacol., 1994, 48, 1310–1313 CrossRef CAS PubMed.
- S. Volpi, U. Cacelli, M. Neri and R. Corradini, Multifunctional Delivery Systems for Peptide Nucleic Acids, Pharmaceuticals, 2021, 14, 14 CrossRef CAS PubMed.
- C. Suparpprom and T. Vilaivan, Perspectives on conformationally constrained peptide nucleic acid (PNA): insights into the structural design, properties and applications, RSC Chem. Biol., 2022, 3, 648–697 RSC.
- For recent reviews, see: (a) V. MacLelland, M. Kravitz and A. Gupta, Therapeutic and diagnostic applications of antisense peptide nucleic acids, Mol. Ther.-Nucleic Acids, 2024, 35, 102086 CrossRef CAS; (b) L. M. Carson and E. E. Watson, Peptide Nucleic Acids: From Origami to Editing, ChemPlusChem, 2024, e202400305 CrossRef CAS; (c) J. Saarbach, P. M. Sabale and N. Winssinger, Peptide nucleic acid (PNA) and its applications in chemical biology, diagnostics, and therapeutics, Curr. Opin. Chem. Biol., 2019, 52, 112–124 CrossRef CAS PubMed.
- (a) S. Cauteruccio, E. Licandro, M. Panigati, G. D'Alfonso and S. Maiorana, Modifying the properties of organic molecules by conjugation with metal complexes: The case of peptide nucleic acids and of the intrinsically chiral thiahelicenes, Coord. Chem. Rev., 2019, 386, 119–137 CrossRef CAS; (b) G. Gasser, A. M. Sosniak and N. Metzler-Nolte, Metal-containing peptide nucleic acid conjugates, Dalton Trans., 2011, 40, 7061–7076 RSC.
- (a) A. Colombo, F. Fiorini, D. Septiadi, C. Dragonetti, F. Nisic, A. Valore, D. Roberto, M. Mauro and L. De Cola, Neutral N^C^N terdentate luminescent Pt(II) complexes: their synthesis, photophysical properties, and bio-imaging applications, Dalton Trans., 2015, 44, 8478–8487 RSC; (b) X. Zhen, R. Qu, W. Chen, W. Wu and X. Jiang, The development of phosphorescent probes for in vitro and in vivo bioimaging, Biomater. Sci., 2021, 9, 285–300 RSC; (c) Y. Ning, G.-Q. Jin, M.-X. Wang, S. Gao and J.-L. Zhang, Recent progress in metal-based molecular probes for optical bioimaging and biosensing, Curr. Opin. Chem. Biol., 2022, 66, 102097–102107 CrossRef CAS; (d) G. De Soricellis, F. Fagnani, A. Colombo, C. Dragonetti, D. Roberto, D. Marinotto, D. H. Hartnell, M. J. Hackett, M. Massi, B. Carboni and V. Guerchais, An attractive family of cyclometalated Ir(III) dyes functionalized with tryptophan for potential neuroimaging applications, Dyes Pigm., 2023, 210, 111012 CrossRef CAS; (e) E. Baggaleya, J. A. Weinstein and J. A. G. Williams, Lighting the way to see inside the live cell with luminescent transition metal complexes, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef.
- (a) G. Gasser, A. Pinto, S. Neumann, A. M. Sosniak, M. Seitz, K. Merz, R. Heumann and N. Metzler-Nolte, Synthesis, characterisation and bioimaging of a fluorescent rhenium-containing PNA bioconjugate, Dalton Trans., 2012, 41, 2304–2313 RSC; (b) S. Cauteruccio, M. Panigati, L. Veronese, N. Zaffaroni, M. Folini and E. Licandro, Luminescent dinuclear rhenium(I)single bondPNA conjugates for microRNA-21 targeting: Synthesis, chemico-physical and biological characterization, J. Organomet. Chem., 2019, 887, 32–39 CrossRef CAS; (c) C. Mari, M. Panigati, L. D'Alfonso, I. Zanoni, D. Donghi, L. Sironi, M. Collini, S. Maiorana, C. Baldoli, G. D'Alfonso and E. Licandro, Luminescent Conjugates between Dinuclear Rhenium Complexes and Peptide Nucleic Acids (PNA): Synthesis, Photophysical Characterization, and Cell Uptake, Organometallics, 2012, 31, 5918–5928 CrossRef CAS; (d) E. Ferri, D. Donghi, M. Panigati, G. Prencipe, L. D'Alfonso, I. Zanoni, C. Baldoli, S. Maiorana, G. D'Alfonso and E. Licandro, Luminescent conjugates between dinuclear rhenium(I) complexes and peptide nucleic acids (PNA) for cell imaging and DNA targeting, Chem. Commun., 2010, 46, 6255–6257 RSC.
