
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diverse reactivity of a cationic N-heterocyclic phosphenium complex towards anionic substrates – substitution vs. reduction†
Christoph M.
Feil
a,
Florian
Goerigk
a,
Yannick
Stöckl
 a,
Martin
Nieger
b and
Dietrich
Gudat
*a
a,
Martin
Nieger
b and
Dietrich
Gudat
*a
aInstitut für Anorganische Chemie, University of Stuttgart, Pfaffenwaldring 55, 70550 Stuttgart, Germany. E-mail: gudat@iac.uni-stuttgart.de
bDepartment of Chemistry, University of Helsinki, P.O Box 55, 00014 University of Helsinki, Finland
First published on 27th December 2024
Abstract
A cationic N-heterocyclic phosphenium (NHP) iron tetracarbonyl complex was synthesised from the free cation and its behaviour towards various anionic reactants studied. Reactions with fluoride, chloride, and hydride sources proceeded under attachment of the anion at phosphorus to yield Fe(CO)4-complexes of neutral diazaphospholenes, while bromide and iodide reacted under addition of the anion at the metal and decarbonylation to yield NHP iron halides. Reactions with amides and organometallics were unselective. At room temperature, predominantly reduction of the cationic complex to yield a spectroscopically detectable Fe-centred radical and its deactivation products was observable. At −78 °C, CH-metalation at the heterocycle was preferred, as evidenced by the structural characterisation of a neutral borane-adduct of the metalation product of a modified NHP complex. The dimer of the Fe-centred radical formed also in reactions of chloro- and bromo-diazaphospholenes with Fe2(CO)9, which proceed not only by complexation of the P-donors as expected, but involve also oxidative addition steps and single electron transfer processes in which excess iron complex acts as the reductant. The title complex and the products isolated in the reaction studies were characterised by spectroscopic data and in many cases by XRD studies. Computational studies were employed to analyse the differing reactivity of the cationic NHP complex towards light and heavy halide ions, and to help in the assignment of the radical intermediate observed. The more diverse reactivity of the cationic NHP complexes compared to their neutral analogues is attributed to their higher electrophilicity.
Introduction
N-heterocyclic phosphenium (NHP) ions (I, Chart 1) are a special manifestation of diaminophosphenium cations, which are long established as carbene-analogue ligands for transition metal complexes.1,2 Known NHP complexes are mostly mono-cationic (IIa, m = 1)3 or neutral molecules (IIb, m = 0) formally assembled from a cationic ligand and a neutral or anionic metal fragment,4 respectively, but it has been noted2a,5,6 that these species may show redox non-innocence spoiling the assignment of defined charge states to individual building blocks in the final molecule (Scheme 1). | ||
| Chart 1 Molecular structures of N-heterocyclic phosphenium ions (I) and their transition metal complexes (IIa,b) referred to in this work (R = aryl, alkyl). Cations I and molecules containing respective fragments will also be labelled as RNHP+ and RNHP-X, respectively. | ||
 | ||
| Scheme 1 Reported7,8 basic reaction modes of an exemplary neutral NHP complex with reactants behaving as nucleophiles (X− = H−, Me−) and bases (Y = NiPr2, tBu; R = Dipp). | ||
Neutral NHP complexes have recently received attention because of their ligand-centred reactivity towards anionic nucleophiles and bases, respectively. The attack of nucleophilic reactants occurs preferably on the metal-bound phosphorus centre to yield anionic phosphine complexes (Scheme 1, (i)), which may be quenched with electrophiles to yield species representing the products of a formal 1,2-addition to the metal–phosphorus double bond in the original NHP complex.6–8 Nucleophilic attack at phosphorus is also thought to play a key role in the dehydrogenation of ammonia borane with a NHP complex of manganese acting as catalyst.7 In contrast, bases instigate the deprotonation of a C–H bond on the heterocycle to give a carbanionic intermediate (Scheme 1, (ii)) whose interception with suitable electrophiles allows post-functionalisation of the initial NHP ligand.8
The selective attachment of anionic nucleophiles to the phosphorus atom of a cationic NHP complex has likewise been demonstrated,9 but it seems that these reactions have not been studied in detail, and attempts to C–H bond deprotonation in a cationic complex IIa, which are considered particularly appealing as they should yield a neutral product, are unknown. We report here on a comprehensive study of the behaviour of a cationic NHP complex of iron (IIa with R = Dipp, MLn = Fe(CO)4) towards various anionic reactants, whose outcome reveals both parallels and differences with respect to analogous reactions of the neutral complexes IIb.
Results and discussion
Synthesis of a cationic NHP complex
On paper, a cationic NHP complex serving as suitable reactant for the planned studies is obtained by formally replacing the nitrosyl in a known complex [(DippNHP)Fe(CO)3(NO)]6 (1, DippNHP = 1,3-bis-(2,6-diisopropylphenyl)-1,3,2-diazaphospholenium) by another carbonyl ligand. Previous syntheses of such cationic diaminophosphenium iron tetracarbonyls were conducted by Lewis acid induced halide abstraction from neutral P-halophosphine precursor complexes,1,10 or by complexation of a pre-formed phosphenium cation using Fe(CO)5 or Fe2(CO)9,1 respectively. By analogy, target complex [3]+ was synthesised by reacting the NHP salt [2]OTf11 with Fe2(CO)9 (Scheme 2).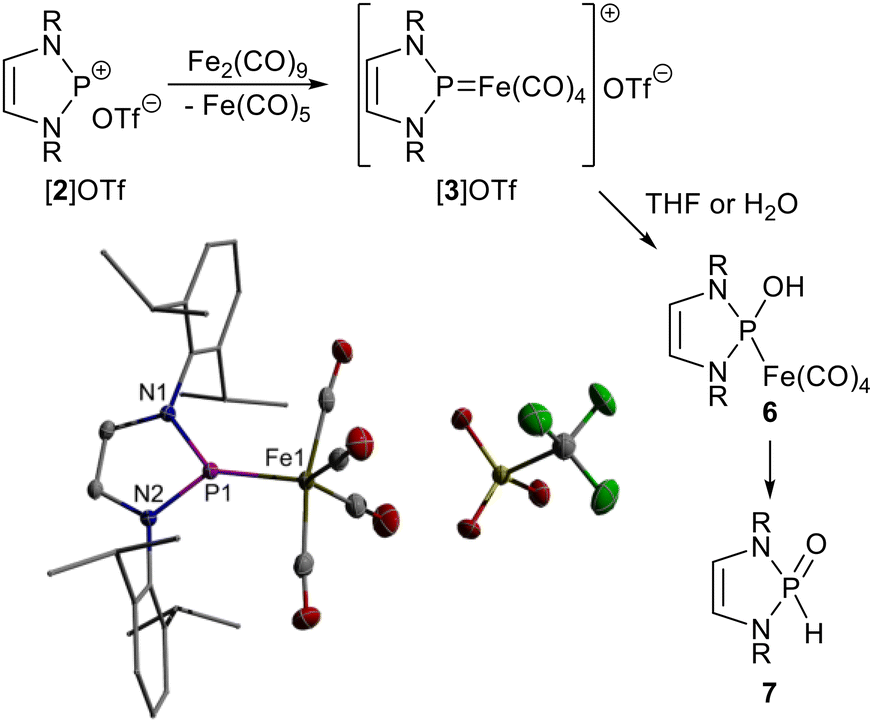 | ||
