
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synergic effect of neutral organophosphorus ligands combined with acidic β-diketones for the extraction and separation of trivalent actinides
Connor K.
Holiski
 a,
Tara
Mastren
a and
Jennifer A.
Shusterman
*b
a,
Tara
Mastren
a and
Jennifer A.
Shusterman
*b
aNuclear Engineering Program, Department of Civil and Environmental Engineering, University of Utah, 110 Central Campus Dr Rm 2000, Salt Lake City, Utah 84112, USA
bNuclear and Chemical Sciences Division, Lawrence Livermore National Laboratory, Livermore, California 94550, USA. E-mail: Shusterman1@llnl.gov; Tel: +1(925) 424-3198
First published on 10th February 2025
Abstract
Separating trivalent f-block elements remains a central challenge due to their similar ionic radii and chemical behaviors. Historically, these separations have been achieved using single extractants, either alone or in combination with ion exchange chromatography. However, recent studies, including this work, have explored the potential of using synergic combinations of multiple extractants to enhance extraction and separation efficiencies for trivalent actinide separations. This study investigated synergic solvent extraction (SX) systems for extracting and separating americium and curium using three neutral organophosphorus ligands: octyl (phenyl)-N,N-diisobutylcarbamoylmethylphosphine (CMPO), dibutyl N,N-diethylcarbamylmethylenephosphonate (DBDECMP), and dihexyl N,N-diethylcarbamylmethylenephosphonate (DHDECMP), combined with either 2-thenoyltrifluoroacetone (HTTA, pKa = 6.25) or 4-benzoyl-3-methyl-1-phenyl-2-pyrazolin-5-one (HP, pKa = 3.95). Distribution ratios (D) were determined for 241Am3+ and 242Cm3+ as functions of nitric acid pH using 1,2-dichloroethane as the solvent. The combination of these ligands resulted in varying degrees of synergy as demonstrated by their synergic extraction enhancement coefficients (SEC). A maximum separation factor (SFAm/Cm) of 2.65 ± 0.21 was achieved with 0.05 M HTTA and 0.05 M DBDECMP at pH 2.50. This extractant combination was impregnated into an inert macroporous support at various ligand ratios using rotary evaporator methods to produce novel extraction chromatographic (EXC) resins. Various parameters affecting the adsorption of 241Am3+ and 242Cm3+ onto EXC resins, such as solution pH, ionic strength, contact time, γ-irradiation dose, and temperature, were studied. Metal extraction and synergism were retained upon conversion to EXC resins, with increasing extraction observed at higher pH levels. Thermodynamic studies showed increased adsorption and decreased Gibbs free energy (ΔG) with rising temperature. Kinetic investigations indicated rapid and consistent uptake after 10 minutes. The EXC resins exhibited excellent metal retention in preliminary column experiments, demonstrating a promising potential to separate americium and curium with a maximum decontamination factor of 88. Overall, this work successfully demonstrated the identification and conversion of synergic SX systems into novel synergic EXC resins for adjacent trivalent actinide separations.
1. Introduction
Trivalent f-block separations have applications across many disciplines, including nuclear fuel reprocessing, nuclear forensics, stockpile stewardship, and nuclear medicine.1–4 Nuclear power remains an integral option in meeting the increasing demand for clean energy. The United States currently operates on a once-through fuel cycle where, after irradiation, the Used Nuclear Fuel (UNF) is destined to decay in a deep geologic repository. Currently, there are no approved repositories in the United States. An alternative to the long-term repository option is recycling UNF; however, one of the main challenges is the High-level Radioactive Liquid Waste (HLW) generated during the reprocessing of UNF. UNF contains a challenging matrix of uranium, plutonium, minor actinides (MA) (Np, Am, and Cm), lanthanides, and fission products. One option for managing the high activity and long-lived nuclear waste is a closed fuel cycle, where partitioning and transmutation are used to recycle valuable uranium and plutonium resources, while addressing the minor actinides’ long-term radiotoxicity and storage concerns.5 While comprising the smallest proportion of UNF by mass, the MA significantly drive long-term radiotoxicity and heat load.6 A variety of well-established separation processes have been developed to recover and isolate uranium (Uranium Extraction, UREX), strontium/cesium (Fission Product Extraction, FPEX), neptunium/plutonium (Neptunium-Plutonium Extraction, NPEX), remaining fission products (Transuranic Extraction, TRUEX), and trivalent actinides/lanthanides (Trivalent Actinide Lanthanide Separation with Phosphorus-Reagent Extraction from Aqueous Komplexes, TALSPEAK, and Actinide Lanthanide Separation Process, ALSEP).7–10 In order to effectively transmute and recycle Am into shorter lived radionuclides, a further separation of the remaining Am and Cm stream is required; however, an appropriate separation method still needs to be developed as the current processes put Am and Cm into the same stream.The trivalent actinides (An3+) are analogous to the trivalent lanthanides (Ln3+) with similar size and charge. Due to similar chemical properties within each series, inter- and intragroup separations of trivalent f-block elements have been a long-standing challenge.11 The challenge in separating americium and curium lies in the trivalent oxidation state that dominates the chemical species in conjunction with similar bonding characteristics and ionic radii.12 The slight decrease in ionic radius with increasing atomic number is often exploited to achieve a separation of Am(III) and Cm(III). Trivalent actinide separations have been explored using a variety of radiochemical techniques such as ion-exchange chromatography, extraction chromatography (EXC), selective oxidation, and solvent extractions (SX).11,13–15 SX has been studied extensively in the separation of actinides.16 In the simplest SX separations, the dissolved actinide is transferred from an aqueous phase to an immiscible organic phase through ligand coordination, with the charge balance maintained to ensure that a neutral actinide complex is extracted.
Previously, neutral bifunctional organophosphorus extractants such as octyl (phenyl)-N,N-diisobutyl carbamoylmethylphosphine (CMPO), dibutyl N,N-diethylcarbamylmethylenephosphonate (DBDECMP), and dihexyl N,N-diethylcarbamylmethylenephosphonate (DHDECMP), have been used alone as efficient extractants of trivalent actinides from acidic solutions.17–21 Other types of extractants, such as acidic β-diketones like 2-thenoyltrifluoroacetone (HTTA, pKa = 6.25) and 4-benzoyl-3-methyl-1-phenyl-2-pyrazolin-5-one (HP, pKa = 3.95), have also been extensively applied to trivalent f-block separations.22–25
Recently, our group reported synergic SX of Gd(III) and Tb(III) with neutral bifunctional organophosphorus (CMPO, DBDECMP, or DHDECMP) and acidic β-diketones (HTTA or HP) extractants (Fig. 1), as a promising technique to enhance extraction and/or separation selectivity of adjacent lanthanides. Several works have explored and characterized the synergism observed when combining bifunctional neutral and acidic extractants, such as CMPO, DBDECMP, DHDECMP, HTTA, and HP for f-element extraction and inter-f-elements separations, but seldom report on the synergic separation of adjacent trivalent actinides.26–31 Numerous studies have proposed the potential utility of synergic extractions using a variety of extractant types for separating f-elements.27,32–37 However, further research is necessary to explore the potential of synergic separations for adjacent actinides using the proposed extractant combinations. This work looks to extend synergic SX techniques to enhance extraction and separation selectivity of Am(III) and Cm(III).