- (a) M. Hibino, Y. Aiba, Y. Watanabe and O. Shoji, Peptide nucleic acid conjugated with Ruthenium–complex stabilizing double–duplex invasion complex even under physiological conditions, ChemBioChem, 2018, 19, 1601–1604 CrossRef CAS PubMed; (b) T. Joshi, G. J. Barbante, P. S. Francis, C. F. Hogan, A. M. Bond, G. Gasser and L. Spiccia, Electrochemiluminescent monomers for solid support syntheses of Ru(II)-PNA bioconjugates: multimodal biosensing tools with enhanced duplex stability, Inorg. Chem., 2012, 51, 3302–3315 CrossRef CAS PubMed; (c) S. Le Gac, M. Foucart, P. Gerbaux, E. Defrancq, C. Moucheron and A. Kirsch-De Mesmaeker, Photo-reactive RuII-oligonucleotide conjugates: influence of an intercalating ligand on the inter- and intra-strand photo-ligation processes, Dalton Trans., 2010, 39, 9672–9683 RSC; (d) J. C. Verheijen, G. A. van der Marel, J. H. van Boom and N. Metzler-Nolte, Transition Metal Derivatives of Peptide Nucleic Acid (PNA) OligomersSynthesis, Characterization, and DNA Binding, Bioconjugate Chem., 2000, 11, 741–743 CrossRef CAS.
- (a) D. W. Dodd, S. Damjanovski and R. H. E. Hudson, Peptide nucleic acid Pt(II) conjugates: A preliminary study of antisense effects in Xenopus laevis, Nucleosides, Nucleotides Nucleic Acids, 2011, 30, 257–263 CrossRef CAS PubMed; (b) K. S. Schmidt, M. Boudvillain, A. Schwartz, G. A. van der Marel, J. H. van Boom, J. Reedijk and B. Lippert, Monofunctionally trans-diammine Platinum(II)-modified peptide nucleic acid oligomers: A new generation of potential antisense drugs, Chem. – Eur. J., 2002, 8, 5566–5570 CrossRef CAS.
- (a) G. De Soricellis, F. Fagnani, A. Colombo, C. Dragonetti and D. Roberto, Exploring the potential of N^C^N cyclometalated Pt(II) complexes bearing 1,3-di(2-pyridyl)benzene derivatives for imaging and photodynamic therapy, Inorg. Chim. Acta, 2022, 541, 121082 CrossRef CAS; (b) E. Clancy, S. Ramadurai, S. R. Needham, K. Baker, T. A. Eastwood, J. A. Weinstein, D. P. Mulvihill and S. W. Botchway, Fluorescence and phosphorescence lifetime imaging reveals a significant cell nuclear viscosity and refractive index changes upon DNA damage, Sci. Rep., 2023, 13, 422 CrossRef CAS; (c) E. Baggaley, S. W. Botchway, J. W. Haycock, H. Morris, I. V. Sazanovich, J. A. G. Williams and J. A. Weinstein, Long-lived metal complexes open up microsecond lifetime imaging microscopy under multiphoton excitation: from FLIM to PLIM and beyond, Chem. Sci., 2014, 5, 879–886 RSC; (d) T. Chatzisideri, S. Thysiadis, S. Katsamakas, P. Dalezis, I. Sigala, T. Lazarides, E. Nikolakaki, D. Trafalis, O. A. Gederaas, M. Lindgren and V. Sarli, Synthesis and biological evaluation of a Platinum(II)-c(RGDyK) conjugate for integrin-targeted photodynamic therapy, Eur. J. Med. Chem., 2017, 141, 221–231 CrossRef CAS PubMed; (e) R. E. Doherty, I. V. Sazanovich, L. K. McKenzie, A. S. Stasheuski, R. Coyle, E. Baggaley, S. Bottomley, J. A. Weinstein and H. E. Bryant, Photodynamic killing of cancer cells by a Platinum(II) complex with cyclometallating ligand, Sci. Rep., 2016, 6, 22668 CrossRef CAS.
- P. E. Nielsen, Peptide Nucleic Acids: Methods and Protocols, Springer Science + Business Media, LLC, part of Springer Nature, Humana New York, NJ, 3rd edn, 2020 Search PubMed.
- P. Wittung, P. E. Nielsen, O. Buchardt, M. Egholm and B. Nordén, DNA-like double helix formed by peptide nucleic acid, Nature, 1994, 368, 561–563 CrossRef CAS PubMed.