| Scheme 2 Synthesis of [3]OTf (R = Dipp), its reaction with THF or H2O, respectively, and representation of the molecular structures of cation and anion in the crystal. For clarity, hydrogen atoms were omitted and the atoms in the Dipp-substituents drawn using a wire model. Thermal ellipsoids are drawn at the 50% probability level. | ||
The triflate salt [3]OTf was isolated after work-up as highly moisture-sensitive crystalline material and characterised by spectroscopic data (see ESI†) and a single-crystal X-ray diffraction (sc-XRD) study. The crystal contains detached complex cations [3]+ (Scheme 2) and triflate anions. The iron centre has a trigonal–bipyramidal (tbp) coordination sphere in which the NHP ligand occupies an equatorial position. The trigonal planar coordination of the phosphorus atom (sum of angles 359.9(2)°) and short P–Fe distance (2.0729(5) Å) are similar as in Cowley's diaminophosphenium complex ([{(Et2N)2P}Fe(CO)4][AlCl4],12 Fe–P 2.10(5) Å) and represent typical characteristics of phosphenium complexes with a Fischer-carbene-analogue13 bonding motif. The Fe–P distance is shorter than in Fe(CO)4-complexes of diaminophosphines (2.15–2.30 Ň) but exceeds those in neutral and anionic complexes with the same NHP ligand (Fe–P 2.0331(5) Å for 1,6 2.011(1) for [(DippNHP)Fe(CO)3H] (4),4 1.989(1) for [(DippNHP)Fe(CO)3]− ([5]−)4). These trends imply that the Fe–P π-bonding is attenuated when the overall charge of the complex increases, but remains even in the cationic species still higher than in phosphine complexes.
Crystalline [3]OTf forms stable solutions in chlorinated solvents, while solutions in THF decay under polymerisation of the solvent and conversion into neutral complex 6, which can also be created deliberately by adding a small quantity of H2O. Isolation of 6 was impeded by its slow decomposition to produce secondary diazaphospholene oxide 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 and other unidentified products, but explicit identification was feasible by NMR spectroscopy and a sc-XRD study of a serendipitously isolated 1:1 co-crystal containing both 6 and 7 (Fig. S1†).
14 and other unidentified products, but explicit identification was feasible by NMR spectroscopy and a sc-XRD study of a serendipitously isolated 1:1 co-crystal containing both 6 and 7 (Fig. S1†).
Reactions with halide and hydridotriethylborate salts
Assuming that, as in the case of neutral NHP complexes,6–8 the attack of nucleophiles on cation 3+ will take place at the phosphorus atom, reactions with halide and borohydride salts (as hydride donor) are expected to yield neutral complexes with P-hydride or P-halogen-substituted N-heterocyclic phosphine ligands, respectively. This synthetic route would be highly welcome as attempts to access 8b and 9 (see Scheme 3) conventionally via complexation of N-heterocyclic phosphines DippNHP-X (X = H,15 Cl) with Fe2(CO)9 turned out problematic: the reactions were unselective, and the expected complexes were spectroscopically detectable, but could not be isolated (vide infra for further discussion of the reaction of DippNHP-Cl). | ||
| Scheme 3 Reactions of [3]OTf with halide salts and LiBH4 to furnish either diazaphospholene complexes (8a,b, 9) or NHP iron halides (10c,d) (R = Dipp, PPN = bis(triphenylphosphine)iminium). | ||
The reactions of 3[OTf] with suitable halide sources and Li[BEt3H] (Scheme 3) were carried out by stirring suspensions of the respective reactants in toluene or benzene. Tetrabutylammonium fluoride ([Bu4N]F) and bis(triphenylphosphine)iminium chloride ([PPN]Cl) reacted within less than one hour at room temperature to furnish yellow solutions of the anticipated complexes 8a,b. Crystalline products were isolated after work-up and characterised by spectroscopic data (see ESI†) and sc-XRD studies. The crystals contain molecular complexes in which the phosphine donors occupy an equatorial position in the distorted tbp coordination sphere of the iron atoms (Fig. 1).
 | ||
| Fig. 1 Representation of the molecular structures of 8a (top left), 8b (top right), 10c (bottom left) and 10d (bottom right) in the crystal. For clarity, hydrogen atoms were omitted and the atoms in the Dipp-substituents drawn using a wire model. Thermal ellipsoids are drawn at the 50% probability level. | ||
The deviating stereochemical disposition with respect to the only other structurally characterised iron complexes of P-halogenated N-heterocyclic phosphines, in which the P-donors occupy axial coordination sites,16,17 reflects obviously the increased steric bulk of the P-ligands in 8a,b (with N-Dipp- rather than NMe-substituents), but the P–Fe distances (8a: 2.1679(5) Å, 8b: 2.1613(4) Å) remain similar as in the reference complexes (Fe–P 2.1726(2) Å,17 2.174(1) Å (ref. 16)) and are at the lower edge of the range of reported distances§ in diaminophosphine complexes. A visible shortening of P–X distances (8a, X = F: 1.6129(11) Å 8b, X = Cl: 2.1878(5) Å) compared to free diazaphospholenes (tBuNHP–F: P–F 1.6544(14) Å,18DippNHP–Cl: P–Cl 2.243(1) Å (ref. 11)) suggests that the electron withdrawing effect associated with the metal coordination strengthens the reactive P–X bonds.
A similar outcome as in the reactions with fluoride and chloride salts was observed upon treatment of [3]OTf with superhydride. The secondary N-heterocyclic phosphine complex 9 (Scheme 3) resulting from hydride transfer to the NHP ligand was isolated as crystalline material after work-up and identified by spectroscopic data and a sc-XRD study. The crystals contain discrete molecules with PHFe(CO)4-units disordered over two positions. The disorder suggests the presence of stereoisomers that have a tbp-coordinated iron atom with the phosphine ligand in either an axial or an equatorial position (see Fig. S2†), but also precludes the further discussion of metric parameters.