 | ||
| Fig. 1 Chemical structures of extractants used in this work. (1) octyl (phenyl)-N,N-diisobutyl carbamoylmethylphosphine, (CMPO), (2) dibutyl N,N-diethylcarbamylmethylenephosphonate (DBDECMP), (3) dihexyl N,N-diethylcarbamylmethylenephosphonate (DHDECMP), (4) 2-thenoyltrifluoroacetone (HTTA), and (5) 4-benzoyl-3-methyl-1-phenyl-5-pyrazolone (HP). | ||
In terms of enhanced extraction, synergism is observed when the combination of extractants results in a distribution ratio (D) that is greater than the sum of the ratios for the individual components (eqn (1)). Conversely, antagonism occurs when the combined effect of the extractants results in a D that is less than the sum of the individual components (eqn (2)). This indicates that the combination is less effective than the individual extractants working independently. When the combined D equals the sum of the individual D (eqn (3)), the effect is neither synergic nor antagonistic, suggesting that the extractants function independently without enhancing each other's performance. Regarding selectivity, synergism is defined when the combined extractants produce a larger separation factor (SF(1,2)) than either of the individual components (SF(1) or SF(2)) or when they significantly improve separation efficiency in scenarios where previous extraction was ineffective.
| D(1,2) > D(1) + D(2) | (1) |
| D(1,2) < D(1) + D(2) | (2) |
| D(1,2) = D(1) + D(2) | (3) |
EXC is a popular separation technique that combines the high selectivity of SX with the potential for straightforward automation, while also eliminating the generation of mixed liquid organic waste through its column-based chromatography procedures.38 EXC is a form of reverse-phase partition chromatography. In this work, the stationary phase is non-polar and comprised of an inert support in which a single or mixture of extractants, modifiers, and/or solvents are physically adsorbed inside the pores of the support. Modifying the mobile phase's acidity and/or including complexing reagents alters the retention affinity of each element differently, facilitating the separation between adjacent elements. SX investigations are often the first step in finding potential extractants for EXC materials.
Effective SX systems with promising separation characteristics can be readily transformed into EXC resins by impregnating an inert support with the extractant system. The preparation and utilization of EXC resins have been thoroughly showcased in the literature by numerous commercial applications.39–43 While synergism in SX has been well investigated, the extension of synergism to EXC resins has rarely been studied.44–46 In 1970, Aly et al. demonstrated one of the first examples of synergism in extraction chromatography using HTTA and DBDECP (dibutyl diethyl carbamoyl phosphonate). In their study, when HTTA and DBDECP were sorbed on Celite to produce an EXC resin, a separation factor of 2.06 was obtained for the adjacent lanthanides, samarium, and europium, compared to a <1 separation factor with either HTTA or DBDECP.47,48 More research is needed on the systematic study of characteristics and translation of synergism between SX and EXC resins. The work presented here aimed to identify and characterize synergism in the SX of Am(III) and Cm(III) by combining neutral organophosphorus compounds (CMPO, DBDECMP, or DHDECMP) with β-diketones (HTTA or HP). The extractant combination yielding the best separation characteristics was later converted to novel synergic EXC resins to investigate selectivity in chromatography columns. By studying the conversion, characterization, and optimization of synergic systems, we can design more efficient and selective methods for adjacent Am(III) and Cm(III) separations.
2. Materials and methods
2.1. Reagents and materials
Concentrated nitric acidic (TraceMetal™ grade, Fisher Chemical™) was used to prepare HNO3 solutions from pH 0.00–3.00 using volumetric flasks. The pH of each solution was measured with an Orion™ Star™ A211 (Thermo Scientific) pH meter and Orion™ PerpHecT™ ROSS™ Combination pH micro electrode (Thermo Scientific) calibrated with pH 1.68, 4.01, and 7.00 buffers. The pH of each solution was adjusted using standardized 1.0 M NaOH (Fisher Chemical™) to ±0.02 units of the desired pH. 1,2 – Dichloroethane (99.8%, Sigma-Aldrich) was used as an organic diluent during solvent extraction. Dodecane (≥99%, TCI America) and methanol (99%, Sigma-Aldrich) were used during resin preparation. 4-Benzoyl-3-methyl-1-phenyl-2-pyrazolin-5-one (HP) (99%, AA Blocks) and 2-thenoyltrifluoroacetone (HTTA) (99%, Sigma-Aldrich) were used as the β-diketone extractants, while N,N-diisobutyl-2-(octylphenylphosphoryl)acetamide (CMPO) (95%, AmBeed), dibutyl N,N-diethylcarbamoylmethylphosphonate (DBDECMP) (97.1%, TCI America), and dihexyl N,N-diethylcarbamylmethylenephosphonate (DHDECMP) (92.7%, TCI America) were used as the neutral organophosphorus extractants. Pre-filter resin (20–50 μm) was purchased from TrisKem International. Sodium nitrate (99%, Sigma-Aldrich) was used to control ionic strength. All reagents were used as received without any additional purification. The radionuclides 241Am and 242Cm were supplied from Lawrence Livermore National Laboratory stocks and dissolved in HNO3 to yield stocks containing ≈555 dpm 241Am3+ per μL (1.0 M HNO3) and ≈82 dpm 242Cm3+ per μL (0.1 M HNO3), respectively. The presence of 238Pu in the 242Cm3+ stock was verified by alpha spectroscopy, and it was found to constitute less than 0.6% of the total activity. All solutions and samples were prepared using Milli-Q® water (Millipore, USA, resistivity >18.2 MΩ cm−1).2.2. Actinide solvent extraction with synergic systems
The distribution ratios (D) of Am(III) and Cm(III) were measured using a standard solvent extraction technique, involving the equilibration of equal volumes of an organic phase and an aqueous phase, followed by phase separation and analysis of metal concentrations in each phase. Diluted 241Am3+ and 242Cm3+ stocks were prepared before each experiment such that the nominal activity was ≈15 dpm 241Am3+ per μL and ≈7.5 dpm 242Cm3+ per μL, respectively. Each dilute stock was adjusted to ±0.02 units of the desired pH, as described above.Weighed amounts of each neutral and β-diketone extractant were dissolved in 1,2-dichloroethane to form 0.2 M stocks by mixing at 1500 RPM for 15 minutes using a digital vortex. Organic extraction solutions were prepared using the stocks such that each extractant had a final concentration of 0.05 M. Extraction solutions were made with each extractant (CMPO, DBDECMP, DHDECMP, HTTA, and HP) alone and combined (CMPO + HTTA, CMPO + HP, DBDECMP + HTTA, DBDECMP + HP, DHDECMP + HTTA, and DHDECMP + HP). All organic extraction solutions were pre-equilibrated with equal volumes of nitric acid at each respective pH value by mixing at 1500 RPM for 30 minutes using a digital vortex, followed by centrifugation at 4500 RPM for 5 minutes. Equal volumes of 500 μL of pre-equilibrated organic phase and adjusted pH diluted metal stocks were mixed at 2000 RPM for 30 minutes at 22 ± 1 °C using a digital vortex, followed by centrifugation at 6000 RPM for 30 seconds.
The extracted aqueous phase was separated from the organic phase and retained, and an aliquot (50–250 μL) was taken for analysis. For extraction with 241Am3+, individual extractants were analyzed in duplicate, while combined extractant solutions were analyzed in triplicate. Conversely, all 242Cm3+ samples were analyzed in duplicate. The 241Am3+ and 242Cm3+ activity was determined using liquid scintillation counting (LSC) with Ultima Gold™ AB scintillation cocktail and Tri-Carb 3110TR LSC analyzer (PerkinElmer). The aqueous samples were each counted for 120 minutes or until a 1% counting uncertainty was attained.
The SX distribution ratios (D) (eqn (4)) were calculated as the ratio of activity between the organic ([An]org) and aqueous ([An]aq) phases.
 | (4) |
The activity in the organic phase was determined as the difference between the initial and final activities. The initial activity was determined from aliquots (50 μL) of standards from the dilute metal stocks. All errors on the distribution ratios are reported as one standard deviation between replicates or the propagated uncertainty in counts per minute (CPM), whichever is greater.