- (a) S. Núñez-Pertíñez, T. R. Wilks and R. K. O'Reilly, Microcalorimetry and fluorescence show stable peptide nucleic acid (PNA) duplexes in high organic content solvent mixtures, Org. Biomol. Chem., 2019, 17, 7874–7877 RSC; (b) T. Ratilainen, A. Holmén, E. Tuite, G. Haaima, L. Christensen, P. E. Nielsen and B. Nordén, Hybridization of peptide nucleic acid, Biochemistry, 1998, 37, 12331–12342 CrossRef CAS PubMed.
- (a) B. J. Pages, K. B. Garbutcheon-Singh and J. R. Aldrich-Wright, Platinum Intercalators of DNA as Anticancer Agents, Eur. J. Inorg. Chem., 2017, 1613–1624 CrossRef CAS; (b) S. Narayanan, J. Gall and C. Richert, Clamping down on weak terminal base pairs: oligonucleotides with molecular caps as fidelity enhancing elements at the 5′- and 3′-terminal residues, Nucleic Acids Res., 2004, 32, 2901–2911 CrossRef CAS PubMed.
- (a) F. Boisten, I. Maisuls, T. Schäfer, C. A. Strassert and J. Müller, Site-specific covalent metalation of DNA oligonucleotides with phosphorescent platinum(II) complexes, Chem. Sci., 2023, 14, 2399–2404 RSC; (b) H. Rao, M. S. Damian, A. Alshiekh, S. K. C. Elmroth and U. Diederichsen, Design, synthesis and DNA interactions of a chimera between a platinum complex and an IHF mimicking peptide, Org. Biomol. Chem., 2015, 13, 11704–11713 RSC; (c) C. Hofr and V. Brabec, Thermal stability and energetics of 15-mer DNA duplex interstrand crosslinked by trans-diamminedichloroplatinum(II), Biopolymers, 2005, 77, 222–229 CrossRef CAS PubMed.
- R. Corradini, T. Tedeschi, S. Sforza and R. Marchelli, Electronic circular dichroism of peptide nucleic acids and their analogues, in Comprehensive Chiroptical Spectroscopy, Volume 2: Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products and Biomolecules, ed. N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, John Wiley & Sons, Inc, 2012, p. 587 Search PubMed.
- C. Hofr and V. Brabec, Thermal and thermodynamic properties of duplex DNA containing site-specific interstrand cross-link of antitumor cisplatin or its clinically ineffective trans isomer, J. Biol. Chem., 2001, 276, 9655–9661 CrossRef CAS PubMed.
- (a) F. Fagnani, G. De Soricellis, A. Colombo, C. Dragonetti, D. Roberto, A. di Biase, S. Fantacci and D. Marinotto, Photophysical investigation of highly phosphorescent N^C^N platinum(II) azido complexes and their triazole derivatives, Dyes Pigm., 2024, 225, 112064 CrossRef CAS; (b) F. Fagnani, A. Colombo, C. Dragonetti, D. Roberto and D. Marinotto, A novel class of cyclometalated Platinum(II) complexes for solution-processable OLEDs, Inorg. Chim. Acta, 2022, 532, 120744 CrossRef CAS.
- Molar extinction coefficients: e260 of adenine = 15400 M−1 cm−1; e260 of guanine = 11700 M−1 cm−1; e260 of thymine = 8800 M−1 cm−1; e260 of cytosine = 7400 M−1 cm−1.
- (a) C. Dragonetti, F. Fagnani, D. Marinotto, A. di Biase, D. Roberto, M. Cocchi, S. Fantacci and A. Colombo, First member of an appealing class of cyclometalated 1,3-di-(2-pyridyl) benzene platinum(II) complexes for solution-processable OLEDs, J. Mater. Chem. C, 2020, 8, 7873–7881 RSC; (b) J. Kalinowski, M. Cocchi, L. Murphy, J. A. G. Williams and V. Fattori, Bi-molecular emissive excited states in platinum(II) complexes for high-performance organic light-emitting diodes, Chem. Phys., 2010, 378, 47–57 CrossRef CAS; (c) A. Lázaro, R. Bosque, J. S. Ward, K. Rissanen, M. Crespo and L. Rodríguez, Toward Near-Infrared Emission in Pt(II)-Cyclometallated Compounds: From Excimers’ Formation to Aggregation-Induced Emission, Inorg. Chem., 2023, 62, 2000–2012 CrossRef PubMed.
- J. B. Birks, D. J. Dyson and I. H. Munro, ‘Excimer’ fluorescence II. Lifetime studies of pyrene solutions, Proc. R. Soc. London, Ser. A, 1963, 275, 575–588 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03366c |
| This journal is © The Royal Society of Chemistry 2025 |