Reaction mixtures obtained upon treating [3]OTf with bromide and iodide salts did not turn yellow as during formation of 8a,b and 9, but developed a deep red colour, and their work-up likewise furnished deep red, crystalline materials. Although only the iodine-containing species was isolated in analytically pure form, spectroscopic data and sc-XRD studies allowed to identify both products as the NHP complexes 10c,d (Scheme 3). The molecules (Fig. 1) contain iron centres with distorted trigonal bipyramidal coordination carrying the NHP unit and the halogen atom in equatorial and three carbonyls in the remaining equatorial and both axial positions. The Fe–Br (10c: 2.4591(4) Å) and Fe–I distances (10d: 2.6478(5) Å) match typical values reported for related complexes (Fe–Br 2.48(3) Å, Fe–I 2.66(3) ŧ). The nearly planar coordination of the phosphorus atom (sum of angles 356.4(3)° for 10c and 358.1(3)° for 10d) and short Fe–P distances (2.0456(7) Å for 10c and 2.0416(9) Å for 10d) add up to the well-known carbene-analogue bonding motif.13 A comparison with other NHP complexes of iron reveals that the Fe–P distances decrease continuously when going from [3]OTf to 10c,d, isosteric hydride [(DippNHP)Fe(CO)3H] (4),4 and anion [(DippNHP)Fe(CO)3]− ([5]−),4 and show a rough correlation with the midmost νCO mode (Fig. S3†). This trend is in accord with the interpretation that increasing electron releasing character of a donor Do (Do = CO, Br−, I−, H−, 2e−) in complexes [(DippNHP)Fe(CO)3(Do)]+ reinforces Fe–P π-bonding and exerts an associated bond shortening. The nature of 10c,d as phosphenium complexes is also in accord with the larger 31P NMR chemical shifts (δ31P 218.1 for 10c and 220.0 for 10d) compared to diazaphospholene complexes 8a (δ31P 161.5) and 8b (δ31P 166.4).
The formation of 10c,d in the reactions of [3]OTf with bromide and iodide marks an alternative to the generation of diazaphospholene complexes that is theoretically also amenable to the reactions with the lighter halides. To help understand the factors governing the selection between both reaction channels and obtain insight into mechanistic details, we analysed the problem computationally (see ESI† for details). To start, we energy optimised the molecular structures of both types of products and computed standard Gibbs free enthalpies ΔG° for the decarbonylation of 8a–d to 10a–d. The optimisation results indicate that the P-donor ligands in 8a–d occupy preferably an axial coordination site in 8a and an equatorial site in the heavier homologues, which is in accord with the conformation of crystalline 8b, but not 8a (X = F, Fig. 1). However, as the pentacoordinate complexes must be viewed as fluxional molecules and energetic differences are small (<2.8 kcal mol−1, see ESI†), we cannot exclude that the conformation in the crystalline phase is enforced by packing effects and does not reflect the true order of stabilities of the individual isomers. Using the more stable isomer in each case as a reference, the thermodynamic driving force for both types of products is forecast to be comparable for X = I, while NHP iron halides 10a–c are increasingly destabilised as the atomic number of the halogen decreases (Fig. 2).
 | ||
| Fig. 2 Minimum Gibbs free energy reaction pathways for the decarbonylation of 8a–d calculated at the COSMO-RS-RI-PBE/def2-TZVP//COSMO-RS-RI-PBE/def2-SVP level of theory. Values of ΔG° refer to stereoisomers with the P-donor ligand in equatorial (8b–d) or axial (8a) position, respectively. Note that we could not locate transition states connecting 10a with 8a or 11a. Colour coding: blue (8a/10a/11a, X = F), black (8b/10b, X = Cl), red (8c/10c, X = Br), green (8d/10d, X = I). | ||
Relaxed potential energy scans indicate that nucleophilic attack of X− at the phosphorus atom of [3]+ to give 8a–d may in all cases proceed as a barrierless reaction, whereas we were unable to establish an analogous pathway leading directly to 10a–d. These results imply that diazaphospholene complexes 8a–d must always be considered the kinetically favoured products while NHP metal halides 10a–d are formed in a subsequent step. In accord with this hypothesis, we could locate transition states for a one-step conversion of 8b–d into 10b–d under decarbonylation and concurrent halide migration (Fig. 2). The process becomes more facile as the atomic number of the halogen increases. Fluorine-containing 8a turned out to be special as its decarbonylation proceeds without halide migration to furnish coordinatively unsaturated complex 11a. Fluoride migration from phosphorus to iron seems to be disfavoured as conversion of 11a into 10a is predicted to be both endothermic (ΔEzpe = 6.6 kcal mol−1) and endergonic (ΔG° = 8.2 kcal mol−1), and we could neither locate a direct transition state for this process.¶
Looking also at the secondary phosphine complex 9, we found that decarbonylation of the preferred axial isomer to yield [DippNHP–Fe(CO)3H] (4) is endergonic (ΔG° = +7.2 kcal mol−1). While this value matches that obtained for chloro-derivatives 8b/10b, the barrier to isomerisation is still higher (ΔG‡ = 43.6 kcal mol−1, Fig. S50†), and we can regard the phosphine complex as the thermodynamically and kinetically favoured product.
All in all, the computational studies suggests that formation of diazaphospholene complexes 8a,b and 9 in the reactions of [3]OTf with fluoride, chloride and hydride sources is kinetically and thermodynamically clearly favoured. The reaction with iodide is presumed to involve 8d as primary intermediate, but the shift of the thermodynamic balance to the side of the NHP metal halide and a comparatively low decarbonylation barrier allow us to rationalise the formation of 10d as the final product. Similar arguments apply to the reaction with bromide; even if the conversion of 8c into 10c is slightly endergonic, it must be kept in mind that the difference comes close to the estimated accuracy of the computational model and the release of CO into the gas phase may well suffice to shift the equilibrium to the side of the NHP complex. We conclude therefore that the reactions of [3]OTf with halides proceed under thermodynamic control, and that the different outcome observed with lighter and heavier halides reflects the different order of stabilities for the two types of products and is no sign that the NHP complex acts as ambident electrophile.
Reactions with amides and organometallics
Sterically demanding organolithium compounds and metal amides act on neutral NHP complexes not as nucleophiles but as bases.6,8 To see if a similar reactivity is also observable for a cationic NHP complex, we studied the reactions of [3]OTf with selected amides (lithium diisopropylamide (LDA), potassium and lithium hexamethyldisilazide), tert-butyl lithium, and sodium hydride in different solvents (Et2O, THF, toluene). Monitoring by NMR spectroscopy revealed that all reactions were unselective but gave similar product mixtures. In the following, we will discuss only the reaction with LDA in THF.Treatment of a freshly prepared solution of [3]OTf in THF with LDA produced a rapid colour change to deep red. The 31P NMR spectrum of the mixture showed several signals, the most intense of which could be assigned to a species later identified (see below) as dimeric complex (12)2 (Scheme 4).