The synergic enhancement coefficient (SEC) that describes the change in distribution ratios between the single extractants and combined extractant system is found by the following equation:
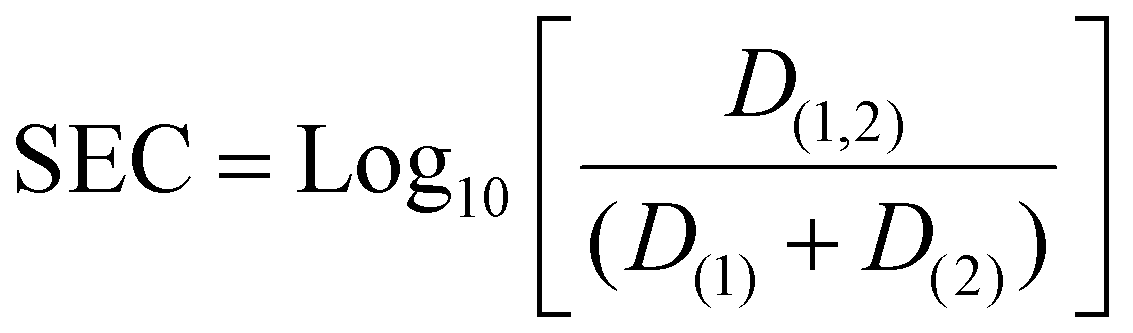 | (5) |
The separation factors (SFAm/Cm) (eqn (6)) for Am(III) and Cm(III) were calculated as the ratio of individual SX distribution ratios obtained from liquid scintillation counting analysis.
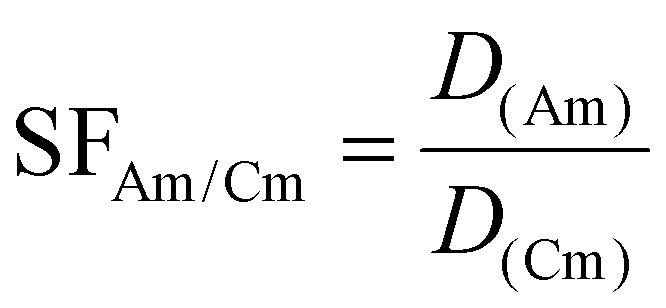 | (6) |
2.3. Optimization of separation behavior for Am/Cm using HTTA-DBDECMP solvent extraction systems
The synergic SX system, utilizing HTTA and DBDECMP, exhibited the most promising separation factors at pH 2.50 and was thus chosen for further optimization. In these studies, the concentration of DBDECMP remained constant at 0.05 M, while the concentration of HTTA varied from 0.0125, 0.025, 0.075, to 0.150 M. Additionally, a separate optimization was conducted by maintaining [HTTA] at 0.05 M while varying the concentration of DBDECMP from 0.0125, 0.025, 0.075, to 0.150 M.2.4. Preparation of HTTA and/or DBDECMP extraction chromatographic (EXC) resins
Based on the findings of the solvent extraction investigation and optimization, the combination of HTTA and DBDECMP at 0.05 M each yielded the highest SFAm/Cm at pH 2.50. This concentration ratio was converted to a mole ratio (g of diketone (Rn) per g of neutral (Rd)) by multiplication of the respective molecular mass.An extractant loading (EL) of 30% (0.3 g of total extractant per g of inert support) was used for all resin preparations. The mass (g) of each extractant for a desired mass of inert support (g) was calculated from eqn (7).
| (T)·(EL) = Rdx + Rnx | (7) |
EXC resins were prepared with 10 wt% dodecane to enhance the retention of the ligands in the inert support. A maximum organic loading of 40 wt% was used based on resin manufacturers’ recommendations. For resin preparation, appropriate masses of each extractant were dissolved separately in 5 mL of 1,2-dichloroethane or methanol and mixed (15 minutes at 2500 RPM, digital vortex). Both organic extractant solutions were transferred to a conical tube containing dodecane and mixed (30 minutes at 2500 RPM, digital vortex). Each extractant tube was rinsed with one, 1 mL fractions of 1,2-dichloroethane or methanol.
The corresponding mass of inert support was slurried with a 30% (v/v) methanol and water solution at liquid-to-solid ratio of 5 mL per g of support. The combined extractant/dodecane solution was transferred to the slurried resin and rinsed with two, 2 mL fractions of 1,2-dichloroethane or methanol. The resin slurry and extractant/dodecane solution were transferred to a 500 mL round-bottom flask and rinsed with two, 5 mL fractions of 30% (v/v) methanol/water. The bulk solvent was removed under vacuum (50–75 Torr) at 45 °C. As the resin approached visual dryness (≈1 hour), the vacuum was decreased (100 Torr) to prevent bumping and continue residual solvent removal. After bumping was no longer observed (≈1 hour), the maximum vacuum (20 Torr) was drawn and held overnight (12 hours). This procedure was repeated for each mass ratio and solvent type studied. Individual extractant EXC resins were prepared in a similar manner. These materials are referred to hereafter as HTTA-DBDECMP resin 1–5. Table 1 summarizes the naming convention, mass ratios, and relevant characteristics for the EXC resins prepared.
| Resin name | Mass ratio (Rd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Rn) :Rn) |
Solvent type | HTTA (g) | DBDECMP (g) | Support (g) | Extractant loading (%) | Organic loading (%) |
|---|---|---|---|---|---|---|---|
| 1 | 1.00:0.88 |
1,2-DCE | 0.4780 | 0.4210 | 3.0044 | 29.9 | 39.9 |
| 2 | 1.00:1.38 |
1,2-DCE | 0.7564 | 1.0454 | 6.0054 | 30.0 | 40.0 |
| 3 | 1.00:1.38 |
Methanol | 0.7322 | 1.0092 | 6.0010 | 29.0 | 39.0 |
| 4 | 0.00:1.00 |
Methanol | — | 0.7510 | 2.5016 | 30.0 | 40.0 |
| 5 | 1.00:0.00 |
Methanol | 0.7496 | — | 2.4996 | 30.0 | 40.0 |
2.5. Determination of the weight distribution ratio (Dw) for HTTA-DBDECMP resins
The weight distribution ratio, Dw (mL g−1), of Am(III) and Cm(III) were measured on HTTA-DBDECMP resins 1–5 from pH 1.50–2.50 solutions of nitric acid via batch extraction experiments. The Dw values were calculated from the following equation: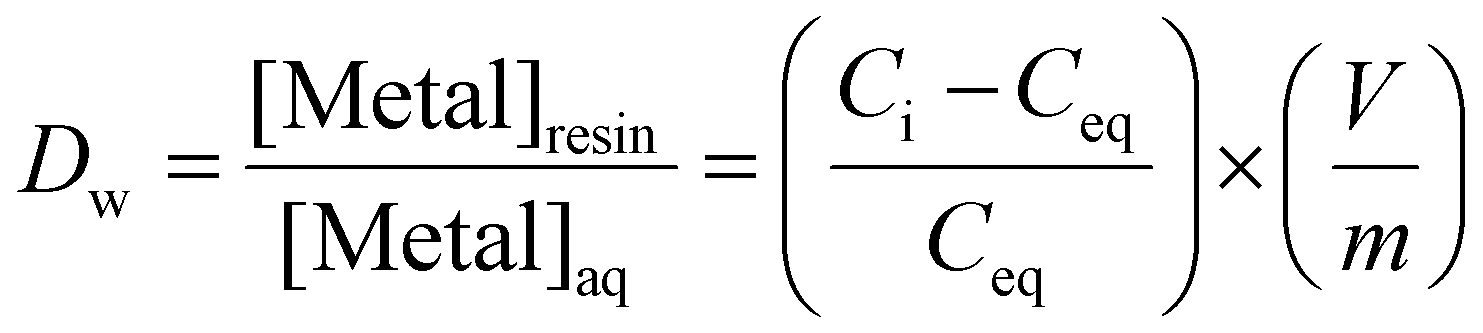 | (8) |
 | (9) |
Microcentrifuge tubes (2.0 mL) were loaded with 50 ± 2 mg of resin and 1.0 mL of HNO3 (pH 1.50–2.50, prepared as described above) with nominal concentrations of ≈14 dpm 241Am3+ μL−1 and ≈5.6 dpm 242Cm3+ per μL. These experiments analyzed each element separately and in duplicate. The tubes were incubated on a digital rotator (30 RPM for 1 hour, 26 ± 1 °C). After equilibration, the resin was filtered using 0.2 μm PTFE syringe filters. For all resin experiments, a 250 μL aliquot of the aqueous filtrate for 241Am3+ and 242Cm3+ for each replicate was counted via LSC as described above. The aqueous samples were each counted for 120 minutes or until a 1% counting uncertainty was attained. The Dw errors for all resin experiments are reported as one standard deviation between replicates or the propagated uncertainty in CPM, whichever is greater.
2.6. Thermodynamics, kinetic, stability, and ionic strength experiments
The pre-heated solutions of 241Am were added to each tube and incubated for 1 hour on a digital shaker (30 RPM). To calculate thermodynamic parameters of adsorption, such as change in Gibb's free energy (ΔG), change of enthalpy (ΔH), and change in the entropy (ΔS), the Van't Hoff equation (eqn (10)) was employed.
The Van't Hoff equation49 can be derived from the fundamental thermodynamic relationship between Gibbs Energy and equilibrium. Through mathematical manipulation, the Van't Hoff equation relates changes in the equilibrium constant to temperature by the following equation:
 | (10) |
The Keq is a dimensionless quantity that provides a measure of the ratio of concentrations of products to reactants at equilibrium for a reversible chemical reaction. The Dw can serve as an analog to Keq, reflecting the equilibrium distribution of the adsorbate between the adsorbent and the surrounding phase. From a plot of ln[Dw × ρwet] versus the reciprocal of temperature (K), the ΔS and ΔH can be calculated. There has been some disagreement over applying the Van't Hoff equation to adsorption data because of the need to have the equilibrium constant with dimensionless units.49 To produce a dimensionless equilibrium constant, the Dw values were multiplied by the wet resin density (ρwet = 0.704 g mL−1).38,42,50
 | (11) |
 is mass energy absorption coefficient for the resin (0.031 cm2 g−1), and
is mass energy absorption coefficient for the resin (0.031 cm2 g−1), and  is mass energy absorption coefficient for the air at 661.7 keV (0.026 cm2 g−1), respectively.
is mass energy absorption coefficient for the air at 661.7 keV (0.026 cm2 g−1), respectively.
Air and individual elemental mass energy absorption coefficient  values were interpolated from National Institute of Standards and Technology (NIST) Standard Reference Database 126.51 These were used to calculate
values were interpolated from National Institute of Standards and Technology (NIST) Standard Reference Database 126.51 These were used to calculate  based on the estimated elemental composition of the prepared resin. Due to the proprietary nature of the macroporous support, the exact elemental composition was difficult to determine. Combined with potential uncertainties in the calculation method, a 15% uncertainty is reported for QResin.
based on the estimated elemental composition of the prepared resin. Due to the proprietary nature of the macroporous support, the exact elemental composition was difficult to determine. Combined with potential uncertainties in the calculation method, a 15% uncertainty is reported for QResin.
Alongside the γ irradiation stability test, an acid stability test was conducted. The resins were exposed to nitric acid for the same duration in both tests. After each test, the acid was decanted, and the resins were dried in a laboratory oven at 50 °C.
2.7. Column separations with HTTA-DBDECMP resin 2
The elution profiles of Am(III) and Cm(III) on HTTA-DBDECMP resin 2 columns were investigated separately via different column types. Each column type was wet-packed with resin slurried in a nitric acid solution at pH 3.00 (60 minutes at 1500 RPM, digital vortex). For all column experiments, a 250 μL aliquot of each fraction was counted via LSC as described above.Following metal loading, each column was washed with approximately 0.5 column volumes of pH 3.00 HNO3, followed by an isocratic elution at pH 1.75 HNO3. After approximately 11 column volumes, the column was stripped with approximately 0.5 column volumes of 1.0 M HNO3.
A relative column decontamination factor for fractions between i and j, DFi,j was determined by eqn (12) for each column type.
 | (12) |
3. Results and discussion
3.1. Synergism in solvent extraction
The investigated pH ranges were tailored for each synergic system based on previously studied extraction efficiencies of lanthanides with these systems from pH 0.00–3.00.52 HTTA and DBDECMP did not display significant extraction (>5%); however, HP, DHDECMP and CMPO demonstrated varying extraction levels over their studied pH regions. HP and DHDECMP displayed increasing extraction from pH 1.50 (D = 0.09 ± 0.01) to 2.00 (D = 0.14 ± 0.02) and pH 2.00 (D = 0.11 ± 0.01) to 3.00 (D = 0.33 ± 0.05), respectively. CMPO displayed decreasing extraction from pH 0.50 (D = 0.15 ± 0.02) to 0.75 (D = 0.05 ± 0.03).Synergic effects were observed for all pairings of neutral extractants with β-diketones. Fig. 2 shows the SEC as a function of acid concentration and demonstrates that as the pH of extraction increased, the SEC value also increased. At high pH values, as much as a thousandfold increase in extraction was observed. As indicated in Fig. 2, at a constant pH of 1.50 CMPO had the largest SEC values among all three neutral ligands, followed by DHDECMP, and DBDECMP. The synergism between the neutral extractants and HP resulted in higher SEC values when compared to HTTA.