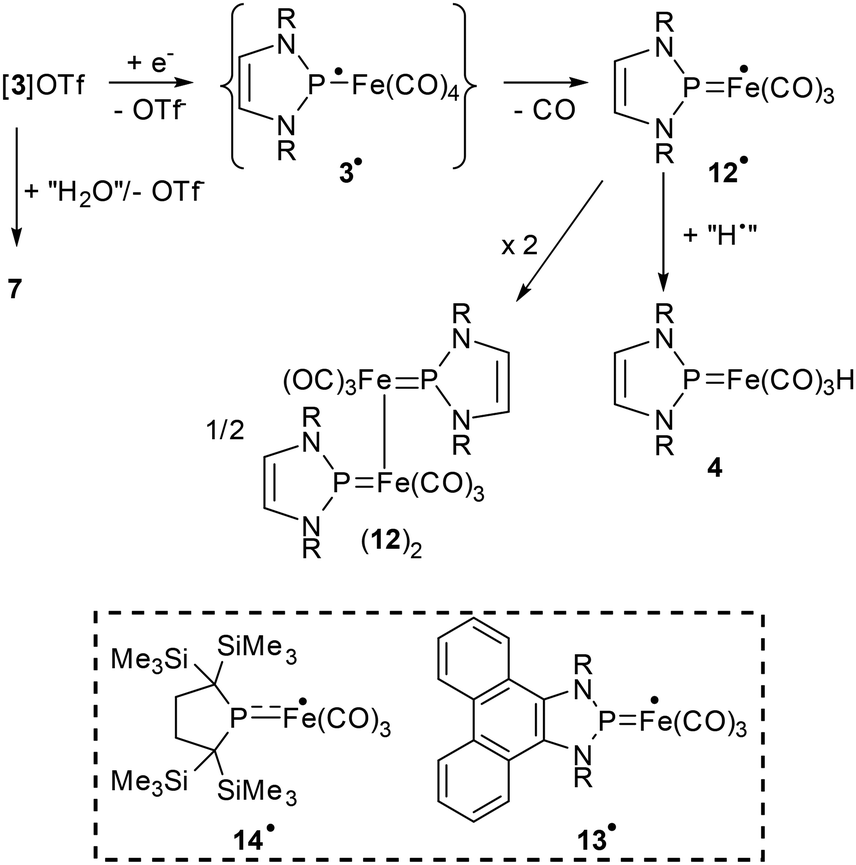 | ||
| Scheme 4 Proposed pathway for the formation of the products of the reactions of [3]OTf with LDA and reductants (Li, Cp2Co) and molecular structures of previously reported radicals 13˙ (ref. 19) and 14˙ (ref. 20) with similar molecular structure as 12˙. Note that 7 can also arise from the hydrolysis of any of the products shown (R = Dipp). | ||
The chemical shifts of the remaining signals indicate the presence of NHP iron hydride 4, further unidentifiable NHP complexes, and diazaphospholene oxide 7 arising presumably from reaction with adventitious water. The generally low signal intensities suggesting that further NMR silent species might have been formed as well, we recorded an EPR spectrum, which showed indeed a strong signal with a doublet splitting arising from hyperfine coupling with a 31P nuclear spin (Fig. S4†). The same product is also formed upon reaction of [3]OTf with reductants like lithium or cobaltocene, but attempts to its isolation failed as these reactions were quite unselective and yielded intricate product mixtures containing (12)2, 4 and 7 as predominant NMR-active constituents. Based on the similarity of the observed EPR data (g = 2.052, A31P = 2.18 mT) with those of previously reported radicals with related molecular structures (13˙,19g = 2.052, A31P = 3.43 mT; 14˙,20g = 2.042, A31P = 3.13 mT, see Scheme 4), we assign the signal to a species 12˙ arising from one-electron reduction and decarbonylation of [3]+. An attribution to a tetracarbonyl radical 3˙ representing the initial reduction product is disfavoured as both a computational study (see ESI†) and experimental findings on related species21 indicate that the EPR data observed is not compatible with a radical of this type.
Cyclovoltammetry studies revealed that the reduction of [3]OTf may also be induced electrochemically and is partially reversible at −78 °C (see Fig. S48†). Monitoring the reaction by IR spectro-electrochemistry (Fig. S49†) confirmed that (partial) re-oxidation of the reduced species is indeed feasible, but gave no further clues about the product itself as decomposition processes occurring even at the low temperature rendered the spectra barely interpretable.
The combination of all findings allows to derive a hypothesis about the reactions taking place. Thus, we propose that both the anion bases and the reductants used react with [3]OTf via single electron transfer (SET) to give a short-lived intermediate 3˙, which rapidly decarbonylates to furnish 12˙. Deactivation of this species by chemical follow-up processes yields (12)2 and 4 as prominent products (Scheme 4). Formation of these species via dimerisation or hydrogen atom abstraction epitomises typical radical reaction modes that are well in accord with the nature of 12˙ as an iron-centred radical (Fig. S53†). If the other products arise from the same mechanism or a different one, in which LDA acts as a nucleophile or base, remains undecided.
A fairly different outcome was observed when [3]OTf and LDA were reacted at −78 °C. A 31P{1H} NMR spectrum recorded directly after mixing of the reactants revealed that the reaction was still unselective but produced an NMR-active main product that had not been detectable at room temperature (Fig. S28†). This is in line with the observation that the signal weakened and finally completely disappeared during warming of the reaction mixture to 0 °C. The deshielding of the 31P NMR signal of the new species (δ31P 297.8) compared to [3]+ (δ31P 238.0) is in accord with the trend observed during the C4-metalation of neutral NHP complexes of manganese and chromium8 and gave a first hint that metalation of [3]+ might have succeeded as well. Even if this surmise was further substantiated by the finding that the same species also detectable upon treatment of [3]OTf with tBuLi, attempts at its further characterisation or interception with boranes, alkylating and silylating agents, or transition metal electrophiles were futile.