 | ||
| Fig. 2 Synergic enhancement coefficients (SEC) for combinations of neutral organophosphorus and β-diketone extractants as a function of pH value (HNO3). | ||
3.2. Solvent extraction separation behavior analysis and optimization
Tables 2–4 summarize the separation factors for each neutral and β-diketone combination. For CMPO and HTTA, separation factors were constant (≈1.90) across the studied pH range, whereas decreased from 1.87 to 1.19 with HP as the pH increased. For DBDECMP and HTTA, the SFAm/Cm increased from 1.05 to 2.65 as pH increased, whereas with HP, decreased from 1.44 to 1.17. The separation potential with DHDECMP was not realized, as low SFAm/Cm were observed with both diketones across the studied pH regions. The highest unoptimized SFAm/Cm was 2.65 ± 0.21 at pH 2.50 with 0.05 M DBDECMP and 0.05 M HTTA with a corresponding SEC value of 2.50 ± 0.26.| pH value (HNO3) | 0.05 M CMPO | |||||
|---|---|---|---|---|---|---|
| 0.05 M HTTA | 0.05 M HP | |||||
| D Cm ± σCm | D Am ± σAm | SFAm/Cm ± σSFAm/Cm | D Cm ± σCm | D Am ± σAm | SFAm/Cm ± σSFAm/Cm | |
| 0.25 | — | — | — | 0.83 ± 0.02 | 1.55 ± 0.03 | 1.87 ± 0.06 |
| 0.50 | 0.22 ± 0.03 | 0.43 ± 0.04 | 1.94 ± 0.29 | 1.58 ± 0.11 | 2.37 ± 0.08 | 1.50 ± 0.12 |
| 1.00 | 0.94 ± 0.02 | 1.83 ± 0.05 | 1.94 ± 0.06 | 13.98 ± 0.34 | 16.70 ± 1.07 | 1.19 ± 0.08 |
| 1.50 | 9.54 ± 0.34 | 17.97 ± 0.38 | 1.88 ± 0.08 | — | — | — |
| pH value (HNO3) | 0.05 M DBDECMP | |||||
|---|---|---|---|---|---|---|
| 0.05 M HTTA | 0.05 M HP | |||||
| D Cm ± σCm | D Am ± σAm | SFAm/Cm ± σSFAm/Cm | D Cm ± σCm | D Am ± σAm | SFAm/Cm ± σSFAm/Cm | |
| 1.00 | — | — | — | 0.27 ± 0.02 | 0.39 ± 0.01 | 1.44 ± 0.10 |
| 1.25 | — | — | — | 1.46 ± 0.02 | 1.80 ± 0.06 | 1.24 ± 0.04 |
| 1.50 | — | — | — | 6.85 ± 0.14 | 8.04 ± 0.41 | 1.17 ± 0.06 |
| 2.00 | 0.42 ± 0.01 | 0.44 ± 0.04 | 1.05 ± 0.10 | — | — | — |
| 2.25 | 1.61 ± 0.02 | 2.25 ± 0.02 | 1.40 ± 0.02 | — | — | — |
| 2.50 | 7.13 ± 0.22 | 18.89 ± 1.37 | 2.65 ± 0.21 | — | — | — |
| pH value (HNO3) | 0.05 M DHDECMP | |||||
|---|---|---|---|---|---|---|
| 0.05 M HTTA | 0.05 M HP | |||||
| D Cm ± σCm | D Am ± σAm | SFAm/Cm ± σSFAm/Cm | D Cm ± σCm | D Am ± σAm | SFAm/Cm ± σSFAm/Cm | |
| 1.00 | — | — | — | 0.47 ± 0.00 | 0.68 ± 0.04 | 1.45 ± 0.08 |
| 1.25 | — | — | — | 2.31 ± 0.15 | 2.57 ± 0.08 | 1.11 ± 0.08 |
| 1.50 | 0.24 ± 0.03 | 0.16 ± 0.01 | 0.69 ± 0.11 | 9.03 ± 0.66 | 11.83 ± 0.28 | 1.31 ± 0.10 |
| 2.00 | 1.77 ± 0.06 | 1.78 ± 0.16 | 1.00 ± 0.10 | — | — | — |
| 2.50 | 15.67 ± 0.84 | 14.60 ± 3.50 | 0.93 ± 0.23 | — | — | — |
Table 5 shows the results of the SX optimization performed with DBDECMP and HTTA at pH 2.50. For the HTTA screen, the concentrations were grouped into two categories: (1) with a higher separation factor at 0.0125–0.0250 M, and (2) with a lower separation factor at 0.0750–0.1500 M. Lower concentrations of HTTA had higher SFAm/Cm than higher HTTA concentrations. Similar conclusions could not be drawn for the DBDECMP screen, where SFAm/Cm was generally within experimental error. Generally, increasing the concentration of HTTA caused a higher increase in the D relative to a similar DBDECMP increase. Previous slope analysis experiments of these extractant combinations using Tb suggest that one neutral extractant is found in the extracted complex, whereas 2 diketone ligands are used.52 This suggests that the increasing the amount of HTTA relative to DBDECMP should produce additional active ligand sites able to extractant metal. The optimization experiments suggested that the highest separation factor was obtained at 0.05 M HTTA and 0.05 M DBDECMP. These concentrations were the basis for converting the solvent extractant system to the EXC resins.
| [Extractant] M | DBDECMP and HTTA extraction at pH = 2.50 | |||||
|---|---|---|---|---|---|---|
| 0.05 M DBDECMP HTTA screen | 0.05 M HTTA DBDECMP screen | |||||
| D Cm ± σCm | D Am ± σAm | SFAm/Cm ± σSFAm/Cm | D Cm ± σCm | D Am ± σAm | SFAm/Cm ± σSFAm/Cm | |
| 0.0125 | 0.37 ± 0.02 | 0.85 ± 0.06 | 2.27 ± 0.21 | 3.35 ± 0.13 | 6.39 ± 0.19 | 1.91 ± 0.09 |
| 0.0250 | 1.65 ± 0.04 | 4.05 ± 0.05 | 2.45 ± 0.06 | 6.64 ± 0.39 | 12.51 ± 0.31 | 1.88 ± 0.12 |
| 0.0750 | 32.09 ± 0.52 | 49.30 ± 2.88 | 1.54 ± 0.09 | 19.53 ± 0.40 | 30.15 ± 0.83 | 1.54 ± 0.05 |
| 0.1500 | 185.0 ± 13.19 | 106.25 ± 7.85 | 0.57 ± 0.06 | 23.72 ± 2.10 | 41.12 ± 0.40 | 1.73 ± 0.15 |
The SFAm/Cm for the solvent extraction systems in this work are low but typical (generally between 2 and 3) for extractant/complexant combinations used in redox-free Am(III)/Cm(III) separations.53–55 While this study did not explore hydrophilic complexing agents with reverse affinity, identifying potential options could provide further improvements in separation selectivity. Although this study did not demonstrate significant improvements in SFAm/Cm compared to existing systems, it revealed that dual ligands, even those with minimal individual extraction efficiency, can be combined to improve both extraction and separation under previously challenging conditions. Previous research on the Lanthaniden und Curium Americum Trennung (LUCA) process—a synergic system combining bis(chlorophenyl)dithiophosphinic acid and tris(2-ethylhexyl)phosphate—achieved an SFAm/Cm value greater than 7, suggesting that synergic systems hold significant promise for enhancing separation.37 This highlights the potential for newer systems, like those investigated in this work, to offer further advancements.
3.3. Preparation of HTTA and/or DBDECMP extraction chromatographic resins
Table 1 describes the combined and individual extractant-loaded resins that were prepared. The unrecovered resin losses (average 22%) are due to transfer loss and material bumping while under vacuum. The possibility of residual 1,2-dichloroethane and methanol in the final resins cannot be eliminated but is unlikely. The final dry density of resin 2 was ρdry = 0.3 g mL−1, whereas for a wet resin it was ρwet = 0.7 g mL−1.3.4. Synergism in solid phase extraction
A corresponding solid-phase SEC was computed for the resin 2 using resin 4 (DBDECMP only) and resin 5 (HTTA only). The solid-phase SEC values of 1.42 ± 0.02 at pH 2.00 and 1.19 ± 0.02 at pH 2.50 demonstrate that synergism is retained upon conversion to an EXC resin. However, as pH increases, resin 5 begins to exhibit a higher affinity for 241Am3+, thereby decreasing the observed synergic effect. While synergism is maintained at higher pH values, a single extractant may become increasingly dominant in the extraction process.3.5. Weight distribution ratio (Dw) and SFAm/Cm of HTTA-DBDECMP resins
The Dw for 241Am3+ for the two studied mass ratios and two solvents are shown in Table 6. Resin 1 had a lower amount of DBDECMP compared to resin 2. Higher Dw for resin 1 suggest that smaller ratios of DBDECMP:HTTA yield higher Dw values. This agrees with the behavior observed in the SX systems. If similar extraction mechanisms are at play in both SX and EXC resins, then increasing the mass of DBDECMP while reducing the amount of HTTA is likely to result in decreased metal extraction. Comparing resin 2 and resin 3 showed that EXC resin made with 1,2-dichloroethane and methanol have similar Dw values with no observed impact on synergism. Overall, the Dw values for all resins showed an upward trend with increasing pH. The observed discrepancies between pH 2.00 and 2.25 are likely due to variations in ionic strength and daily environmental variables that can impact pH, not captured in daily calibrations.
| pH value (HNO3) | HTTA-DBDECMP resins | ||
|---|---|---|---|
| Resin 1 | Resin 2 | Resin 3 | |
| D w, Am ± σAm | D w, Am ± σAm | D w, Am ± σAm | |
| 1.75 | 12.7 ± 0.3 | 9.0 ± 0.2 | 9.0 ± 0.5 |
| 2.00 | 317.7 ± 6.0 | 219.4 ± 5.1 | 184.4 ± 1.5 |
| 2.25 | 203.4 ± 1.6 | 138.0 ± 2.1 | 122.2 ± 1.7 |
| 2.50 | 1546.4 ± 50.9 | 1143.1 ± 22.4 | 927.1 ± 18.2 |
Table 7 shows extraction results done at constant ionic strength and in one day to avoid any effects of temperature on the pH calibrations. The highest separation factor achieved was 1.38 ± 0.02. Curium exhibited higher Dw values at pH 1.75 and 2.00, while americium displayed higher Dw and at pH 2.25 and 2.50. Further studies are needed to determine if the switch in apparent preference is a flip in affinity or if other experimental conditions are at play. Overall, the separation factors between Am and Cm appears to increase with increasing pH. Further increasing pH might result in increasing separation factors, but at reduced utility with high Dw values. Evaluating Dw values of EXC resins at higher metal loading might reveal renewed possibilities.
| pH value (HNO3) | HTTA-DBDECMP resin 2 | ||
|---|---|---|---|
| D w, Cm ± σCm | D w, Am ± σAm | SFAm/Cm ± σAm | |
| 1.75 | 131.4 ± 1.3 | 94.9 ± 1.1 | 0.72 ± 0.01 |
| 2.00 | 163.7 ± 5.1 | 142.7 ± 1.4 | 0.87 ± 0.03 |
| 2.25 | 341.5 ± 6.3 | 396.5 ± 3.2 | 1.16 ± 0.02 |
| 2.50 | 878.8 ± 13.1 | 1204.1 ± 17.0 | 1.37 ± 0.03 |
3.6. Thermodynamics, kinetic, stability, and ionic strength experiments
 | ||
| Fig. 3 Ln(Dw × ρwet) versus 1/T (K) for HTTA-DBDECMP resin 2 at pH 2.00 HNO3. | ||
| Resin | pH, HNO3 | ΔS (kJ mol−1 K−1) | ΔH (kJ mol−1) | ΔG (kJ mol−1) | ||
|---|---|---|---|---|---|---|
| Temperature (K) | ||||||
| 303 ± 1 | 318 ± 1 | 333 ± 1 | ||||
| HTTA-DBDECMP Resin 2 | 2.00 | 0.25 ± 0.03 | 65.72 ± 8.96 | −11.19 ± 0.29 | −14.99 ± 0.30 | −18.80 ± 0.61 |
Extraction increases in synergic SX systems are driven by a gain in configurational entropy.56 This entropy increase arises because synergic systems expand the number of possible extraction configurations for a metal ion. Whether a similar phenomenon occurs in synergic EXC resins remains unclear without further investigation. However, the observed trend of increasing extraction with temperature suggests that additional conformational states become energetically favorable at higher temperatures. If this theory holds, future experiments should explore less sterically hindered yet sufficiently hydrophobic neutral donors to access additional extraction configurations. Reduced steric hindrance could enhance binding at lower temperatures, which may be advantageous for high-loading applications.
 | ||
| Fig. 4 Kinetic Uptake of 241Am3+ adsorption on HTTA-DBDECMP Resin 2 (20–50 μm) at pH 2.00 HNO3. | ||
| pH value (HNO3) | HTTA-DBDECMP resin 2 | |||
|---|---|---|---|---|
| Dry resin | γ-Irradiation resin | Acid contact | Pre-filter support | |
| D w, Am ± σAm | D w, Am ± σAm | D w, Am ± σAm | D w, Am ± σAm | |
| 2.00 | 71.90 ± 0.78 | 511.90 ± 9.65 | 14.48 ± 1.48 | ≅MDA95 |
| pH value (HNO3) | HTTA-DBDECMP resin 2 | |||
|---|---|---|---|---|
| 0.05 M | 0.10 M | 0.50 M | 1.00 M | |
| D w, Am ± σAm | D w, Am ± σAm | D w, Am ± σAm | D w, Am ± σAm | |
| 2.00 | 82.94 ± 4.67 | 69.89 ± 3.69 | 58.62 ± 0.25 | 61.11 ± 1.36 |
3.7. Preliminary column separation studies
In both column types, americium was more strongly retained on the column compared to curium. This is opposite as what was observed in the batch Dw studies. Dw may differ between batch and column studies due to dynamic flow conditions, contact time variations, resin bed packing, concentration gradients, and changes in ionic strength and pH.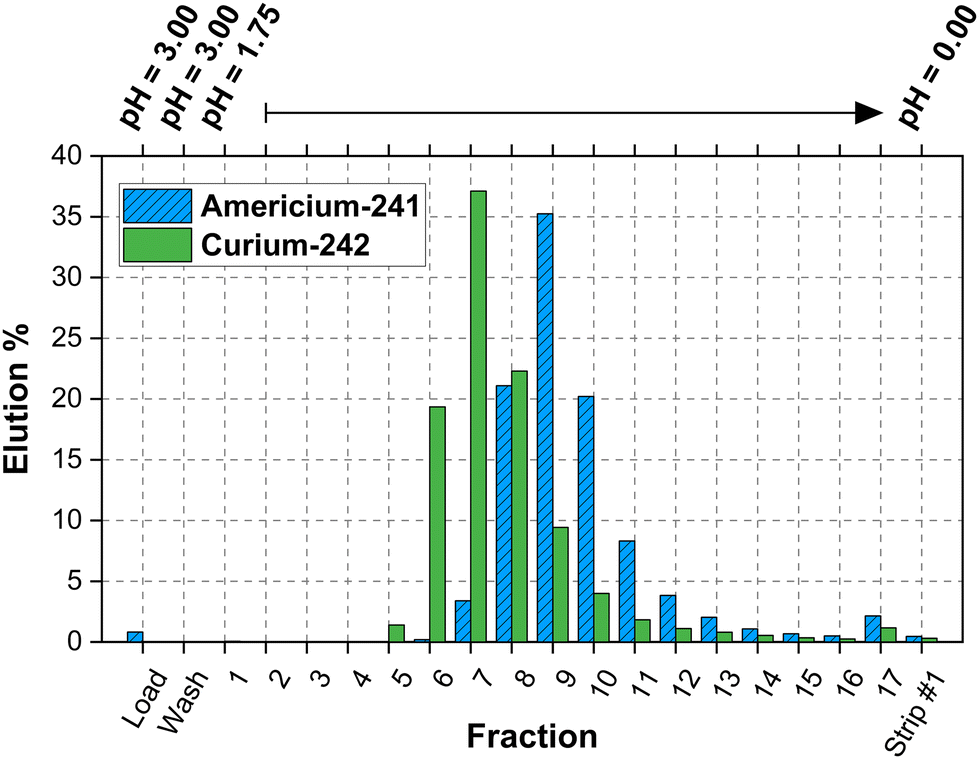 | ||
| Fig. 5 Elution profile for 241Am3+ and 242Cm3+ using column type 1 (inner diameter: 4.5 mm, length: 45 mm, column volume: ≈800 μL, and fraction volume: ≈500 μL). | ||
 | ||
| Fig. 6 Elution profile for 241Am3+ and 242Cm3+ using column type 2 (inner diameter: 2.5 mm, length: 60 mm, column volume: ≈400 μL, and fraction volume: ≈300 μL). | ||
The raffinates from UNF reprocessing schemes present challenging matrices for chromatographic separations, which explains the limited literature on Am/Cm separations and a stronger focus on solvent extraction.16 Most column chromatography efforts have centered on ion exchange,57,58 with extraction chromatography being studied less frequently.59–61 However, Usuda et al. investigated the use of single lipophilic extractant-impregnated adsorbents eluted with a hydrophilic complexant in nitric acid. In their study involving a TODGA-based adsorbent system with N,N,N′,N′-tetraethyl-3,6-dioxaoctanediamide (DOODA(C2)) in dilute HNO3, they achieved relatively high-purity fractions, with the curium fraction containing 97.8% Cm and only 2.62% Am.62 In contrast, our preliminary column system yielded a lower overall Cm recovery of 27%, with no detectable Am. Another popular system that avoids the use of complexing agents altogether is a tertiary pyridine impregnated silica resin and a nitric acid/methanol elution mixture. This is a simple method to separate nearly all Am from Cm but poses safety concerns over the use of nitric acid and methanol. Suzuki et al. do not report a DF or % recovery/contamination in their work, but column separation factor of 2.54.63
These proof of principle column separations served as preliminary attempts to demonstrate that synergic EXC resins can facilitate effective separations. Despite facing chromatographic challenges, future investigations could enhance chromatographic performance and achieve more refined separations. This work establishes a foundational approach for synergic EXC resins, highlighting the potential for identifying synergic ligand combinations that could lead to improved separations of americium and curium.