Assuming that the bad chemical selectivity is related to the high electrophilicity and steric accessibility of the Fe–P unit in [3]+, we posited that replacing an Fe-bound carbonyl by a bulkier and more electron rich ligand might help to block this reactive site and direct the attack of anion bases to the carbon atoms in the NHP ring. An appropriate target complex [15]OTf generated by treating [3]OTf with one equivalent of PPh3 in toluene at 80 °C (Scheme 5) was isolated in reasonable yield after work-up and identified spectroscopically (see ESI†). The splitting of the νCO modes in the FTIR spectrum suggests the presence of a trans-disubstituted complex with local C3v symmetry. The 31P{1H} NMR signals of [15]OTf appear as an AX-type pattern with a coupling constant (2JPP = 47 Hz) slightly larger than in heteroleptic bis-phosphine complexes of iron(0), which is in harmony with the trend that the magnitude of the coupling in these complexes is known to increase when the electron donating power of the phosphine ligands decreases.22
 | ||
| Scheme 5 Synthesis of [15]OTf, its reaction with LDA under different conditions, and representation of the molecular structure of 16 in the crystal. For clarity, hydrogen atoms were omitted and the atoms in peripheral substituents drawn using a wire model. Thermal ellipsoids are drawn at the 50% probability level (R = Dipp). | ||
The reaction of [15]OTf with LDA in THF at room temperature furnished once more a product mixture. The most prominent constituents, PPh3 and (12)2, arise presumably as in the case of [3]OTf from reduction of the starting material, cleavage of PPh3, and dimerisation of the resulting radical 12˙. Further species could not be identified. Again, the reaction took a different course when carried out at −78 °C and with addition of triethyl borane. Even if it was still not entirely selective (see ESI† for details), work-up allowed to acquire a precipitate consisting of a mixture of a complex 16 (approx. 85% by NMR) and [Fe(CO)3(PPh3)2]23 (approx. 8–10%, formed during work-up), PPh3, and traces of additional unidentified side products. Although further purification proved unfeasible, the identity of 16 was established by NMR spectroscopy and a sc-XRD study of a crystal picked from the inhomogeneous solid. Characteristic spectroscopic data include the presence of an AX-type pattern in the 31P{1H} NMR spectrum (δ31P 256.8, 78.2, 2JPP = 72 Hz), a singlet (δ11B −7.4) with a chemical shift typical for alkyl borates24 in the 11B NMR spectrum, and 1H NMR signals indicating the presence of a single NHP ring proton, two inequivalent NDipp-substituents, and the ethyl groups of a BEt3 unit. The XRD study confirms the presence of a complex with a tbp-coordinated iron centre that carries a PPh3 and a formally zwitterionic, borane-stabilised NHP unit in mutual trans-positions (Scheme 5). The trigonal planar coordination of the NHP phosphorus atom is similar as in [3]OTf, while the P1–Fe distance (2.1191(11) Å) is longer than in [3]OTf (2.0729(5) Å) but still shorter than the P2–Fe distance to the PPh3 ligand (2.2354(11) Å). This trend is in accord with the interpretation that the P–Fe bond displays still partial double bond character but the zwitterionic donor is a weaker π-acceptor than the cationic NHP ligand in [3]OTf.
The characterisation of 16 confirms that metalation of a CH-bond at the heterocycle of a cationic NHP complex is indeed feasible, but implies as well that the reactions of [15]OTf (and [3]OTf) with amides or organometallics take a different course at low temperatures than at room temperature. We explain this switch by assuming that CH-metalation product formed at low temperature is re-protonated (e.g. by proton abstraction from the solvent or the amine formed in the previous step) upon warming, and the resulting anion (amide or deprotonated solvent) can then react with the cationic complex under SET to furnish radicals such as 3˙ and 12˙, or, as with halides, via nucleophilic attack at phosphorus. The accessibility of multiple reaction channels is not only in accord with the low thermal stability of the metalated NHP complexes in the absence of a stabilising borane, but allows us as well to understand the unselective reactions of the cationic NHP complexes with the anion bases. It should be noted that the postulated scenario has precedence in the competition between polar and electron transfer mechanisms in the reactions of certain carbanions25 and carbenes26 with electrophiles, and that a related rivalry between SET and donor–acceptor reactivity is also discussed in the chemistry of (frustrated) Lewis pairs.27
Formation of a dinuclear bis-NHP complex
Seeking to access a chlorine-containing analogue of 10c,d, we posited that reaction of DippNHP–Cl (17b)28 with [(ba)Fe(CO)3]29 (ba = benzylidene acetone) might yield a transient complex [(DippNHP–Cl)Fe(CO)3] (11b) that could then rearrange via 1,2-Cl-shift to furnish the target complex without having to pass a high barrier for CO-cleavage.¶ Highly selective (>85%) conversion of 17b into a phosphorus-containing product with the signature of a NHP complex (δ31P 239.1 ppm) was indeed observable by 31P NMR spectroscopy when the reaction was performed at 40 °C in toluene using a 1.5-fold excess of the iron compound (Fig. S27†). Even if the work-up was complicated by the poor separability of ba, which has a similar solubility to the product in all solvents, we managed to isolate a low yield (5%) of crystalline material allowing for characterisation by spectroscopic data and a sc-XRD study. Surprisingly, the product was identified as (12)2 rather than the expected NHP metal chloride 10b (Scheme 6).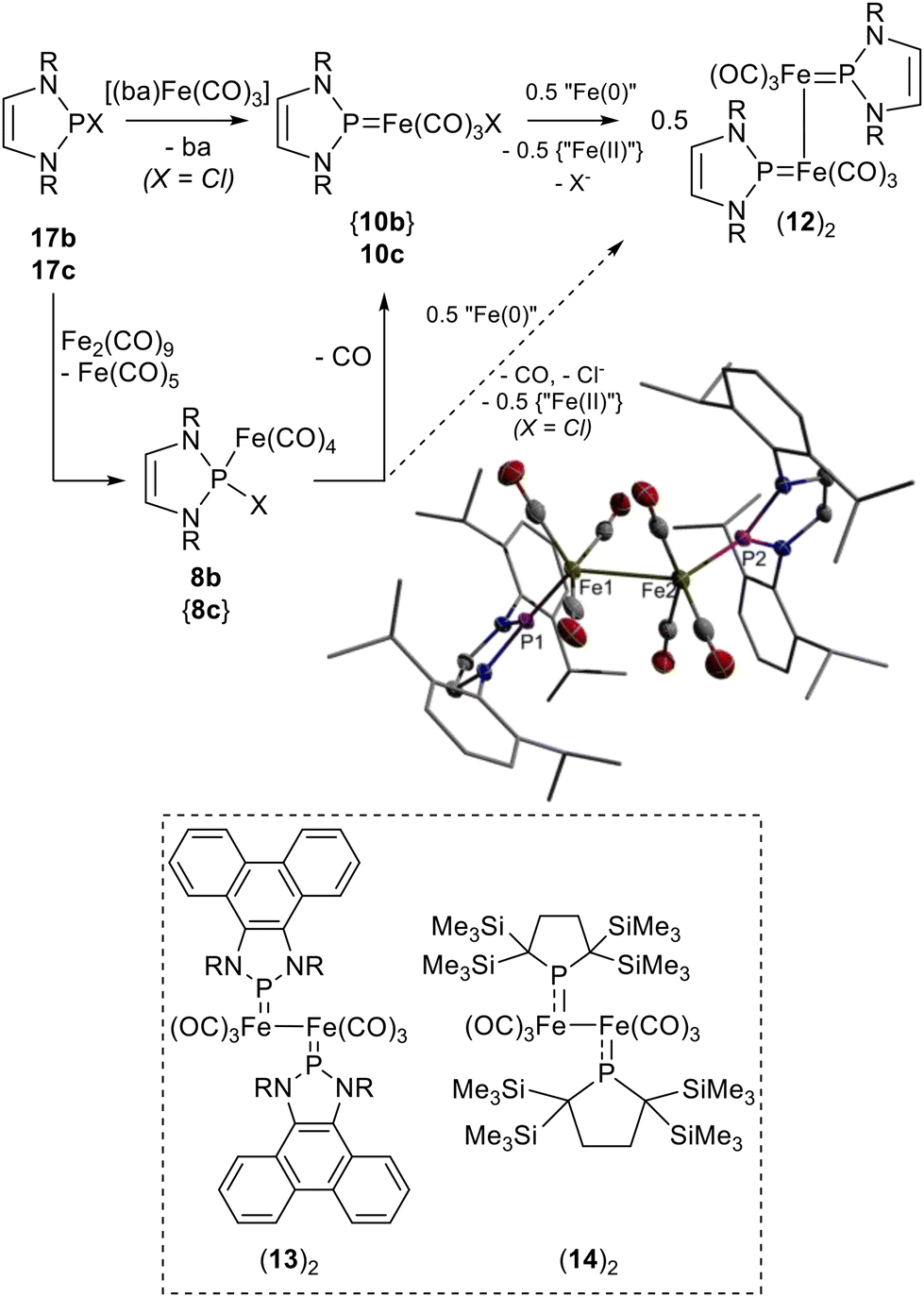 | ||
| Scheme 6 Reactions of 17b,c with Fe(0) complexes, representation of the molecular structure of (12)2 in the crystal (for clarity, hydrogen atoms are omitted and the atoms in peripheral substituents drawn using a wire model; thermal ellipsoids are drawn at the 50% probability level), and molecular formulae of (13)219 and (14)2.20 Species in curly brackets were not explicitly observed (R = Dipp, L = benzylidene acetone/ba; “Fe(0)” and “Fe(II)” denote any of the Fe(0) complexes present ([(dba)Fe(CO)3], Fe2CO9 or Fe(CO)5) and the oxidation product formed, which is assumed to be a Fe(II) compound). | ||
The chemically symmetrical molecule features two penta-coordinate iron centres with a coordination geometry that is somewhat closer to sp-4 than tbp (geometrical parameter30τ5 = 0.38). The metal atoms are linked by an unsupported Fe–Fe bond and each one carries three terminal CO and one terminal NHP ligand. Even if the NHP units in multinuclear complexes seem to prefer a μ2-bridging binding mode,5,19,31 the assembly observed here has precedence in (13)219 and (14)220 (Scheme 6) arising from dimerization of the radicals 13˙ and 14˙, respectively. The Fe–P (2.0292(5)/2.0268(5) Å) and Fe–Fe (2.7164(4) Å) distances in (12)2 are shorter than in (14)2 (Fe–P 2.0934(12)/2.1047(13) Å, Fe–Fe 2.7374(10) Å) but closely match those in (13)2 (Fe–P 2.0298(8)/2.0370(8) Å, Fe–Fe 2.7166(5) Å) where the NHP unit is part of an annulated π-electron system, the likeness underpinning that both complexes exhibit very similar electronic structures characterised by a marked Fe–P π-bonding contribution. The trend in Fe–P distances observed for [3]+ (2.0729(5) Å), (12)2, (avg. 2.028 Å) and [(NHP)Fe(CO)3]− ([5]−) (1.989(2) Å)4 correlates with the charge states of the complexes, reflecting the effect of stronger π-back donation from the metal fragment as the negative charge increases.
The outcome of the reaction of 17b with [(ba)Fe(CO)3] implies that NHP metal chloride 10b is generated as expected and is then reduced by unreacted iron complex to give 12˙, which subsequently dimerises to furnish (12)2 as one final product. The associated oxidation product is assumed to be a chloro-iron(II) species, although this has not been identified.
Trying to avoid the tedious separation of the ba co-ligand, we also explored the use of Fe2(CO)9 as alternative iron source. Although its reaction with 17b at room temperature is messy (vide supra), NMR spectroscopic survey allowed us to identify 8b as a major and (12)2 as a minor component in the product mixture. Envisaging to aid the decarbonylation needed to produce (12)2, the reaction was carried out at 60 °C. A 31P NMR spectroscopic assay revealed that (12)2 is now indeed the major NMR-active product detected (48% by integration), but its isolation proved still unfeasible. A cleaner reaction than with 17b took place with the heavier homologue, 17c. Treatment with a 1.25-fold excess of Fe2(CO)9 at room temperature resulted in selective conversion to 10c within 30 minutes (Scheme 6), and the product was isolable in a decent yield after work-up. If the reaction was carried out with a larger excess of Fe2(CO)9 at 65 °C, quantitative conversion of 10c into secondary products was detectable after 7 hours. NMR spectroscopy confirmed that (12)2 was again formed as the dominant product (70% by integration of the 31P{1H} NMR spectrum) along with smaller amounts of 4 (16%) and [(DippNHP)2Fe(CO)2]4 (18, 14%), but its isolation was once more precluded by the similar solubility of all three components.||
The course of the reactions of 17b and 17c with Fe2(CO)9 suggests that the formation of (12)2 is in both cases set off by a Lewis acid/base step producing diazaphospholene complexes 8b,c (Scheme 6). As in the reaction of [3]+ with Br− and in accord with our computational studies (vide supra), 8c is supposed to immediately decarbonylate with formation of 10c. Similar reductive dehalogenations of iron-bound halogenophosphine ligands have been reported previously.32 The reduction of 10c to yield (12)2 as the final product is then induced by one of the iron(0) complexes. The by-products 4 and 18 arising presumably from abstraction of a hydrogen atom or a diazaphospholenyl unit by radical 12˙, we presume that this redox step proceeds via a radical mechanism. The fate of chloro-diazaphospholene complex 8b is less clear. Its further reaction may proceed as that of 8cvia conversion into an elusive NHP iron chloride 10b and subsequent reduction, but we cannot exclude that reaction with another iron(0) precedes the decarbonylation to bypass the formation of 10b. Redox reactions involving SET steps may in this case produce either 3˙ or 12˙ as transient radicals.
Even if the reactions of 17b,c with Fe2(CO)9 did not bring any synthetic benefit, they make it clear that the failure in cleanly accessing complexes 8b,c from the halogeno-diazaphospholene precursors is owed to an exceptional ability of the complexes to undergo intra- or intermolecular reductive P–X bond activation with low-valent iron species. The inability to selectively address the different reaction channels by controlling the stoichiometry as in other cases32 is presumably owed to the fact that the ionic bond polarization in halogeno-diazaphospholenes33 facilitates P–X activation processes and formation and decay of 8b,c may thus occur more or less parallel. Moreover, it seems that the ability of the NHP unit to support Fe–P π-bonding favours the generation of radicals like 3˙ and 12˙, which provide an entry point into radical chemistry that is unavailable in other cases.
Conclusions
The reactivity of a newly synthesised cationic NHP complex of iron towards various anionic reactants is more diverse than that reported6–8 for its neutral analogues. Basic reaction channels include nucleophilic attack of the anion at phosphorus as in neutral NHP complexes to yield diazaphospholene-Fe(CO)4-complexes (preferred with fluoride, chloride and hydride sources), decarbonylation and attachment of a heavier halide (with bromide or iodide) to iron to furnish NHP iron halides, or finally reductive dehalogenation and CO cleavage to produce a radical intermediate that is then deactivated in an unselective manner to produce complex product mixtures (with amides and organometallics). Reactions with amides were further found to be temperature dependent, proceeding at low temperatures preferably under metalation of a CH-bond on the NHP ring rather than reduction. A borane adduct of the resulting thermolabile zwitterionic product was in one case structurally characterised. Computational model studies suggest that the preference for attaching a halide ion to phosphorus or iron does not reflect ambident Lewis acid behaviour; the attack of the anion occurs in all cases at phosphorus, but the primary adduct may rearrange if the NHP iron halide is thermodynamically more stable. Both this rearrangement, which is unknown for the adducts of neutral NHP complexes with anionic nucleophiles,6–8 and the enhanced tendency of the cationic NHP complex to undergo one-electron reduction are attributable to its increased electrophilicity induced by the higher positive charge.The synthesis of a dinuclear NHP complex observed as product in some reactions was also accomplished by treatment of P-halogeno-diazaphospholenes with an excess of iron(0) species, which act in this case both as complexing agent and reductant. The route to the final product seems to involve not only 2-electron oxidative addition steps as had previously been noted in reactions of iron(0) species with chlorophosphines,32 but also one-electron transfer events yielding radical intermediates. The unique predestination of halogeno-diazaphospholenes for P–X bond activation processes33 and the unspecific deactivation of the radical intermediates are held responsible for the failure to synthesise the Fe(CO)4-complex of a chloro-diazaphospholene via direct complexation of the free ligand.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for [3]OTf, (6·7), 8a,b, 9, 10c,d, 16, and (12)2 has been deposited at the CCDC under 2394830 ((12)2), 2394831 ([3]OTf), 2394832 (10d), 2394833 (9), 2394834 (8a), 2394835 (10c), 2394836 (6·7), 2394838 (8b), and 2394839 (16)† and can be obtained from https://www.ccdc.cam.ac.uk/data_request/cif.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Institut für Anorganische Chemie for financial support. We further thank B. Förtsch for elemental analyses, Dr K. Beyer, Dr S. Bauer and Dr C. Sondermann for cyclic voltammetry and IR-spectro-electrochemistry measurements, and Dr W. Frey (Institut für Organische Chemie) for collecting the X-ray data sets. The computational studies were supported by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no. INST 40/575-1 FUGG (JUSTUS 2 cluster).References
- R. G. Montemayor, D. T. Sauer, S. Fleming, D. W. Bennett, M. G. Thomas and R. W. Parry, J. Am. Chem. Soc., 1978, 100, 2231 CrossRef CAS.
- For reviews, see: (a) A. H. Cowley and R. A. Kemp, Chem. Rev., 1985, 85, 367 CrossRef CAS; (b) M. Sanchez, M.-R. Mazières, L. Lamandé and R. Wolf, Phosphenium Cations, in Multiple Bonds and Low Coordination in Phosphorus Chemistry, ed. M. Regitz and O. J. Scherer, Thieme, Stuttgart, 1990, 129ff Search PubMed; (c) D. Gudat, Coord. Chem. Rev., 1997, 163, 71 CrossRef CAS; (d) H. Nakazawa, J. Organomet. Chem., 2000, 611, 349 CrossRef CAS; (e) H. Nakazawa, Adv. Organomet. Chem., 2004, 50, 107 CrossRef; (f) D. Gudat, Low-Coordinate Main Group Compounds - Group 15, in Compreh. Inorg. Chem. II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Oxford, 2013, vol. 1, 587ff Search PubMed.
- (a) D. Gudat, A. Haghverdi and M. Nieger, J. Organomet. Chem., 2001, 617–618, 383 CrossRef CAS; (b) C. A. Caputo, A. L. Brazeau, Z. Hynes, J. T. Price, H. M. Tuononen and N. D. Jones, Organometallics, 2009, 28, 5261 CrossRef CAS; (c) J. T. Price, M. Lui, N. D. Jones and P. J. Ragogna, Inorg. Chem., 2011, 50, 12810 CrossRef CAS PubMed.
- For an anionic NHP complex, see: B. Stadelmann, J. Bender, D. Förster, W. Frey, M. Nieger and D. Gudat, Dalton Trans., 2015, 44, 6023 RSC.
- B. Pan, Z. Xu, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2012, 51, 4170 CrossRef CAS PubMed.
- C. M. Feil, Th. D. Hettich, K. Beyer, C. Sondermann, S. H. Schlindwein, M. Nieger and D. Gudat, Inorg. Chem., 2019, 58, 6517 CrossRef CAS PubMed.
- M. Gediga, C. M. Feil, S. H. Schlindwein, J. Bender, M. Nieger and D. Gudat, Chem. – Eur. J., 2017, 23, 11560 CrossRef CAS PubMed.
- M. Gediga, S. H. Schlindwein, J. Bender, M. Nieger and D. Gudat, Angew. Chem., Int. Ed., 2017, 56, 15718 CrossRef CAS PubMed.
- H. Nakazawa, M. Ohta, K. Miyoshi and H. Yoneda, Organometallics, 1989, 8, 638 CrossRef CAS.
- A. H. Cowley, R. A. Kemp and J. C. Wilburn, Inorg. Chem., 1981, 20, 4219 CrossRef.
- S. Burck, D. Gudat, K. Nättinen, M. Nieger, M. Niemeyer and D. Schmid, Eur. J. Inorg. Chem., 2007, 5112 CrossRef CAS.
- A. H. Cowley, R. A. Kemp, E. A. V. Ebsworth, D. W. H. Rankin and M. D. Walkinshaw, J. Organomet. Chem., 1984, 265, C19 CrossRef CAS.
- C. A. Caputo, M. C. Jennings, H. M. Tuononen and N. D. Jones, Organometallics, 2009, 28, 990 CrossRef CAS.
- Z. Li, X. Chen, M. Bergeler, M. Reiher, C.-Y. Su and H. Grützmacher, Dalton Trans., 2015, 44, 6431 RSC.
- B. Stadelmann, PhD Thesis, University of Stuttgart, Stuttgart, 2015.
- D. W. Bennett, R. J. Neustadt, R. W. Parry and F. W. Cagle, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 1978, 34, 3362 CrossRef.
- P. C. Brehm, G. Schnakenburg and R. Streubel, Dalton Trans., 2024, 53, 13201 RSC.
- D. Gudat, A. Haghverdi, H. Hupfer and M. Nieger, Chem. – Eur. J., 2000, 6, 3414 CrossRef CAS PubMed.
- G. G. Kazakov, N. O. Druzhkov, R. V. Rumyantcev, G. K. Fukin, A. G. Starikov, A. V. Piskunov and V. K. Cherkasov, New J. Chem., 2023, 47, 5953 RSC.
- Y. Sunada, S. Ishida, F. Hirakawa, Y. Shiota, K. Yoshizawa, S. Kanegawa, O. Sato, H. Nagashima and T. Iwamoto, Chem. Sci., 2016, 7, 191 RSC.
- B. Ndiaye, S. Bhat, A. Jouaiti, T. Berclaz, G. Bernardinelli and M. Geoffroy, J. Phys. Chem. A, 2006, 110, 9736 CrossRef CAS PubMed.
- R. L. Keiter, J. W. Benson, E. A. Keiter, T. A. Harris, M. W. Hayner, L. L. Mosimann, E. E. Karch, C. A. Boecker, D. M. Olson, J. VanderVeen, D. E. Brandt, A. L. Rheingold and G. P. A. Yap, Organometallics, 1997, 16, 2246 CrossRef CAS.
- T. Li, A. J. Lough and R. H. Morris, Chem. – Eur. J., 2007, 13, 3796 CrossRef CAS PubMed.
- M. G. Crestani, M. Muñoz-Hernández, A. Arévalo, A. Acosta-Ramírez and J. J. García, J. Am. Chem. Soc., 2005, 127, 18066 CrossRef CAS PubMed.
- (a) F. G. Bordwell and C. A. Wilson, J. Am. Chem. Soc., 1987, 109, 5470 CrossRef CAS; (b) R. E. Gawley, E. Low, Q. Zhang and R. Harris, J. Am. Chem. Soc., 2000, 122, 3344 CrossRef CAS.
- (a) Z. Dong, C. Pezzato, A. Sienkiewicz, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Chem. Sci., 2020, 11, 7615 RSC; (b) A. Maiti, B. J. Elvers, S. Bera, F. Lindl, I. Krummenacher, P. Ghosh, H. Braunschweig, C. B. Yildiz, C. Schulzke and A. Jana, Chem. – Eur. J., 2022, 28, e202104567 CrossRef CAS PubMed.
- (a) L. Liu, L. L. Cao, Y. Shao, G. Ménard and D. W. Stephan, Chem, 2017, 3, 259 CrossRef CAS; (b) A. Merk, H. Großekappenberg, M. Schmidtmann, M.-P. Luecke, C. Lorent, M. Driess, M. Oestreich, H. F. T. Klare and T. Müller, Angew. Chem., Int. Ed., 2018, 57, 15267 CrossRef CAS PubMed; (c) F. Holtrop, A. R. Jupp, B. J. Kooij, N. P. van Leest, B. de Bruin and J. C. Slootweg, Angew. Chem., Int. Ed., 2020, 59, 22210 CrossRef CAS PubMed; (d) F. Holtrop, A. R. Jupp, N. P. van Leest, M. Paradiz Dominguez, R. M. Williams, A. M. Brouwer, B. de Bruin, A. W. Ehlers and J. C. Slootweg, Chem. – Eur. J., 2020, 26, 9005 CrossRef CAS PubMed. Review (e) L. J. C. van der Zee, S. Pahar, E. Richards, R. L. Melen and J. C. Slootweg, Chem. Rev., 2023, 123, 9653 CrossRef CAS PubMed.
- S. Burck, D. Gudat, M. Nieger and W. W. du Mont, J. Am. Chem. Soc., 2006, 128, 3946 CrossRef CAS PubMed.
- J. A. S. Howell, B. F. G. Johnson, P. L. Josty and J. Lewis, J. Organomet. Chem., 1972, 39, 329 CrossRef CAS.
- W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- (a) L. D. Hutchins, R. W. Light and R. T. Paine, Inorg. Chem., 1982, 21, 266 CrossRef CAS; (b) D. Förster, J. Nickolaus, M. Nieger, Z. Benkő, A. W. Ehlers and D. Gudat, Inorg. Chem., 2013, 52, 7699 CrossRef PubMed; (c) J. Nickolaus, J. Bender, M. Nieger and D. Gudat, Eur. J. Inorg. Chem., 2014, 3030 CrossRef CAS; (d) D. A. Evers-McGregor, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Dalton Trans., 2016, 45, 1918 RSC; (e) J. Nickolaus, D. A. Imbrich, S. H. Schlindwein, A. H. Geyer, M. Nieger and D. Gudat, Inorg. Chem., 2017, 56, 3071 CrossRef CAS PubMed; (f) A. M. Poitras, M. W. Bezpalko, C. E. Moore, D. A. Dickie, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2020, 59, 4729 CrossRef CAS PubMed.
- (a) H. Lang, L. Zsolnai and G. Huttner, J. Organomet. Chem., 1985, 282, 23 CrossRef CAS; (b) F. Bitterer, D. J. Brauer, F. Dörrenbach, F. Gol, P. C. Knüppel, O. Stelzer, C. Krüger and Y.-H. Tsay, Z. Naturforsch., B, 1991, 46, 1131 CrossRef CAS.
- D. Gudat, Acc. Chem. Res., 2010, 43, 1307 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, XRD data, computational studies, NMR and IR spectra. CCDC 2394830–2394836, 2394838 and 2394839. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03352c |
| ‡ Result of a query in the CSD database for complexes [{(R2N)2PR′}Fe(CO)4] with R, R′ = any substituent. |
| § Mean value and standard deviation returned by a query in the CSD database for bromo and iodo complexes of iron containing at least one phosphine and one carbonyl co-ligand. |
| ¶ The heavier homologues (with X = Cl) show a reversed order of stabilities, with the Gibbs free energy of 11b 16.6 kcal mol−1 above that of 10b. |
| || Formation of (12)2 as the main NMR-active product was also spectroscopically detectable in reactions of bis-diazaphospholenyl (DippNHP)2 with excess Fe2(CO)9, and of azido-diazaphospholene DippNHP-N3 with equimolar [(ba)Fe(CO)3]. In both cases, separation of pure (12)2 from the product mixtures formed was unfeasible. Co-photolysis of DippNHP-N3 and Fe(CO)5 gave a product mixture from which a low yield (5%) of (12)2 was isolated after work-up. |
| This journal is © The Royal Society of Chemistry 2025 |