4. Conclusions
The study presented here focused on identifying synergic extractant combinations to improve separation efficiency of americium and curium, aiming to fill a gap in the existing EXC resin compendium. Enhanced metal extraction was observed for all SX ligand combinations, though most showed limited selectivity between Am(III) and Cm(III). Although establishing clear relationships between synergism and selectivity is challenging, increased synergism generally correlates with decreased selectivity. An important exception was observed with DBDECMP and HTTA, which showed an increase in selectivity with increasing synergism. Subsequently, this ligand combination was translated into synergic EXC resins and evaluated under several experimental conditions. Once converted, the resulting resins exhibited acid dependency comparable to that of the SX experiments, while maintaining synergic behavior. The resulting Dw ratio for Am(III) and Cm(III) decreased compared to SX, suggesting reduced selectivity when translated and evaluated in EXC form. In preliminary column evaluations, the EXC resins exhibited promising metal retention and separation under mildly acidic conditions. Despite a low Dw ratio, the resin demonstrated the potential to separate Am(III) and Cm(III) with a DF8,10 = 88. However, further systematic investigations are necessary to enhance chromatographic performance and evaluate the technical readiness of this separation method.This work demonstrates that a synergic EXC resin improves the extraction and separation of Am(III) and Cm(III) compared to individual ligand EXC resins; however, its performance does not surpass that of state-of-the-art ion-exchange chromatography. While not suitable for large-scale industrial applications, synergic EXC resins could enable the production of small-scale, high-purity Am and Cm samples for research and development. Overall, this study advances the development of EXC materials by demonstrating that synergic ligand combinations enhance the extraction and separation efficiency of trivalent actinides. Additionally, it highlights the feasibility of converting SX systems into EXC resins. These findings underscore the importance of exploring dual-ligand systems for the effective separation of Am(III) and Cm(III).
Author contributions
C. H. writing – original draft, writing – review & editing, visualization, validation, methodology, investigation, formal analysis, data curation, conceptualization, funding acquisition. T. M. Writing – review & editing, validation, supervision, resources, methodology, investigation formal analysis, conceptualization. J. S. writing – review & editing, validation, supervision, resources, project administration, methodology, investigation, funding acquisition, formal analysis, data curation, conceptualization.Data availability
The authors confirm that the processed data supporting the findings of this study are included in the article. Raw counting data are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
(1) This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Workforce Development for Teachers and Scientists, Office of Science Graduate Student Research (SCGSR) program. The SCGSR program is administered by the Oak Ridge Institute for Science and Education for the DOE under contract number DE-SC0014664.(2) This research was also supported by the U.S. Department of Energy Isotope Program, managed by the Office of Science for Isotope R&D and Production (DE-SC0020955 and SCW1759).
(3) This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. (IM # LLNL-JRNL-864828)
(4) This work was supported in part by the United States Nuclear Regulatory Commission Fellowship [NRC-HQ-84-15-G-0041].
References
- A. Leoncini, J. Huskens and W. Verboom, Chem. Soc. Rev., 2017, 46, 7229–7273 RSC.
- L. Rodríguez-Penalonga and B. Y. Moratilla Soria, Energies, 2017, 10, 1235 CrossRef.
- M. D. Straub, J. Arnold, J. Fessenden and J. L. Kiplinger, Anal. Chem., 2021, 93, 3–22 CrossRef CAS PubMed.
- N. Gharibyan, Ph.D. Thesis, University of Las Vegas, Nevada, 2011.
- Implications of Partitioning and Transmutation in Radioactive Waste Management, International Atomic Energy Agency, Vienna, 2004 Search PubMed.
- G. A. Office, Nuclear Fuel Cycle Options: DOE Needs to Enhance Planning for Technology Assessment and Collaboration with Industry and Other Countries, United States Government Accountability Office, Washington, D.C., 2011 Search PubMed.
- I. Kumari, B. V. R. Kumar and A. Khanna, Nucl. Eng. Des., 2020, 358, 110410 CrossRef CAS.
- C. L. Riddle, J. D. Baker, J. D. Law, C. A. McGrath, D. H. Meikrantz, B. J. Mincher, D. R. Peterman and T. A. Todd, Solvent Extr. Ion Exch., 2005, 23, 449–461 CrossRef CAS.
- A. V. Gelis, G. F. Vandegrift, A. Bakel, D. L. Bowers, A. S. Hebden, C. Pereira and M. Regalbuto, Radiochim. Acta, 2009, 97, 231–232 CrossRef CAS.
- A. V. Gelis and G. J. Lumetta, Ind. Eng. Chem. Res., 2014, 53, 1624–1631 CrossRef CAS.
- K. L. Nash, Solvent Extr. Ion Exch., 1993, 11, 729–768 CrossRef CAS.
- H. C. Aspinall, Chemistry of the f-Block Elements, Routledge, 2018 Search PubMed.
- K. L. Nash, C. Madic, J. N. Mathur and J. Lacquement, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer Netherlands, Dordrecht, 2006, pp. 2622–2798, DOI:10.1007/1-4020-3598-5_24.
- E. P. Horwitz, R. Chiarizia, M. L. Dietz, H. Diamond and D. M. Nelson, Anal. Chim. Acta, 1993, 281, 361–372 CrossRef CAS.
- H. Kurosaki and S. B. Clark, Radiochim. Acta., 2011, 99, 65–69 CrossRef CAS.
- P. Zsabka, A. Wilden, K. Van Hecke, G. Modolo, M. Verwerft and T. Cardinaels, J. Nucl. Mater., 2023, 581, 154445 Search PubMed.
- E. P. Horwitz, A. C. Muscatello, D. G. Kalina and L. Kaplan, Sep. Sci. Technol., 1981, 16, 417–437 CrossRef CAS.
- G. Eshima, A. C. Muscatello and J. D. Navratil, J. Less-Common Met., 1983, 94, 327–332 Search PubMed.
- K. L. Nash, in Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 1994, vol. 18, pp. 197–238 Search PubMed.
- A. Sengupta, M. S. Murali, S. K. Thulasidas and P. K. Mohapatra, Hydrometallurgy, 2014, 147–148, 228–233 CrossRef CAS.
- J. N. Mathur, M. S. Murali and P. R. Natarajan, J. Radioanal. Nucl. Chem., 1992, 162, 171–178 CrossRef CAS.
- J. N. Mathur and P. K. Khopkar, Polyhedron, 1987, 6, 2099–2102 CrossRef CAS.
- V. M. Jordanov, M. Atanassova and I. L. Dukov, Sep. Sci. Technol., 2002, 37, 3349–3356 CrossRef CAS.
- D. M. Czakis-Sulikowska, B. Kuźnik and A. Malinowska, Monatsh. Chem., 1991, 122, 789–794 CrossRef CAS.
- R. B. Gujar, S. A. Ansari, A. Sengupta, M. S. Murali and P. K. Mohapatra, Inorg. Chem. Commun., 2016, 73, 72–76 CrossRef CAS.
- S. A. Pai, K. V. Lohithakshan, P. D. Mithapara and S. K. Aggarwal, Radiochim. Acta, 2002, 90, 13–18 CrossRef CAS.
- G. J. Lumetta, A. V. Gelis and G. F. Vandegrift, Solvent Extr. Ion Exch., 2010, 28, 287–312 CrossRef CAS.
- S. A. Pai, K. V. Lohithakshan, P. D. Mithapara and S. K. Aggarwal, J. Radioanal. Nucl. Chem., 2000, 245, 623–628 CrossRef CAS.
- D. J. Pruett, M. C. Clark and D. D. Ensor, Sep. Sci. Technol., 1990, 25, 1777–1783 CrossRef CAS.
- A. N. Turanov, V. K. Karandashev, A. V. Kharlamov and N. A. Bondarenko, Solvent Extr. Ion Exch., 2014, 32, 492–507 CrossRef CAS.
- L. Rao, Y. Xia, B. M. Rapko and P. F. Martin, Solvent Extr. Ion Exch., 1998, 16, 913–929 CrossRef CAS.
- A. C. Muscatello, J. D. Navratil and M. E. Killion, Solvent Extr. Ion Exch., 1983, 1, 127–139 CrossRef CAS.
- J. N. Mathur, Solvent Extr. Ion Exch., 1983, 1, 349–412 CrossRef CAS.
- X. Yang, L. Xu, A. Zhang and C. Xiao, Chem. – Eur. J., 2023, 29(33) DOI:10.1002/chem.202300456.
- S. Sujatha, M. L. P. Reddy, T. R. Ramamohan, A. D. Damodaran, J. N. Mathur, M. S. Murali, M. S. Nagar and R. H. Iyer, Radiochim. Acta, 1994, 65, 167–172 CrossRef CAS.
- M. Atanassova and I. Dukov, J. Solution Chem., 2009, 38, 289–301 CrossRef CAS.
- G. Modolo, P. Kluxen and A. Geist, Radiochim. Acta, 2010, 98, 193–201 CAS.
- E. P. Horwitz, D. R. McAlister and M. L. Dietz, Sep. Sci. Technol., 2006, 41, 2163–2182 CrossRef CAS.
- A. M. Tamang, N. Singh, S. K. Chandraker and M. K. Ghosh, Curr. Res. Green Sustainable Chem., 2022, 5, 100232 CrossRef CAS.
- N. Kabay, J. L. Cortina, A. Trochimczuk and M. Streat, React. Funct. Polym., 2010, 70, 484–496 CrossRef CAS.
- J. L. Cortina and A. Warshawsky, Developments in Solid-Liquid Extraction by Solvent-Impregnated Resins, Ion Exchange and Solvent Extraction, 1st edn, 1997, vol. 13 Search PubMed.
- E. P. Horwitz, D. R. McAlister, A. H. Bond and R. E. Barrans, Solvent Extr. Ion Exch., 2005, 23, 319–344 CrossRef CAS.
- E. R. Bertelsen, J. A. Jackson and J. C. Shafer, Solvent Extr. Ion Exch., 2020, 38, 251–289 CrossRef CAS.
- K. Onishi, T. Nakamura, S. Nishihama and K. Yoshizuka, Ind. Eng. Chem. Res., 2010, 49, 6554–6558 CrossRef CAS.
- J. L. Cortina, N. Miralles, A. M. Sastre and M. Aguilar, React. Funct. Polym., 1997, 32, 221–229 CrossRef CAS.
- K. Svoboda and M. Kyrš, J. Radioanal. Nucl. Chem., 1997, 220, 245–247 CrossRef CAS.
- H. F. Aly and M. Raieh, Anal. Chim. Acta, 1971, 54, 171–176 CrossRef CAS.
- H. F. Aly and M. A. El-Haggan, Radiochem. Radioanal. Lett., 1970, 3, 249–253 CAS.
- E. C. Lima, A. Hosseini-Bandegharaei, J. C. Moreno-Piraján and I. Anastopoulos, J. Mol. Liq., 2019, 273, 425–434 CrossRef CAS.
- M. D. Gott, B. D. Ballard, L. N. Redman, J. R. Maassen, W. A. Taylor, J. W. Engle, F. M. Nortier, E. R. Birnbaum, K. D. John, D. S. Wilbur, C. S. Cutler, A. R. Ketring, S. S. Jurisson and M. E. Fassbender, Radiochim. Acta., 2014, 102, 325–332 CrossRef CAS.
- J. H. Hubbell and S. M. Seltzer, Tables of X-Ray Mass Attenuation Coefficients and Mass Energy-Absorption Coefficients 1 keV to 20 MeV for Elements Z = 1 to 92 and 48 Additional Substances of Dosimetric Interest, National Institute of Standards and Technology, 1995, DOI:10.18434/T4D01F.
- C. K. Holiski, L. Hoekstra, N. Becker, H. M. Hennkens, M. F. Embree, M.-J. Wang, G. E. Sjoden and T. Mastren, J. Chromatogr., A, 2025 Search PubMed , submitted.
- S. Chapron, C. Marie, V. Pacary, M. T. Duchesne, G. Arrachart, S. Pellet-Rostaing and M. Miguirditchian, Procedia Chem., 2016, 21, 133–139 CrossRef.
- S. Lange, A. Wilden, G. Modolo, F. Sadowski, M. Gerdes and D. Bosbach, Solvent Extr. Ion Exch., 2017, 35, 161–173 CrossRef CAS.
- C. Wagner, U. Müllich, A. Geist and P. J. Panak, Solvent Extr. Ion Exch., 2016, 34, 103–113 CrossRef CAS.
- M. Špadina, K. Bohinc, T. Zemb and J.-F. Dufrêche, ACS Nano, 2019, 13, 13745–13758 CrossRef PubMed.
- H. L. Smith and D. C. Hoffman, J. Inorg. Nucl. Chem., 1956, 3, 243–247 CrossRef CAS.
- G. R. Choppin and R. J. Silva, J. Inorg. Nucl. Chem., 1956, 3, 153–154 CrossRef CAS.
- K. Street Jr. and G. T. Seaborg, J. Am. Chem. Soc., 1950, 72, 2790–2792 CrossRef.
- W. H. Hale and J. T. Lowe, Inorg. Nucl. Chem. Lett., 1969, 5, 363–368 CrossRef CAS.
- R. M. Diamond, K. Street Jr. and G. T. Seaborg, J. Am. Chem. Soc., 1954, 76, 1461–1469 CrossRef CAS.
- S. Usuda, K. Yamanishi, H. Mimura, Y. Sasaki, A. Kirishima, N. Sato and Y. Niibori, J. Radioanal. Nucl. Chem., 2015, 303, 1351–1355 CrossRef CAS.
- T. Suzuki, K. Otake, M. Sato, A. Ikeda, M. Aida, Y. Fujii, M. Hara, T. Mitsugashira and M. Ozawa, J. Radioanal. Nucl. Chem., 2007, 272, 257–262 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |